Determination of Uric Acid in Co-Presence of Dopamine and Ascorbic Acid Using Cuprous Oxide Nanoparticle-Functionalized Graphene Decorated Glassy Carbon Electrode

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
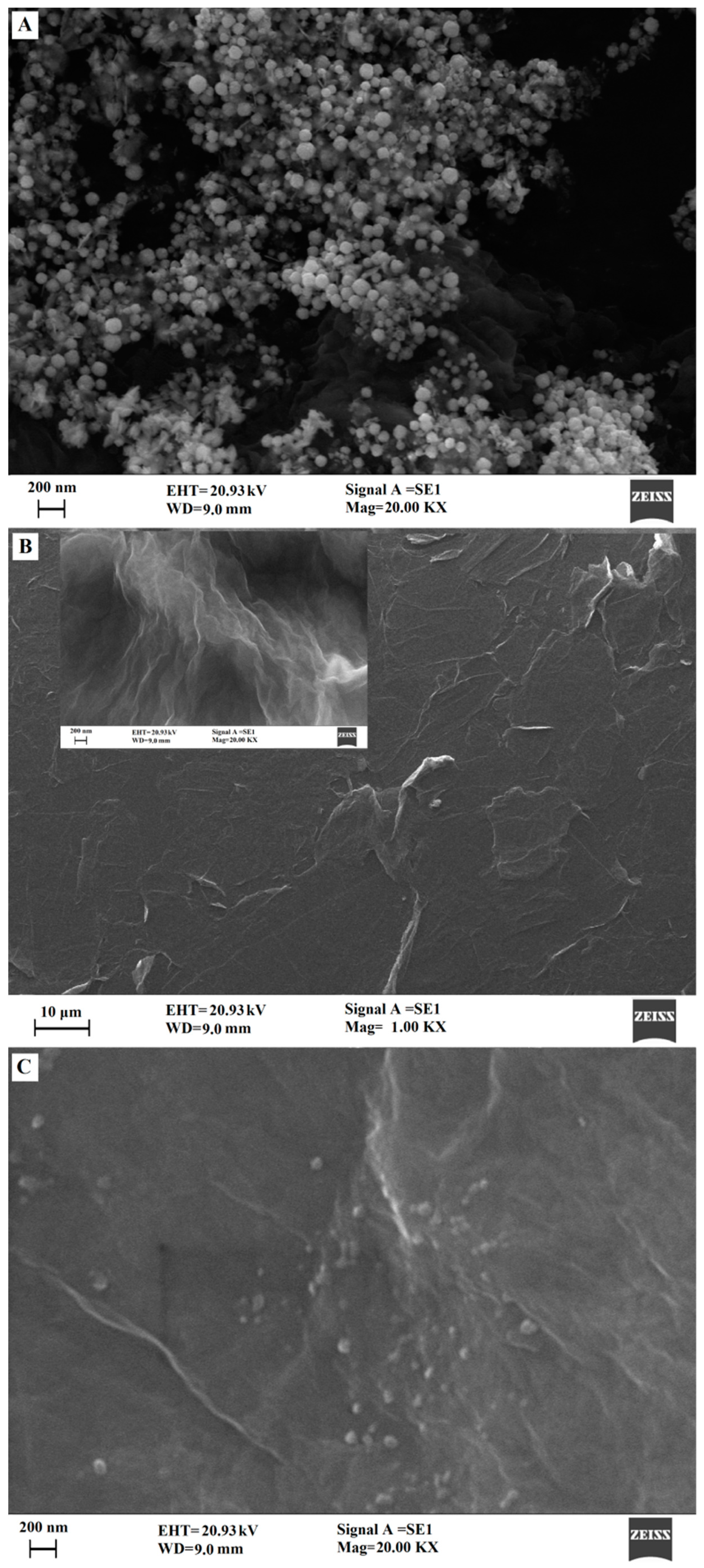
2.1. Characterization of Surface Morphologies
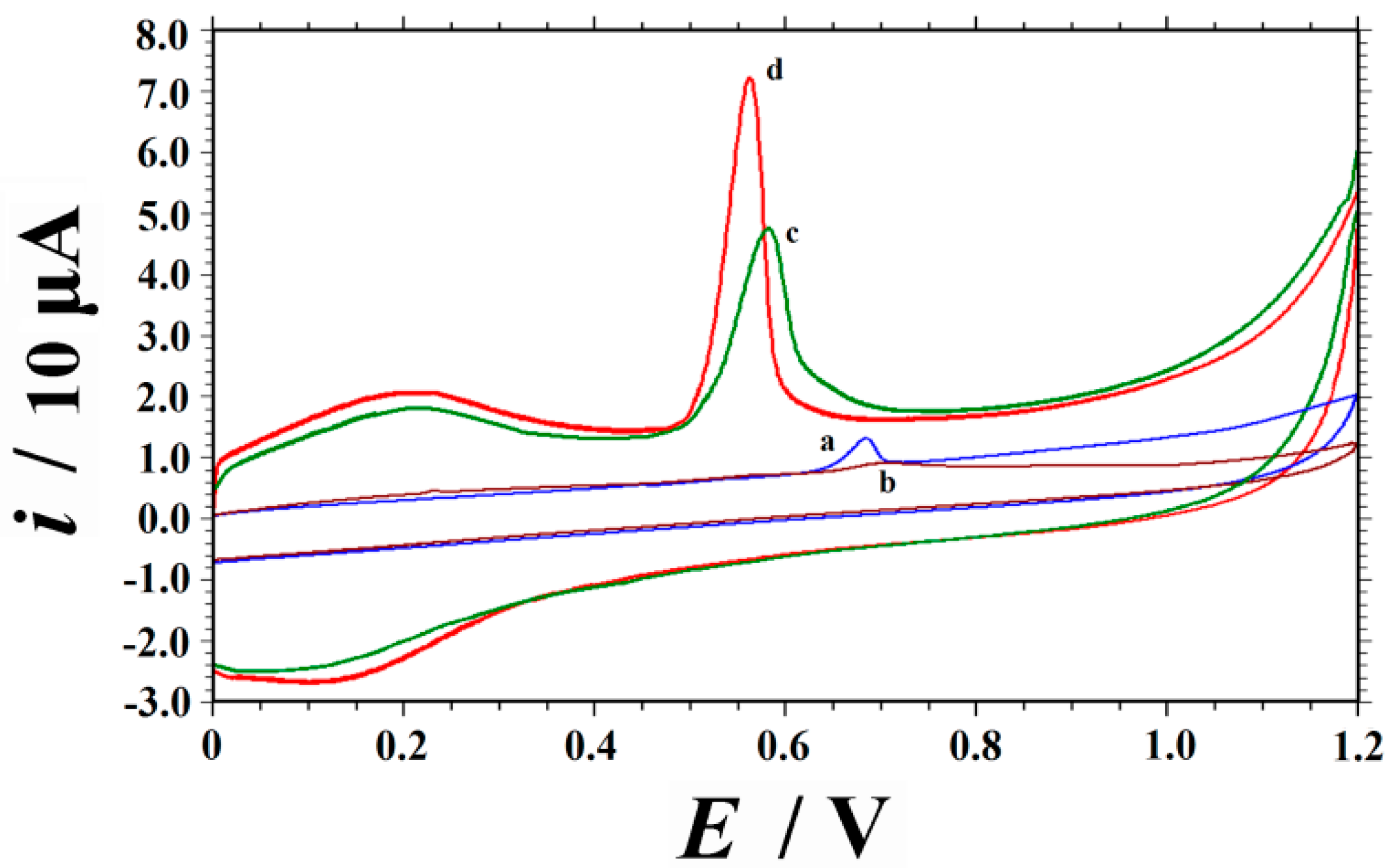
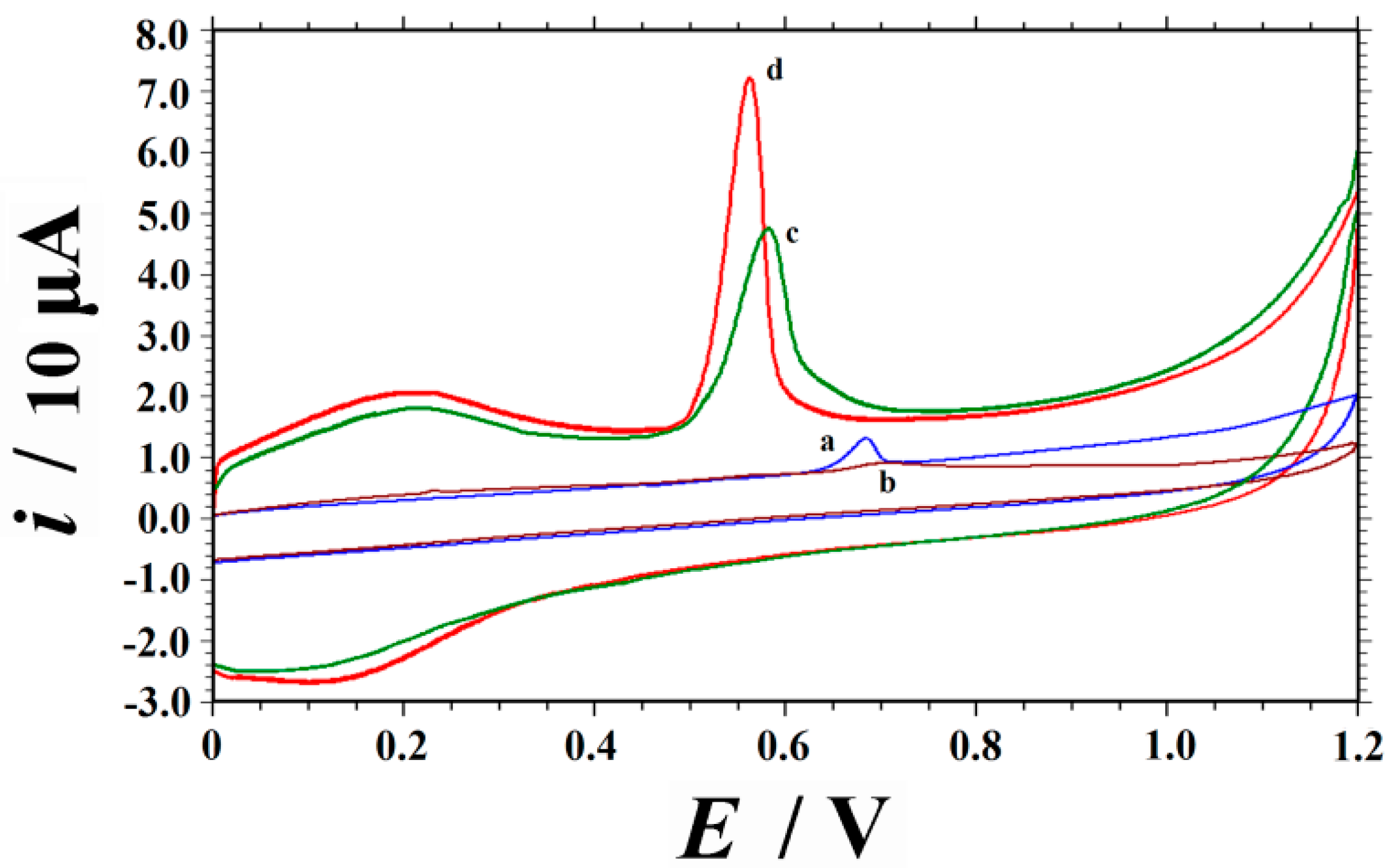
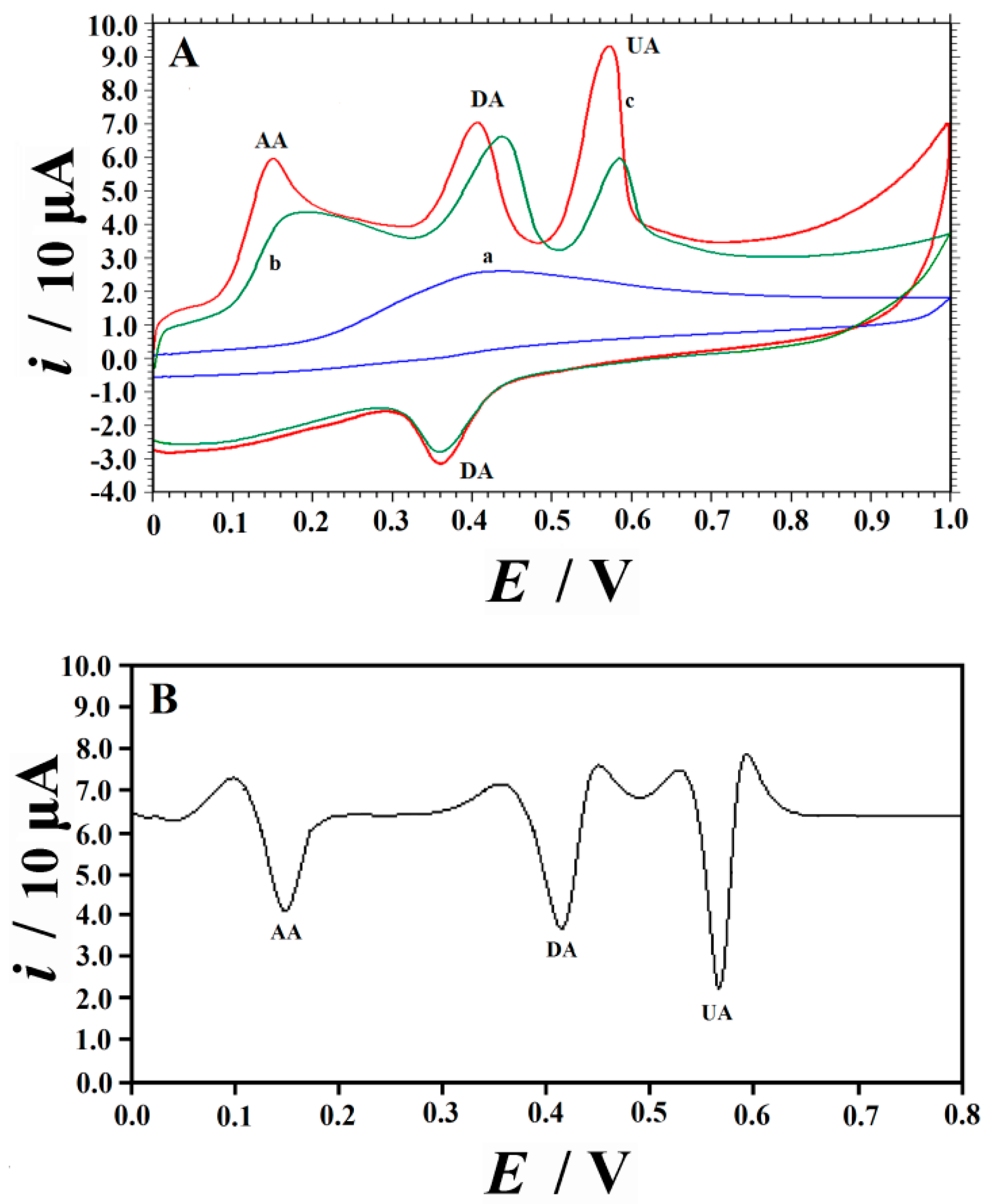
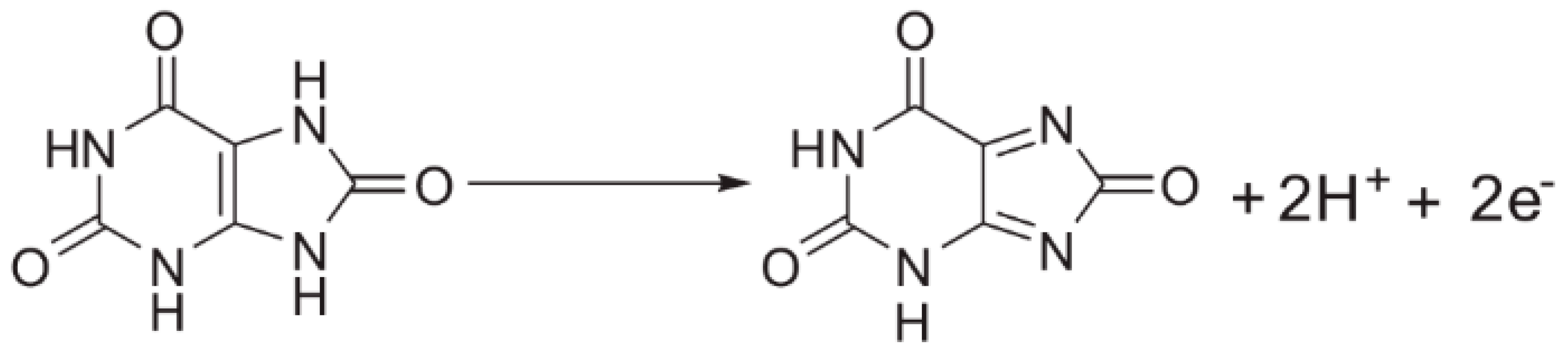
2.2. Voltammetric Responses of UA on Different Electrodes
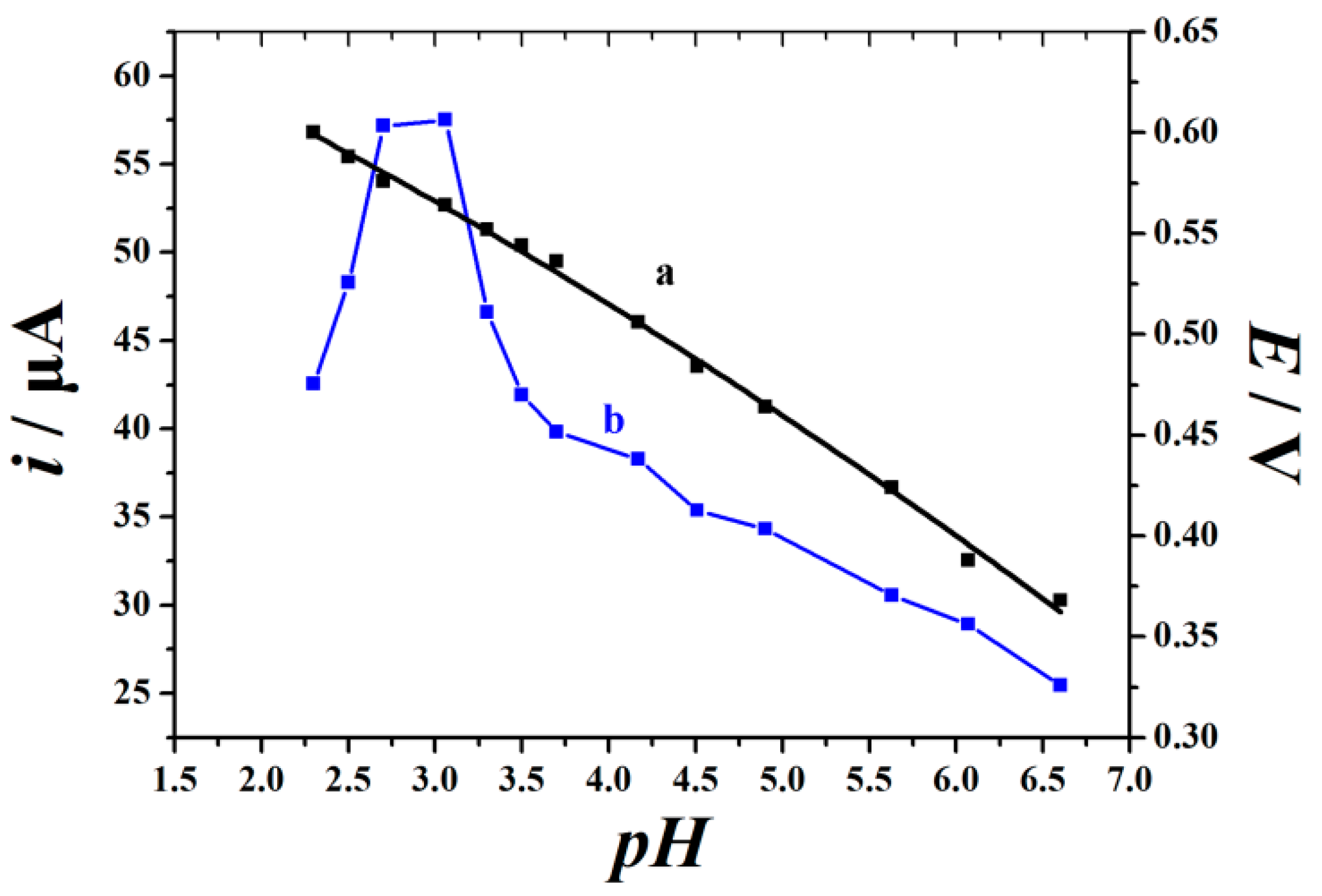
2.3. pH-Dependent Electrochemical Response
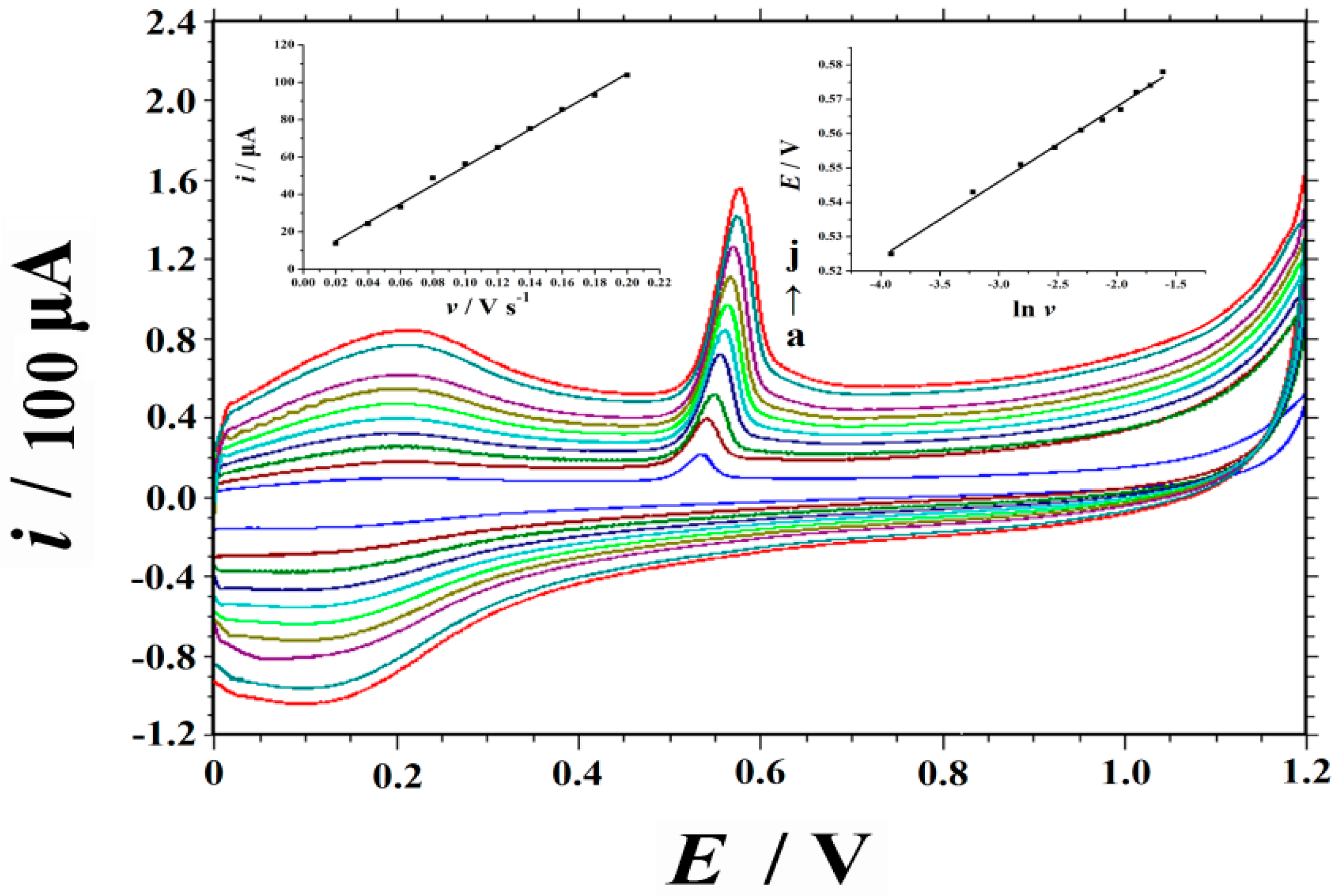
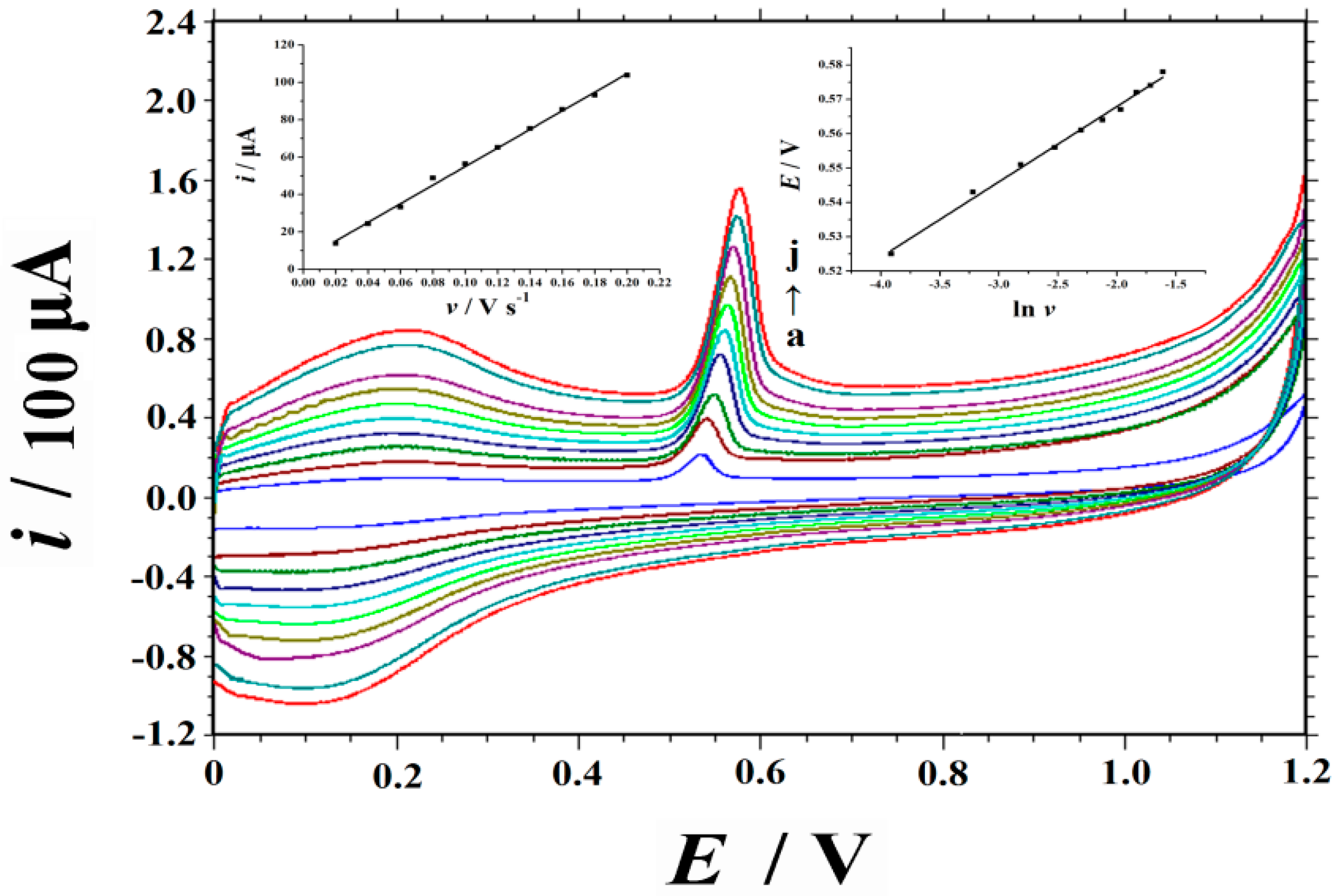
2.4. Sweep Rate and Electrochemical Kinetics
2.5. Optimization of the Analytical Conditions
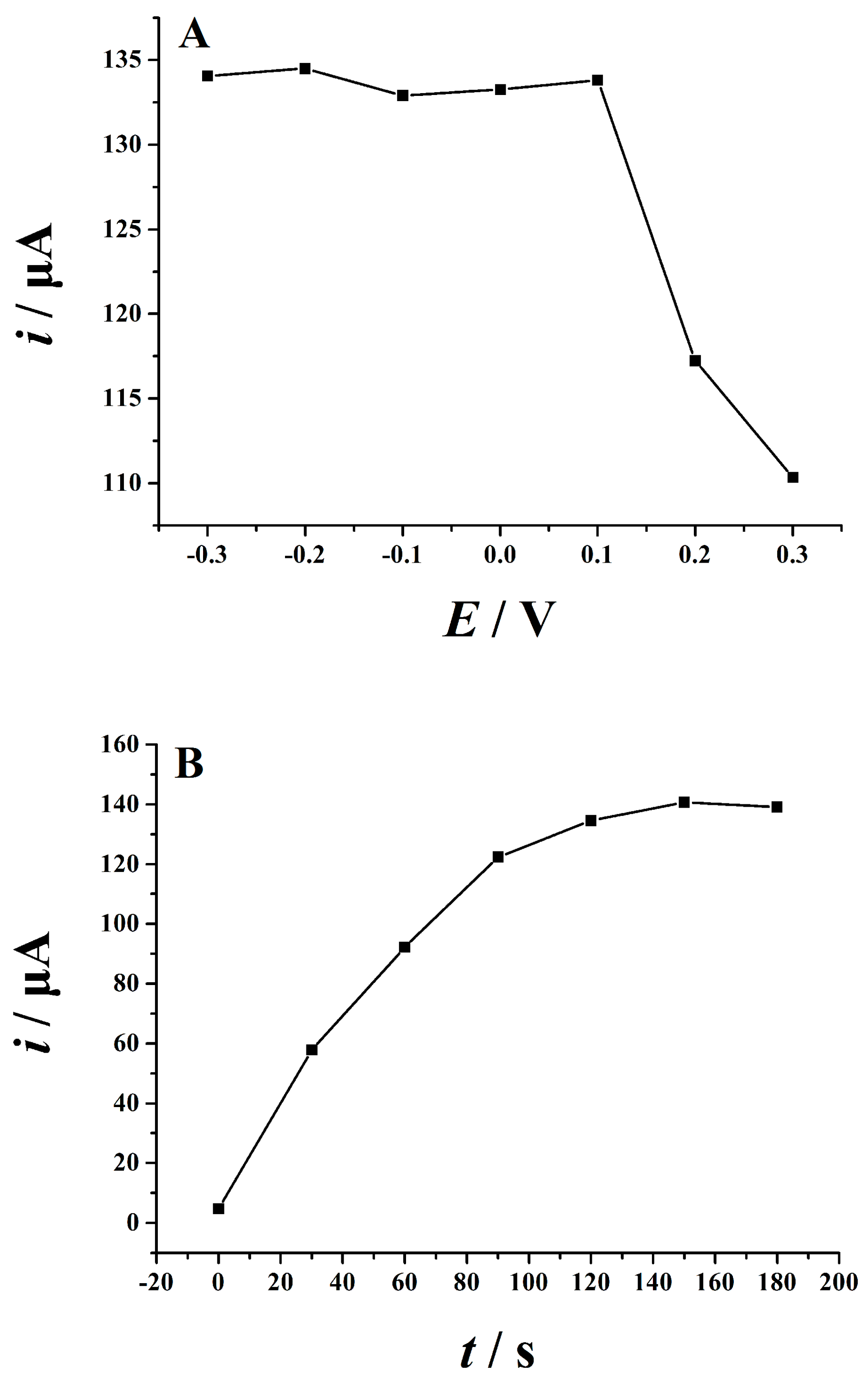
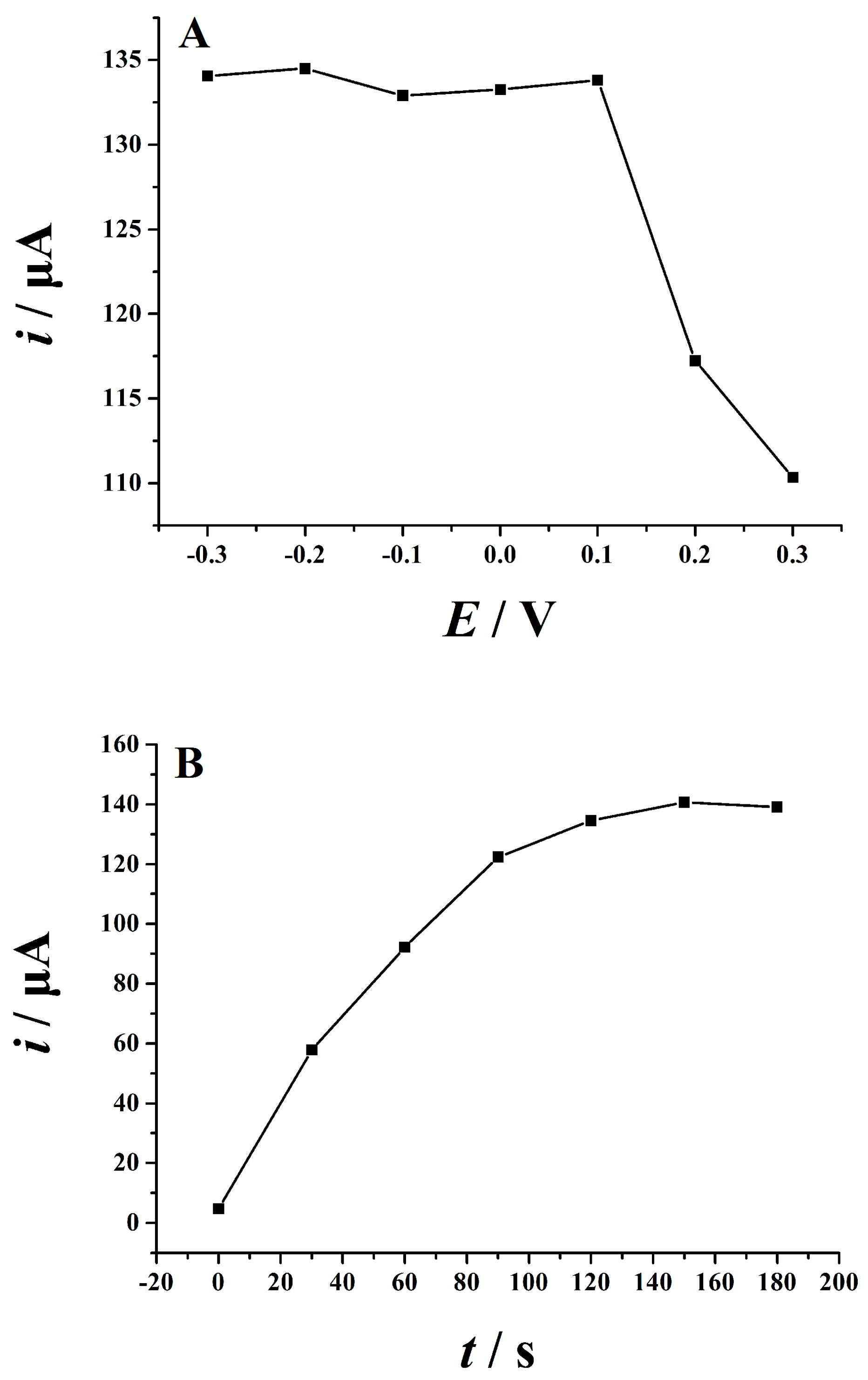
2.5.1. Optimization of Supporting Electrolytes
2.5.2. Optimization of Accumulation Conditions
2.6. Analytic Properties
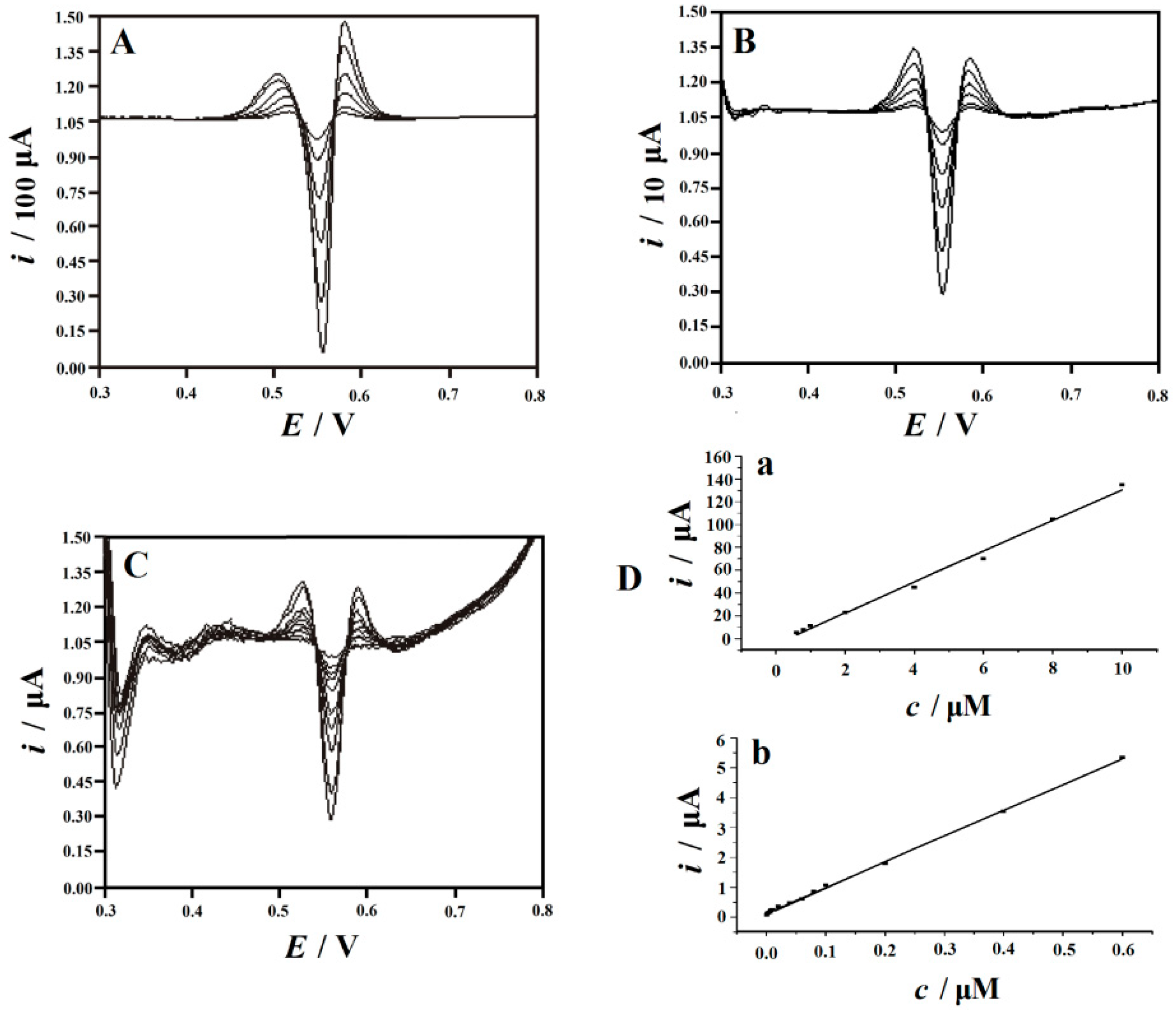
2.6.1. Calibration Curves, Dynamical Response Regions and Detection Limit
2.6.2. Interference
2.6.3. Repeatability, Reproducibility and Stability
2.6.4. Real Sample Analysis
3. Experimental
3.1. Reagents and Solutions
3.2. Synthesis of Cu2O Nanoparticles
3.3. Preparation of GO and Cu2O–GO Composite
3.4. Fabrication of Cu2O-ErGO/GCE
3.5. Characterization and Measurements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alderman, M.; Aiyer, K.J.V. Uric acid: Role in cardiovascular disease and effects of losartan. Curr. Med. Res. Opin. 2004, 20, 369–379. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Li, W.; Liu, Z.; Ding, Z.; Tuo, D. Towards Improvements for Penetrating the Blood–Brain Barrier—Recent Progress from a Material and Pharmaceutical Perspective. Cell 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, R.R. A simple spectrophotometric method for the direct determination of uric acid in avian excreta. Poult. Sci. 1983, 62, 2106–2108. