
Dynamic Processes on Gold-Based Catalysts Followed by Environmental Microscopies


Abstract
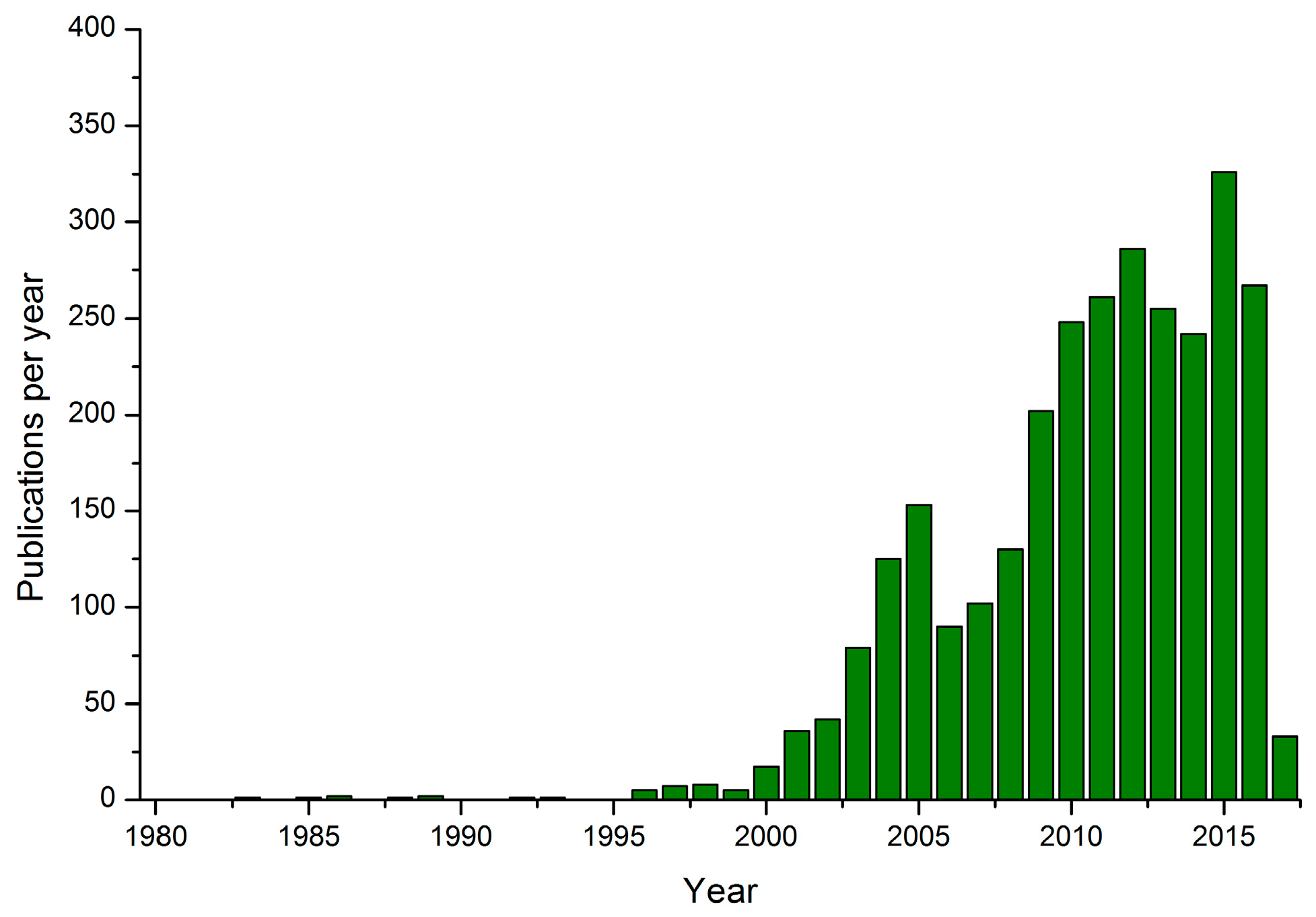
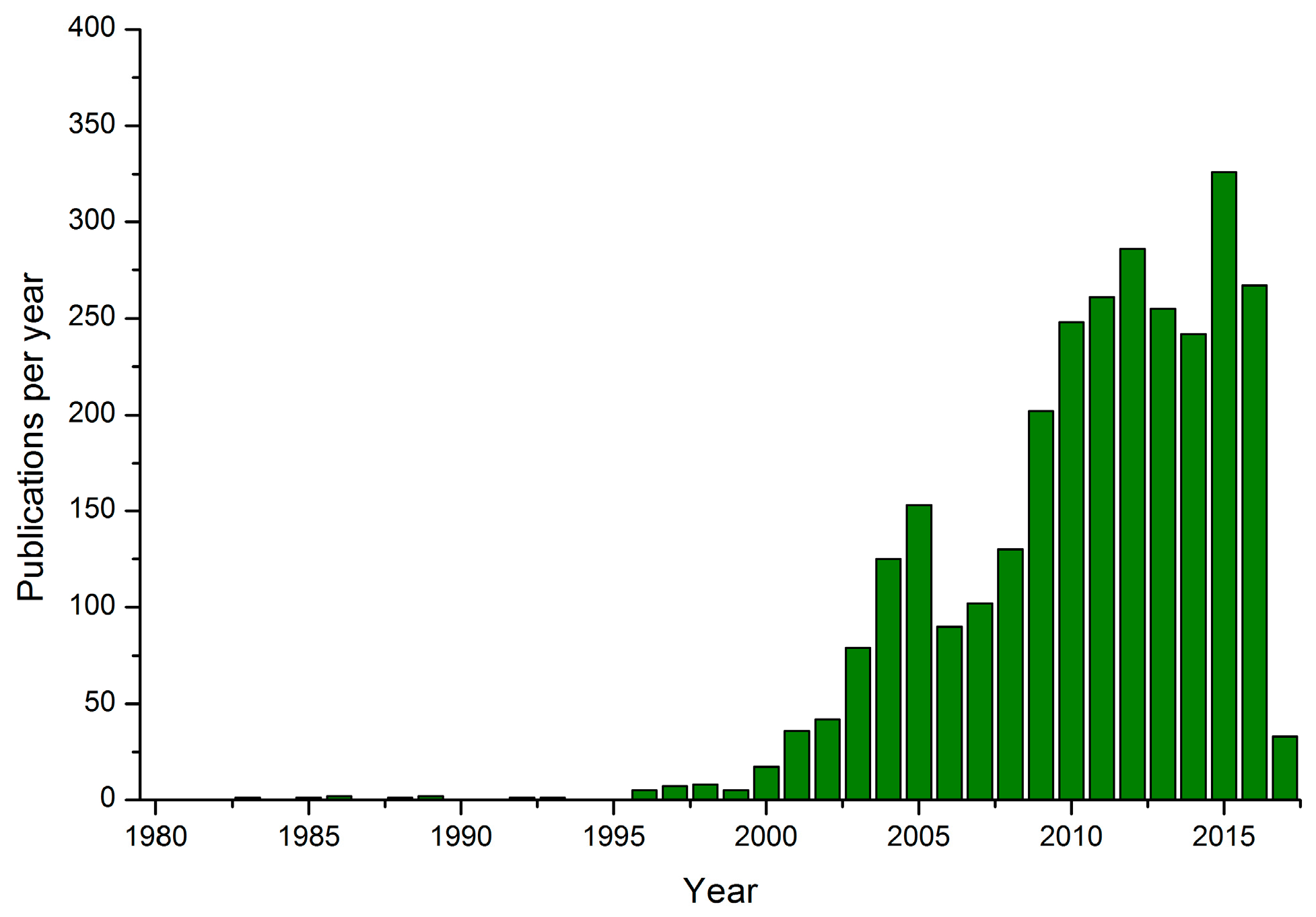
:1. Introduction
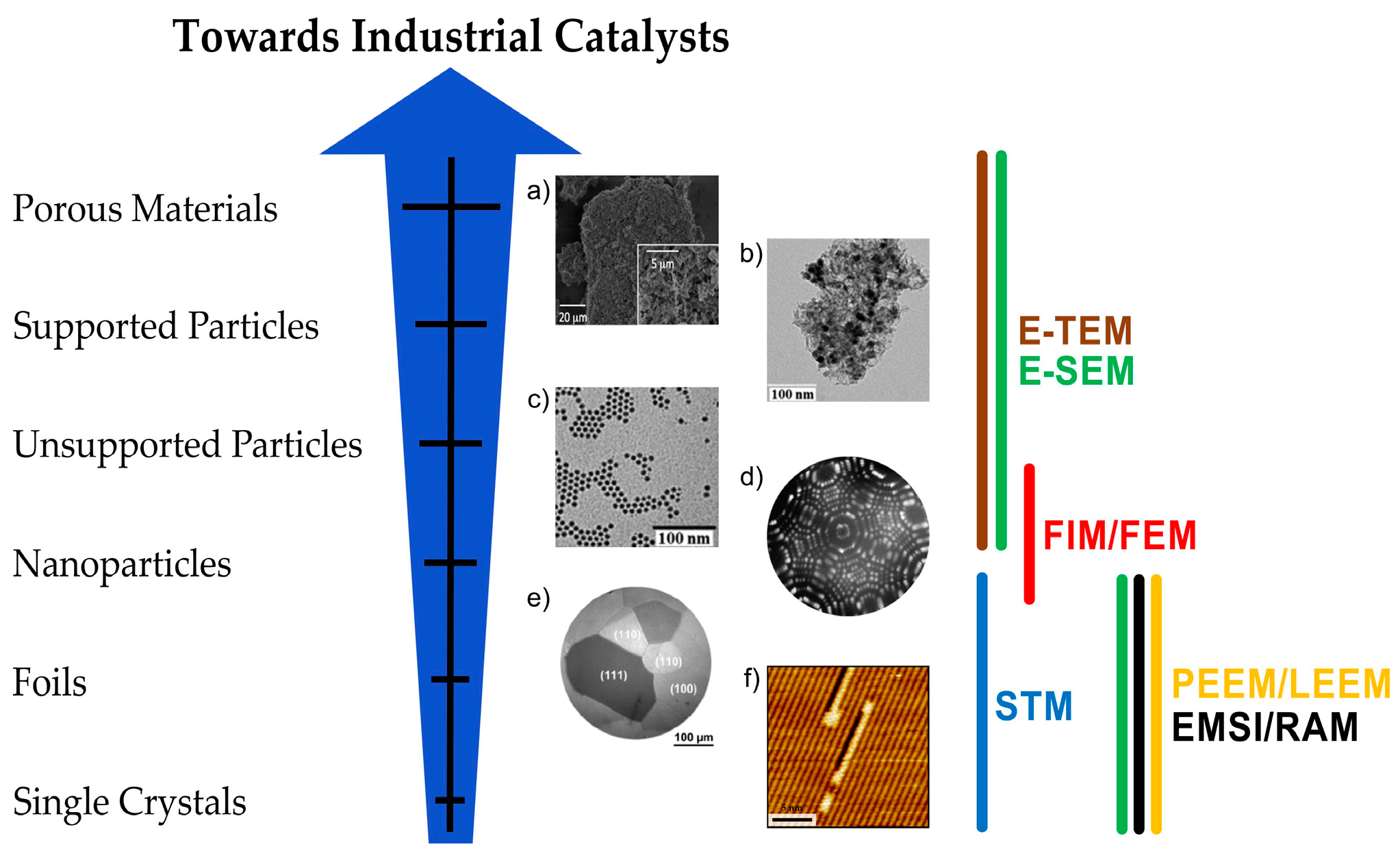
