Coordinated “Naked” Pnicogenes and Catalysis †


1
Research Group for Petrochemistry of the Hungarian Academy of Sciences, H-8200 Veszprém, Egyetem u. 10, Hungary
2
Department of Organic Chemistry, University of Pannonia, H-8200 Veszprém, Egyetem u. 10, Hungary
3
Department of Life Sciences, University of Modena and Reggio Emilia, I-4125 Modena, Via Campi, 103, Italy
*
Author to whom correspondence should be addressed.
†
This paper is dedicated to the 90th birthday of László Markó, one of the pioneers of transition metal cluster chemistry and long time friend of A.V.-O. and G.P.
Catalysts 2018, 8(12), 583; https://doi.org/10.3390/catal8120583
Submission received: 25 October 2018
/
Revised: 13 November 2018
/
Accepted: 16 November 2018
/
Published: 26 November 2018
(This article belongs to the Special Issue Coordination Chemistry and Catalysis)
Abstract
:Diphosphorous (P2) side-on coordinated to a dicobalt (Co–Co) moiety was described 45 years ago. This discovery had several links to actual problems of homogeneous molecular catalysis. The new type of organometallic complexes induced several ingenious new ramifications in main-group/transition metal cluster chemistry in the last decades. The present review traces the main lines of these research results and their contacts to actual problems of industrial catalysis.
1. Introduction
(μ2-P2)Co2(CO)6(Co–Co) (1) (Figure 1A) and (μ3-S=P)Co3(CO)9(3Co–Co) (2) (Figure 2A) were reported [1,2] in 1973. The structure of 1 was confirmed by X-ray crystal diffraction of its triphenylphosphane derivative (μ2-P2)Co2(CO)5P(C6H5)3(Co–Co) (3) (Figure 1B) a few years later [3]; while the structure of 2 was based on infrared ν(C–O) spectroscopic analogy [4] (Figure 2B). These compounds, even if new at that time, were prepared as a part of important research trends, which were and are also today intimately linked to very practical problems of homogeneous molecular catalysis. Since then, reference [1] was cited 69 times and these 69 papers were cited more than 2300 times (Web of Science, 12 September 2018). The following discussion will be based on references selected from these and on some additional related publications.
One of the related challenges was the (at that time) recent discovery of the possibility of dinitrogen fixation by transition metal-based systems [6], and the related preparation of monometallic dinitrogen complexes [7] as well as the hypothesis (which later became proved) that the biological nitrogen fixation proceeds through side-on coordination of the N2 molecule [8,9,10,11]. At that time such μ-coordinated N2-complexes had not yet been described. Obviously, the side-on μ2-coordinated P2 moiety in complexes 1 and 3 counted as a “next” model of the nitrogen-fixation problem.
Another viewpoint came from the mechanistic studies of transition metal catalyzed petrochemical reactions [12,13,14,15,16]. It has been found that unsaturated hydrocarbon molecules, or fragments of these, get coordinated to one or more transition metal atoms in the catalytic (and side reaction) processes and in the latter case the transition metal atoms are often linked by metal–metal bonds [17]. These intermediate (or side reaction) compounds could also be regarded as 3D, heteronuclear “cluster” molecules of transition metals and (main group) carbon atoms [5,18]. It became obvious to extend this “Bauprinzip” to the introduction of main group elements other than carbon into the 3D skeleton of transition metal cluster complexes too. Compounds 1 to 3 were early representatives of this research goal.
Another background aspect of this new chemistry was the research concerning the petrochemical catalytic transformations of sulfur containing mineral oils. In the 1960ies it was found that the sulfur content in raw mineral oil gets (in part) transformed during transition metal catalyzed processes, to various S-containing transition metal compounds, for example paramagnetic (μ3-S)Co3(CO)9(3Co–Co) (4) [19,20] (Figure 3A) and diamagnetic (μ3-S)Co2Fe(CO)9(Co–Co, 2Co–Fe) (5) [21] (Figure 3B), as well as several other, higher nuclearity complexes [22].
2. Isolobal Principle and Small Clusters
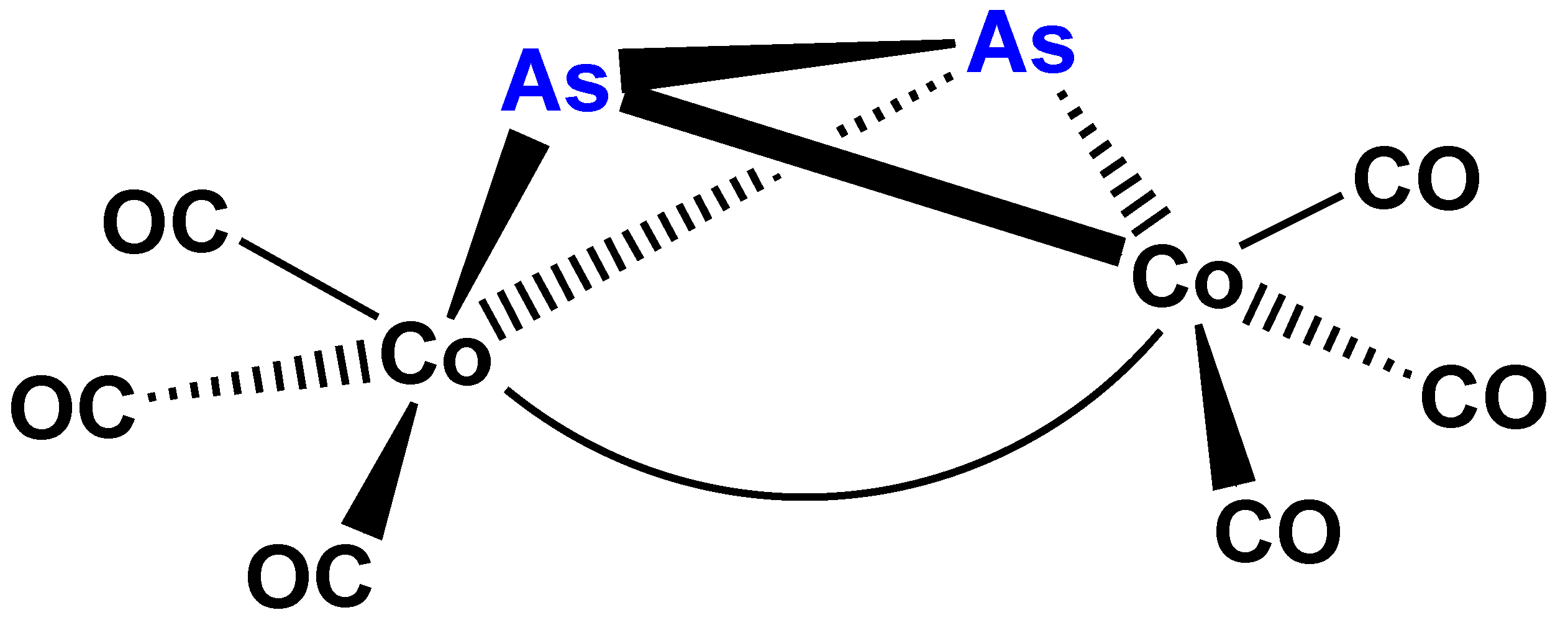
Complexes (1) to (4) can easily be described with the general formula En[Co(CO)3]4−n where E = P, S and n = 1, 2. The question, whether this formula could be extended to additional main group elements (E) and/or other values of n, emerged evidently. Particularly, since (μ2-As2)Co2(CO)6(Co–Co) (6) (Figure 4) was reported contemporaneously by Larry Dahl’s group [29]. At the same time appeared Ron Hoffmann’s brilliant idea about “isolobality”, that is organic or organometallic groups can “substitute” each other in molecules, if these groups have a similar electron donor (or acceptor) orbitals with right symmetry behavior [30,31,32]. These main group elements vs. transition metal complexes appeared to be ideal models for the preparative realization of this principle.
The “lacking” (μ3-P)Co3(CO)9(3Co–Co) (7) (Figure 5) and (η-P3)Co(CO)3 (8) (Figure 6) derivatives could be prepared from phosphorous trihalogenides and Na[Co(CO)4] following the experience with the carbon analogs [4,5], as well as from white phosphorous (P4) and dicobalt octacarbonyl [33]. The μ3-P derivative (7), however, was proved to be fairly instable against the formation of a dark crystalline fraction, which turned out to be P3Co9(CO)24(9Co–Co) (9) [34]. Compound 9 was identified as the cyclic trimer of complex 7 where the “monomeric” 7 unit substituted one of the carbonyl ligands on one of the Co atoms in the “next” 7 unit, giving an unexpected derivative of a 1,3,5-triphospha-2,4,6-tricobalta-cyclo-hexatriyne-1,3,5 with the triple bonds linked by μ2-coordination to Co2(CO)6 units, as in compound 1.
The next step in this kind of research could have been done in several directions: (a) to introduce E elements other than P in structures like 1, 2, 7, 8, and 9, or (b) to try metals other than cobalt or to study the reactivity of the known heterometallic complexes.
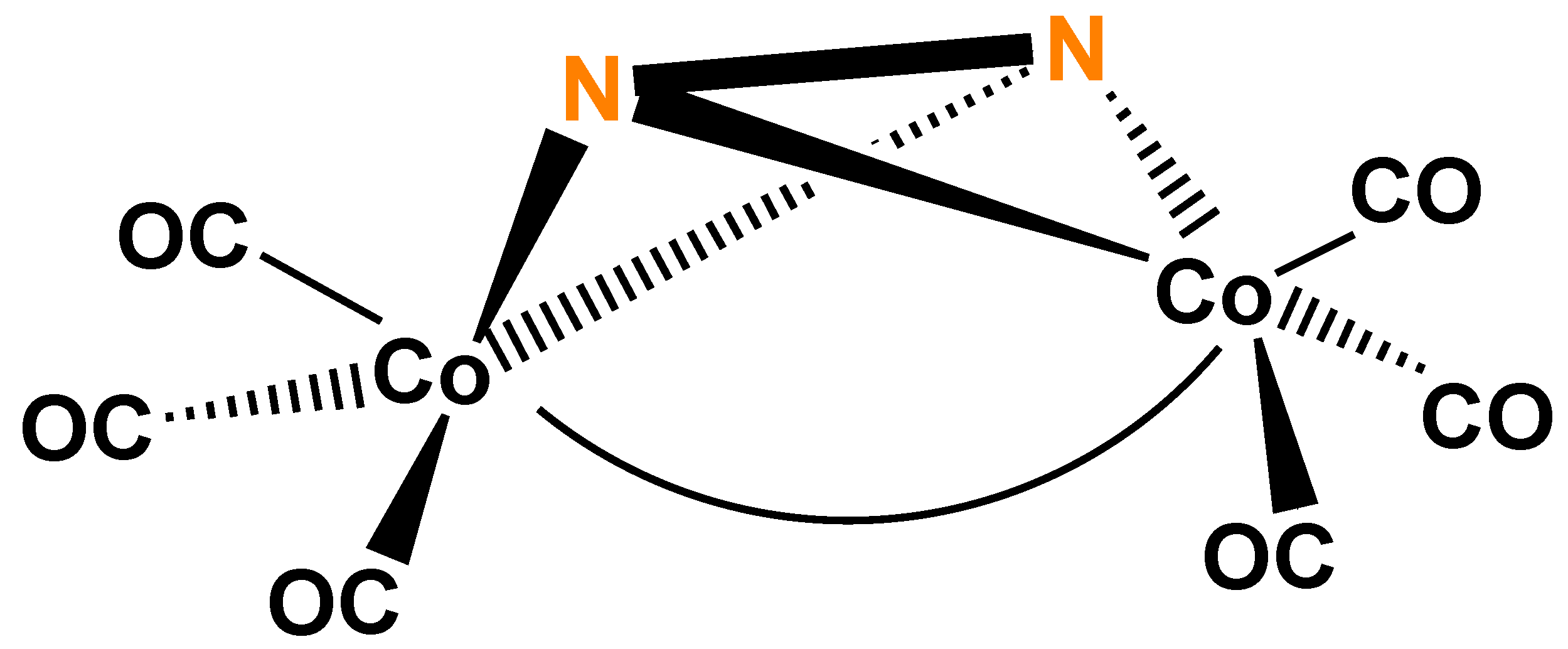
A successful attempt to prepare the arsenic analog of complex 1 was reported contemporaneously, as mentioned earlier in this paper, resulting the side-on bridging As2 complex 6 [29]. Other group 15 E2 derivatives have not yet (?) been described. Theoretical studies [35,36] of the highly interesting side-on coordination of N2 were published, but the hypothetic (μ2-N2)Co2(CO)6(Co–Co) (Figure 7) had no preparative realization to the best of our knowledge.
The arsenic analog of 7, (μ3-As)Co3(CO)9(3Co–Co) (10), could easily be prepared from AsI3 and Co2(CO)8, however the reaction of AsX3 (X = Cl, Br, I) with Na[Co(CO)4] yielded beside the expected 10 also 6 and a deep green fraction with 12 ν(C-O) IR bands, which turned out to be the As analog of the cyclic complex 9, with the formula As3Co9(CO)24(9Co–Co) (11) [34] (Figure 8), which again can be regarded as a fully inorganic cyclo-hexatriyne stabilized by alternating Co2(CO)6 moieties coordinated to the triple bonds. The overall structure of complex 11 was confirmed also by an X-ray diffraction study [37,38] but details were not yet published.
