Electrocarboxylation of Dichlorobenzenes on a Silver Electrode in DMF

 ,
,
Abstract
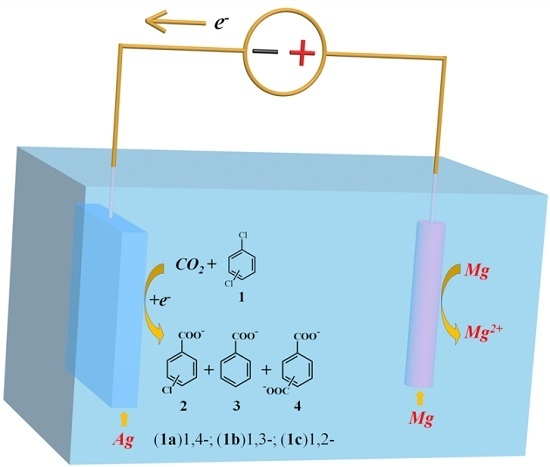
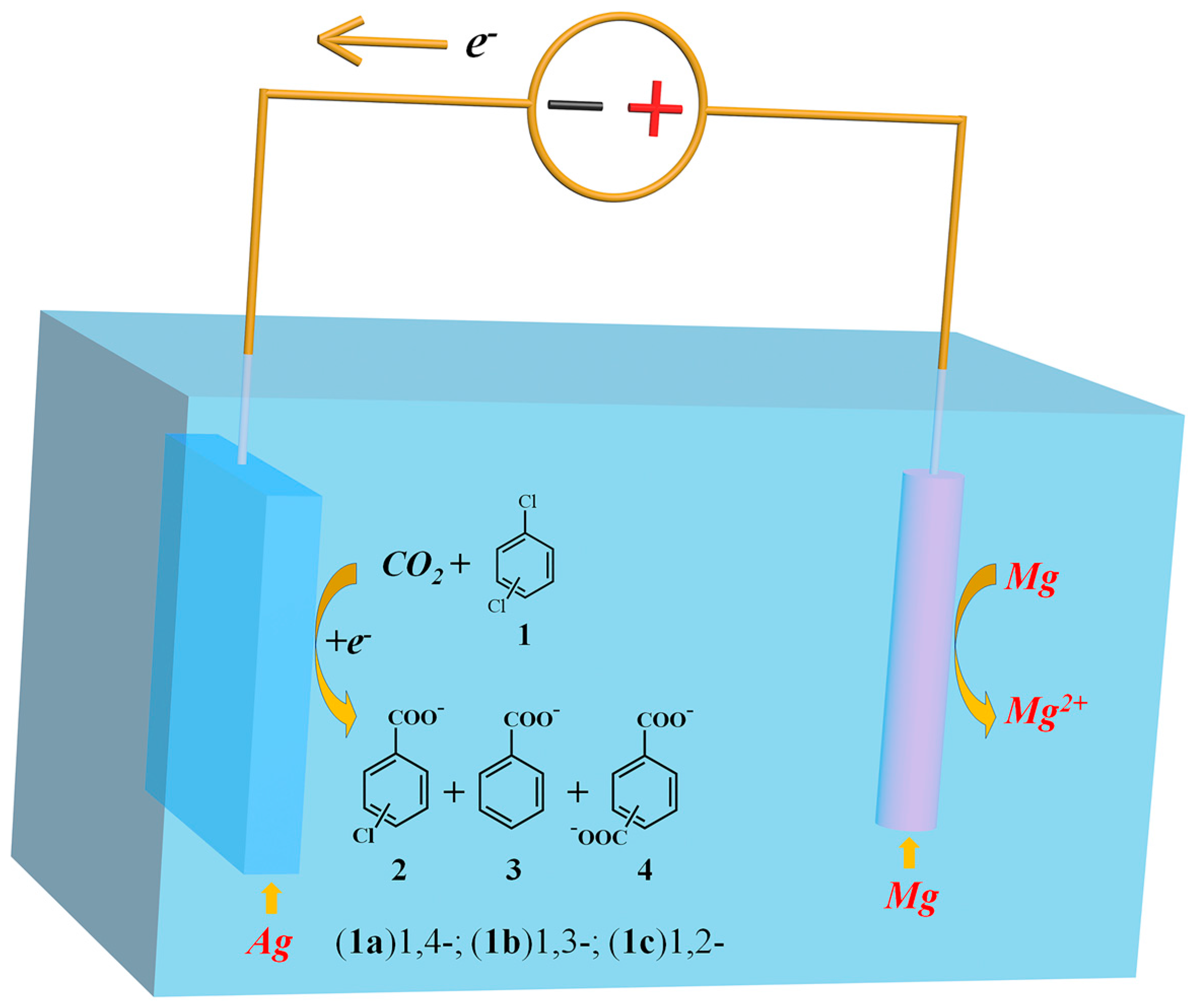
:
1. Introduction
2. Results and Discussion
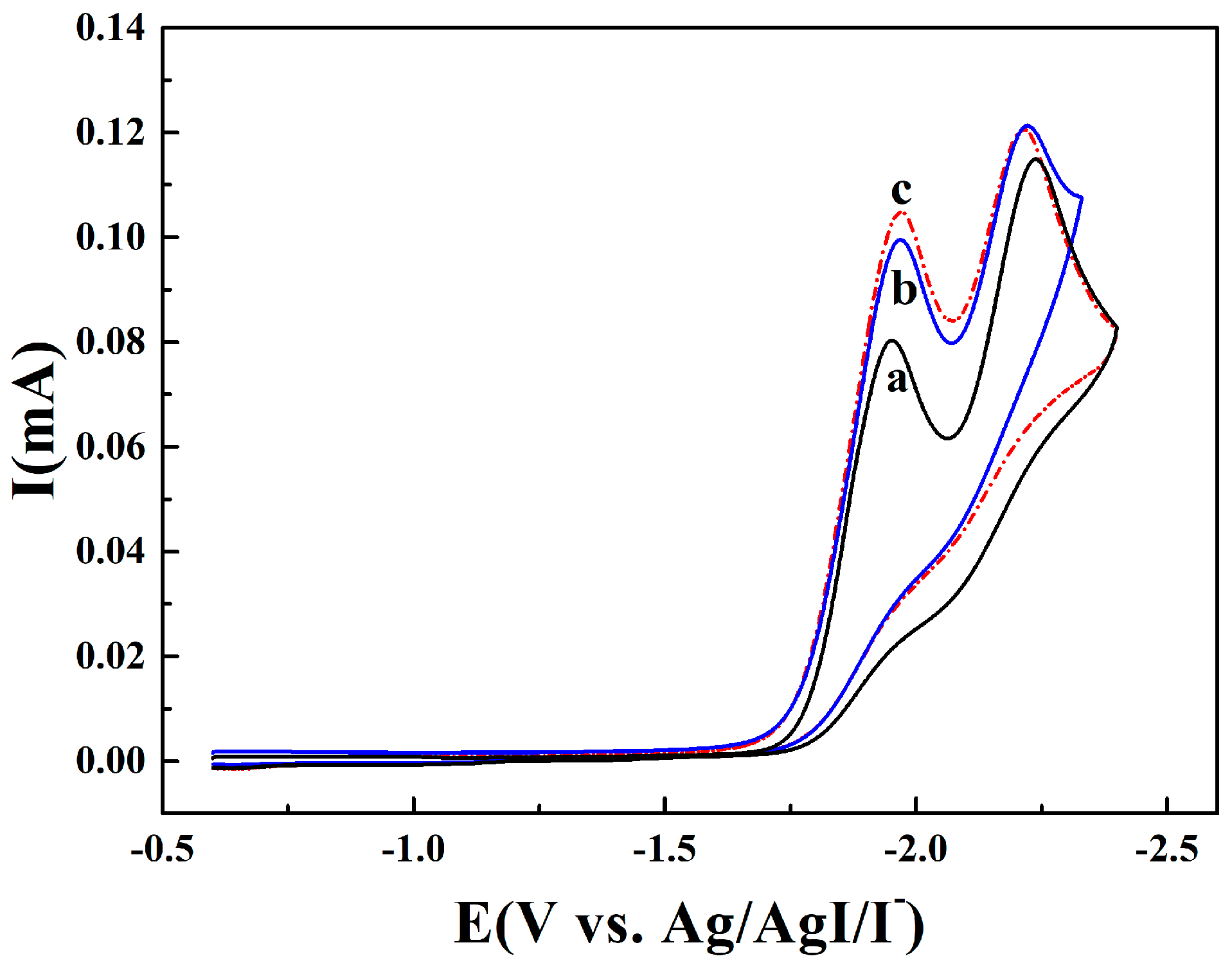
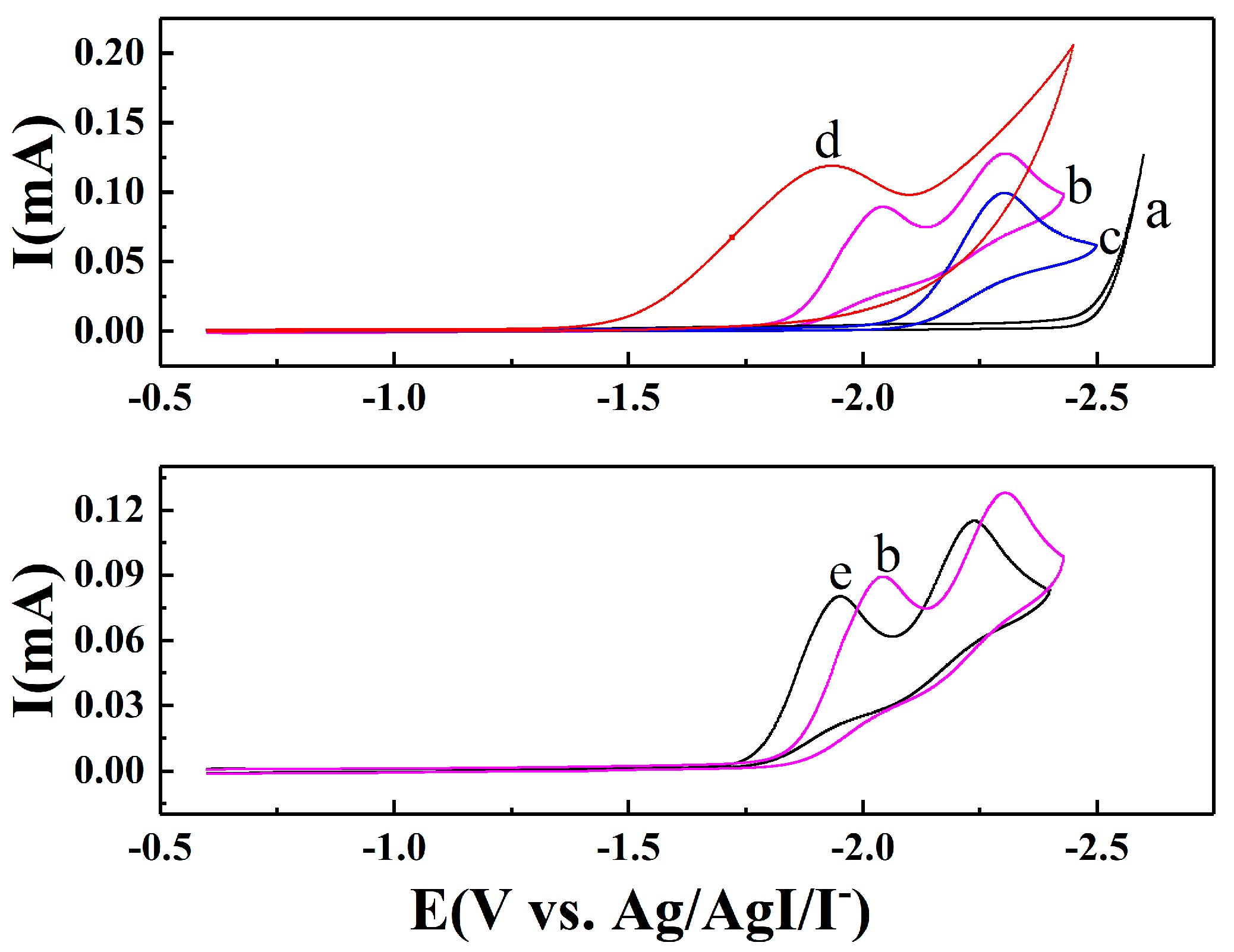
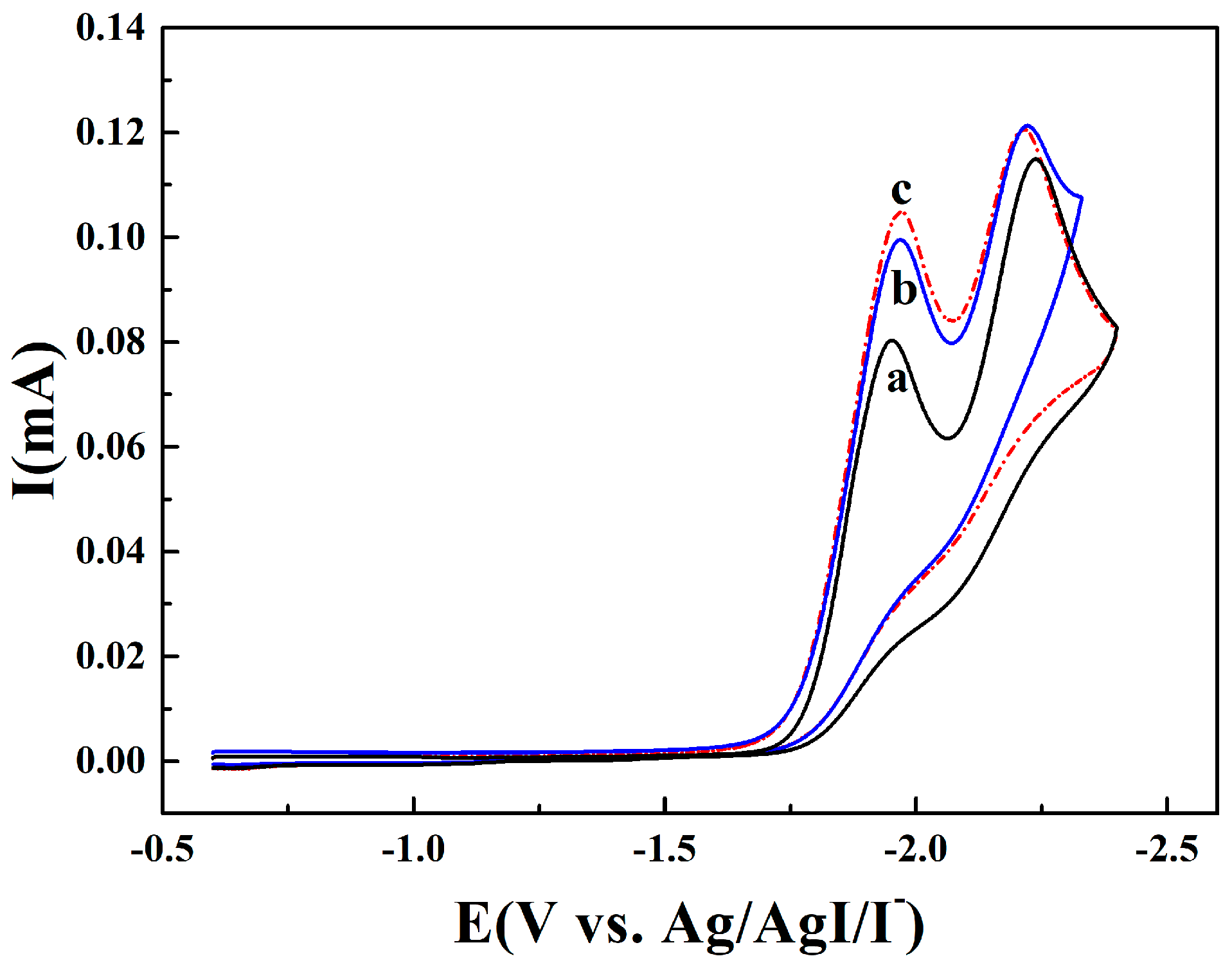
2.1. Electroanalytical Measurement of 1,4-Dichlorobenzene
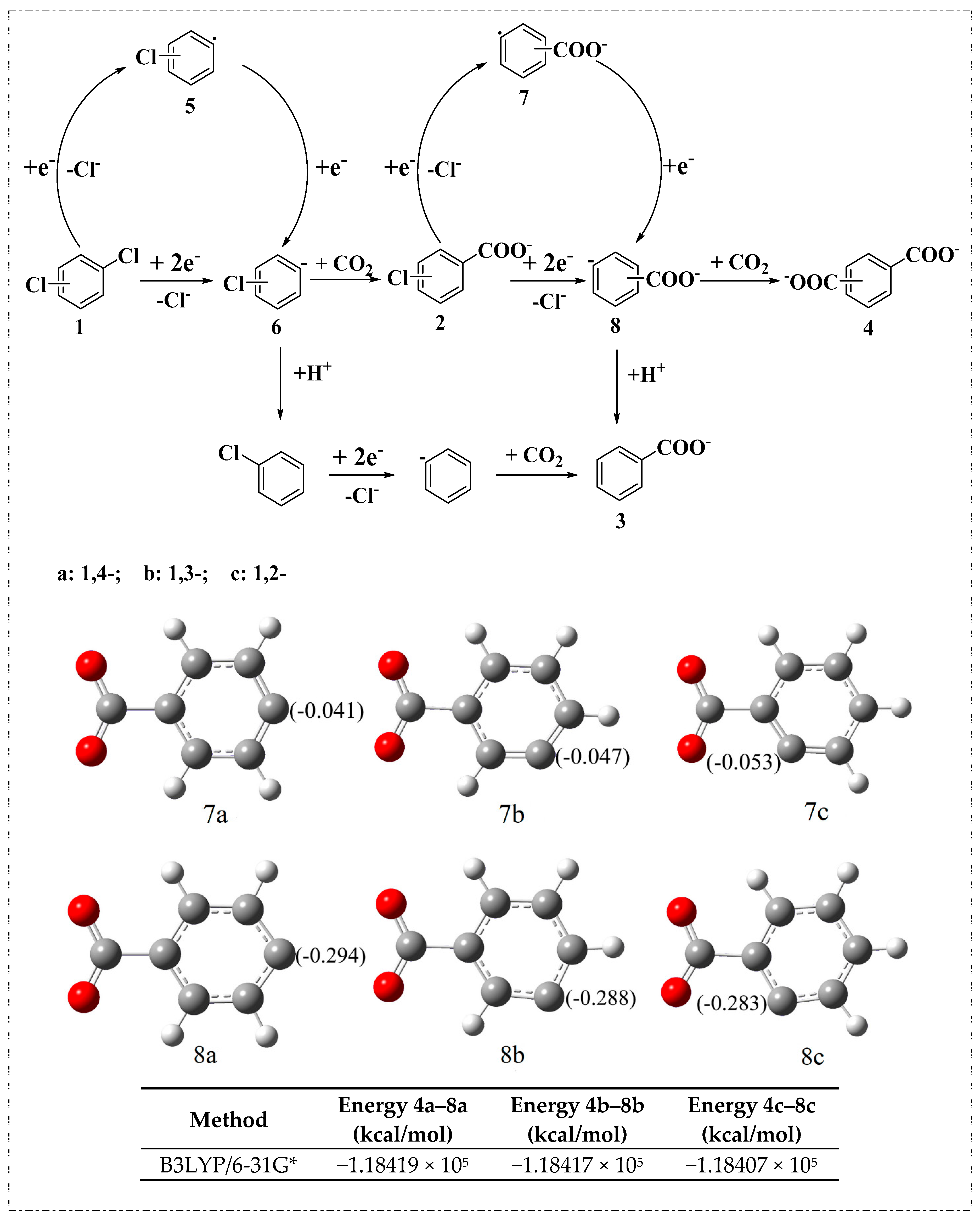
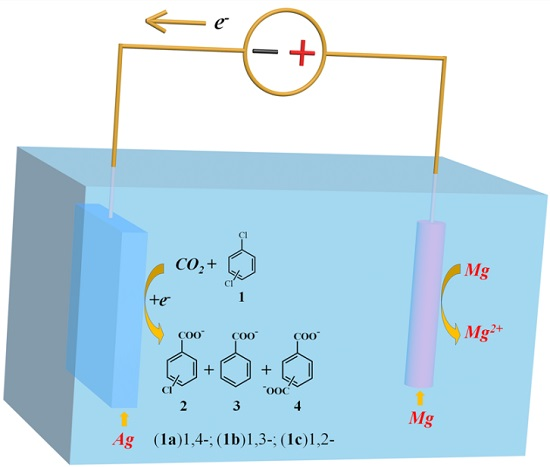
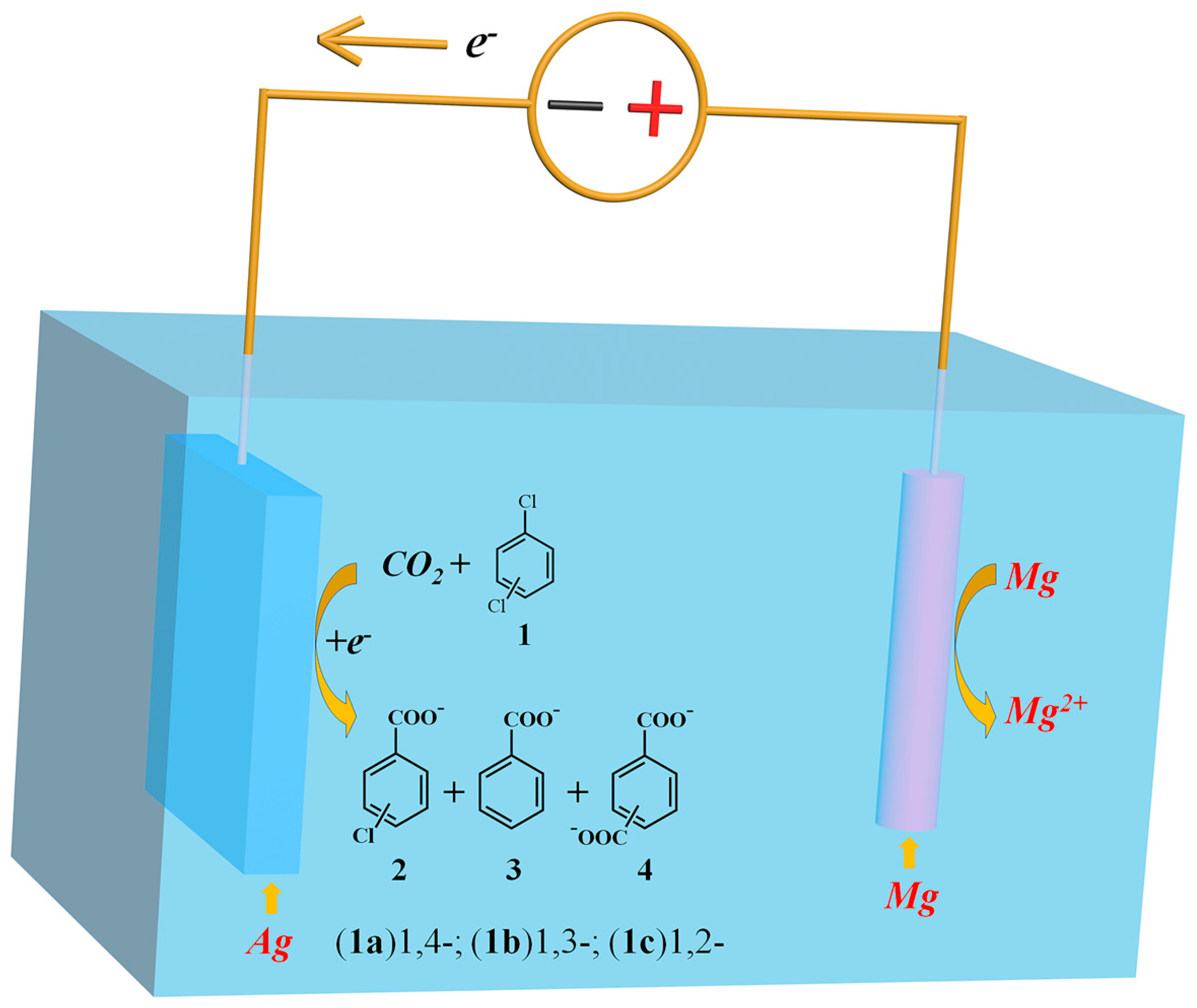
2.2. Electrocarboxylation of Dichlorobenzene
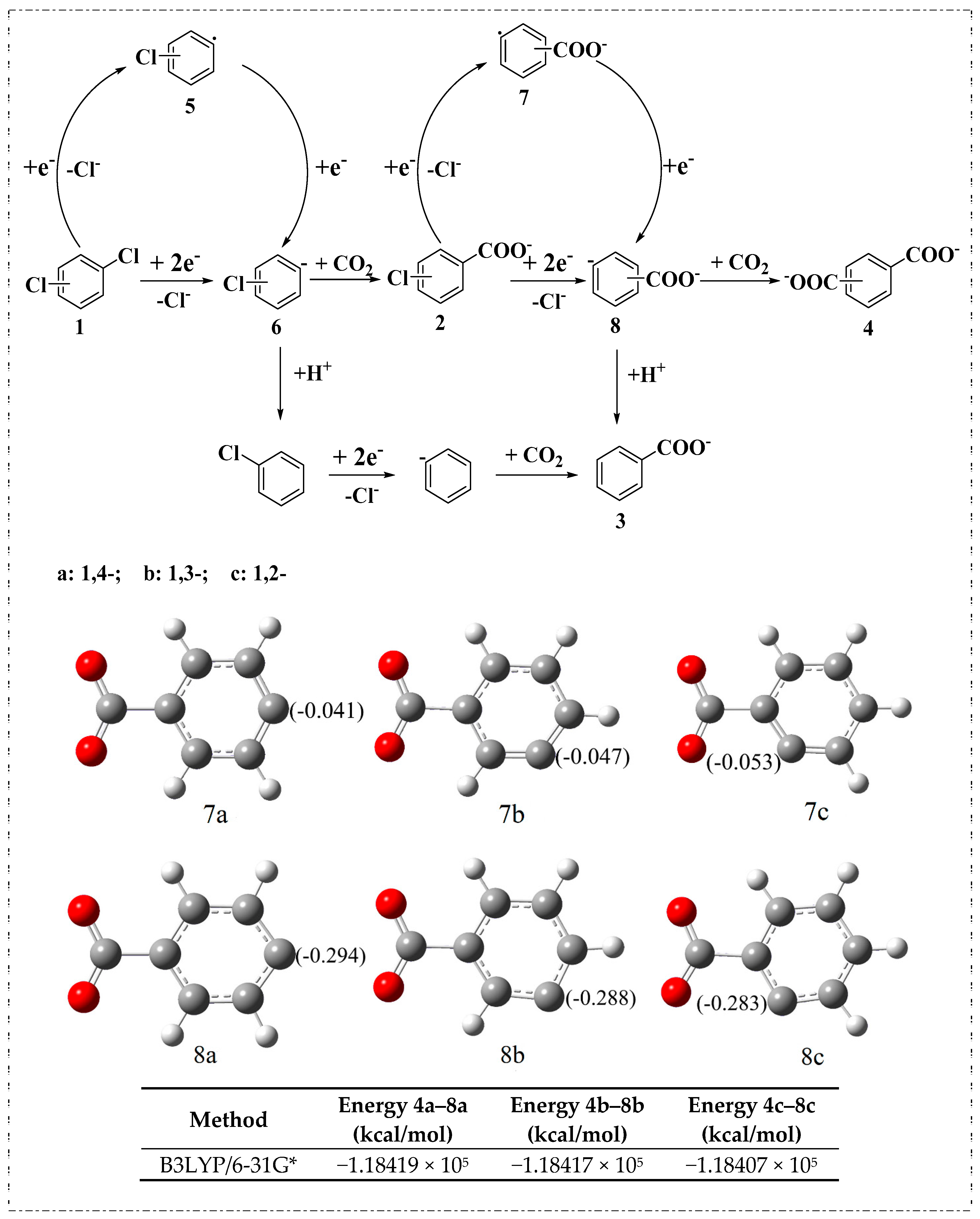
2.3. Electrocarboxylation of Other Dichlorobenzenes
3. Materials and Methods
3.1. Materials and Product Measurement
3.2. General Electroanalytical Procedure
3.3. General Electrolysis Procedure
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karl, T.R.; Trenberth, K.E. Modern global climate change. Science 2003, 302, 1719. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic reduction of CO2 on TiO2 and other semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yu, B.; Yang, Z.; Zhang, H.; Hao, L.; Gao, X.; Liu, Z. A protic ionic liquid catalyzes Co(2) conversion at atmospheric pressure and room temperature: Synthesis of quinazoline-2,4(1 h,3 h)-diones. Angew. Chem. Int. Ed. 2014, 53, 5922–5925. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Kanan, M.W. Pd-catalyzed electrohydrogenation of carbon dioxide to formate: High mass activity at low overpotential and identification of the deactivation pathway. J. Am. Chem. Soc. 2015, 137, 4701–4708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kang, P.; Meyer, T.J. Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J. Am. Chem. Soc. 2014, 136, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, L.; Guo, R.R.; Zeng, S.; Wang, H.; Lu, J.X. Electroreduction of CO2 into ethanol over an active catalyst: Copper supported on titania. Catalysts 2017, 7, 220. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Lin, Y.; Jiao, X.; Sun, Y.; Luo, Q.; Zhang, W.; Li, D.; Yang, J.; Xie, Y. Partially oxidized atomic cobalt layers for carbon dioxide electroreduction to liquid fuel. Nature 2016, 529, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.L.; Xiao, Y.; Xu, X.M.; Yang, H.P.; Wang, H.; Lu, J.X. Alkaloid induced enantioselective electroreduction of acetophenone. Electrochim. Acta 2013, 107, 320–326. [Google Scholar] [CrossRef]
- Chen, B.L.; Tu, Z.Y.; Zhu, H.W.; Sun, W.W.; Wang, H.; Lu, J.X. CO2 as a C1-organic building block: Enantioselective electrocarboxylation of aromatic ketones with CO2 catalyzed by cinchona alkaloids under mild conditions. Electrochim. Acta 2014, 116, 475–483. [Google Scholar] [CrossRef]
- Chen, B.L.; Zhu, H.W.; Xiao, Y.; Sun, Q.L.; Wang, H.; Lu, J.X. Asymmetric electrocarboxylation of 1-phenylethyl chloride catalyzed by electrogenerated chiral [CoI(salen)]− complex. Electrochem. Commun. 2014, 42, 55–59. [Google Scholar] [CrossRef]
- Lan, Y.C.; Wang, H.; Wu, L.X.; Zhao, S.F.; Gu, Y.Q.; Lu, J.X. Electroreduction of dibromobenzenes on silver electrode in the presence of CO2. J. Electroanal. Chem. 2012, 664, 33–38. [Google Scholar] [CrossRef]
- Wu, L.X.; Wang, H.; Xiao, Y.; Tu, Z.Y.; Ding, B.B.; Lu, J.X. Synthesis of dialkyl carbonates from CO2 and alcohols via electrogenerated N-heterocyclic carbenes. Electrochem. Commun. 2012, 25, 116–118. [Google Scholar] [CrossRef]
- Xiao, Y.; Chen, B.L.; Yang, H.P.; Wang, H.; Lu, J.X. Electrosynthesis of enantiomerically pure cyclic carbonates from CO2 and chiral epoxides. Electrochem. Commun. 2014, 43, 71–74. [Google Scholar] [CrossRef]
- Khoshro, H.; Zare, H.R.; Namazian, M.; Jafari, A.A.; Gorji, A. Synthesis of cyclic carbonates through cycloaddition of electrocatalytic activated CO2 to epoxides under mild conditions. Electrochim. Acta 2013, 113, 263–268. [Google Scholar] [CrossRef]
- Feroci, M.; Inesi, A.; Rossi, L. The reaction of amines with an electrogenerated base. Improved synthesis of arylcarbamic esters. Tetrahedron Lett. 2000, 41, 963–966. [Google Scholar] [CrossRef]