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Fang, X.; Zhang, C.; Xu, R.; Xu, B. Determination of serum uric acid using high-performance liquid chromatography (hplc)/isotope dilution mass spectrometry (id-ms) as a candidate reference method. J. Chromatogr. B 2007, 857, 287–295. [Google Scholar] [CrossRef] [PubMed]
- George, S.K.; Dipu, M.T.; Mehra, U.R.; Singh, P.; Verma, A.K.; Ramgaokar, J.S. Improved hplc method for the simultaneous determination of allantoin, uric acid and creatinine in cattle urine. J. Chromatogr. B 2006, 832, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Gjorup, S.; Poulsen, H.; Prætorius, E. The uric acid concentration in serum determined by enzymatic spectrophotometry. Scin. J. Clin. Lab. Investig. 1955, 7, 201–203. [Google Scholar] [CrossRef]
- Prætorius, E.; Poulsen, H. Enzymatic Determination of Uric Acid with Detailed Directions. Scand. J. Clin. Lab. Investig. 1953, 5, 273–280. [Google Scholar] [CrossRef]
- Yu, J.; Wang, S.; Ge, L.; Ge, S. A novel chemiluminescence paper microfluidic biosensor based on enzymatic reaction for uric acid determination. Biosens. Bioelectron. 2011, 26, 3284–3289. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiao, S.; Li, M. Determination of uric acid in human urine by eliminating ascorbic acid interference on copper (ii)-polydopamine immobilized electrode surface. Electrochim. Acta 2014, 121, 233–239. [Google Scholar] [CrossRef]
- He, Q.; Li, G.; Liu, X.; Liu, J.; Deng, P.; Chen, D. Morphologically tunable MnO2 nanoparticles fabrication, modelling and their influences on electrochemical sensing performance toward dopamine. Catalysts 2018, 8, 323. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liang, J.; Liu, X.; Tuo, D.; Li, W. Chemically Surface Tunable Solubility Parameter for Controllable Drug Delivery—An Example and Perspective from Hollow PAA-Coated Magnetite Nanoparticles with R6G Model Drug. Materials 2018, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Deng, P.; Liang, J.; Chen, D. Sensitive and selective detection of tartrazine based on TiO2-electrochemically reduced graphene oxide composite-modified electrodes. Sensors 2018, 18, 1911. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Xiao, H.; Xie, Q.; Liu, J.; Sun, L.; Zhou, Y.; Zhang, Y.; Chao, L.; Chen, C.; Yao, S. Simultaneous electroanalysis of isoniazid and uric acid at poly(sulfosalicylic acid)/electroreduced carboxylated graphene modified glassy carbon electrode. Sens. Actuators B Chem. 2015, 207, 167–176. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, X.; Ji, X.; Lin, R.; Lin, W. Simultaneous determination of ascorbic acid, dopamine and uric acid using poly(4-aminobutyric acid) modified glassy carbon electrode. Sens. Actuators B Chem. 2013, 178, 359–365. [Google Scholar] [CrossRef]
- Lian, Q.; He, Z.; He, Q.; Luo, A.; Yan, K.; Zhang, D.; Lu, X.; Zhou, X. Simultaneous determination of ascorbic acid, dopamine and uric acid based on tryptophan functionalized graphene. Anal. Chim. Acta 2014, 823, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Du, J.; Wang, H.; Zou, C.; Jiang, F.; Yang, P.; Du, Y. A facile electrochemical sensor based on reduced graphene oxide and au nanoplates modified glassy carbon electrode for simultaneous detection of ascorbic acid, dopamine and uric acid. Sens. Actuators B Chem. 2014, 204, 302–309. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, Z.; Zhang, B.; Li, X.; Pan, J. Selective and sensitive determination of uric acid in the presence of ascorbic acid and dopamine by pdda functionalized graphene/graphite composite electrode. Talanta 2013, 112, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bi, C. Simultaneous electrochemical determination of ascorbic acid, dopamine and uric acid using poly (tyrosine)/functionalized multi-walled carbon nanotubes composite film modified electrode. J. Mol. Liq. 2013, 177, 26–31. [Google Scholar] [CrossRef]