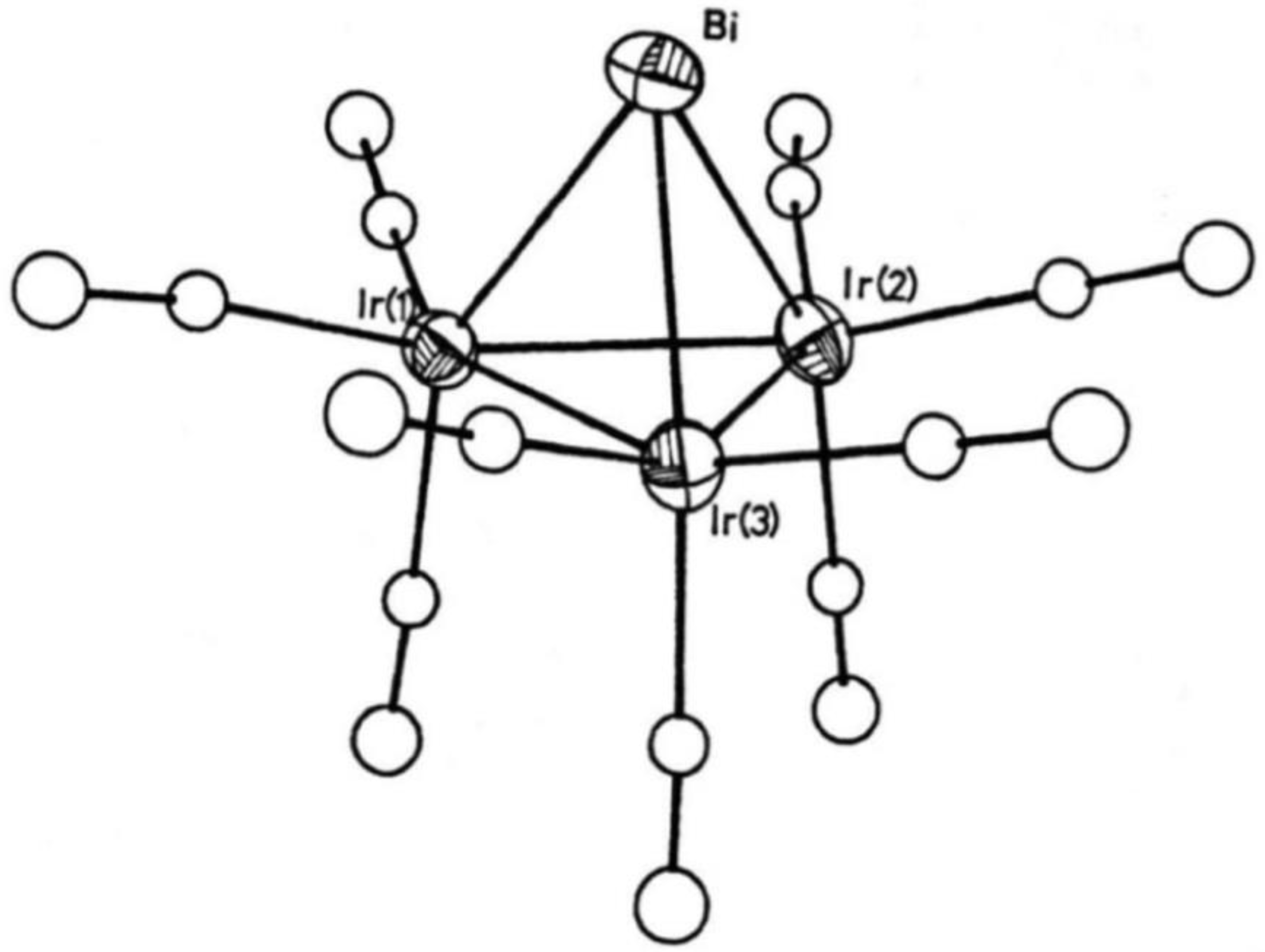
Attempts have been made at preparing antimony and bismuth analogs of 7 and 10; however, the products obtained were not exactly that what was expected. Antimony gave an octanuclear, cubane-like structure, (μ3-Sb)4Co4(CO)12 (12) (Figure 9) where each Sb atom was linked to three Co atoms, but where no Co–Co bonds are present (average Co–Co distance 411.5 (4) pm) [39]. An additional attempt at preparing Sb-analogs of the previously discussed P- and As-complexes by a tertiary phosphorous ligand containing cobalt reagent, K[Co(CO)3PR3] (R = OPh, OMe, Ph, Tolyl, R3 = Ph2Me) was made and the corresponding open Sb[Co(CO)3PR3]3 (13) complexes were obtained [40]. This structural difference can be attributed to the relatively long Sb–Co distances (~260 pm). The first attempts at preparing bismuth analogs of complexes 7 and 10 appeared to show the same tendency: Bi[Co(CO)4]3 (14) could be prepared from BiCl3 or elementary Bi and [Co(CO)4]- reagent [41]. The X-ray diffraction structure of 14 showed again a fairly long Bi-Co distance (~280 pm), suggesting that the tetrahedral cluster skeleton could not be closed because of the large size of the Bi atom. In an elegant additional attempt at clearing up this point Günter Schmid and Coworkers prepared the iridium/bismuth analog of 7 and 10, (μ3-Bi)Ir3(CO)9(3Ir–Ir) (15) (Figure 10), which showed the “closed” BiIr3 trigonal pyramidal structure [42]. (μ3-Bi)Co3(CO)9(3Co–Co) could also be prepared under somewhat more favorable conditions and characterized by X-ray single crystal diffraction [43].
One of the main goals of these experiments was to test analogies and differences to the hypothetic (μ2-N2)Co2(CO)6(Co–Co) (Figure 7) and its presumed role in molecular nitrogen activation. Experiments did not result in preparative success (reaction of NH3·NCl3 with Na[Co(CO)4] failed [36], yet(?)), theoretical calculations (EH [35], CNDO/2 [36,44,45]) indicate that the ambition in this direction is not hopeless. It is an important conclusion of these calculations that the 3d orbitals of P in complex 1 contribute significantly to the bonding strength, while this is absent at the hypothetical N2 analog [35]. This could be the reason why this latter complex could not be prepared.
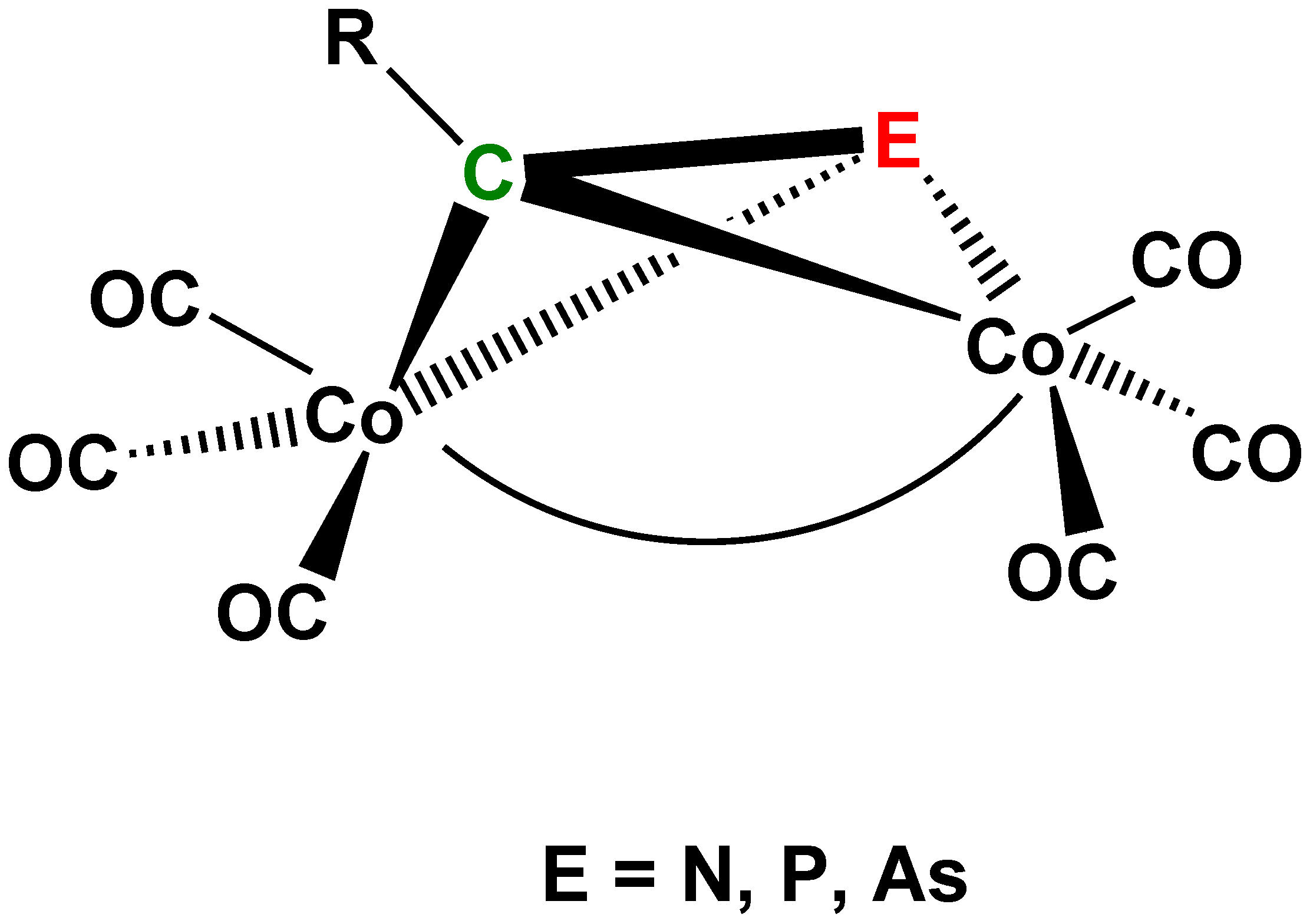
The above-discussed complexes 1 and 6 show a striking similarity to the (μ2-acetylene)Co2(CO)6(Co–Co) (18) complex family [15,16,17,18]. This similarity gave the brilliant idea to Dietmar Seyferth and his coworkers trying to prepare dicobalt hexacarbonyl derivatives of phospha- (μ2-RCP)Co2(CO)6(Co–Co), (R = Me, Ph, tBu, Me3Si) (16) (Figure 11, E = P) and arsaacetylenes (μ2-RCAs)Co2(CO)6(Co–Co) (R = H, Me, Ph, Me3Si) (17) (Figure 11, E = As) [46,47,48]. These very sensitive molecules got remarkably stabilized by the coordination to the Co2(CO)6 moiety and the complexes could be isolated and characterized. The bonding properties and photoelectron spectra indicate that at these complexes the stability (or even the existence) of these molecules is considerably assisted by the participation of the d orbitals of P and (presumably also) of As in bonding [49]. This, however, this is not possible at the hypothetic analog (μ2-RCN)Co2(CO)6(Co–Co) (Figure 11 E = N) complexes, which have been supposed as intermediates (or models thereof) in the cocyclization of acetylenes and alkyl cyanides in the cocatalyzed synthesis of pyridines [50,51,52].
3. Substituting CO Ligands in Heteronuclear Clusters
Carbonylation reactions (hydroformylation, alkoxycarbonylation, etc.) catalyzed prevalently by cobalt and rhodium complexes are favorably influenced by tertiary organic derivatives of phosphane (or phosphine, PH3) [24,53,54,55,56]. As a consequence of this industrially important fact in mechanistic studies on suspected intermediates of these reactions the tertiary phosphane derivatives [57,58,59,60,61,62] were almost always prepared. The presence of the tertiary phosphane ligand according to laboratory and industrial experience remarkably contributed to the catalytic activation of the substrate to be carbonylated and of CO. Such influence was found also in the catalytic carbonylation of acetylenes in the presence of cobalt carbonyl catalysts [63,64]. In this reaction the (μ2-R1C2R2)Co2(CO)6 (Co–Co) (18) (Figure 12) complexes, analogs of 1 and 6, are proved to act as intermediates of bifurandione synthesis (Figure 13) [65,66]. The activation of the μ2-acetylene ligand in complex 18 was supported also by CNDO/2 calculations [67]. An X-ray structural proof for the activation of the bridging As2 unit in complex 6 by its mono- and bis-substitution with triphenylphosphane was found by lengthening of the the As–As bond [29].
Beyond these viewpoints from catalytic chemistry there was also a very practical reason for preparing derivatives with soft Lewis bases (mostly tertiary organophosphane ligands). Ultimate structural information of chemical compounds can be obtained primarily by single crystal X-ray diffraction. For the preparation of suitable crystals of organometallic compounds, as those discussed above, it is usual to introduce large rigid groups as triphenylphosphane, P(C6H5)3 and related ligands. This technique was used in several occasions also in the experiments cited above.
