Copper-Catalyzed Synthesis, Bio-Evaluation, and in Silico Studies of 2-Aryl-N-alkylbenzimidazoles as Neuroprotective Agents

Abstract
:1. Introduction
2. Results and Discussion
2.1. Copper-Catalyzed Synthesis of 2-Aryl-N-alkylbenzimidazoles
2.2. Neuroprotective Performances of 2-Aryl-N-alkylbenzimidazoles against MPP+
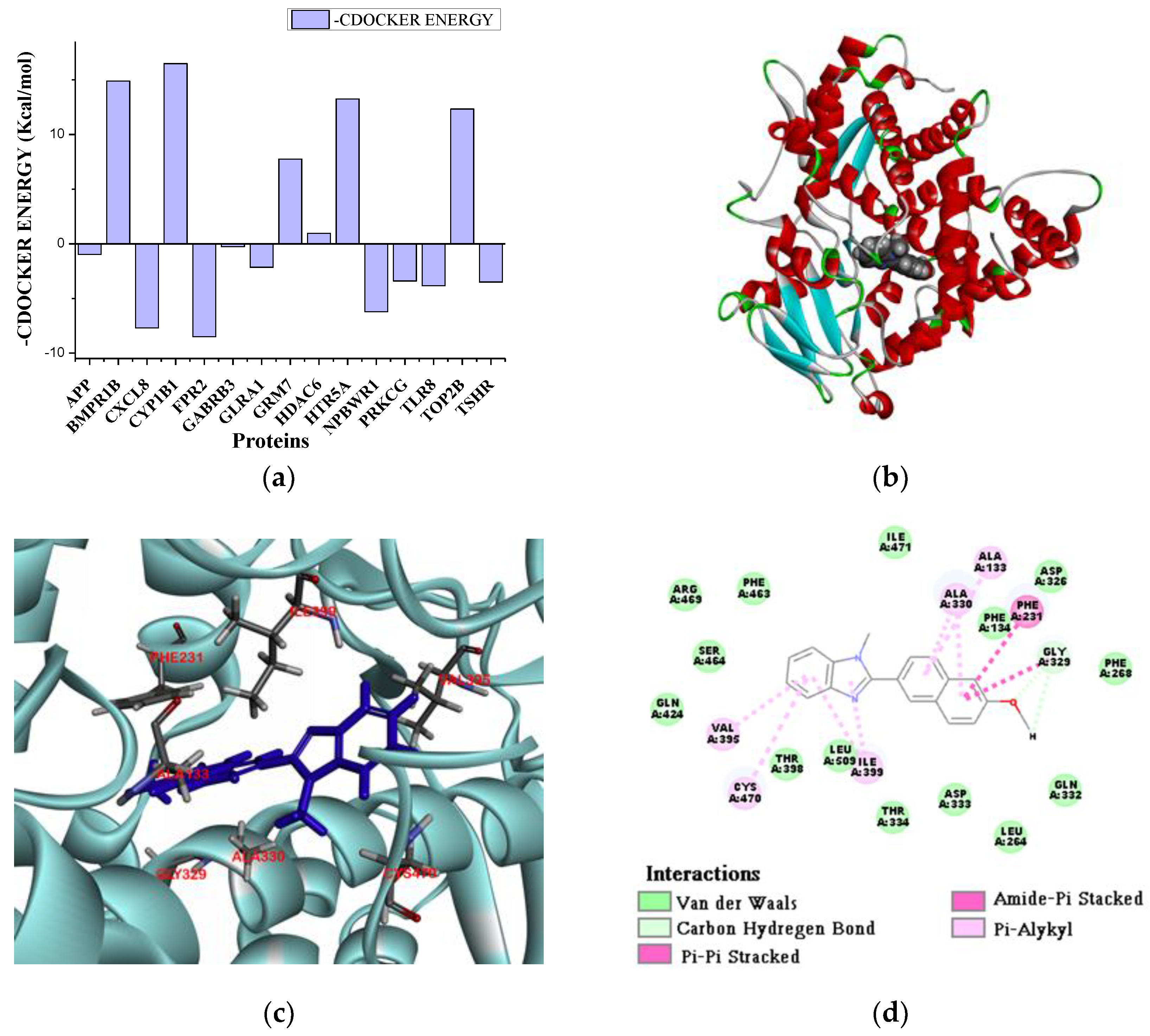
2.3. The Computer Anaysis of Neuroprotective Targets Metablising 3g and Docking Analysis
2.3.1. The Preparation of Neuroprotective Targets
2.3.2. The Molecular Docking Simulation of 3g and Neuroprotective Targets
3. Materials and Methods
3.1. General Methods
3.2. A Typical Procedure for the Cu-Catalyzed Arylation of Benzoimidazoles
3.3. Preparation of Compounds 3a–3t
3.4. Determination of Protective Rate by MTT Assay
3.5. A Procedure for the Preparation of Neuroprotective Targets
3.6. A Procedure for Molecular Docking Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Briguglio, I.; Piras, S.; Corona, P.; Gavini, E.; Nieddu, M.; Boatto, G.; Carta, A. Benzotriazole: An overview on its versatile biological behavior. Eur. J. Med. Chem. 2015, 97, 612–648. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.D.; Palomino, H.L.; Brondstetter, T.I.; Kanelakis, K.C.; Wu, X.; Haug, P.V.; Yan, W.; Young, A.; Hua, H.; Hart, J.C.; et al. Pharmacological characterization of 1-(5-chloro-6-(trifluoromethoxy)-1H-benzoimidazol-2-yl)-1H-pyrazole-4-carboxylic acid (JNJ-42041935), a potent and selective hypoxia-inducible factor prolyl hydroxylase inhibitor. Mol. Pharmacol. 2011, 79, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Conn, J.P.; Lindsley, C.W.; Hopkins, C.R.; Weaver, C.D.; Niswender, C.M.; Cheung, Y. Substituted Benzoimidazolesulfonamides and Substituted Indolesulfonamides as mGluR4 Potentiators. U.S. Patent 13/386,651, 19 July 2012. [Google Scholar]
- Duroux, R.; Renault, N.; Cuelho, J.E.; Agouridas, L.; Blum, D.; Lopes, L.V.; Melnyk, P.; Yous, S. Design, synthesis and evaluation of 2-aryl benzoxazoles as promising hit for the A2A receptor. J. Enzyme Inhib. Med. Chem. 2017, 32, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A. Adenosine A2A Receptor Antagonists in Parkinson’s Disease: Progress in Clinical Trials from the Newly Approved Istradefylline to Drugs in Early Development and Those Already Dscontinued. CNS Drugs 2014, 28, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.; Jung, K.; Kim, M.; Park, Y.; Ahn, H.; Kwon, Y.H.; Hah, J. Syntheses and biological evaluation of 1-heteroaryl-2-aryl-1H-benzimidazole. Bioorg. Med. Chem. 2013, 21, 2271–2285. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Ryu, J.-S.; Hah, J.