- Noroozifar, M.; Khorasani-Motlagh, M.; Jahromi, F.Z.; Rostami, S. Sensitive and selective determination of uric acid in real samples by modified glassy carbon electrode with holmium fluoride nanoparticles/multi-walled carbon nanotube as a new biosensor. Sens. Actuators B Chem. 2013, 188, 65–72. [Google Scholar] [CrossRef]
- Wang, Z.; Xia, J.; Zhu, L.; Zhang, F.; Guo, X.; Li, Y.; Xia, Y. The fabrication of poly (acridine orange)/graphene modified electrode with electrolysis micelle disruption method for selective determination of uric acid. Sens. Actuators B Chem. 2012, 161, 131–136. [Google Scholar] [CrossRef]
- Wang, G.; Meng, J.; Liu, H.; Jiao, S.; Zhang, W.; Chen, D.; Fang, B. Determination of uric acid in the presence of ascorbic acid with hexacyanoferrate lanthanum film modified electrode. Electrochim. Acta 2008, 53, 2837–2843. [Google Scholar] [CrossRef]
- Ojani, R.; Raoof, J.-B.; Maleki, A.A.; Safshekan, S. Simultaneous and sensitive detection of dopamine and uric acid using a poly(l-methionine)/gold nanoparticle-modified glassy carbon electrode. Chin. J. Catal. 2014, 35, 423–429. [Google Scholar] [CrossRef]
- Chen, K.; Wang, M.; Li, G.; He, Q.; Liu, J.; Li, F. Spherical α-MnO2 Supported on N-KB as Efficient Electrocatalyst for Oxygen Reduction in Al–Air Battery. Materials 2018, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, K.; Liu, J.; He, Q.; Li, G.; Li, F. Efficiently Enhancing Electrocatalytic Activity of α-MnO2 Nanorods/N-Doped Ketjenblack Carbon for Oxygen Reduction Reaction and Oxygen Evolution Reaction Using Facile Regulated Hydrothermal Treatment. Catalysts 2018, 8, 138. [Google Scholar] [CrossRef]
- Akimoto, K.; Ishizuka, S.; Yanagita, M.; Nawa, Y.; Paul, G.K.; Sakurai, T. Thin film deposition of Cu2O and application for solar cells. Sol. Energy 2006, 80, 715–722. [Google Scholar] [CrossRef]