2. Environmental Microscopies
2.1. Environmental Microscopies in Catalysis
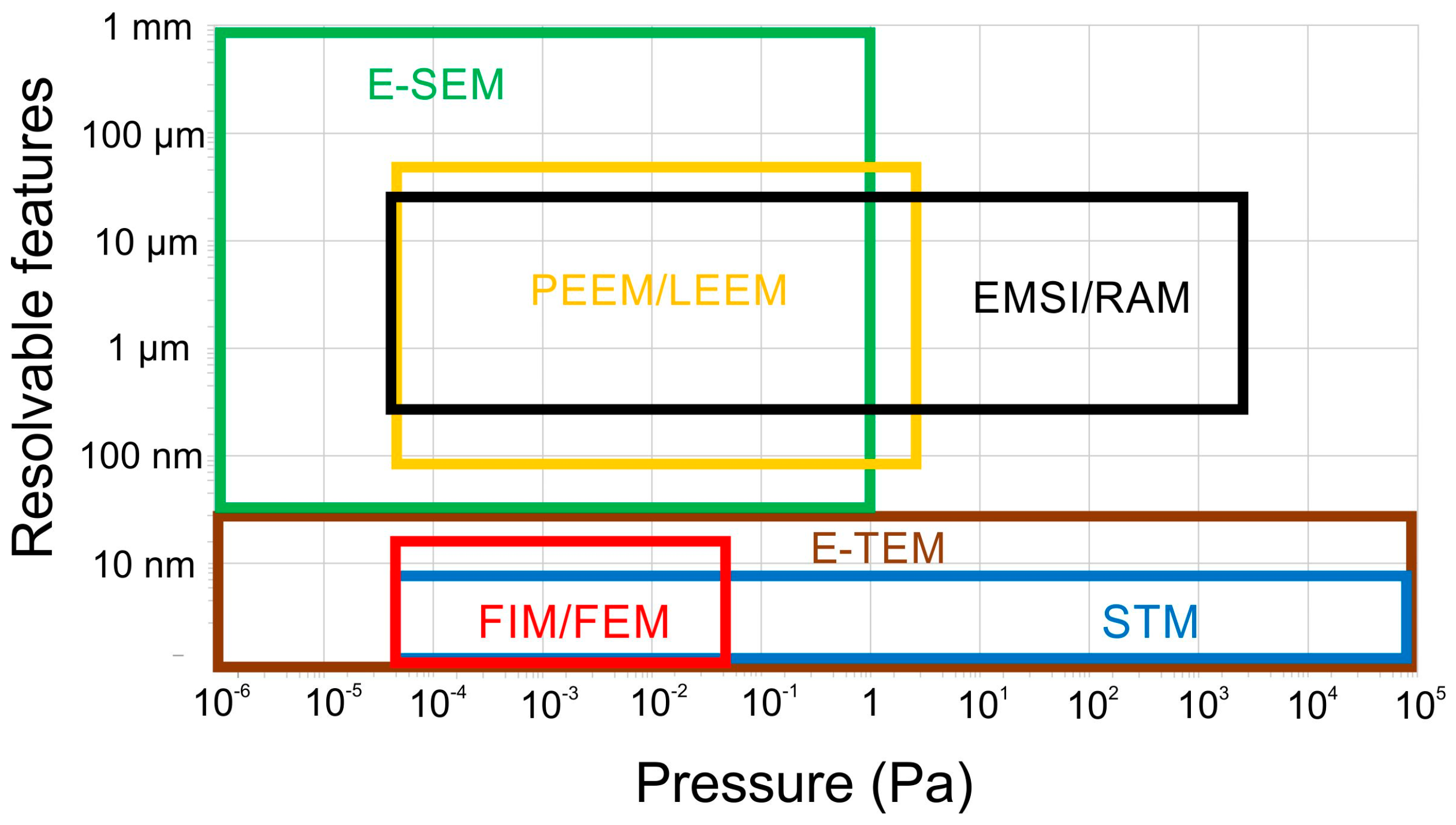
2.2. Basic Principles of Relevant Techniques
2.2.1. Field Emission Microscopy FEM/Field Ion Microscopy FIM