In the field of transition metal pnicogenic complexes most of the efforts in this direction have been made with complexes 1 and 6 (both are oily substances at room temperature). One of these reports on the substitution of complex 6 with phosphane and phosphite ligands [68]. It turned out that up to 2 PMe3 but up to 4 P(OMe)3 ligands could be introduced in this molecule by substitution of CO ligands and without structural changes in the starting As2Co2 tetrahedral cluster unit. The phosphane/phosphite substitution increased the basicity of the As atoms as soft Lewis bases for transition metals different from cobalt. The molecular s structure of [(CO)5W]2As2Co2(CO)4[P(OMe)3]2 (Co–Co) (19) could be determined by single crystal X-ray diffraction.
Formally an exhaustive substitution of the CO ligands in complex 8 (Figure 6) was achieved by a “tripod” ligand, leading to (η3-P3)Co[(Ph2PCH2)3CCH3 (20) [69], but this complex was prepared not through 8, but by a different approach which does not belong to the scope of the present review.
A systematic study of the comparison of monosubstitution of the structurally related (μ2-L)2Co2(CO)6 complex series, including L2 = acetylene, P2 (a), As2 (b), and CO plus but-2-en-4-olid-4-ylidene (c) (21) (Figure 14) bridging units with trivalent phosphorous, PR3 and P(OR)3 (R = alkyl, aryl) was also reported [70]. This study allows two important conclusions. First, the introduced P-ligand appears to occupy always an axial position that is the X-ray structural results on 3 and its As2 analog (22) can be generalized. The second important point is that the very sensitive position of the ν1(C–O) band indicates P(nBu)3 or P(cHex)3 as the most electron donor (activating) ligand, which is in accordance with trends of observations in catalytic carbonylations. It should be noted, that the addition of a non-CO ligand to only one of the cobalts in complexes 21 generates a center of chirality on the 4-C atom of the butenolide ring, but the enantiomers could not be resolved [65,66].
4. Pnicogen/Cobalt Clusters as Ligands
The pnicogenic elements in the En[Co(CO)3]4−n clusters discussed above are in (trivalent) bonding conditions where the formation of a “free” donor orbital (derived from an sp3 hybrid, with more-less d contribution) should be present. This theoretical consideration prompted to explore this possibility also experimentally. In a systematic study, the more readily accessible complex 10 was used as ligand for substituting CO in low valent transition metal carbonyls. Complex 10, as ligand was reacted either in apolar (n-hexane) solvent under UV irradiation or in more polar environment (THF) at room temperature with M(CO)6 (M = Cr, Mo, W) or with Fe2(CO)9 which resulted the formation of (CO)5MAsCo3(CO)9(3Co–Co) (M = Cr, Mo, W) (23) (Figure 15) and (CO)4FeAsCo3(CO)9(3Co–Co) (24) (Figure 15 adducts which could be isolated and characterized by analyses, characteristic ν(C-O), and mass spectra. The ligand/complex 10 reacted also with the hard Lewis acid AlCl3 but the product could not be isolated only characterized by IR spectra [71]. A few years later, these experiments were successfully repeated and supplemented by similar substitutions with complex 7 and the preparation of (η5-C5H5)(CO)2MnECo3(CO)9 (3Co–Co) (E = P, As) (25) derivatives [72].
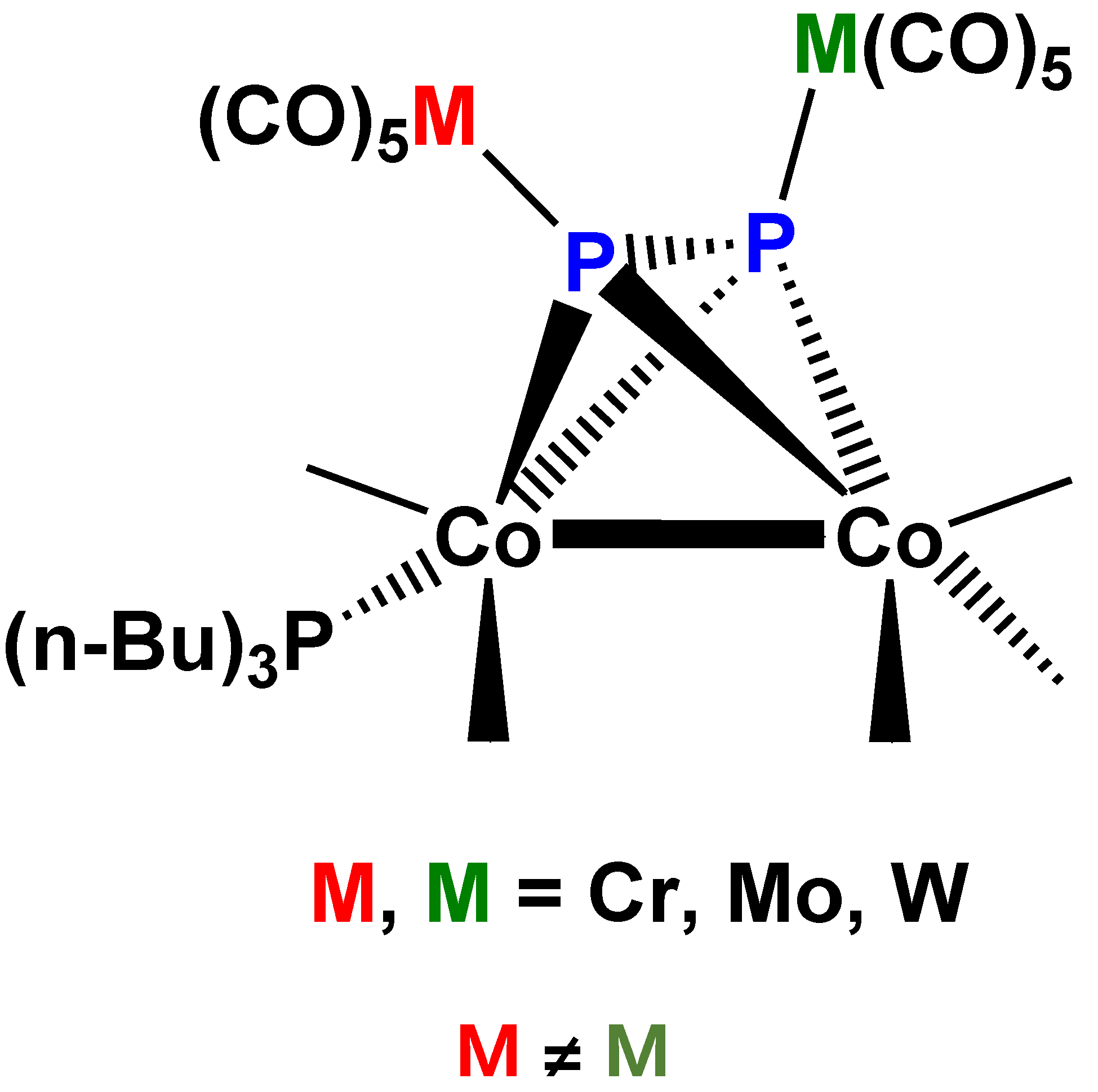
An analogous systematic study has been performed with compound 1, as “ligand” and mononuclear transition metal carbonyls (Cr, Mo, W, and Fe) [73]. In this series of preparative experiments complex 1 could be used both as monodentate and bidentate ligand, yielding [(CO)nM]P2Co2(CO)6 (Co–Co) (M = Cr, Mo, W, n = 5; M = Fe, n = 4] (26) (Figure 16A) as well as [(CO)nM]2P2Co2(CO)6 (Co–Co) (M = Cr, Mo, W, n = 5; M = Fe, n = 4] (27) (Figure 16B), or even “mixed” [(CO)5M][(CO)5M’]P2Co2(CO)6 (Co–Co) (M = Cr, M’ = W) (28) derivatives. A few mono-tercier phosphane substituted derivatives (by P(nBu)3 or PPh3) were also reported. The X-ray diffraction structure of complex 28 was determined (Figure 17). It is interesting that the two bulky M(CO)5 groups do not significantly alter the structure of the “ligand” fragment in complex 28, but the P2 unit undergoes additional activation, as indicated by the length of the P–P bond: P2 in gas phase 189.3 pm [74], in complex 3 201.0 pm [3], in the homobimetallic 26 (M = Cr, n = 5) 206.6 pm [26] and in the heterobimetallic complex 28, 206.1 pm [73]. It should be observed that the phosphane derivative of complex 28, [(CO)5Cr][(CO)5W]P2Co2(CO)5[P(nBu)3] (Co–Co) (29) (Figure 18) has a center of chirality in the center of the P2Co2 pseudotetrahedral unit. The separation and characterization of the enantiomers due to this particular case of chirality represents an interesting research challenge for the future.
Coordinated or noncoordinated E (E = P, As, Sb) and E2 (E = P, As) atoms or molecules behave as donor groups also in transition metal complexes other than Co, giving cluster or open structures, e.g., [27,28,75,76,77,78,79,80,81,82]. These interesting reports are beyond the scope of the present review.
5. Interconversions of Pnicogen/Cobalt Clusters
Studies on interconversions of cluster compounds have been surprisingly rare [5,23,83,84,85,86,87] and one could say that the rules governing these processes are not yet fully understood even today. On the other hand, clusters could be important catalysts and (as we have seen above) models of industrially important processes. It was the goal to contribute to these problems by starting a systematic study, asking how do the relatively small, tetrahedral pnicogene/cobalt clusters En[Co(CO)3]4−n (E = P, As) undergo interconversion processes [88].
The interconversion processes have been found accidentally in an attempt at preparing transition metal derivatives from complex 6 using its As atom(s) as Lewis base(s) and adding UV-excited M(CO)6 (M = Cr, Mo W) carbonyls as precursors of expected Lewis acid M(CO)5 intermediates. The reaction, however, led to trinuclear derivatives 11 and 23. Adding also Co2(CO)8 increased the yields of the more Co-rich trinuclear complexes. Starting from complex 1 the reaction gave essentially similar results. This transformation, which could be written simplified as E2Co2 => ECo3 is analogous to the reaction of (μ2-IC2I)Co2(CO)6 (aided with excess Co2(CO)8) providing [CCo3(CO)9]2 [85], with the exception, that in the latter case the “last” C–C bond from the triple bond of the starting acetylene remains unaltered (Figure 19).
Treatment of the trinuclear complex 10 with P(OPh)3 yielded in apolar solvent a mixture of its monosubstituted derivative, AsCo3(CO)8[P(OPh)3] (30) and that of the dinuclear 6, As2Co2(CO)5[P(OPh)3], indicating an ECo3 => E2Co2 transformation, that is, the opposite of the former.
These cluster transformations are in accordance with the observation that the diphosphorous P–P bridge in complex 1 gets cleaved under hydroformylation conditions [87].
6. Outlook and Conclusions
The above-outlined results induced several additional research efforts. We cannot list here all, but some interesting preparative developments should be mentioned.
Coordination was proved to stabilize such phosphorous molecules which are not known in the free state, that is P5 [89] (Figure 20A) and P8, which appears only in the lattice of Hittorf’s monoclinic phosphorous allotrope [90] (Figure 20B).
Preparative efforts at combining experiences of the chemistry of large clusters [84] and the above outlined results enabled the synthesis of clusters with interstitial pnicogene atoms in transition metal cages, e.g., [91].
These outstanding results might indicate the way to new preparative research activities.
From catalytic point of view two aspects should be underlined:
(1) The above-mentioned catalytic synthesis of bifurandiones aided by tertiary phosphorous compounds [65,65] proceeds through a C2Co2 cluster (18) is one of the “greenest” carbonylation reactions (100% atom utilization). The synthesis in the presence of tertiary phosphines resulted a dramatic diminution of the necessary reaction pressure, but this is not yet enough to make it industrially economic. Additional research in this direction appears to be promising, probably for green fine-chemical industry.
(2) The really great challenge in this field is to achieve progress in the field of atmospheric N2 fixation. The world’s production of ammonia by high pressure/high temperature (essentially Haber–Bosch, 15–25 MPa, 400–500 °C) synthesis amounts 176 Mt/year (2014) [92], while the coordination-based, transition metal-catalyzed biological nitrogen fixation proceeds at cca. room temperature and atmospheric pressure, yielding approximately the same amount of fixed N as the industrial process does. Finding the possibility of a lower reaction temperature and lower pressure would mean enormous energy savings. For this goal it is really worth to continue the above outlined efforts on pnicogenic complexes, which are close relatives of the molecular events displaying in the enzymatic nitrogen fixation. The first outstanding results in this direction have been reported recently [93].
Author Contributions
All three authors contributed equally to the preparation of the present review. The original experimental material, which is reviewed here was the result of the cooperation of A.V.-O. and G.P., also with other scientists, as it can be seen from the reference list.