- Niu, D.F.; Xiao, L.P.; Zhang, A.J.; Zhang, G.R.; Tan, Q.Y.; Lu, J.X. Electrocatalytic carboxylation of aliphatic halides at silver cathode in acetonitrile. Tetrahedron 2008, 64, 10517–10520. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A. Electrocatalytic carboxylation of benzyl chlorides at silver cathodes in acetonitrile. Chem. Commun. 2002, 2798–2799. [Google Scholar] [CrossRef]
- Isse, A.A.; Durante, C.; Gennaro, A. One-pot synthesis of benzoic acid by electrocatalytic reduction of bromobenzene in the presence of CO2. Electrochem. Commun. 2011, 13, 810–813. [Google Scholar] [CrossRef]
- Isse, A.A.; De Giusti, A.; Gennaro, A.; Falciola, L.; Mussini, P.R. Electrochemical reduction of benzyl halides at a silver electrode. Electrochim. Acta 2006, 51, 4956–4964. [Google Scholar] [CrossRef]
- Scialdone, O.; Galia, A.; Errante, G.; Isse, A.A.; Gennaro, A.; Filardo, G. Electrocarboxylation of benzyl chlorides at silver cathode at the preparative scale level. Electrochim. Acta 2008, 53, 2514–2528. [Google Scholar] [CrossRef]
- Chen, D.; Fabre, P.L.; Reynes, O. Electrocarboxylation of chloroacetonitrile by a cobalt(I) complex of terpyridine. Electrochim. Acta 2011, 56, 8603–8610. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Huang, K.; Liu, S.; Wang, X. Electrocatalytic carboxylation of 2-amino-5-bromopyridine with CO2 in ionic liquid 1-butyl-3-methyllimidazoliumtetrafluoborate to 6-aminonicotinic acid. Electrochim. Acta 2010, 55, 5741–5745. [Google Scholar] [CrossRef]
- Tateno, H.; Nakabayashi, K.; Kashiwagi, T.; Senboku, H.; Atobe, M. Electrochemical fixation of CO2 to organohalides in room-temperature ionic liquids under supercritical CO2. Electrochim. Acta 2015, 161, 212–218. [Google Scholar] [CrossRef]
- Wang, H.M.; Sui, G.J.; Wu, D.; Feng, Q.; Wang, H.; Lu, J.X. Selective electrocarboxylation of bromostyrene at silver cathode in dmf. Tetrahedron 2016, 72, 968–972. [Google Scholar] [CrossRef]