2.2.2. Photoemission Electron Microscopy PEEM/Low Energy Electron Microscopy LEEM
2.2.3. Environmental Transmission Electron Microscopy E-TEM
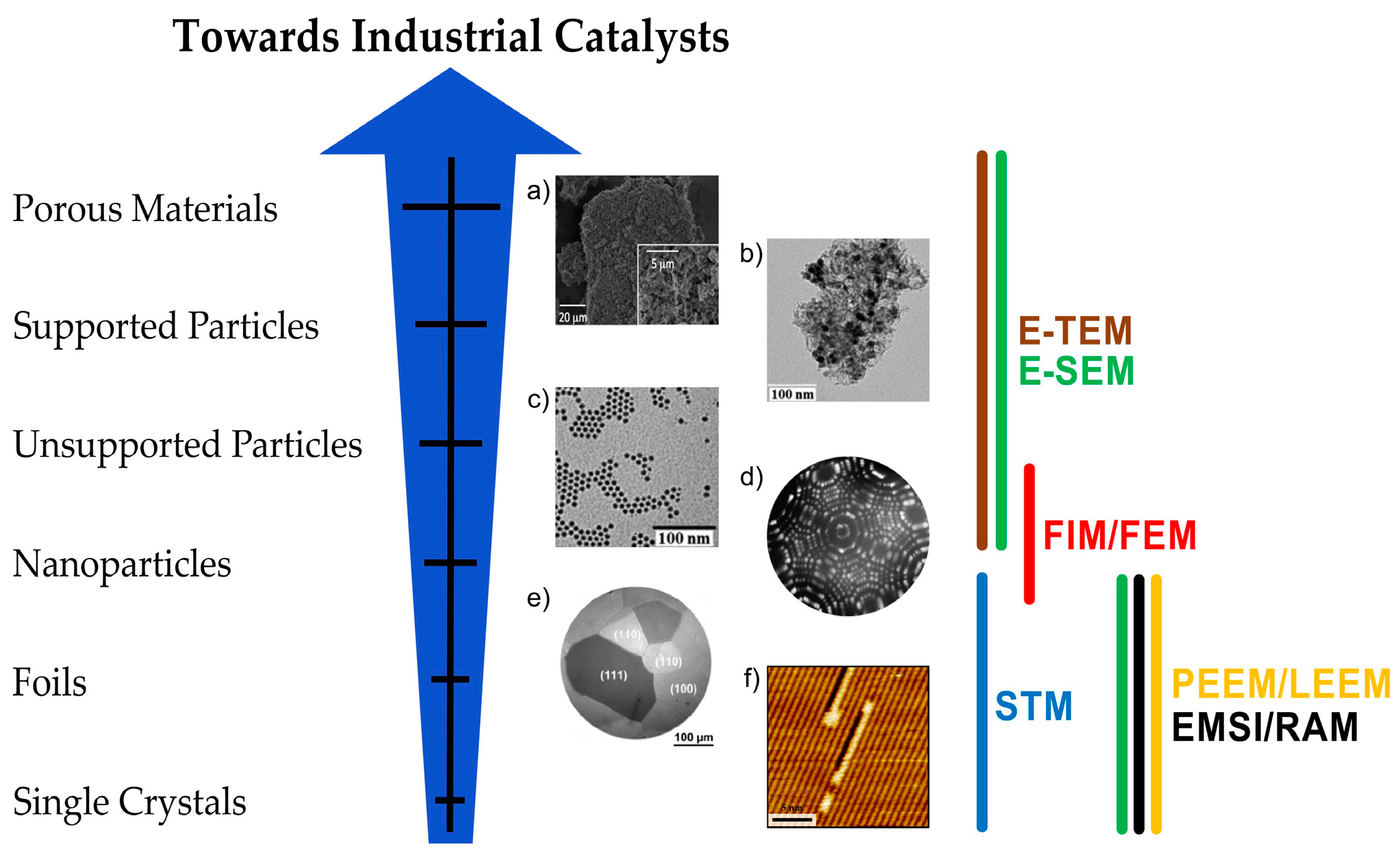
3. Case Studies
3.1. Study of Processes on Single Crystals by PEEM/LEEM