Funding
No special funding for the present review was obtained or used.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Vízi-Orosz, A.; Pályi, G.; Markó, L. Phosphido cobalt carbonyl clusters: Co2(CO)6P2 and Co3(CO)9PS. J. Organomet. Chem. 1973, 60, C25–C26. [Google Scholar] [CrossRef]
- Vízi-Orosz, A.; Pályi, G.; Markó, L. (μ2-Diphosphorous)hexacarbonyldicobalt (Co–Co) and derivatives. In Organometallic Syntheses; King, R.B., Eisch, J.J., Eds.; Elsevier: Amsterdam, The Netherlands, 1988; pp. 272–277. [Google Scholar]
- Campana, C.F.; Vízi-Orosz, A.; Pályi, G.; Markó, L.; Dahl, L.F. Structural characterization of Co2(CO)5[P(C6H5)3](μ-P2): A tricyclic complex containing bridging P2 ligand. Inorg. Chem. 1979, 18, 3054–3059. [Google Scholar] [CrossRef]
- Bor, G.; Markó, L.; Markó, B. Kobaltcarbonylderivate des Tetrachlorkohlenstoffs und organischer gem.-Trihalogen Verbindungen: Trikobaltenneacarbonylkohlenstoff Verbindungen. Chem. Ber. 1962, 95, 333–340. [Google Scholar] [CrossRef]
- Pályi, G.; Piacenti, F.; Markó, L. Methinyltricobalt enneacarbonyl compounds: Preparation, structure and properties. Inorg. Chim. Acta Rev. 1970, 4, 109–121. [Google Scholar] [CrossRef]
- Volpin, M.E.; Shur, V.B. Nitrogen fixation by transition metal complexes. Nature 1966, 209, 1236. [Google Scholar] [CrossRef]
- Allen, A.D.; Senoff, C.V. Nitrogenopentammineruthenium(II) complexes. Chem. Commun. 1965, 24, 621–622. [Google Scholar] [CrossRef]
- Brinzinger, H. Formation of ammonia by insertion of molecular nitrogen into metal-hydride bonds, III. Considerations on the properties of enzymatic nitrogen fixing systems and proposal of a general mechanism. Biochemistry 1966, 5, 3947–3950. [Google Scholar] [CrossRef]
- Newton, W.E. Chemical approaches to nitrogen fixation. Adv.Chem. [Bio. Chem.] 1980, 191, 351–377. [Google Scholar]
- Hu, Y.; Ribbe, M.W. Nitrogenase and homologs. J. Biol. Inorg. Chem. 2015, 20, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Shilov, A.E. Intermediate complexes in chemical and biological nitrogen fixation. Pure Appl. Chem. 1992, 64, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.W. The metal carbonyls. Quart. Rev. (Lond.) 1963, 17, 133–159. [Google Scholar] [CrossRef]
- Markó, L. Catalytic properties of the transition metal carbonyls and of their organic π-complexes. In Chemistry of Metal Carbonyls and Their Derivatives; Bor, G., Ed.; Akadémiai Kiadó: Budapest, Hungary, 1966; pp. 138–170. (In Hungarian) [Google Scholar]
- Fischer, E.O. Coordination compounds of unsaturated hydrocarbons with metals. Chem. Soc. (Lond.) Spec. Publ. 1959, 13, 73–92. [Google Scholar]
- Sternberg, H.W.; Wender, I. Metal carbonyls and related compounds as catalytic intermediates in organic syntheses. Chem. Soc. (Lond.) Spec. Publ. 1959, 13, 35–55. [Google Scholar]
- Pauson, P.L. Hydrocarbon derivatives of transition metals. Endeavour 1962, 21, 175–182. [Google Scholar]
- Sternberg, H.W.; Greenfield, H.; Friedel, R.A.; Wotiz, J.H.; Markby, R.; Wender, I. A new type of metallo-organic complex derived from dicobalt octacarbonyl and acetylenes. J. Am. Chem. Soc. 1954, 76, 1457–1458. [Google Scholar] [CrossRef]
- Pályi, G.; Váradi, G.; Markó, L. Stereochemistry of acetylenes coordinated to cobalt. In Stereochemistry of Organometallic and Inorganic Compounds; Bernal, I., Ed.; Elsevier: Amsterdam, The Netherlands, 1986; Volume 1, pp. 358–410. [Google Scholar]
- Markó, L.; Bor, G.; Almásy, G. Tricobaltenneacarbonylsulphide. Chem. Ind. 1961, 1491–1492. [Google Scholar]
- Wei, C.H.; Dahl, L.F. Organometallic complexes, VI., Molecular structure of a tricyclic complex, tricobalt enneacarbonyl sulfide. Inorg. Chem. 1967, 6, 1229–1236. [Google Scholar] [CrossRef]
- Khattab, S.A.; Markó, L.; Bor, G. Sulphur containing metal carbonyls. VI., A mixed cobalt-iron carbonyl sulphide. J. Organomet. Chem. 1964, 1, 373–376. [Google Scholar] [CrossRef]
- Bor, G. Chemistry of Metal Carbonyls and Their Derivatives; Akadémiai Kiadó: Budapest, Hungary, 1966; pp. 155–159. (In Hungarian) [Google Scholar]
- Markó, L. Tetrahedral cobalt carbonyl clusters with heteroatoms. Gazz. Chim. Ital. 1979, 109, 247–253. [Google Scholar]
- Markó, L.; Ungváry, F.; Pályi, G.; Sisak, A.; Vízi-Orosz, A. Synthesis and catalytical applications of cobalt and iron carbonyls—Hydroformylation and related reactions. Magyar Kém. Folyóirat 1994, 100, 385–393. (In Hungarian) [Google Scholar]
- Issleib, K. Coordination chemistry of tetravalent phosphorous. Pure Appl. Chem. 1975, 44, 237–267. [Google Scholar] [CrossRef]
- Lang, H.; Zsolnai, L.; Huttner, G. Diphosphorous, -P=P-, as 8-electron ligand. Angew. Chem. Int. Ed. 1983, 22, 976–977. [Google Scholar]
- Caporali, M.; Gonsalvi, L.; Rossin, A.; Petruzzini, M. P4-activation by late transition metal complexes. Chem. Rev. 2010, 110, 4178–4235. [Google Scholar] [CrossRef] [PubMed]
- Whitmire, K.H. Transition metal complexes of the pnictide elements. Coord. Chem. Rev. 2018, 376, 114–195. [Google Scholar] [CrossRef]
- Foust, A.S.; Campana, C.F.; Sinclair, J.D.; Dahl, L.F. Preparation and structural characterization of the mono(triphenylphosphine) and bis(triphenylphosphine) substituted derivatives of Co2(CO)6(μ-As2)—Effect of phosphine ligand substitution on the Co2As2 metal cluster system. Inorg. Chem. 1979, 18, 3047–3054. [Google Scholar] [CrossRef]
- Elian, M.; Chen, M.M.-L.; Mingos, D.M.P.; Hoffmann, R. Comparative bonding study of conical fragments. Inorg. Chem. 1976, 15, 1148–1155. [Google Scholar] [CrossRef]
- De Kock, R.L.; Fehlner, T.P. On the validity of the isolobal principle. Polyhedron 1982, 1, 521–523. [Google Scholar] [CrossRef]
- Hoffmann, R. Building bridges between inorganic and organic chemistry. Angew. Chem. Int. Ed. 1982, 21, 711–724. [Google Scholar] [CrossRef]
- Vízi-Orosz, A. Phosphido cobalt carbonyl clusters Pn[Co(CO)3]4-n (n = 1, 2, 3). J. Organomet. Chem. 1976, 111, 61–64. [Google Scholar] [CrossRef]
- Vízi-Orosz, A.; Galamb, V.; Pályi, G.; Markó, L.; Bor, G.; Natile, G. AsCo3(CO)9, its cyclic trimer, As3Co9(CO)24 and the phosphorous-containing analog P3Co9(CO)24. J. Organomet. Chem. 1976, 107, 235–240. [Google Scholar] [CrossRef]
- Goldberg, K.I.; Hoffmann, D.M.; Hoffmann, R. Existence of binuclear π-bonded dinitrogen complexes. Inorg. Chem. 1982, 21, 3863–3868. [Google Scholar] [CrossRef]
- Bán, M.I.; Dömötör, G.; Vízi-Orosz, A.; Pályi, G. Electronic structure of (μ2-X2)Co2(CO)6 (X = CH, N, P, As) complexes. Atti Accad. Sci. Bologna Cl. Sci. Fis. 1983, 271, 217–225. [Google Scholar]
- Foust, A.S. Synthetic and Structural Studies of the Coordinative Versatility of Pnicogen Atoms in Metal Carbonyl Compounds. Ph.D. Thesis, University of Wisconsin, Madison, WI, USA, 1970. [Google Scholar]
- Gall, R.S.; Foust, A.S.; Pollick, P.J.; Woiciki, A.; Dahl, L.F. In Proceedings of the Abstracts, American Crystallogrphic Association, Meeting, Berkeley, CA, USA, 24–28 March 1974.
- Dahl, L.F.; Foust, A.S. Organometallic pnicogen complexes, V., Preparation, structure and bonding of the tetrameric antimony-cobalt cluster system, Co4(CO)12Sb4: The first known (main group element)(metal carbonyl) cubane type structure. J. Am. Chem. Soc. 1970, 92, 7337–7341. [Google Scholar] [CrossRef]
- Norman, N.C.; Webster, P.M.; Farrugia, L.J. Further synthetic and structural studies on cobalt carbonyl containing antimony complexes. J. Organomet. Chem. 1992, 430, 205–219. [Google Scholar] [CrossRef]
- Etzrodt, G.; Boese, R.; Schmid, G. Heteronucleare Clustersysteme, XVI., Darstellung und Untersuchung von Tris(tetracarbonylcobaltio)bismutan—Ein Beitrag zur Frage der Existenz Bismuthaltiger Nonacarbonyltricobalt Clusterverbindungen. Chem. Ber. 1979, 112, 2574–2580. [Google Scholar] [CrossRef]
- Kruppa, W.; Blaser, D.; Boese, R.; Schmid, G. Heteronuclear cluster systems, XX [1] μ3-Bismutio-cyclo-tris(tricarbonyliridium) (3Ir-Ir), BiIr3(CO)9—Synthesis and structural investigation of a novel iridium cluster. Z. Naturforsch. B 1982, 37, 209–213. [Google Scholar] [CrossRef]
- Martinengo, S.; Ciani, G. Bismuth-cobalt heteronuclear carbonyl cluster compounds. Synthesis and X-ray characterization of the neutral [BiCo3(CO)9] and of the paramagnetic anion [Bi2Co4(CO)11]−. J. Chem. Soc. Chem. Commun. 1987, 20, 1589–1591. [Google Scholar] [CrossRef]
- Bán, M.I.; Dömötör, G.; Vízi-Orosz, A.; Pályi, G. Electronic structures of the hetreonuclear cobalt clusters Pn[Co(CO)3]4-n. J. Mol. Struct. THEOCHEM 1986, 31, 193–196. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. Side-on binding of the nitrogen molecule to first-row transition metal dimers. J. Chem. Phys. 1991, 95, 364–372. [Google Scholar] [CrossRef]
- Seyferth, D.; Henderson, R.S. Phosphaacetylenehexacarbonyldicobalt complexes—New cluster Lewis bases. J. Organomet. Chem. 1978, 162, C35–C38. [Google Scholar] [CrossRef]
- Seyferth, D.; Merola, J.S. Arsaacetylenes, RC≡As, as ligands in dicobalt hexacarbonyl complexes: Novel main group element—Transition metal hybrid cluster compounds. J. Am. Chem. Soc. 1978, 100, 6783–6784. [Google Scholar] [CrossRef]
- Seyferth, D.; Merola, J.S.; Henderson, R.S. Organocobalt cluster complexes, 34., Preparation properties and chemical reactivity of phospha- and arsaacetylenedicobalt hexacarbonyl complexes, (RCM)Co2(CO)6 (M = P, As). Organometallics 1982, 1, 859–866. [Google Scholar] [CrossRef]
- Bán, M.I.; Dömötör, G.; Vízi-Orosz, A.; Pályi, G. Electronic structures, bonding properties and photoelectron spectra of (μ2-diphosphorous)- and (μ2-1-alkyl-2-phosphaacetylene)hexacarbonyldicobalt (Co–Co). J. Mol. Struct. THEOCHEM 1990, 209, 399–410. [Google Scholar] [CrossRef]