-M. 3D-QSAR studies of 1, 2-diaryl-1H-benzimidazole derivatives as JNK3 inhibitors with protective effects in neuronal cells. Bioorg. Med. Chem. 2013, 23, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Ozadali-Sari, K.; Küçükkılınç, T.T.; Ayazgok, B.; Balkan, A.; Unsal-Tan, O. Novel multi-targeted agents for alzheimer’s disease: Synthesis, biological evaluation, and molecular modeling of novel 2-[4-(4-substitutedpiperazin-1-yl)phenyl]benzimidazoles. Bioorg. Chem. 2017, 72, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.E.; Bonnefous, C.; Symons, K.T.; Nguyen, P.M.; Sablad, M.; Rozenkrants, N.; Zhang, Y.; Wang, L.; Yazdani, N.; Shiau, A.K.; et al. Discovery of dual inducible/neuronal nitric oxide synthase (iNOS/nNOS) inhibitor development candidate 4-((2-cyclobutyl-1H-imidazo[4,5-b]pyrazin-1-yl)methyl)-7,8-difluoroquinolin-2(1H)-one (KD7332) part 2: Identification of a novel, potent, and selectives. J. Med. Chem. 2010, 53, 7739–7755. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Xiong, S.; Xie, Y.; Dong, X.; Zhou, L. Copper-catalyzed synthesis of benzazoles via aerobic oxidative condensation of o-amino/mercaptan/hydroxyanilines with benzylamines. RSC Adv. 2013, 3, 15592. [Google Scholar] [CrossRef]
- Sharma, H.; Singh, N.; Jang, D.O. A ball-milling strategy for the synthesis of benzothiazole, benzimidazole and benzoxazole derivatives under solvent-free conditions. Green Chem. 2014, 16, 4922–4930. [Google Scholar] [CrossRef]
- Yang, D.; Zhu, X.; Wei, W.; Sun, N.; Yuan, L.; Jiang, M.; You, J.; Wang, H. Magnetically recoverable and reusable CuFe2O4 nanoparticle-catalyzed synthesis of benzoxazoles, benzothiazoles and benzimidazoles using dioxygen as oxidant. RSC Adv. 2014, 45, 17832–17839. [Google Scholar] [CrossRef]
- Peng, J.; Ye, M.; Zong, C.; Hu, F.; Feng, L.; Wang, X.; Wang, Y.; Chen, C. Copper-catalyzed intramolecular C−N bond formation: A straightforward synthesis of benzimidazole derivatives in water. J. Org. Chem. 2011, 76, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zong, C.; Ye, M.; Chen, T.; Gao, D.; Wang, Y.; Chen, C. Direct transition-metal-free intramolecular C–O bond formation: Synthesis of benzoxazole derivatives. Org. Biomol. Chem. 2011, 4, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Khemnar, A.B.; Bhanage, B.M. Iron catalyzed efficient synthesis of 2-arylbenzothiazoles from benzothiazole and olefins using environmentally benign molecular oxygen as oxidant. RSC Adv. 2014, 4, 8939–8942. [Google Scholar] [CrossRef]
- Gao, Y.; Song, Q.; Cheng, G.; Cui, X. KI-catalyzed arylation of benzothiazoles from the boupling of aryl aldehydes with benzothiazoles in neat water. Org. Biomol. Chem. 2014, 7, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Yoshizumi, T.; Hirano, K.; Satoh, T.; Miura, M. Copper-mediated direct arylation of 1, 3, 4-oxadiazoles and 1, 2, 4-triazoles with aryl iodides. Org. Lett. 2009, 11, 3072–3075. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Guo, Q.; Cheng, Y.; Lan, J.; You, J. Palladium-catalyzed desulfitative C-H arylation of heteroarenes with sodium sulfinates. Chem. Eur. J. 2011, 17, 13415–13419. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, S.; Hirano, K.; Satoh, T.; Miura, M. Synthesis of triarylmethanes by palladium-catalyzed C–H/C–O coupling of oxazoles and diarylmethanol derivatives. J. Org. Chem. 2014, 79, 5401–5411. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.-B.; Zhang, Y.; Chen, W.-X.; Xiao, Z.-K.; Hu, T.-T.; Shao, L.