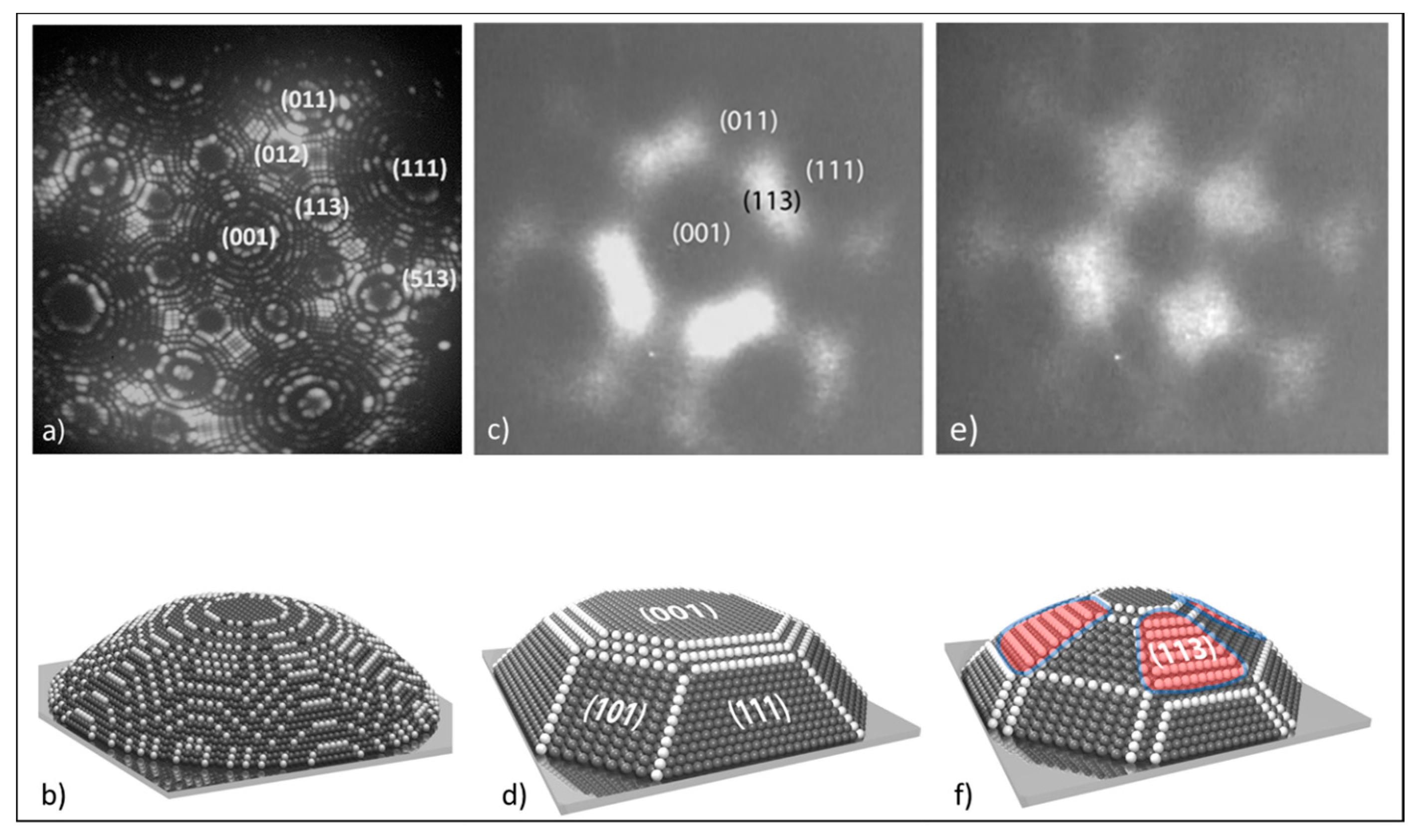
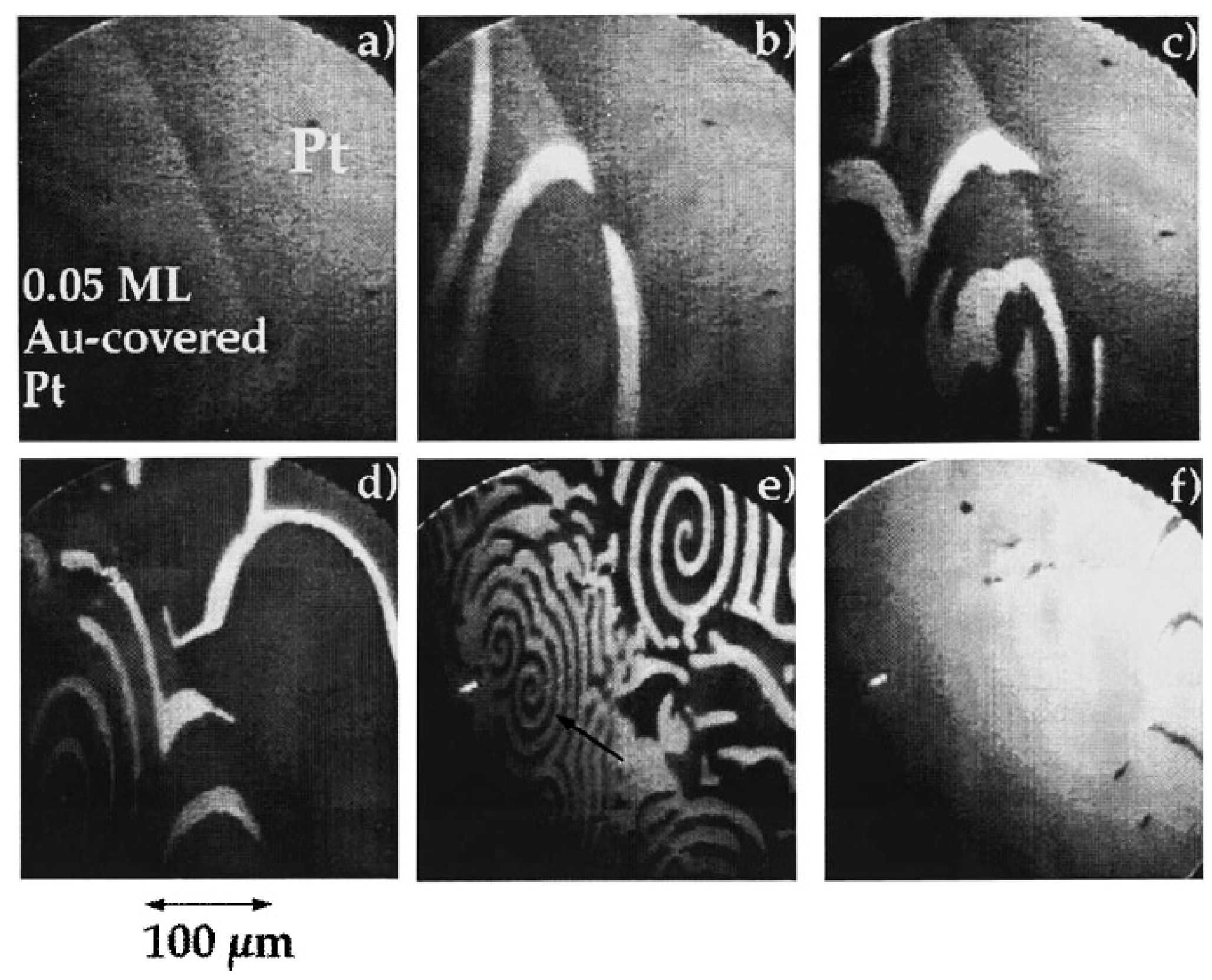
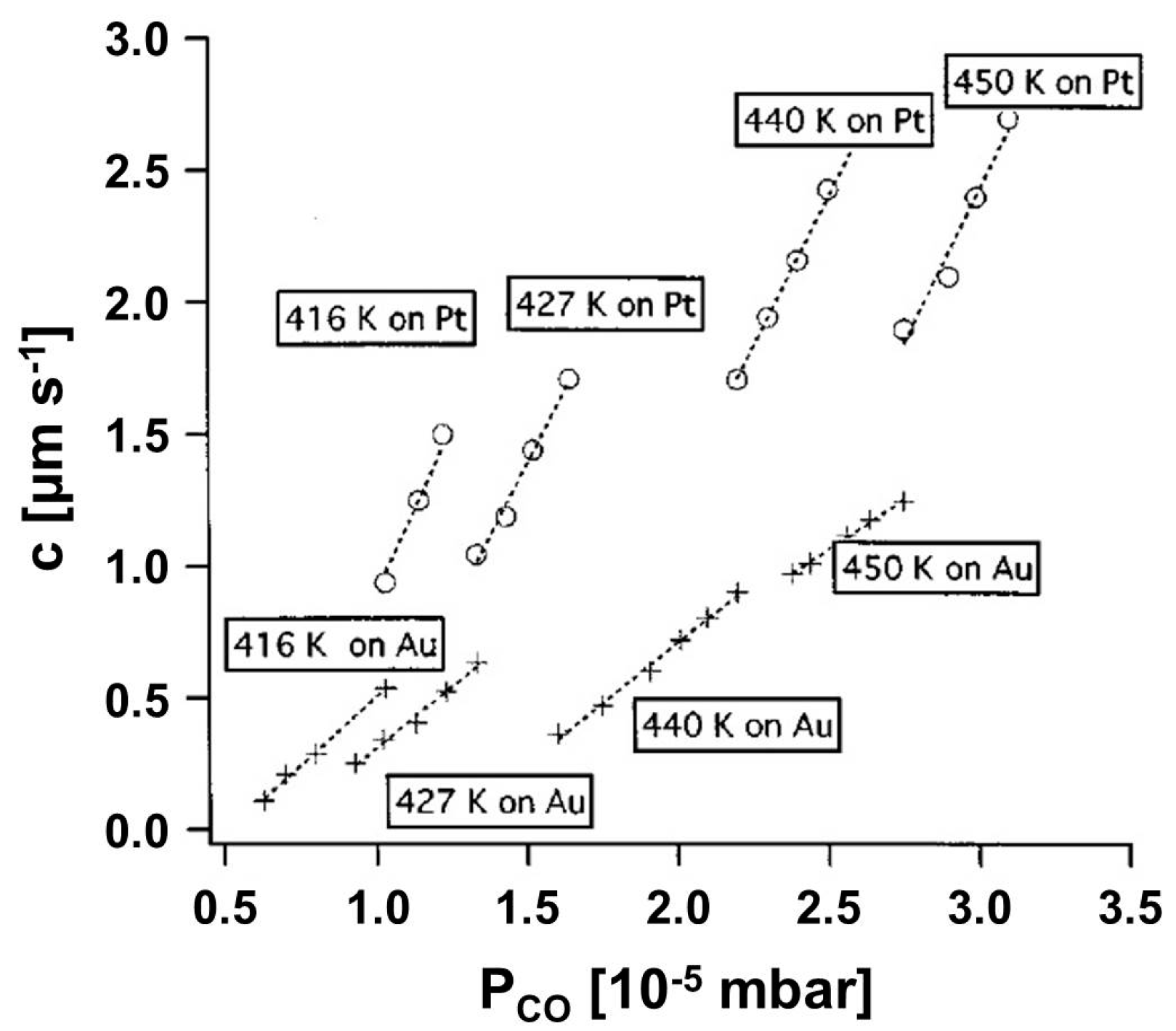
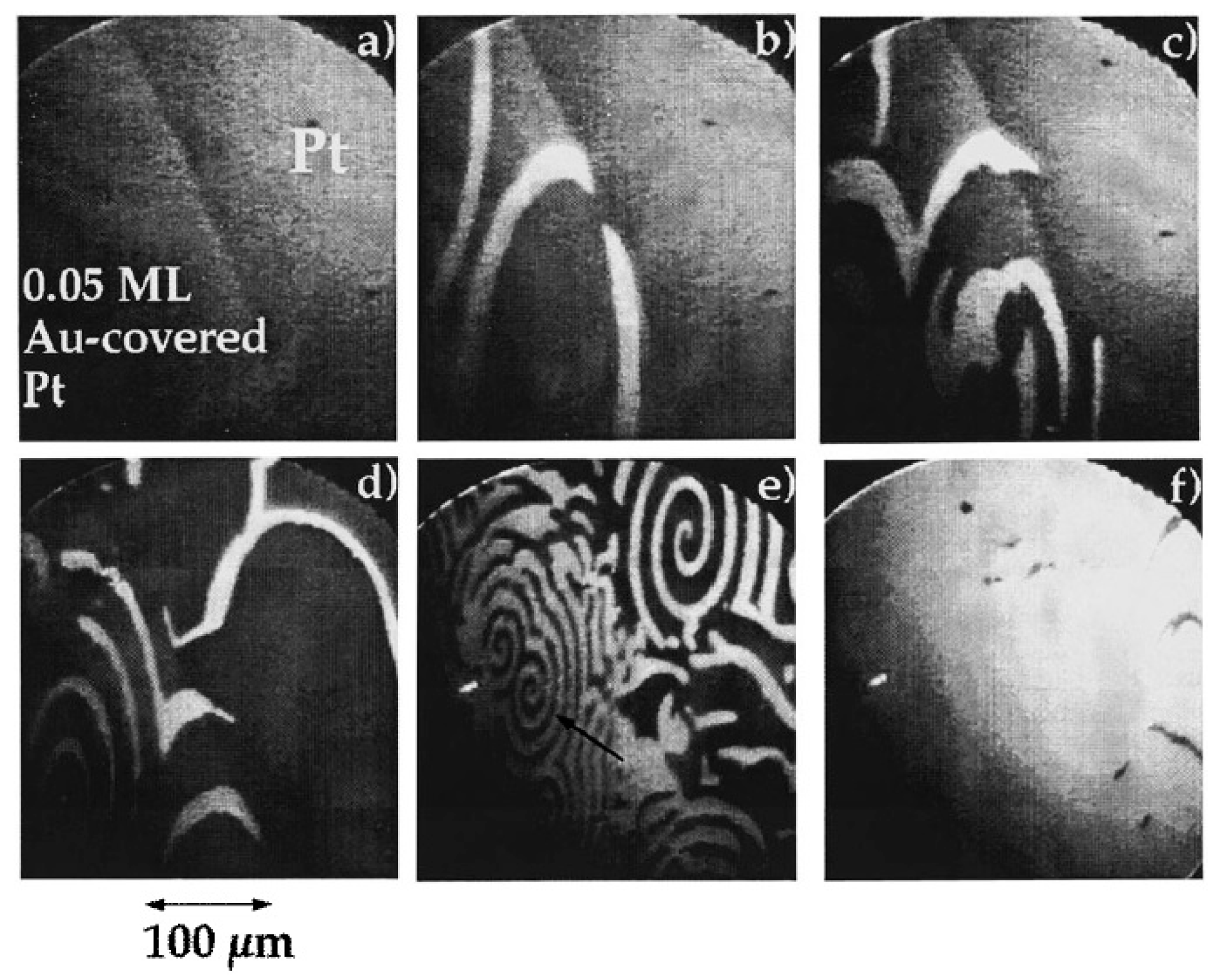
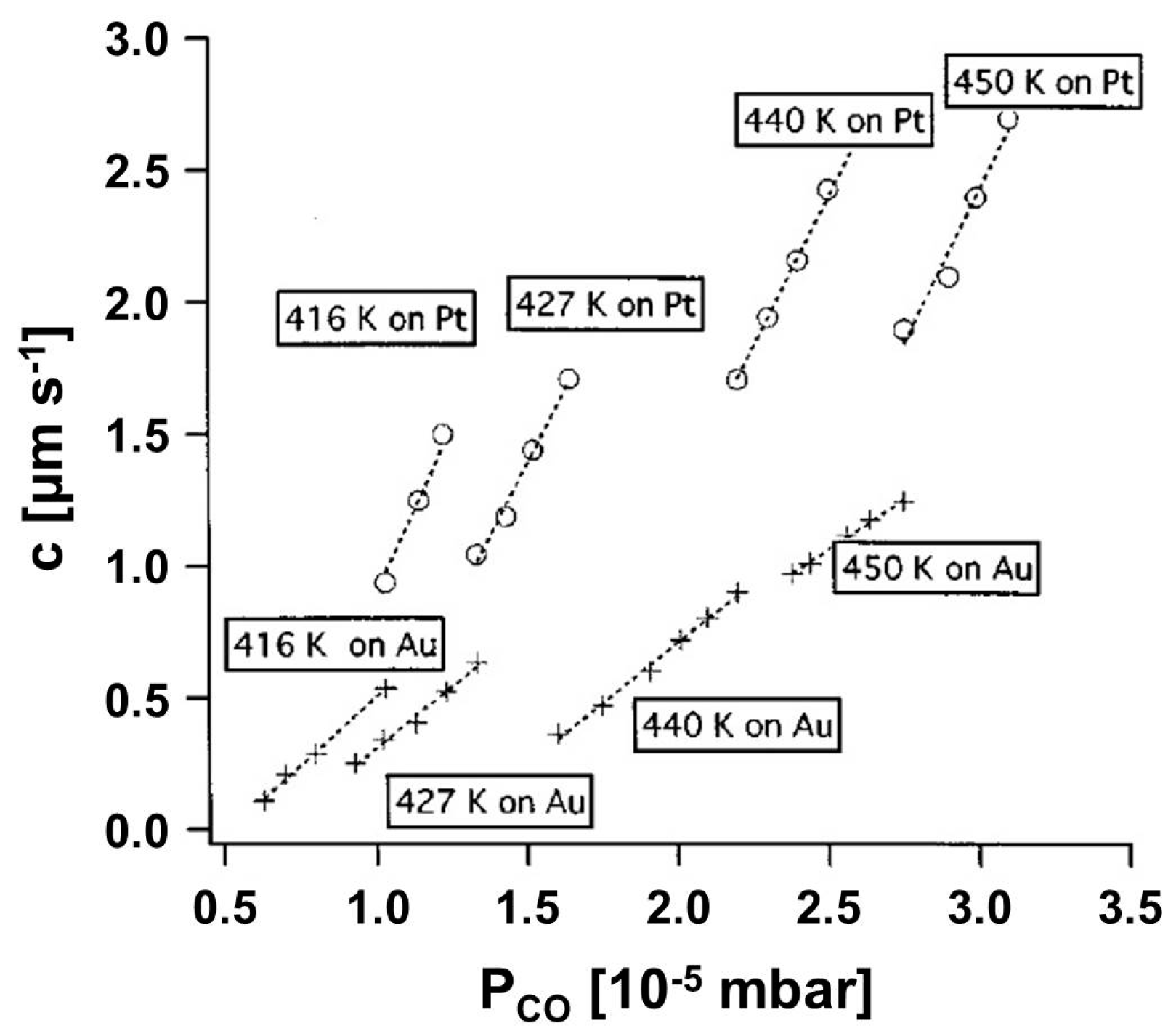