- Zhao, S.F.; Wang, H.; Lan, Y.C.; Liu, X.; Lu, J.X.; Zhang, J. Influences of the operative parameters and the nature of the substrate on the electrocarboxylation of benzophenones. J. Electroanal. Chem. 2012, 664, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, L.; Sui, G.J.; Lu, J.X. Electrocatalytic reduction of PhCH2Br on a Ag–Y zeolite modified electrode. RSC Adv. 2015, 5, 42663–42665. [Google Scholar] [CrossRef]
- Sui, G.J.; Sun, Q.L.; Wu, D.; Meng, W.J.; Wang, H.; Lu, J.X. Electrocatalytic reduction of PhCH2Cl on Ag-zsm-5 zeolite modified electrode. RSC Adv. 2016, 6, 63493–63496. [Google Scholar] [CrossRef]
- Scialdone, O.; Sabatino, M.A.; Belfiore, C.; Galia, A.; Paternostro, M.P.; Filardo, G. An unexpected ring carboxylation in the electrocarboxylation of aromatic ketones. Electrochim. Acta 2006, 51, 3500–3505. [Google Scholar] [CrossRef]
- Feng, Q.; Huang, K.; Liu, S.; Yu, J.; Liu, F. Electrocatalytic carboxylation of aromatic ketones with carbon dioxide in ionic liquid 1-butyl-3-methylimidazoliumtetrafluoborate to α-hydroxy-carboxylic acid methyl ester. Electrochim. Acta 2011, 56, 5137–5141. [Google Scholar] [CrossRef]
- Saravanan, K.R.; Chandrasekaran, M.; Suryanarayanan, V. Efficient electrocarboxylation of benzophenone on silver nanoparticles deposited boron doped diamond electrode. J. Electroanal. Chem. 2015, 757, 18–22. [Google Scholar] [CrossRef]
- Doherty, A.P. Electrochemical reduction of butyraldehyde in the presence of CO2. Electrochim. Acta 2002, 47, 2963–2967. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, G.; Liu, Y.; Luo, Y.; Lu, J. Electrocarboxylation of activated olefins in ionic liquid BMIMBF4. Electrochem. Commun. 2007, 9, 2235–2239. [Google Scholar] [CrossRef]
- Zhang, K.; Xiao, Y.; Lan, Y.; Zhu, M.; Wang, H.; Lu, J. Electrochemical reduction of aliphatic conjugated dienes in the presence of carbon dioxide. Electrochem. Commun. 2010, 12, 1698–1702. [Google Scholar] [CrossRef]
- Li, J.; Inagi, S.; Fuchigami, T.; Hosono, H.; Ito, S. Selective monocarboxylation of olefins at 12CaO·7Al2O3 electride cathode. Electrochem. Commun. 2014, 44, 45–48. [Google Scholar] [CrossRef]