-X. Direct C–H bond arylation of (benzo)oxazoles with aryl chlorides catalyzed by N-heterocyclic carbene–palladium(II)–1-methylimidazole complex. Org. Lett. 2014, 16, 1984–1987. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Z.-X. Palladium-catalyzed coupling of azoles or thiazoles with aryl thioethers via C–H/C–S activation. Org. Lett. 2015, 17, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Tao, J.-L.; Wang, Z.-X. Palladium-catalyzed C–H arylation of (benzo)oxazoles or (benzo)thiazoles with aryltrimethylammonium triflates. Org. Lett. 2015, 17, 4926–4929. [Google Scholar] [CrossRef] [PubMed]
- Do, H.Q.; Daugulis, O. Copper-catalyzed arylation of heterocycle C-H bonds. J. Am. Chem. Soc. 2007, 41, 12404–12405. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Tsurugi, H.; Satoh, T.; Miura, M. Copper-mediated direct arylation of benzoazoles with aryl iodides. Tetrahedron Lett. 2008, 49, 1598–1600. [Google Scholar] [CrossRef]
- Phipps, R.J.; Grimster, N.P.; Gaunt, M.J. Cu (II)-catalyzed direct and site-selective arylation of indoles under mild conditions. J. Am. Chem. Soc. 2008, 130, 8172–8174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tian, Y.; Zhao, N.; Wang, Y.; Li, J.; Wang, Z. Nano CuO-catalyzed C–H functionalization of 1,3-azoles with bromoarenes and bromoalken es. Tetrahedron 2014, 70, 6120–6126. [Google Scholar] [CrossRef]
- Kim, D.; Yoo, K.; Kim, S.E.; Cho, H.J.; Lee, J.; Kim, Y.; Kim, M. Copper-catalyzed selective arylations of benzoxazoles with aryl iodides. J. Org. Chem. 2015, 80, 3670–3676. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Koeller, J.; Ackermann, L. Photoinduced copper-catalyzed C−H arylation at room temperature. Angew. Chem. Int. Ed. 2016, 55, 4759–4762. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Tian, X.; Qu, X.; Chen, X.; Cao, Y.; Yao, Y.; Gao, F.; Zhou, X. Copper-catalyzed direct 2-arylation of benzoxazoles and benzoimidazoles with aryl bromides and cytotoxicity of products. Sci. Rep. 2017, 7, 43758. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotech. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modeling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. Swiss-model: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Peitsch, M.C. Protein Modeling by E-mail. Biotechnology 1995, 13, 658–660. [Google Scholar] [CrossRef]
- Wu, G.; Robertson, D.H.; Brooks III, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.; Marchand-Geneste, N.; Giraudel, J.L.; Shimada, T. Docking and QSAR comparative studies of polycyclic aromatic hydrocarbons and other procarcinogen interactions with cytochromes P450 1A1 and 1B1. SAR QSAR Environ. Res. 2012, 23, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.M.H.; Largeron, M. Catalytic oxidative coupling of primary amines under air: A flexible route to benzimidazole derivatives. Eur. J. Org. Chem. 2016, 2016, 1025–1032. [Google Scholar] [CrossRef]