- Bönnemann, H.; Brinkmann, R. Single-step cobalt-catalyzed synthesis of pyridines. Synthesis 1975, 9, 600–602. [Google Scholar] [CrossRef]
- Bönnemann, H. Cobalt-catalyzed pyridine syntheses from alkynes and nitriles. Angew. Chem. Int. Ed. 1978, 17, 505–515. [Google Scholar] [CrossRef]
- Bönnemann, H. Organocobalt compounds in the synthesis of pyridines—An example of structure-effectivity relationships in homogeneous catalysis. Angew. Chem. Int. Ed. 1985, 24, 248–262. [Google Scholar] [CrossRef]
- Falbe, J. Synthesen mit Kohlenmonoxid; Springer: Berlin/Heidelberg, Germany, 1967. [Google Scholar]
- Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpainter, C.W. Progress in hydroformylation and carbonylation. J. Mol. Catal. A 1995, 104, 17–85. [Google Scholar] [CrossRef]
- Klinger, R.J.; Chen, M.J.; Rathke, J.W.; Kramarz, K.W. Effect of phosphines on the thermodynamics of the cobalt-catalyzed hydroformylation system. Organometallics 2007, 26, 352–357. [Google Scholar] [CrossRef]
- Falbe, J. (Ed.) New Syntheses with Carbon Monoxide; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Bor, G.; Markó, L. Monosubstituted derivative of dicobalt octacarbonyl with triphenylphosphine: Co2(CO)7P(C6H5)3. Chem. Ind. (Lond.) 1963, 22, 912–913. [Google Scholar]
- Piacenti, F.; Bianchi, M.; Benedetti, E.; Frediani, P. Hydroformylation of propene in the presence of Co2(CO)6[P(C4H9)3]2. J. Organomet. Chem. 1970, 23, 257–264. [Google Scholar] [CrossRef]
- Szabó, P.; Fekete, L.; Nagy-Magos, Z.; Bor, G.; Markó, L. Phosphorous containing metal carbonyls, III., Monosubstituted derivatives of dicobalt octacarbonyl with phosphines and phosphites. J. Organomet. Chem. 1968, 12, 245–248. [Google Scholar] [CrossRef]
- Bartik, T.; Krümmling, T.; Happ, B.; Seiker, A.; Markó, L.; Boese, R.; Ugo, R.; Zucchi, C.; Pályi, G. Intermediates and isomers in the substitution of cobalt carbonyl hydride with thertiary phosphorous ligands. Catal. Lett. 1993, 19, 383–389. [Google Scholar] [CrossRef]
- Galamb, V.; Pályi, G.; Cser, F.; Furmanova, M.G.; Struchkov, Y.T. Stable alkylcobalt carbonyls—[(alkoxycarbonyl)methyl]cobalt tetracarbonyl compounds. J. Organomet. Chem. 1981, 209, 183–195. [Google Scholar] [CrossRef]
- Galamb, V.; Pályi, G.; Ungváry, F.; Markó, L.; Boese, R.; Schmid, G. (η1-Benzyl)cobalt, (η3-benzyl)cobalt and (η1-phenylacetyl)cobalt carbonyls. J. Am. Chem. Soc. 1986, 108, 3344–3351. [Google Scholar] [CrossRef]
- Váradi, G.; Horváth, I.T.; Palágyi, J.; Bak, T.; Pályi, G. On the reactivity of acetylenes coordinated to cobalt, IV., Influence of tertiary phosphorous compounds on the catalytic synthesis of bifurandione. J. Mol. Catal. 1980, 9, 457–460. [Google Scholar] [CrossRef]
- Pályi, G.; Váradi, G.; Horváth, I.T. Activation of carbon monoxide and acetylenes by cobalt carbonyls. J. Mol. Catal. 1981, 13, 61–70. [Google Scholar] [CrossRef]
- Pályi, G.; Váradi, G.; Vízi-Orosz, A.; Markó, L. Reactivity of μ2-acetylenes coordinated to cobalt: Stereochemistry of the formation of the butene-2-olide-4 complexes Co2(CO)7(RR’C4O2). J. Organomet. Chem. 1975, 90, 85–91. [Google Scholar] [CrossRef]
- Váradi, G.; Vecsei, I.; Ötvös, I.; Pályi, G. Reactivity of acetylenes coordinated to cobalt, 2., Regiospecifity of carbonylation of internal acetylenes. J. Organomet. Chem. 1979, 182, 415–423. [Google Scholar] [CrossRef]
- Bán, M.I.; Révész, M.; Bálint, I.; Váradi, G.; Pályi, G. A CNDO/2 study of (μ2--acetylene)hexacarbonyldicobalt (Co–Co) complexes. J. Mol. Struct. THEOCHEM 1982, 88, 357–370. [Google Scholar] [CrossRef]
- Müller, M.; Vahrenkamp, H. Darstellung von Mehrkernkomplexen aus As2Co2(CO)6. J. Organomet. Chem. 1983, 252, 95–104. [Google Scholar] [CrossRef]
- Di Varia, M.; Ghilardi, C.A.; Midollini, S.; Sacconi, L. cyclo-Triphosphorus (Δ-P3) as ligand in cobalt and nickel complexes with 1,1,1-tris(diphenylphosphinomethyl)ethane. Formation and structure. J. Am. Chem. Soc. 1978, 100, 2550–2551. [Google Scholar] [CrossRef]
- Váradi, G.; Vízi-Orosz, A.; Vastag, S.; Pályi, G. Preparation and infrared spectra of monosubstituted PR3 and P(OR)3 (R = alkyl, aryl) derivatives of (μ2-L)2Co2(CO)6 compounds. J. Organomet. Chem. 1976, 108, 225–233. [Google Scholar] [CrossRef]
- Vízi-Orosz, A.; Galamb, V.; Ötvös, I.; Pályi, G.; Markó, L. The behaviour of the AsCo3(CO)9 cluster as a ligand for Cr, Mo, W and Fe carbonyls and AlCl3. Transit. Met. Chem. 1979, 4, 415–423. [Google Scholar] [CrossRef]
- Lang, H.; Huttner, G.; Sigwarth, B.; Jibril, I.; Zsolnai, L.; Orama, O. μ3-P und μ3-As verbrückte Cluster als Liganden. J. Organomet. Chem. 1986, 304, 137–155. [Google Scholar] [CrossRef]
- Vízi-Orosz, A.; Pályi, G.; Markó, L.; Boese, R.; Schmid, G. Reaction of coordinated diphosphorous with metal carbonyl fragments. Crystal structure of [(CO)5Cr][(CO)5W]P2Co2(CO)6. J. Organomet. Chem. 1985, 288, 179–187. [Google Scholar] [CrossRef]
- Douglas, A.E.; Rao, K.S. A new band system of the P2 molecule analogous to the Lyman-Birge-Hopfield bands of N2. Can. J. Phys. 1958, 36, 565–570. [Google Scholar] [CrossRef]
- Malisch, W.; Panster, P. Tris[dicarbonyl(η-cyclopentadienyl)ferrio]stibane—A 5B element base with total transition metal substitution. Angew. Chem. Int. Ed. 1976, 15, 618–619. [Google Scholar] [CrossRef]
- Schmid, G.; Kempny, H.P. The use of elemental phosphorus as ligand in iron carbonyls. Z. Anorg. Allg. Chem. 1977, 432, 160–166. [Google Scholar] [CrossRef]
- Huttner, G.; Sigwarth, B.; Scheidsteger, O.; Zsolnai, L.; Orama, O. Diarsenic, As2, as four-, six- or eight-electron donor ligand. Organometallics 1985, 4, 326–332. [Google Scholar] [CrossRef]
- Scherer, O.J. Phosphorous, arsenic, antimony and bismuth multiply bonded systems with low coordination number—Their roles as complex ligands. Angew. Chem. Int. Ed. 1985, 24, 924–943. [Google Scholar] [CrossRef]
- Lang, H.; Huttner, G.; Zsolnai, L.; Mohr, G.; Sigwarth, B.; Weber, U.; Orama, O.; Jibril, I. Diphosphor, Phosphor-, Arsen- und Antimonatome als Clusterbaugruppen. J. Organomet. Chem. 1986, 304, 157–179. [Google Scholar] [CrossRef]
- Di Varia, M.; Stoppioni, P.; Peruzzini, M. Naked phosphorous atoms and units in transition metal compounds. Polyhedron 1987, 6, 351–382. [Google Scholar]
- Barr, A.M.; Kerlogue, M.D.; Norman, N.C.; Webster, P.M.; Farrugia, L.J. The synthetic and structural characterization of some organotransition metal complexes incorporating antimony. Polyhedron 1989, 8, 2495–2505. [Google Scholar] [CrossRef]
- Scherer, O.J. Complexes with substituent-free acyclic and cyclic phosphorous, arsenic, antimony and bismuth ligands. Angew. Chem. Int. Ed. 1990, 29, 1104–1122. [Google Scholar] [CrossRef]
- Seyferth, D. Chemistry of carbon-functional alkylidynetricobalt nonacarbonyl cluster complexes. Adv. Organomet. Chem. 1976, 14, 97–144. [Google Scholar]
- Chini, P.; Longoni, G.; Albano, V.G. High nuclearity metal carbonyl clusters. Adv. Organomet. Chem. 1976, 14, 285–344. [Google Scholar]
- Váradi, G.; Pályi, G. (μ2-Dihaloacetylene)hexacarbonyldicobalt (Co–Co) compounds and their reaction with cobalt carbonyls. Inorg. Chim. Acta 1976, 20, L33–L34. [Google Scholar] [CrossRef]
- Váradi, G.; Galamb, V.; Palágyi, J.; Pályi, G. On the reactivity of acetylenes coordinated to cobalt, V., Unexpected formation of trinuclear carbyne derivatives from acetylene mono- and dicarboxylic acid esters. Inorg. Chim. Acta 1981, 53, L29–L30, L222. [Google Scholar]
- Harley, A.D.; Guskey, G.J.; Geoffroy, G.L. Interconversion of phosphido-bridged polynuclear cobalt carbonyl complexes—Clevage of the phosphido bridge during hydroformylation catalysis. Organometallics 1983, 2, 53–59. [Google Scholar] [CrossRef]
- Vízi-Orosz, A.; Galamb, V.; Pályi, G.; Markó, L. Transformations of the heteronuclear clusters En[Co(CO)3]4-n (E = P, As; n = 1-3) when treated with soft Lewis acids and bases. J. Organomet. Chem. 1981, 216, 105–111. [Google Scholar] [CrossRef]
- Scheer, M.; Friedrich, G.; Schuster, K. [Cr(CO)5PCl3]—A P1 building block for the formation of complexes with cyclo-Px (X = 3,5) ligands. Angew. Chem. Int. Ed. 1993, 32, 593–594. [Google Scholar] [CrossRef]
- Barr, M.E.; Adams, B.R.; Weller, R.R.; Dahl, L.F. Synthesis and structural-bonding analysis of (η5-C5H4Me)4Fe4(CO)6P8 and (η5-C5H4Me)4Fe6(CO)13P8, two unprecedented transition-metal complexes containing cage-like P8 subunit of Hittorf’s monoclinic phosphorous allotrope. J. Am. Chem. Soc. 1991, 113, 3052–3060. [Google Scholar] [CrossRef]
- Fremoni, C.; Bussoli, G.; Ciabatti, I.; Ermini, M.; Hayatifar, M.; Iapalucci, M.C.; Zacchini, S. Interstitial bismuth atoms in icosahedral rhodium cages: Syntheses and molecular structures of the [Bi@Rh12(CO)27Bi2]3−, [(Bi@Rh12(CO)26)2Bi]5−, [Bi@Rh14(CO)27Bi2]3− and [Bi@Rh17(CO)33Bi2]4− carbonyl clusters. Inorg. Chem. 2017, 56, 6343–6351. [Google Scholar] [CrossRef] [PubMed]
- Sloveichik, G. Future of Ammonia Production: Improvement of Haber-Bosch Process or Electrochemical Synthesis. Available online: https://nh3fuelassociation.org/2017/10/01/future-of-ammonia-production-improvement-of-haber-bosch-process-or-electrochemical-synthesis/ (accessed on 18 September 2018).
- Nishibayashi, Y. Development of the catalytic nitrogen fixation using transition metal-dinitrogen complexes under mild reaction conditions. Dalton Trans. 2018, 47, 11290–11297. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic view of the structure (A) of complex 1 [1,2]; Schematic view of the structure (B) of complex 3 [3].