- Yuan, G.Q.; Jiang, H.F.; Lin, C.; Liao, S.J. Efficient electrochemical synthesis of 2-arylsuccinic acids from CO2 and aryl-substituted alkenes with nickel as the cathode. Electrochim. Acta 2008, 53, 2170–2176. [Google Scholar] [CrossRef]
- Steinmann, S.N.; Michel, C.; Schwiedernoch, R.; Wu, M.; Sautet, P. Electro-carboxylation of butadiene and ethene over Pt and Ni catalysts. J. Catal. 2016, 343, 240–247. [Google Scholar] [CrossRef]
- Yuan, G.Q.; Jiang, H.F.; Lin, C. Efficient electrochemical dicarboxylations of arylacetylenes with carbon dioxide using nickel as the cathode. Tetrahedron 2008, 64, 5866–5872. [Google Scholar] [CrossRef]
- Damodar, J.; Krishna Mohan, S.R.; Jayarama Reddy, S.R. Synthesis of 2-arylpropionic acids by electrocarboxylation of benzylchlorides catalysed by PdCl2(PPh3)2. Electrochem. Commun. 2001, 3, 762–766. [Google Scholar] [CrossRef]
- Fabre, P.L.; Reynes, O. Electrocarboxylation of chloroacetonitrile mediated by electrogenerated cobalt(I) phenanthroline. Electrochem. Commun. 2010, 12, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Bessel, C.A.; Rolison, D.R. Electrocatalytic reactivity of zeolite-encapsulated Co(salen) with benzyl chloride. J. Am. Chem. Soc. 1997, 119, 12673–12674. [Google Scholar] [CrossRef]
- Isse, A.A.; Falciola, L.; Mussini, P.R.; Gennaro, A. Relevance of electron transfer mechanism in electrocatalysis: The reduction of organic halides at silver electrodes. Chem. Commun. 2006, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Scialdone, O.; Galia, A.; Filardo, G.; Isse, A.A.; Gennaro, A. Electrocatalytic carboxylation of chloroacetonitrile at a silver cathode for the synthesis of cyanoacetic acid. Electrochim. Acta 2008, 54, 634–642. [Google Scholar] [CrossRef]
- Fiori, G.; Rondinini, S.; Sello, G.; Vertova, A.; Cirja, M.; Conti, L. Electroreduction of volatile organic halides on activated silver cathodes. J. Appl. Electrochem. 2005, 35, 363–368. [Google Scholar] [CrossRef]