3.1.1. Processes on Au-Covered Pt Single Crystals: Influence of Au on the Presence of Kinetic Instabilities on Pt(110)
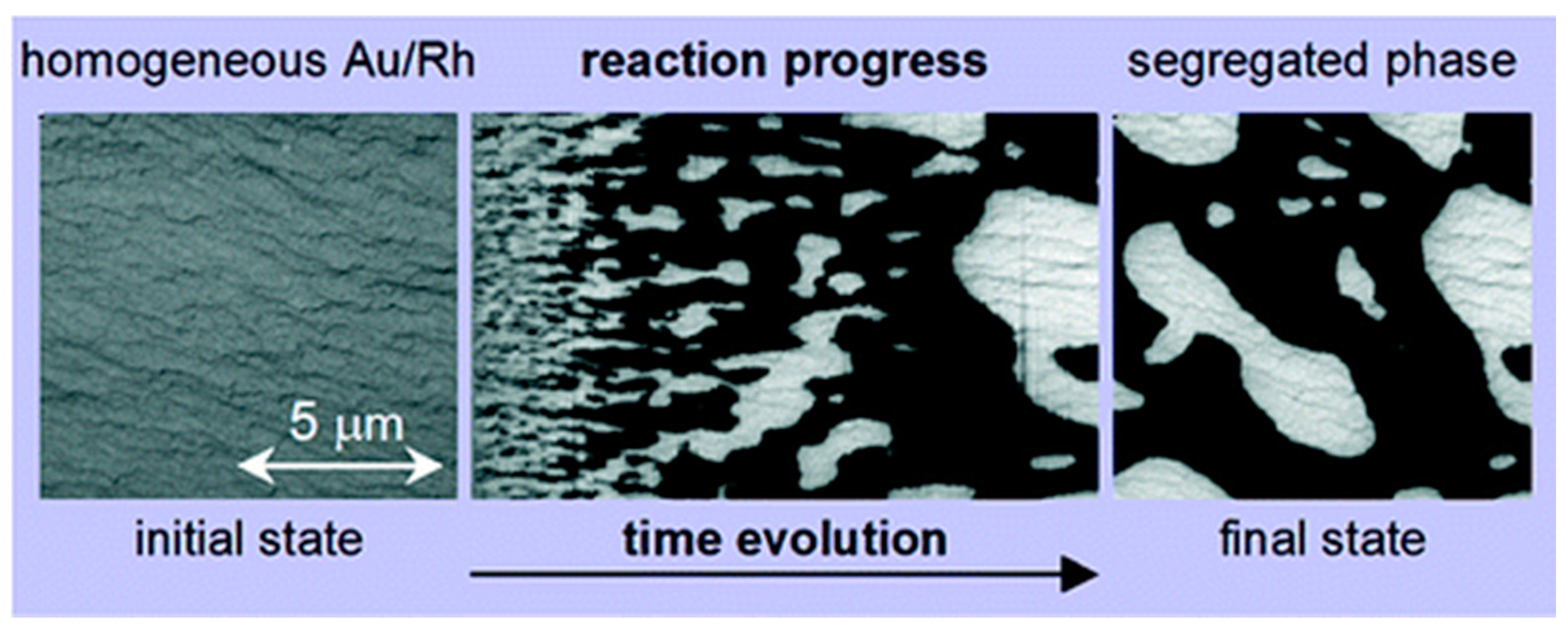
3.1.2. Processes on Au-Covered Rh Single Crystals
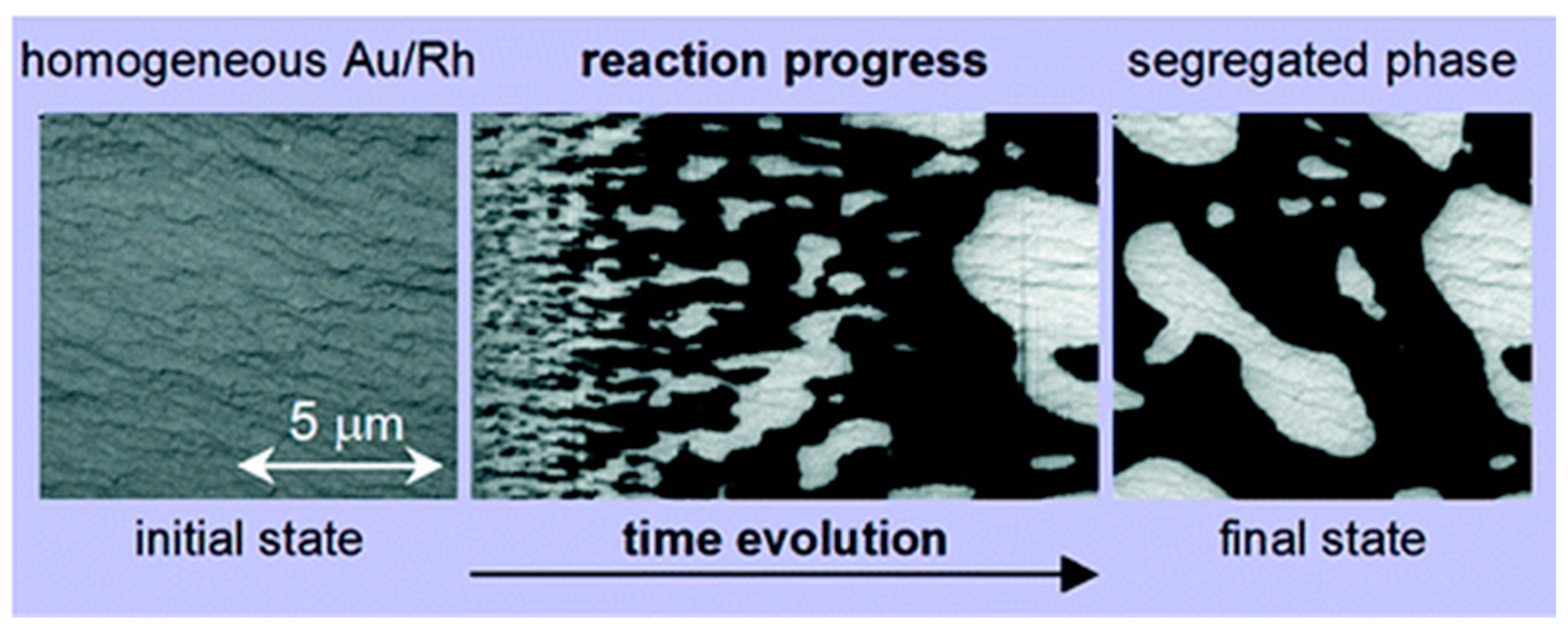