- Poizot, P.; Laruelle, S.; Grugeon, S.; Dupont, L.; Tarascon, J.-M. Cheminform abstract: Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. ChemInform 2001, 32, 496–499. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Q.; Zhang, Y.; Wang, Y.; Zhao, L.; Yu, B. Inside front cover: One-pot synthesis and hierarchical assembly of hollow Cu2O microspheres with nanocrystals-composed porous multishell and their gas-sensing properties. Adv. Funct. Mater. 2007, 17, 2766–2771. [Google Scholar] [CrossRef]
- Hua, Q.; Shang, D.; Zhang, W.; Chen, K.; Chang, S.; Ma, Y.; Jiang, Z.; Yang, J.; Huang, W. Morphological evolution of Cu2O nanocrystals in an acid solution: Stability of different crystal planes. Langmuir 2011, 27, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Deng, M.; Li, G.; Chen, S.; Wang, L. Electrochemical behavior of cuprous oxide–reduced graphene oxide nanocomposites and their application in nonenzymatic hydrogen peroxide sensing. Electrochim. Acta 2013, 88, 59–65. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, Y.; Zhang, Y.; Weng, W.; Li, S. Carbon quantum dots/octahedral Cu2O nanocomposites for non-enzymatic glucose and hydrogen peroxide amperometric sensor. Sens. Actuators B Chem. 2015, 206, 735–743. [Google Scholar] [CrossRef]
- Cao, H.; Yang, A.; Li, H.; Wang, L.; Li, S.; Kong, J.; Bao, X.; Yang, R. A non-enzymatic glucose sensing based on hollow cuprous oxide nanospheres in a nafion matrix. Sens. Actuators B Chem. 2015, 214, 169–173. [Google Scholar] [CrossRef]
- Yuan, B.; Xu, C.; Liu, L.; Zhang, Q.; Ji, S.; Pi, L.; Zhang, D.; Huo, Q. Cu2O/NiOx/graphene oxide modified glassy carbon electrode for the enhanced electrochemical oxidation of reduced glutathione and nonenzyme glucose sensor. Electrochim. Acta 2013, 104, 78–83. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Y.; Gu, Y.; Wang, Z.; Wang, C. One-pot solvothermal synthesis of a Cu2O/graphene nanocomposite and its application in an electrochemical sensor for dopamine. Microchim. Acta 2011, 173, 103–109. [Google Scholar] [CrossRef]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Deng, P.; Liang, J. Preparation of Cu2O-Reduced Graphene Nanocomposite Modified Electrodes towards Ultrasensitive Dopamine Detection. Sensors 2018, 18, 199. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Li, G.; Chen, D.; Deng, P.; Liang, J. Fabrication of Amine-Modified Magnetite-Electrochemically Reduced Graphene Oxide Nanocomposite Modified Glassy Carbon Electrode for Sensitive Dopamine Determination. Nanomaterials 2018, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Gu, Y.; Wang, C. Fabrication of the Cu2O/polyvinyl pyrrolidone-graphene modified glassy carbon-rotating disk electrode and its application for sensitive detection of herbicide paraquat. Sens. Actuators B Chem. 2012, 173, 530–539. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Gu, A.; Wu, H.; Fang, B. Preparation of porous Cu2O octahedron and its application as l-tyrosine sensors. Solid State Commun. 2008, 148, 525–528. [Google Scholar] [CrossRef]
- Ning, J.; He, Q.; Luo, X.; Wang, M.; Liu, D.; Wang, J. Rapid and Sensitive Determination of Vanillin Based on a Glassy Carbon Electrode Modified with Cu2O-Electrochemically Reduced Graphene Oxide Nanocomposite Film. Sensors 2018, 18, 2762. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, J.; Liu, X.; Xia, Y.; Li, G.; Deng, P.; Chen, D. Novel Electrochemical Sensors Based on Cuprous Oxide-Electrochemically Reduced Graphene Oxide Nanocomposites Modified Electrode toward Sensitive Detection of Sunset Yellow. Molecules 2018, 23, 2130. [Google Scholar] [CrossRef] [PubMed]
- Shishiyanu, S.T.; Shishiyanu, T.S.; Lupan, O.I. Novel NO2 gas sensor based on cuprous oxide thin films. Sens. Actuators B Chem. 2006, 113, 468–476. [Google Scholar] [CrossRef]