- Isse, A.A.; Sandonà, G.; Durante, C.; Gennaro, A. Voltammetric investigation of the dissociative electron transfer to polychloromethanes at catalytic and non-catalytic electrodes. Electrochim. Acta 2009, 54, 3235–3243. [Google Scholar] [CrossRef]
- Durante, C.; Isse, A.A.; Sandonà, G.; Gennaro, A. Electrochemical hydrodehalogenation of polychloromethanes at silver and carbon electrodes. Appl. Catal. B Environ. 2009, 88, 479–489. [Google Scholar] [CrossRef]
- Huang, B.; Isse, A.A.; Durante, C.; Wei, C.; Gennaro, A. Electrocatalytic properties of transition metals toward reductive dechlorination of polychloroethanes. Electrochim. Acta 2012, 70, 50–61. [Google Scholar] [CrossRef]
- Peverly, A.A.; Karty, J.A.; Peters, D.G. Electrochemical reduction of (1R,2R,3S,4R,5R,6S)-hexachlorocyclohexane (lindane) at silver cathodes in organic and aqueous–organic media. J. Electroanal. Chem. 2013, 692, 66–71. [Google Scholar] [CrossRef]
- Mubarak, M.S.; Peters, D.G. Electrochemical reduction of di-, tri-, and tetrahalobenzenes at carbon cathodes in dimethylformamide evidence for a halogen dance during the electrolysis of 1,2,4,5-tetrabromobenzene. J. Electroanal. Chem. 1997, 435, 47–53. [Google Scholar] [CrossRef]
- Gennaro, A.; Isse, A.A.; Vianello, E. Solubility and electrochemical determination of CO2 in some dipolar aprotic solvents. J. Electroanal. Chem. 1990, 289, 203–215. [Google Scholar] [CrossRef]
- Rondinini, S.; Mussini, P.R.; Muttini, P.; Sello, G. Silver as a powerful electrocatalyst for organic halide reduction: The critical role of molecular structure. Electrochim. Acta 2001, 46, 3245–3258. [Google Scholar] [CrossRef]
- Bellomunno, C.; Bonanomi, D.; Falciola, L.; Longhi, M.; Mussini, P.R.; Doubova, L.M.; Di Silvestro, G. Building up an electrocatalytic activity scale of cathode materials for organic halide reductions. Electrochim. Acta 2005, 50, 2331–2341. [Google Scholar] [CrossRef]