- Liao, L.-Y.; Liu, K.-M.; Duan, X.-F. Unified protocol for cobalt-catalyzed oxidative assembly of two aryl metal reagents using oxygen as an oxidant. J. Org. Chem. 2015, 80, 9856–9867. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.M.; Liao, L.Y.; Duan, X.F. Iron catalyzed oxidative assembly of N-heteroaryl and aryl metal reagents using oxygen as an oxidant. Chem. Commun. 2015, 51, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Haneda, S.; Gan, Z.; Eda, K.; Hayashi, M. Ligand effects of 2-(2-pyridyl)benzazole−pd complexes on the X-ray crystallographic structures, 1 h nmr spectra, and catalytic activities in mizoroki-heck reactions. Organometallics 2007, 26, 6551–6555. [Google Scholar] [CrossRef]
- Guru, M.M.; Ali, M.A.; Punniyamurthy, T. Copper-mediated synthesis of substituted 2-aryl-N-benzylbenzimidazoles and 2-arylbenzoxazoles via C–H functionalization/C–N/C–O bond formation. J. Org. Chem. 2011, 76, 5295–5308. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.W.; Steffens, L.D.; Sigman, M.S. On the mechanism of the palladium-catalyzed tert-butylhydroperoxide-mediated wacker-type oxidation of alkenes using quinoline-2-oxazoline ligands. J. Am. Chem. Soc. 2011, 133, 8317–8325. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Joo, J.C.; Song, B.K.; Yoo, Y.J.; Kim, Y.H. Engineering a horseradish peroxidase C stable to radical attacks by mutating multiple radical coupling sites. Biotechnol. Bioeng. 2015, 112, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}

| Entry | Base | Solvent | Tem. (°C) | Time (h) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | KOH | DMF | 140 | 24 | Trace |
| 2 | NaOH | DMF | 140 | 24 | Trace |
| 3 | t-BuOK | DMF | 140 | 24 | Trace |
| 4 | K2CO3 | DMF | 140 | 24 | 60 |
| 5 | Na2CO3 | DMF | 140 | 24 | 38 |
| 6 | KH2PO4 | DMF | 140 | 24 | 30 |
| 7 | K2CO3 | DMF | 120 | 24 | 45 |
| 8 | K2CO3 | DMF | 150 | 24 | 64 |
| 9 | K2CO3 | Xylene | 130 | 24 | 40 |
| 10 | K2CO3 | Xylene | 140 | 24 | 75 |
| 11 | K2CO3 | DMF | 150 | 40 | 69 |
| 12 c | K2CO3 | Xylene | 150 | 40 | 78 |
 |
 |
 |
 |
| Conc. (μM) | 3b | 3g | 3h | 3i | 3j | 3k | 3o | 3q | 3s | 3t | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protective rate (%) | 50 | −42.99 | −10.2 | 0.79 | 2.23 | −2.23 | 1.97 | −17.82 | −2.72 | 2.04 | 0.95 |
| 10 | −4.72 | 16.19 | 0.13 | 0.92 | −4.19 | 5.64 | 0.41 | 0.44 | 2.99 | 3.13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Y.-X.; Jia, N.-N.; Cao, Y.-N.; Chen, X.-X.; Gao, F.; Liang, X.-X. Copper-Catalyzed Synthesis, Bio-Evaluation, and in Silico Studies of 2-Aryl-N-alkylbenzimidazoles as Neuroprotective Agents. Catalysts 2018, 8, 433. https://doi.org/10.3390/catal8100433
Yao Y-X, Jia N-N, Cao Y-N, Chen X-X, Gao F, Liang X-X. Copper-Catalyzed Synthesis, Bio-Evaluation, and in Silico Studies of 2-Aryl-N-alkylbenzimidazoles as Neuroprotective Agents. Catalysts. 2018; 8(10):433. https://doi.org/10.3390/catal8100433
Chicago/Turabian StyleYao, Yun-Xin, Nan-Nan Jia, Ya-Nan Cao, Xing-Xiu Chen, Feng Gao, and Xiao-Xia Liang. 2018. "Copper-Catalyzed Synthesis, Bio-Evaluation, and in Silico Studies of 2-Aryl-N-alkylbenzimidazoles as Neuroprotective Agents" Catalysts 8, no. 10: 433. https://doi.org/10.3390/catal8100433