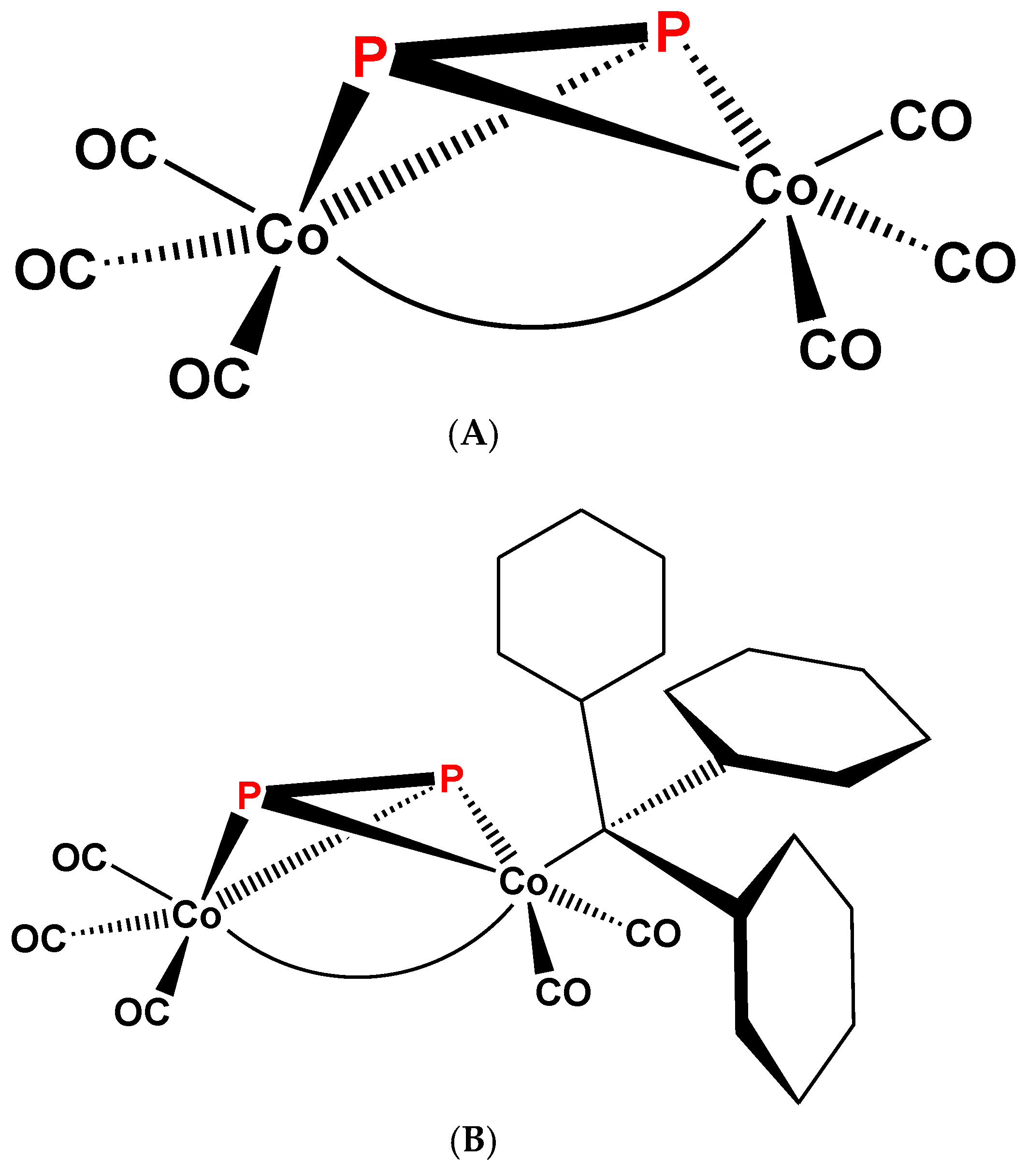
Figure 2.
Schematic views of the structures of complex 2 (A) [1,2] and of its organic analog, (μ3-RC)Co3 (CO)9 (3Co–Co), R = organic groups, halogens, etc. (B) [4,5].
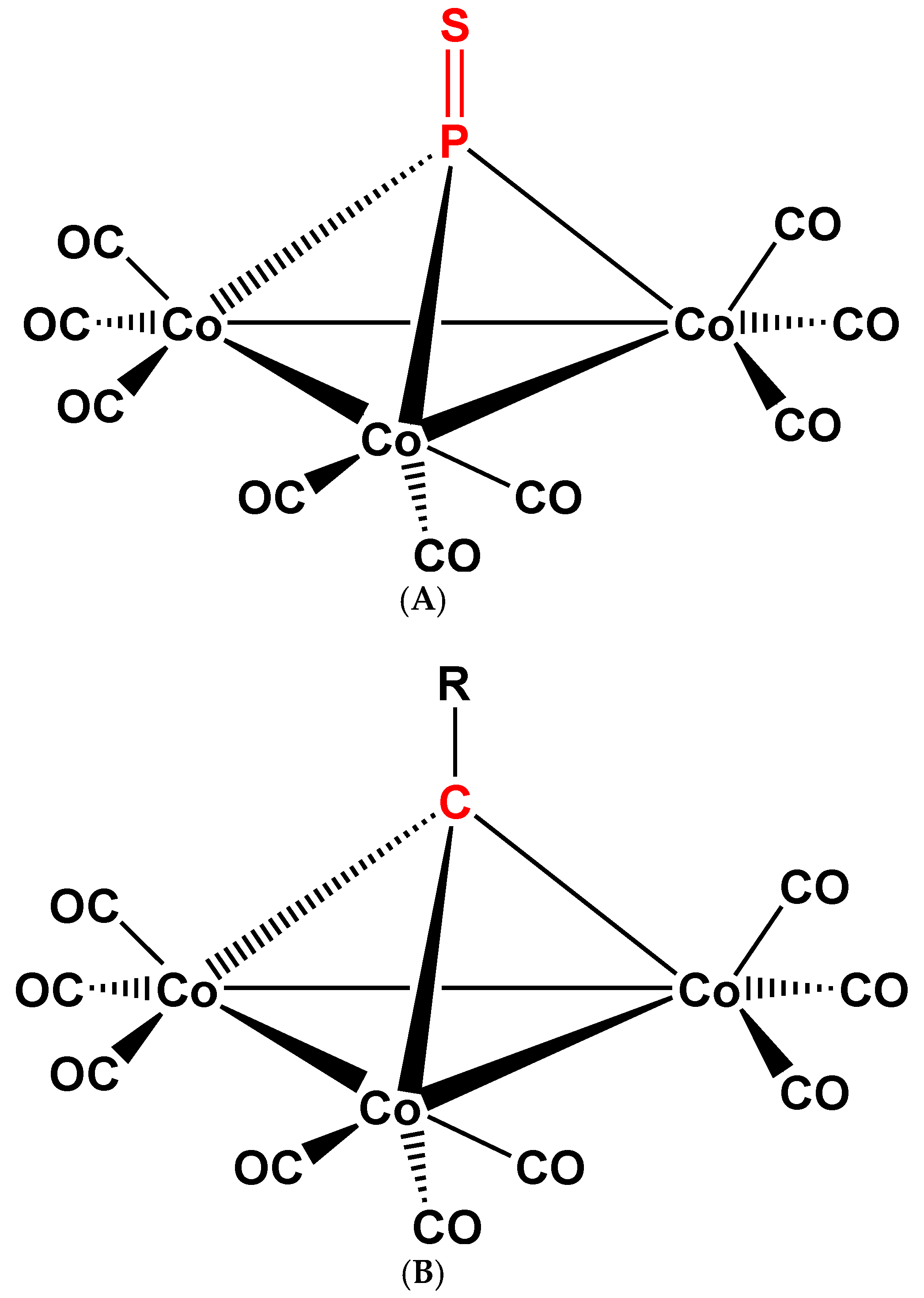
Figure 3.
Schematic view of the structure (A) of complex 4 [19,20]; Schematic view of the structure (B) of complex 5 [21].
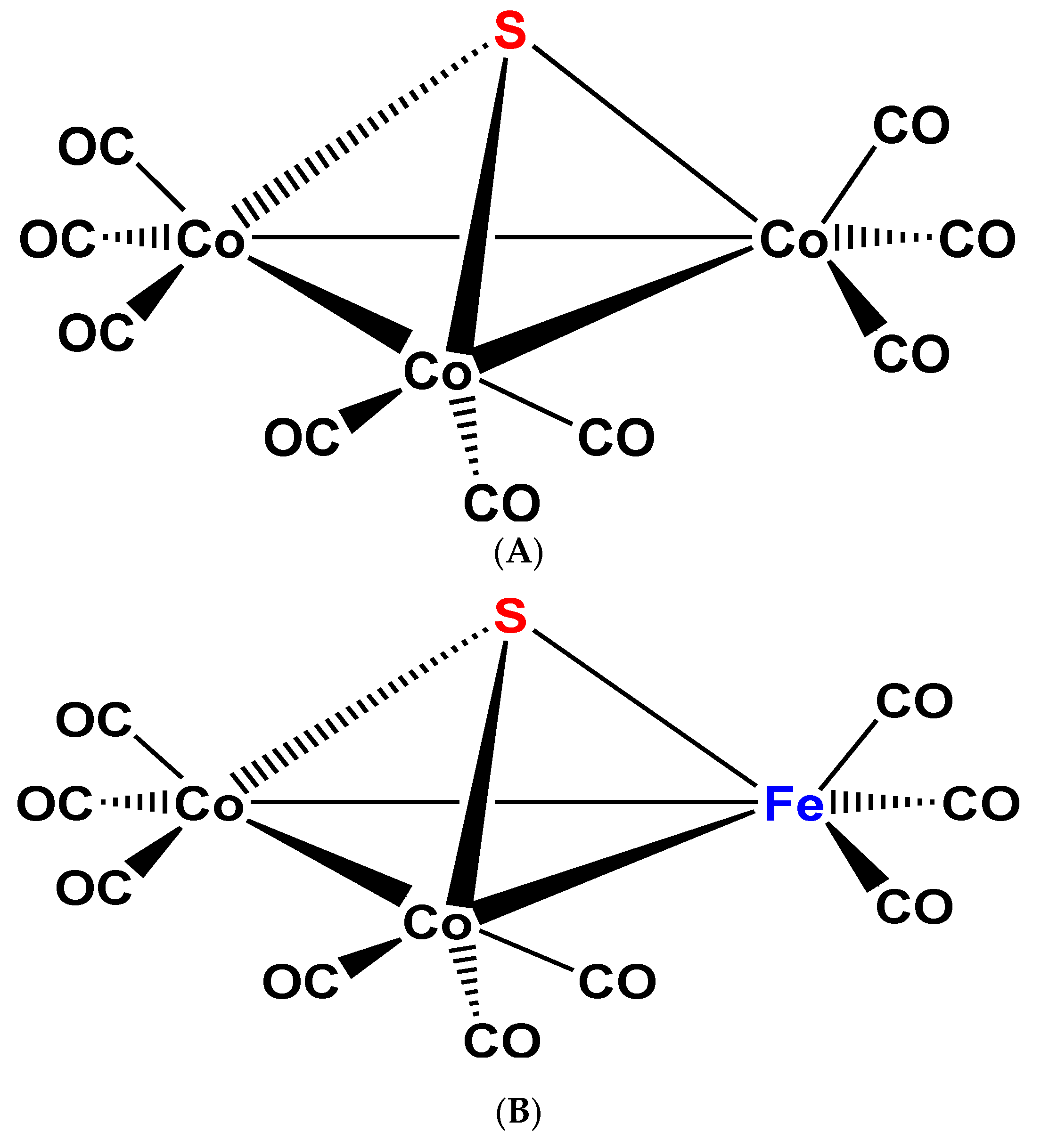
Figure 4.
Schematic view of the structure of complex 6 [29].
Figure 4.
Schematic view of the structure of complex 6 [29].
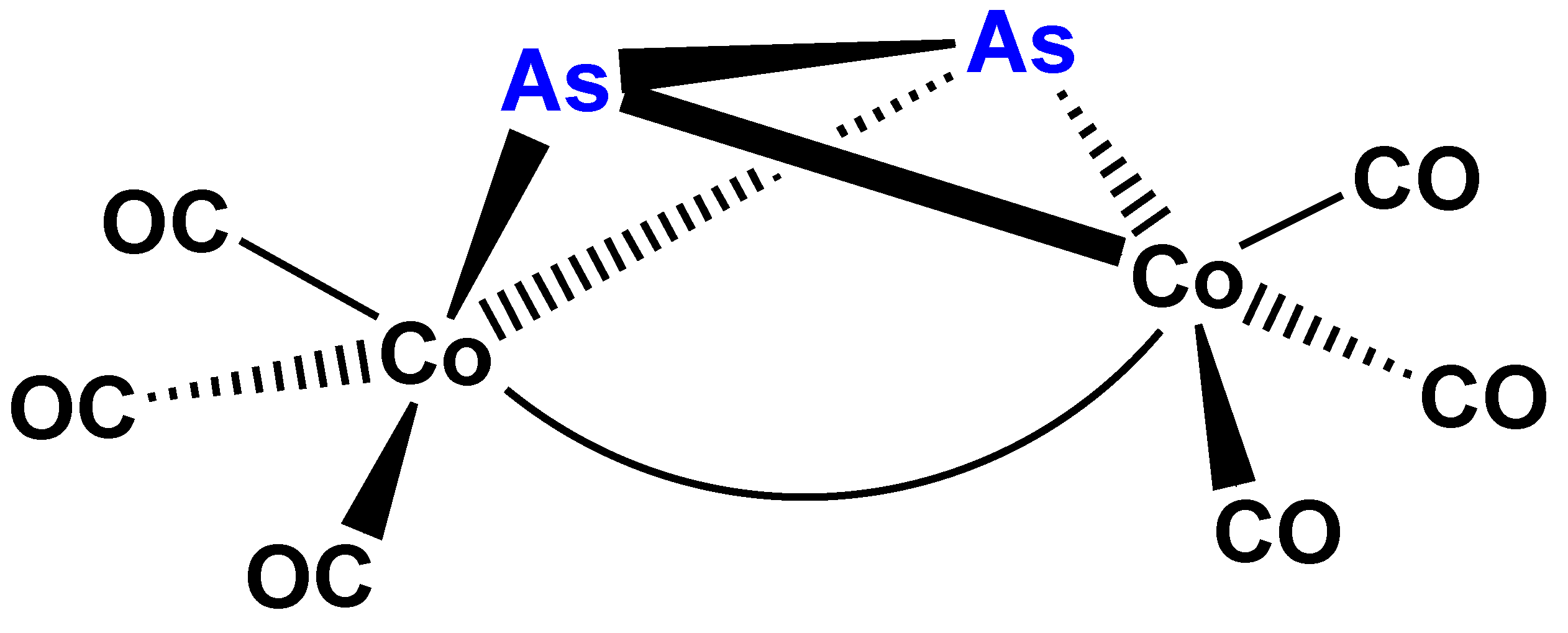
Figure 5.