- Fedurco, M.; Coppex, L.; Augustynski, J. Ab initio and electrochemical studies on the reductive bond dissociation in haloethanols. J. Phys. Chem. B 2002, 106, 2625–2633. [Google Scholar] [CrossRef]
- Rondinini, S.B.; Mussini, P.R.; Crippa, F.; Sello, G. Electrocatalytic potentialities of silver as a cathode for organic halide reductions. Electrochem. Commun. 2000, 2, 491–496. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, H.; Zhao, S.F.; Niu, D.F.; Lu, J.X. Asymmetric electrochemical carboxylation of prochiral acetophenone: An efficient route to optically active atrolactic acid via selective fixation of carbon dioxide. J. Electroanal. Chem. 2009, 630, 35–41. [Google Scholar] [CrossRef]
- Aishah, A.J.; Hartini, M.A.; Normala, S.; Norhuda, A.M.; Hanis, H.H.N.; Razif, H.M.; Sugeng, T. Carbon dioxide fixation method for electrosynthesis of benzoic acid from chlorobenzene. J. Nat. Gas Chem. 2007, 16, 273–277. [Google Scholar] [CrossRef]
- Isse, A.A.; Berzi, G.; Falciola, L.; Rossi, M.; Mussini, P.R.; Gennaro, A. Electrocatalysis and electron transfer mechanisms in the reduction of organic halides at ag. J. Appl. Electrochem. 2009, 39, 2217–2225. [Google Scholar] [CrossRef]
- Zhao, S.F.; Wu, L.X.; Wang, H.; Lu, J.X.; Bond, A.M.; Zhang, J. A unique proton coupled electron transfer pathway for electrochemical reduction of acetophenone in the ionic liquid [BMIM][BF4] under a carbon dioxide atmosphere. Green Chem. 2011, 13, 3461–3468. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01, Gaussian, Inc.: Wallingford, CT, USA, 2010.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | SE | J (mA/cm2) | Q (F/mol) | T (°C) | Carboxylation Product Yields (%) b | Total Yield (%) b | ||
|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | ||||||
| 1 | TEACl | 10 | 4 | 0 | 3.1 | trace | 1.0 | 4.1 |
| 2 | TEABr | 10 | 4 | 0 | 2.1 | trace | 1.0 | 3.1 |
| 3 | TEAI | 10 | 4 | 0 | 1.2 | trace | 1.0 | 2.2 |
| 4 | TEABF4 | 10 | 4 | 0 | 2.3 | trace | trace | 2.3 |
| 5 | TBACl | 10 | 4 | 0 | 31.2 | 3.2 | 2.3 | 36.7 |
| 6 | TBABr | 10 | 4 | 0 | 24.3 | 3.4 | 2.4 | 30.1 |