- Geim, A.K. Graphene: Status and prospects. Science 2009, 324, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Dong, J.; Wang, X.; Ding, Z.; Zhang, Z.; Wu, L.; Li, Z.; Fu, X. Photochemical synthesis of submicron- and nano-scale Cu2O particles. J. Colloid Interface Sci. 2009, 333, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Laviron, E. Adsorption, autoinhibition and autocatalysis in polarography and in linear potential sweep voltammetry. J. Electroanal. Chem. Interfacial Electrochem. 1974, 52, 355–393. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrodes | Techniques | Supporting Electrolyte | Working Potential | Linear Range/μM | Detection Limit/μM | Repeatability (RSD%) | Stability | Ref. |
|---|---|---|---|---|---|---|---|---|
| a PD-Cu(II)/GCE | l DPV | 0.1 M o PBS (pH 7.5) | +0.4 V vs. SCE | 60–1680 | 24.6 | 1.6 | 5.8% loss after 28 days | [12] |
| b PSA-ERCG/GCE | DPV | 0.1 M NH3-NH4Cl (pH 9.0) | +0.016 V vs. SCE | 0.02–15 | 0.012 | 2.8 | 10% loss after 2 weeks | [13] |
| c P-4-ABA/GCE | DPV | 0.2 M PBS (pH 4.5) | +0.576 V vs. Ag/AgCl | 1.0–80.0 | 0.5 | 3.1 | 2.3% loss after 1 week | [14] |
| d Trp-GR/GCE | DPV | 0.1 M PBS (pH 7.0) | +0.355 V vs. Ag/AgCl | 10–1000 | 1.24 | 5.92 | 2.77% loss after 1 week and 6.91% in 2 weeks | [15] |
| e Au-RGO/GCE | DPV | 0.1 M PBS (pH 7.0) | +0.26 V vs. SCE | 8.8–53 | 1.8 | 2.0 | 5% loss after 1 week | [16] |
| f PDDA-G/CPE | DPV | 0.1 M PBS (pH 7.0) | +0.18 V vs. Ag/AgCl | 0.5–20 | 0.08 | 3.6 | 10.1% loss after 20 days | [17] |
| g poly (Tyr)/MWCNTs-COOH/GCE | DPV | 0.1 M PBS (pH 7.4) | +0.30 V vs. SCE | 1.0–350 | 0.30 | 3.0 | Not mentioned | [18] |
| h MWCNT–HoFNPs–CH/GCE | m CV | 0.1 M p DCAABS (pH 1.0) | +0.605 V vs. SCE | 0.2–500.0 | 0.16 | 1.19 | No significant change after 6 months | [19] |
| i PAO/GH/GCE | DPV | 0.1 M PBS (pH 6.5) | +0.260 V vs. SCE | 0.1–1000 | 0.02 | 2.2 | 7.0% loss after 1 month | [20] |