Schematic view of the structure of complex 7 [33].
Figure 5.
Schematic view of the structure of complex 7 [33].

Figure 6.
Schematic view of the structure of complex 8 [33].
Figure 6.
Schematic view of the structure of complex 8 [33].
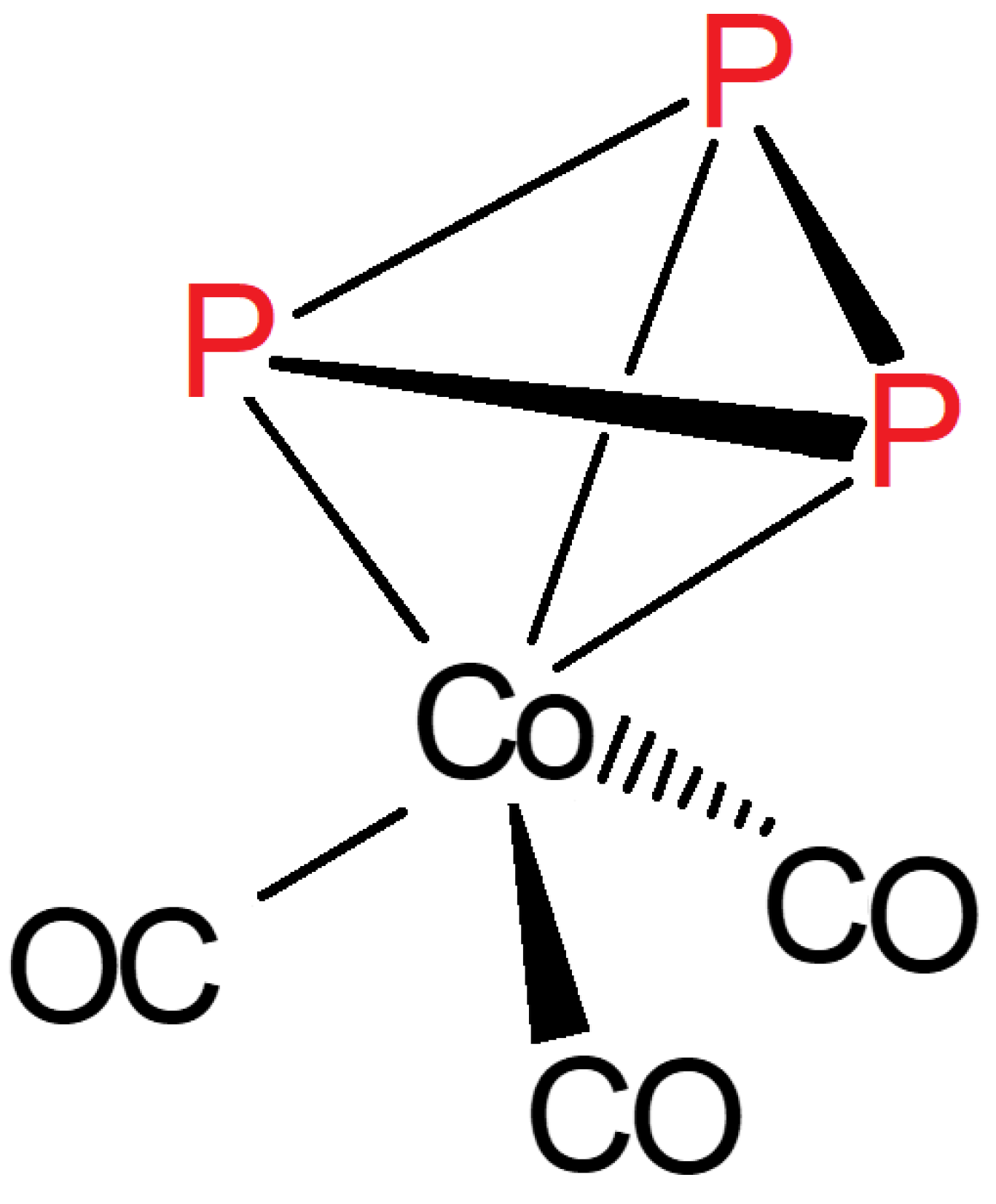

Figure 8.
(A): Schematic view of the structure of complex 9, E = P [34] and of complex 11, E = As [34,37,38]. (B): Schematic view of the 1,3,5-triphospha- and 1,3,5-triarsa-2,4,6-tricobalta-cyclo-hexatriyne rings in complexes 9 (E = P) and 11 (E = As).
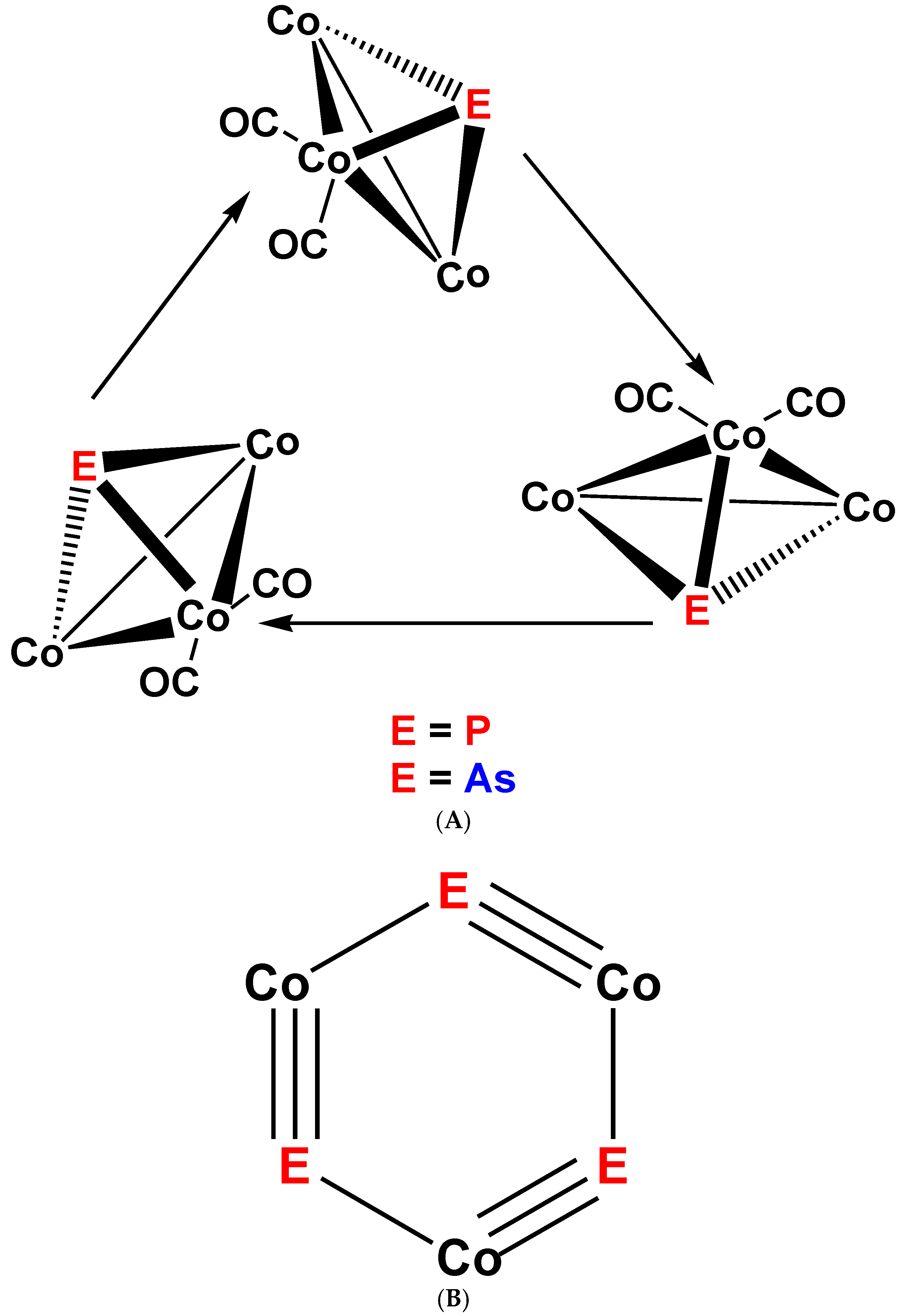
Figure 9.
Schematic view of the structure of complex 12 [39].
Figure 9.
Schematic view of the structure of complex 12 [39].
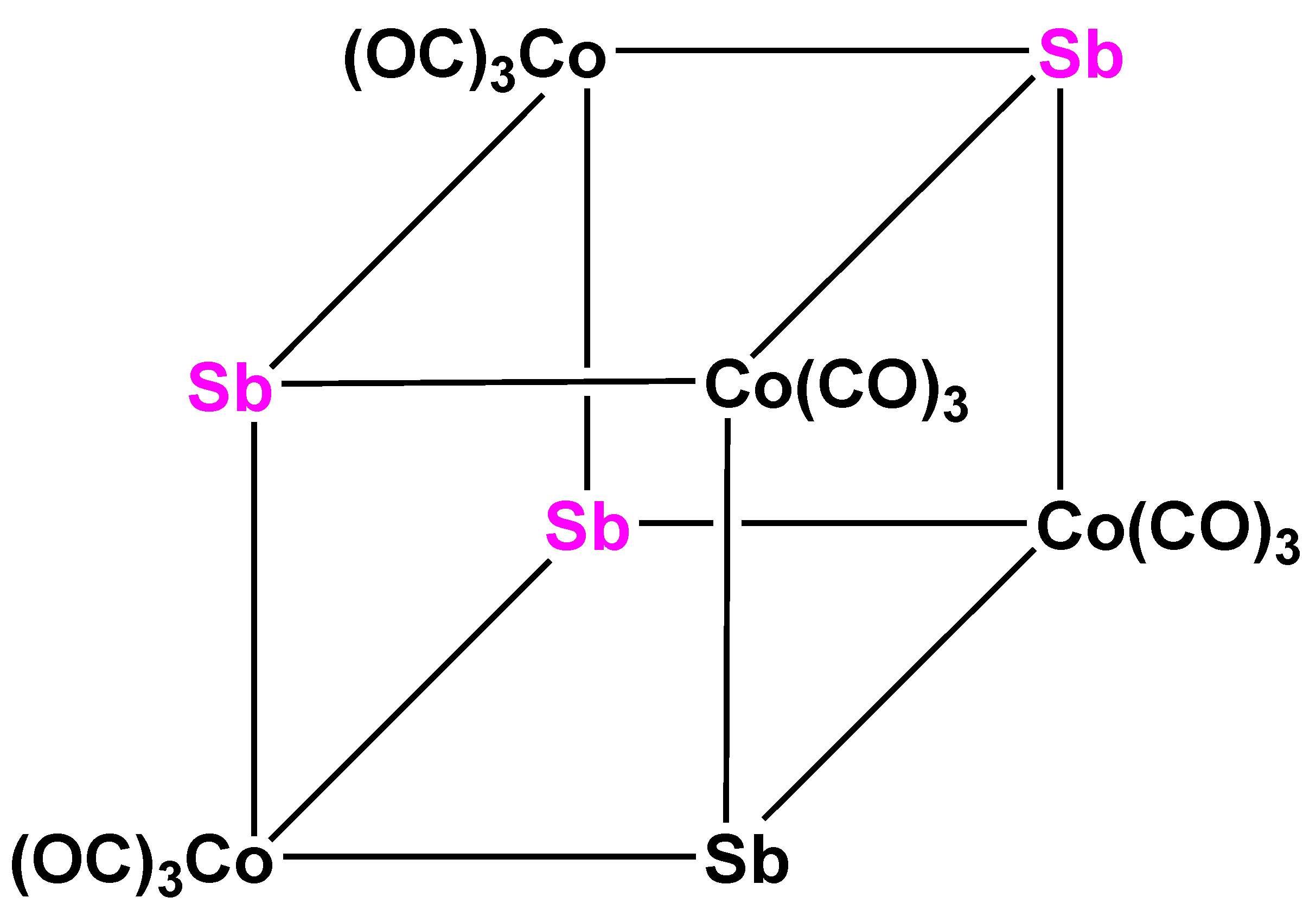
Figure 10.
Schematic view of the X-ray structure of complex 15 [42].
Figure 10.
Schematic view of the X-ray structure of complex 15 [42].

Figure 11.
Schematic view of the structures of complexes 16 (E = P), 17 (E = As) [46,47,48,49] and of the related hypothetic E = N [50,51,52] derivative.

Figure 12.