| 7 | TBAI | 10 | 4 | 0 | 25.3 | trace | 1.0 | 26.3 |
| 8 | TBACl | 8 | 4 | 0 | 16.1 | 4.2 | 2.2 | 22.5 |
| 9 | TBACl | 13 | 4 | 0 | 35.7 | 4.3 | 2.9 | 42.9 |
| 10 | TBACl | 14 | 4 | 0 | 37.6 | 3.3 | 2.7 | 43.6 |
| 11 | TBACl | 15 | 4 | 0 | 34.4 | 4.9 | 3.3 | 42.6 |
| 12 | TBACl | 16 | 4 | 0 | 26.8 | 2.6 | 2.2 | 31.6 |
| 13 | TBACl | 14 | 2 | 0 | 14.9 | 2.6 | 1.7 | 19.2 |
| 14 | TBACl | 14 | 3 | 0 | 23.0 | 2.6 | 1.6 | 27.2 |
| 15 | TBACl | 14 | 4.2 | 0 | 37.2 | 4.7 | 3.4 | 45.3 |
| 16 | TBACl | 14 | 4.5 | 0 | 26.8 | 3.9 | 2.3 | 33.0 |
| 17 | TBACl | 14 | 5 | 0 | 24.8 | 3.5 | 2.2 | 30.5 |
| 18 | TBACl | 14 | 4.2 | −10 | 41.6 | 1.0 | 1.0 | 43.6 |
| 19 | TBACl | 14 | 4.2 | −5 | 40.7 | 1.9 | 1.8 | 44.4 |
| 20 | TBACl | 14 | 4.2 | 5 | 38.4 | 4.6 | 3.3 | 46.3 |
| 21 | TBACl | 14 | 4.2 | 10 | 38.6 | 4.1 | 3.1 | 45.8 |
| 22 | TBACl | 14 | 4.2 | 25 | 24.2 | 6.7 | 3.1 | 34.0 |
| Entry | Substrate | Carboxylation Product Yields (%) b | Total Yield (%) b | ||
|---|---|---|---|---|---|
| 2 | 3 | 4 | |||
| 1 | 1,4-dichlorobenzene (1a) | 38.4 | 4.6 | 3.3 | 46.3 |
| 2 | 1,3-dichlorobenzene (1b) | 41.4 | 7.8 | 0 | 49.2 |
| 3 | 1,2-dichlorobenzene (1c) | 43.7 | 5.8 | 0 | 49.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, P.-P.; Zhang, Y.-T.; Chen, B.-L.; Yu, S.-X.; Zhou, H.-W.; Qu, K.-G.; Kong, Y.-X.; Huang, X.-Q.; Zhang, X.-X.; Lu, J.-X. Electrocarboxylation of Dichlorobenzenes on a Silver Electrode in DMF. Catalysts 2017, 7, 274. https://doi.org/10.3390/catal7090274
Luo P-P, Zhang Y-T, Chen B-L, Yu S-X, Zhou H-W, Qu K-G, Kong Y-X, Huang X-Q, Zhang X-X, Lu J-X. Electrocarboxylation of Dichlorobenzenes on a Silver Electrode in DMF. Catalysts. 2017; 7(9):274. https://doi.org/10.3390/catal7090274
Chicago/Turabian StyleLuo, Pei-Pei, Ying-Tian Zhang, Bao-Li Chen, Shu-Xian Yu, Hua-Wei Zhou, Kong-Gang Qu, Yu-Xia Kong, Xian-Qiang Huang, Xian-Xi Zhang, and Jia-Xing Lu. 2017. "Electrocarboxylation of Dichlorobenzenes on a Silver Electrode in DMF" Catalysts 7, no. 9: 274. https://doi.org/10.3390/catal7090274