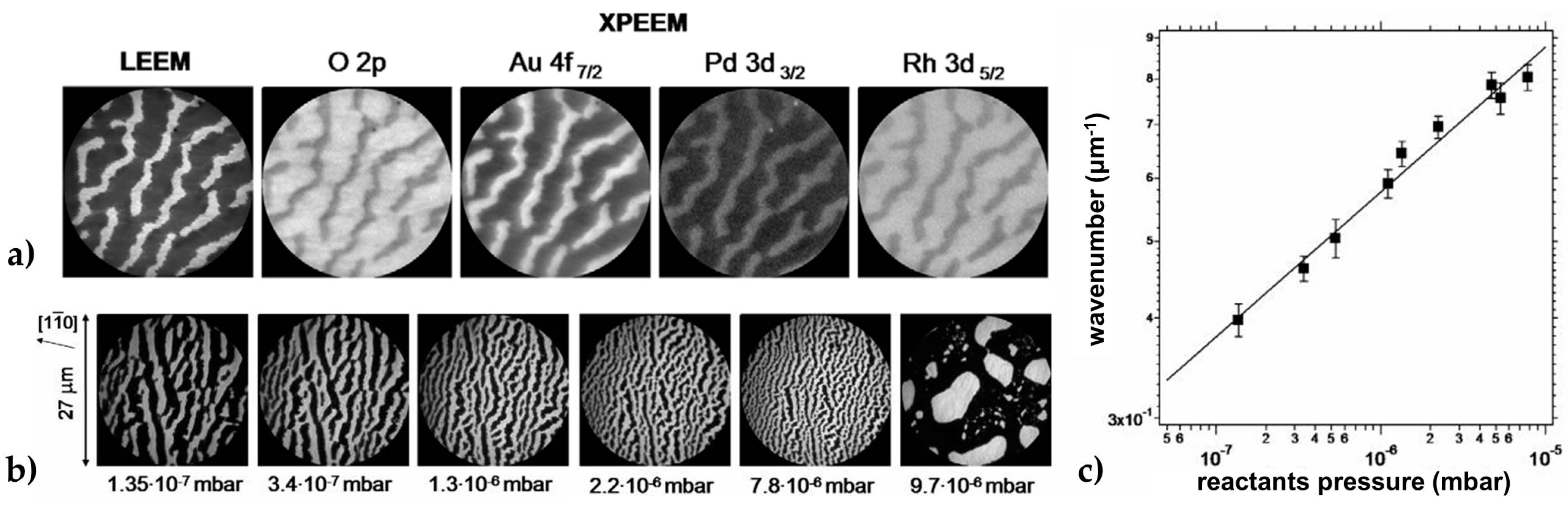
Reaction-Induced Patterning of Au/Rh(110) Surfaces
Chemically Frozen Phase Separation
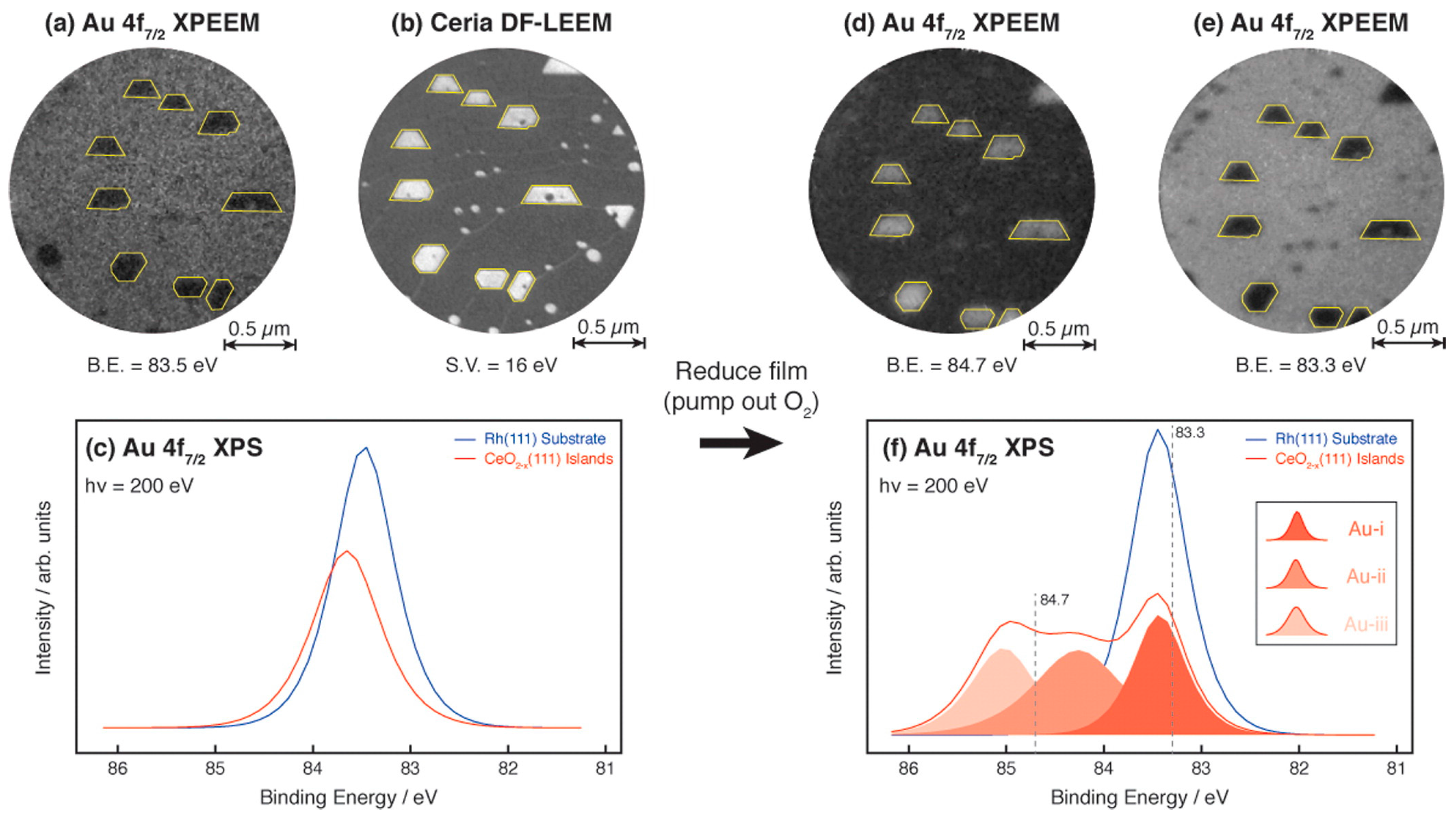
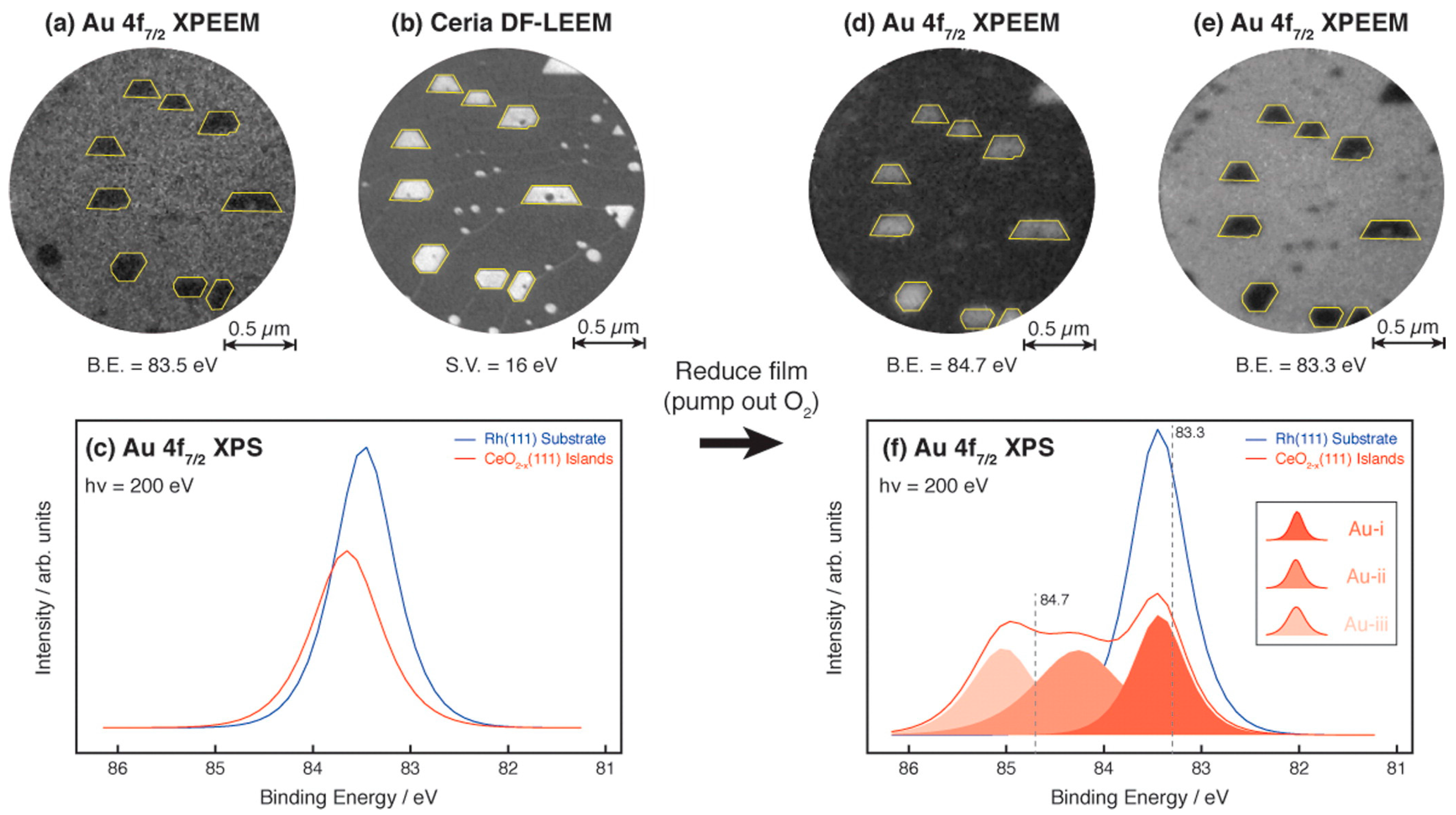
3.1.3. Spectromicroscopy of Au-Supported Nanoparticles