| j LaHCF/GCE | DPV | 0.1 M PBS (pH 7.0) | +0.432 V vs. SCE | 0.2–100 | 0.1 | Not mentioned | Not mentioned | [21] |
| k Nano-Au-PMT/GCE | DPV | 0.1 M PBS (pH 7.0) | +0.37 V vs. SCE | 0.07–1.0 | 0.045 | Not mentioned | 12.0% loss after 2 weeks | [22] |
| Cu2O-ErGO/GCE | n LSV | 0.1 M PBS (pH 3.0) | 0.561 V vs. SCE | 0.002–0.6, 0.6–10 | 0.001 | 2.2 | 7.6% loss after 2 weeks | This work |
| Samples | a Detected/μM | Added/μM | Total Found/μM | Recovery/% | b Content Detected/mM |
|---|---|---|---|---|---|
| urine 1 | 3.488 (±0.084) | 3.0 | 6.527 (±0.144) | 101.3 | 1.744 (±0.042) |
| urine 2 | 3.637 (±0.087) | 4.0 | 7.535 (±0.165) | 97.5 | 1.819 (±0.044) |
| urine 3 | 5.142 (±0.118) | 5.0 | 10.046 (±0.210) | 98.1 | 2.571 (±0.059) |
| serum 1 | 2.483 (±0.065) | 2.0 | 4.522 (±0.108) | 102.0 | 0.248 (±0.006) |
| serum 2 | 4.336 (±0.095) | 4.0 | 8.446 (±0.186) | 102.8 | 0.434 (±0.010) |
| serum 3 | 3.210 (±0.067) | 3.0 | 6.162 (±0.142) | 98.4 | 0.321 (±0.007) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, J.; He, Q.; Luo, X.; Wang, M.; Liu, D.; Wang, J.; Li, G.; Liu, J. Determination of Uric Acid in Co-Presence of Dopamine and Ascorbic Acid Using Cuprous Oxide Nanoparticle-Functionalized Graphene Decorated Glassy Carbon Electrode. Catalysts 2018, 8, 407. https://doi.org/10.3390/catal8100407
Ning J, He Q, Luo X, Wang M, Liu D, Wang J, Li G, Liu J. Determination of Uric Acid in Co-Presence of Dopamine and Ascorbic Acid Using Cuprous Oxide Nanoparticle-Functionalized Graphene Decorated Glassy Carbon Electrode. Catalysts. 2018; 8(10):407. https://doi.org/10.3390/catal8100407
Chicago/Turabian StyleNing, Jingheng, Quanguo He, Xin Luo, Min Wang, Donglin Liu, Jianhui Wang, Guangli Li, and Jun Liu. 2018. "Determination of Uric Acid in Co-Presence of Dopamine and Ascorbic Acid Using Cuprous Oxide Nanoparticle-Functionalized Graphene Decorated Glassy Carbon Electrode" Catalysts 8, no. 10: 407. https://doi.org/10.3390/catal8100407