Schematic view of the structure of complexes 18 (R1, R2 = H or organic group(s)) [65,66,67].

Figure 13.
Example of the favorable effect of the PR3 ligand additive in catalysis: Carbonylation of acetylenes to bifurandiones without [14,16] and with [63,64,65,66,67] additive.
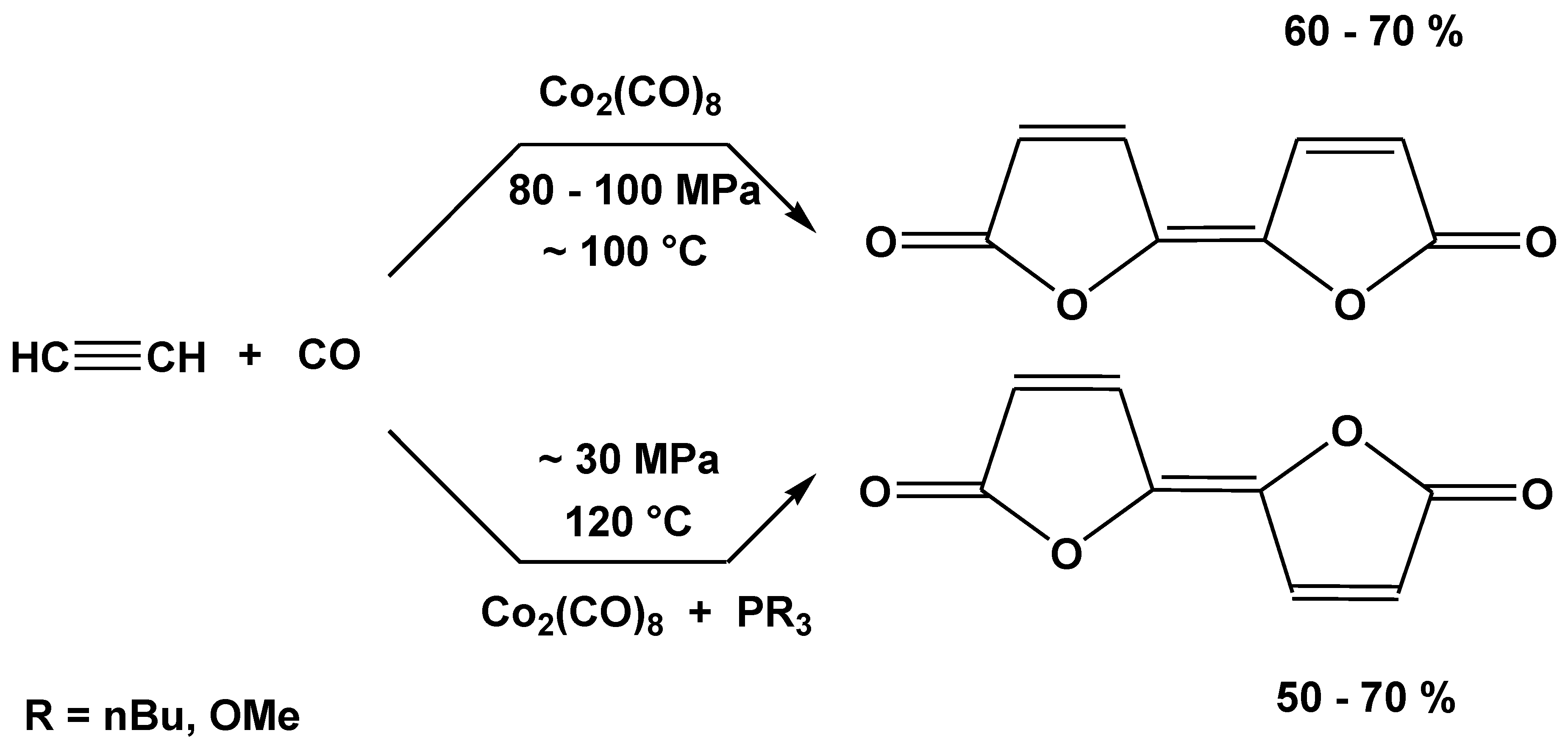
Figure 14.
Schematic view of the structure of complex 21c [65,66]. For 21a and 21b see Figure 1 and Figure 2, respectively.
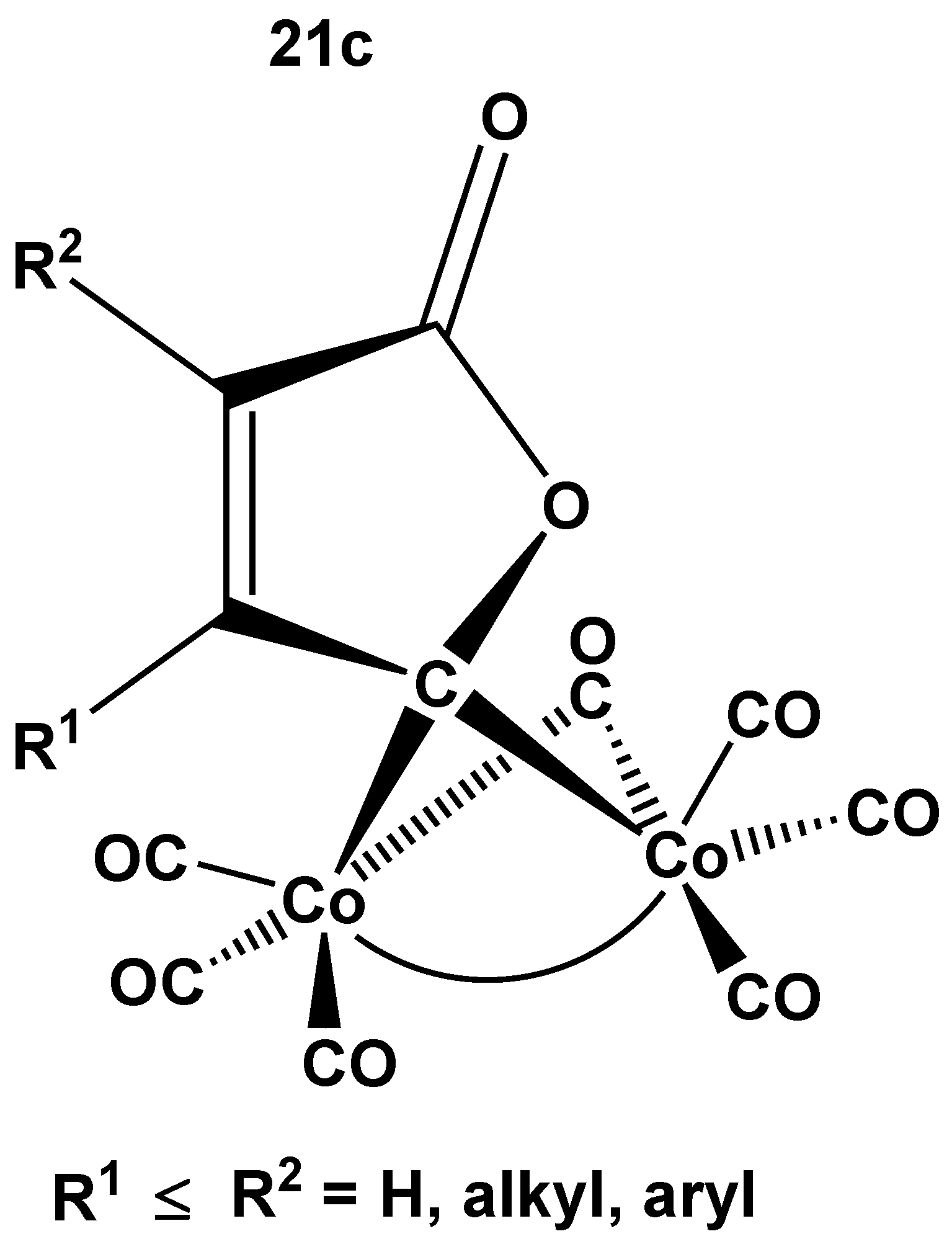
Figure 15.
Schematic view of the structures of complexes 23 (M = Cr, Mo, W) and of complex 24 [71].
Figure 15.
Schematic view of the structures of complexes 23 (M = Cr, Mo, W) and of complex 24 [71].

Figure 16.
Schematic views of the structures (A) of complexes 26 and (B) of complexes 27 [73].
Figure 16.
Schematic views of the structures (A) of complexes 26 and (B) of complexes 27 [73].
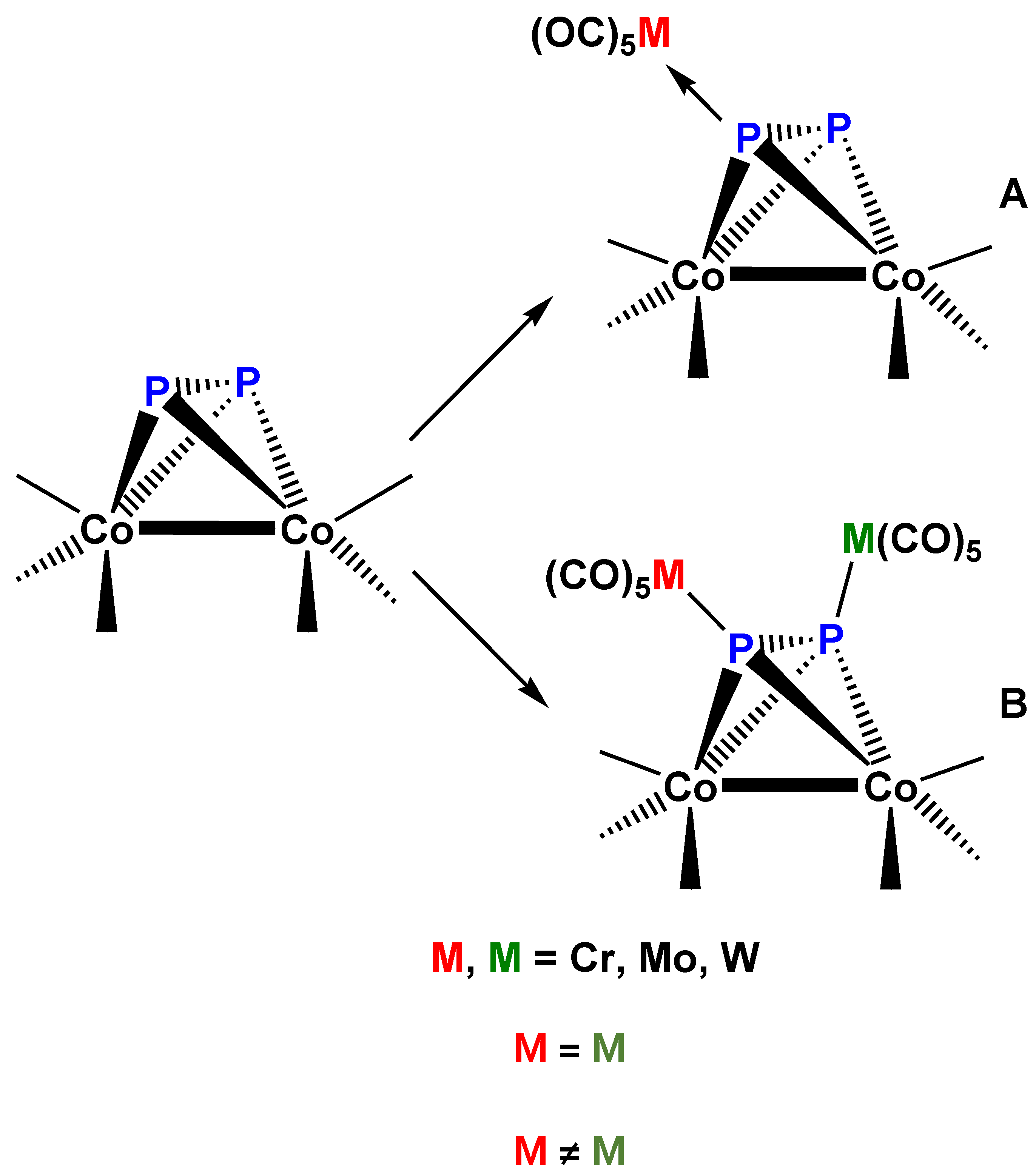
Figure 17.
X-ray structure of complex 28 [73].
Figure 17.
X-ray structure of complex 28 [73].

Figure 18.
Schematic view of the structure of complex 29 [73].
Figure 18.
Schematic view of the structure of complex 29 [73].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vízi-Orosz, A.; Berzeviczy, G.; Pályi, G. Coordinated “Naked” Pnicogenes and Catalysis. Catalysts 2018, 8, 583. https://doi.org/10.3390/catal8120583
AMA Style
Vízi-Orosz A, Berzeviczy G, Pályi G. Coordinated “Naked” Pnicogenes and Catalysis. Catalysts. 2018; 8(12):583. https://doi.org/10.3390/catal8120583
Chicago/Turabian StyleVízi-Orosz, Anna, Gergely Berzeviczy, and Gyula Pályi. 2018. "Coordinated “Naked” Pnicogenes and Catalysis" Catalysts 8, no. 12: 583. https://doi.org/10.3390/catal8120583
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.