3.1.4. Perspectives
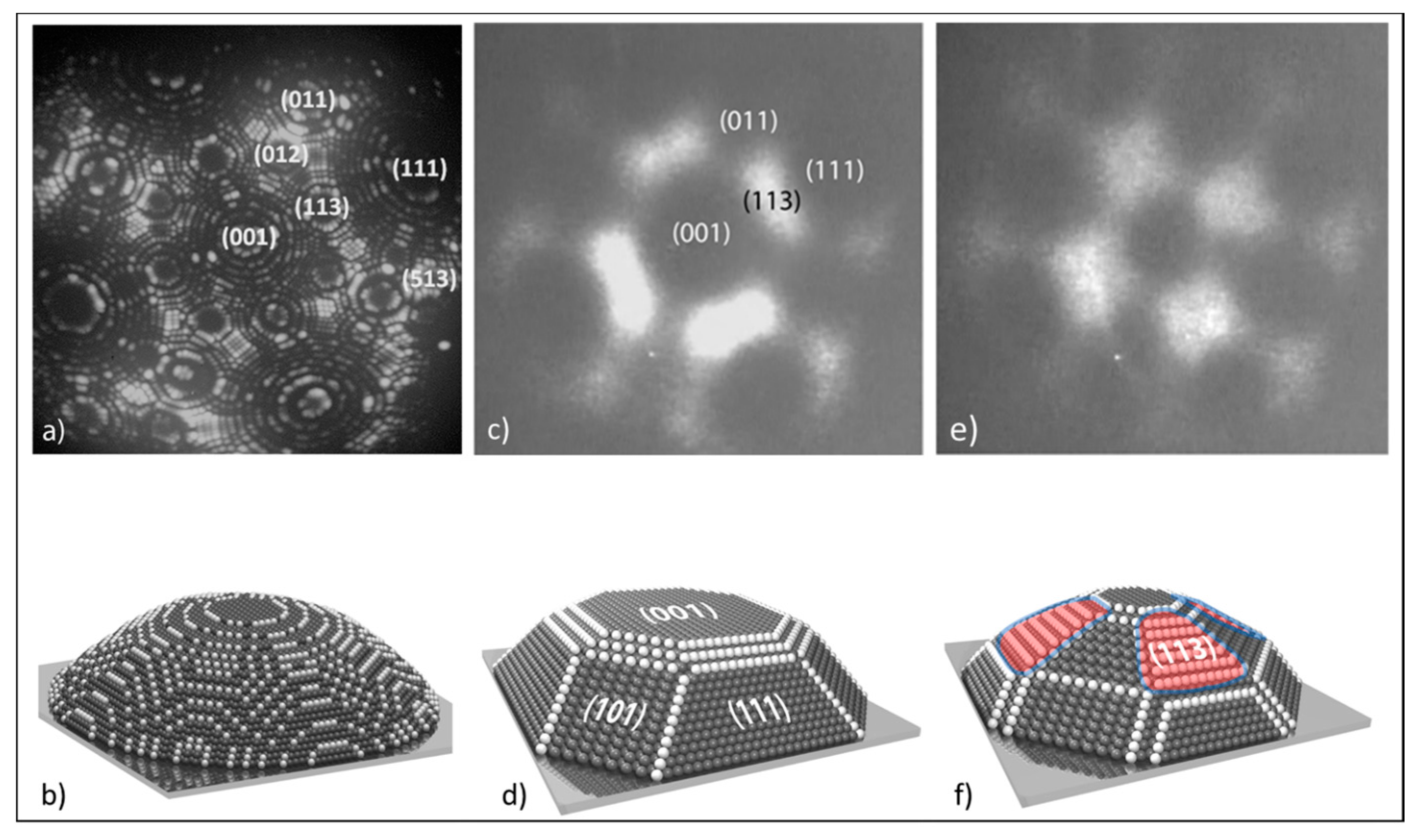
3.2. Study of Processes on a Single Grain of Catalyst by FEM/FIM
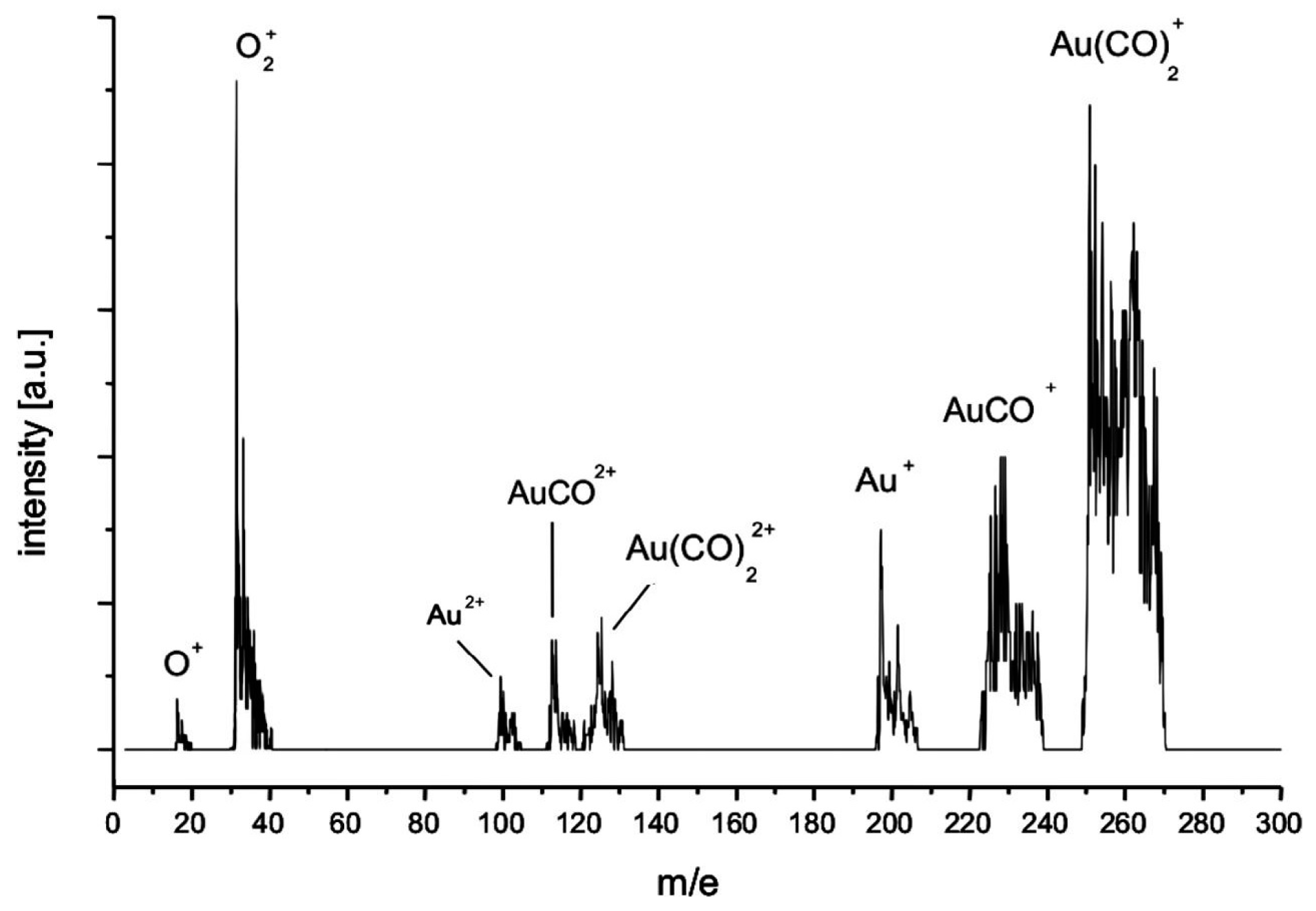
3.2.1. Processes on Au Tips
Water Gas Shift Reaction (WGSR)
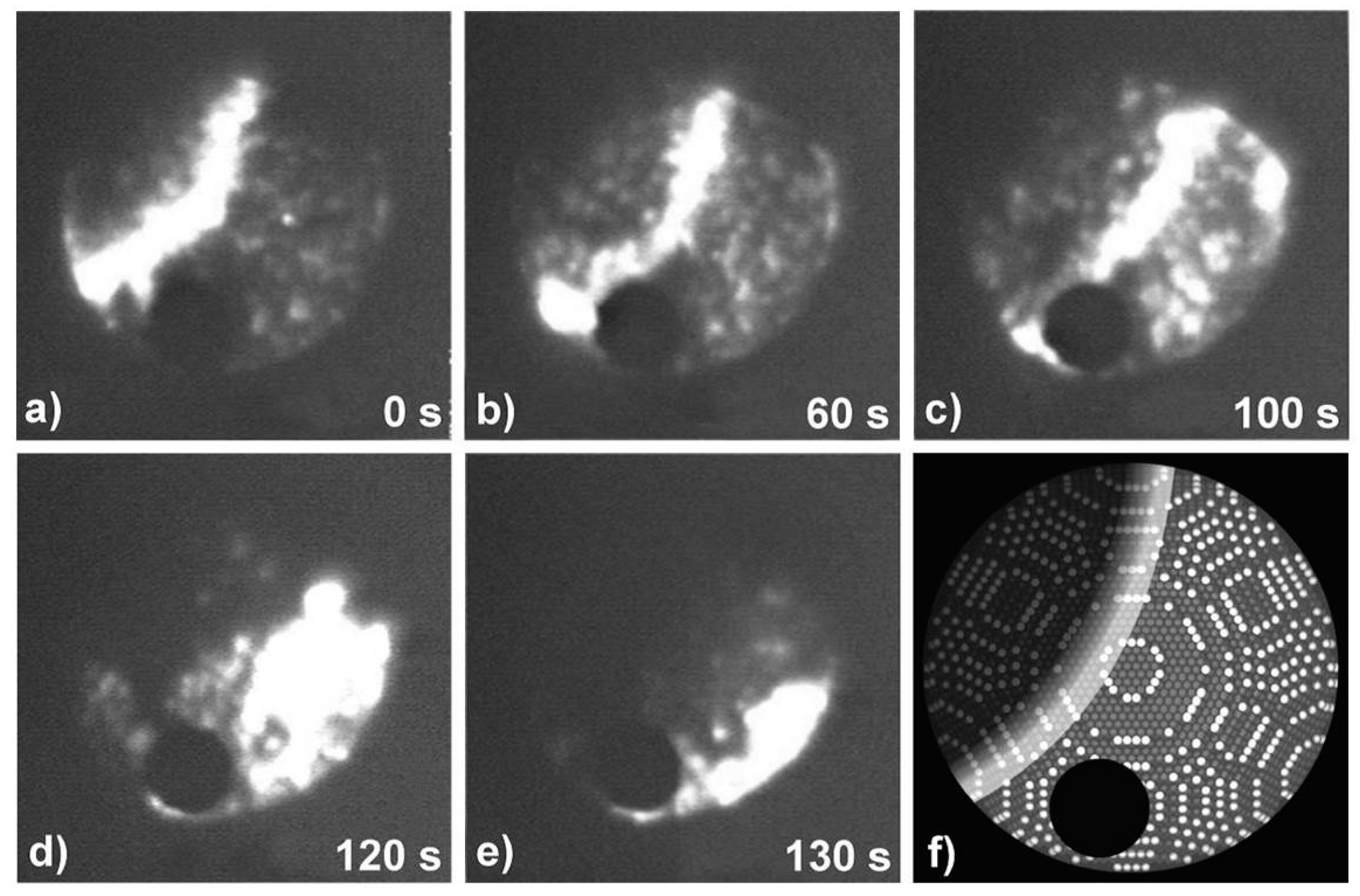
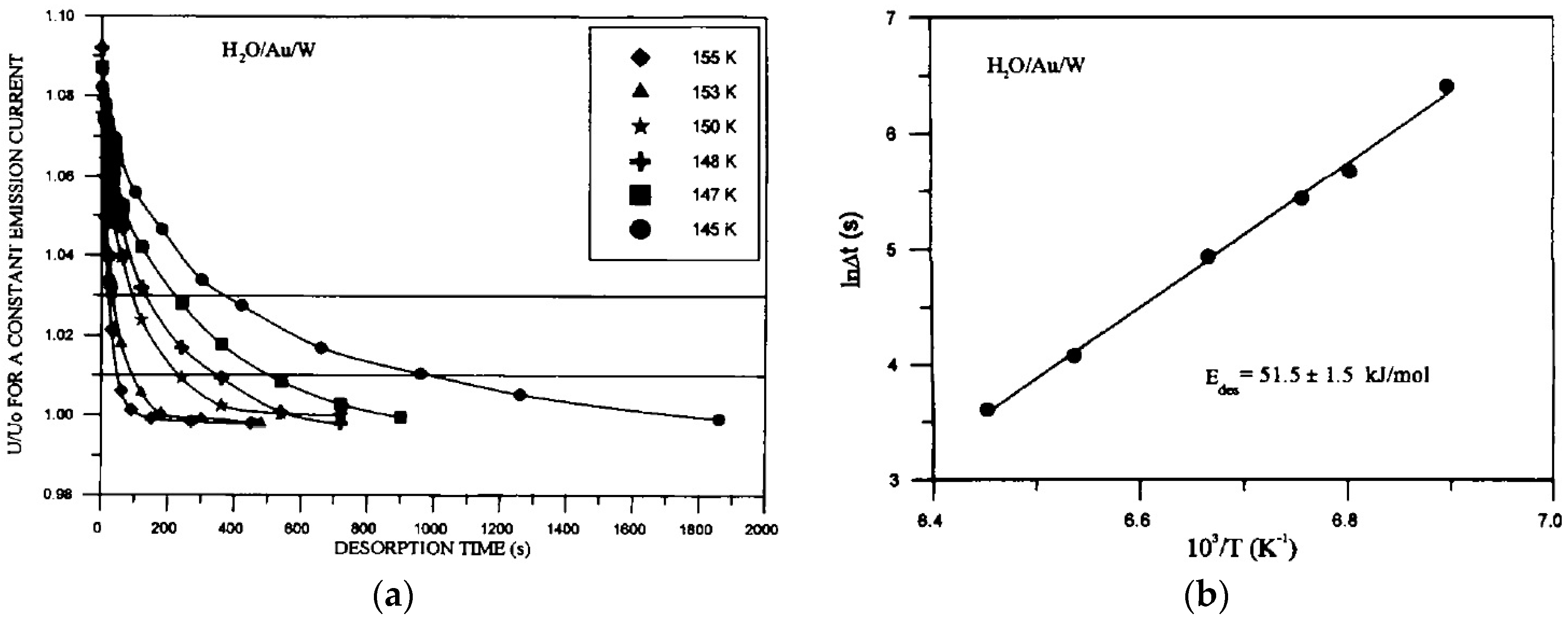
3.2.2. Processes on Au-Covered Tips: Determining H2O Adsorption/Desorption/Diffusion Energies by FEM
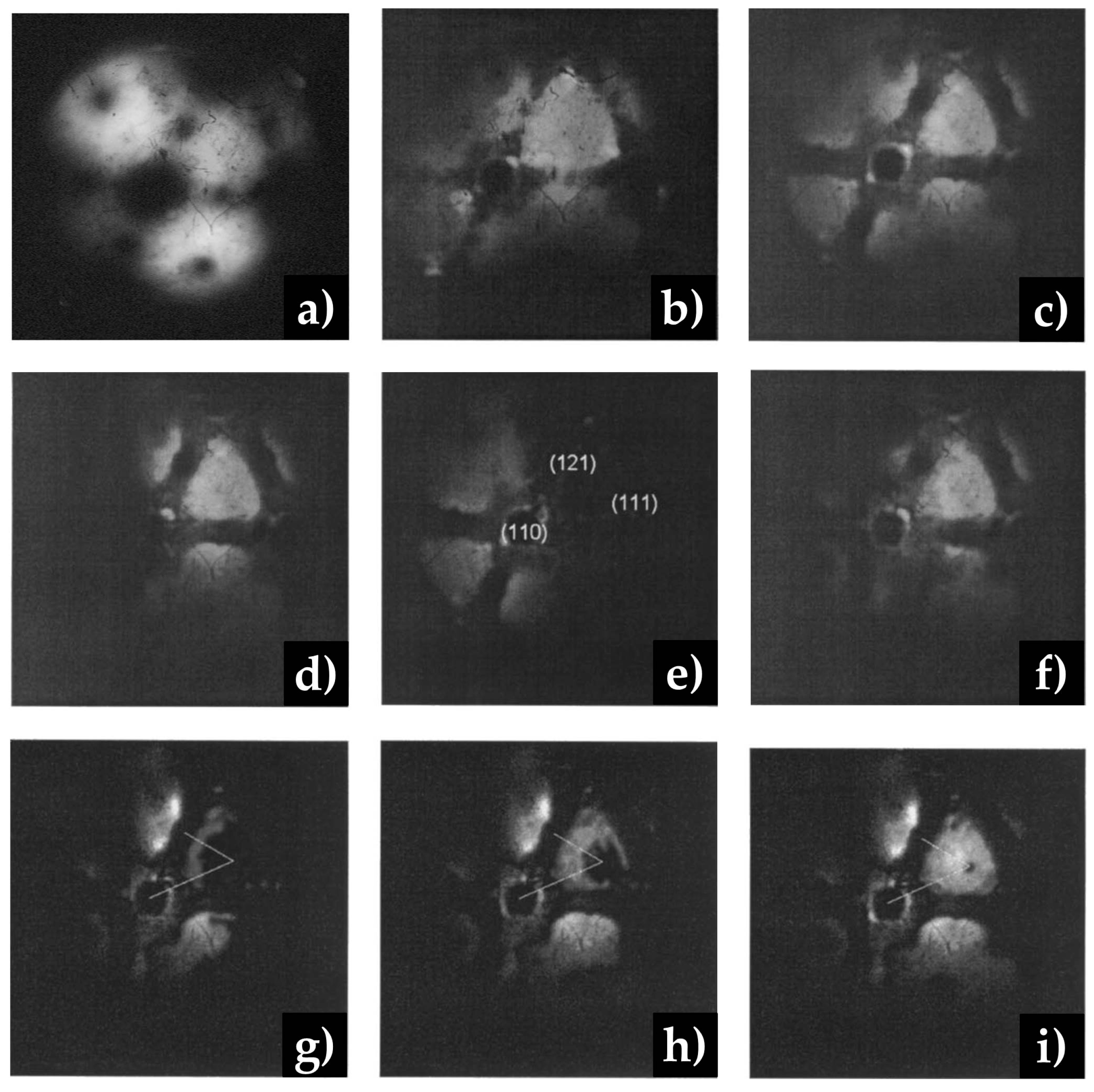
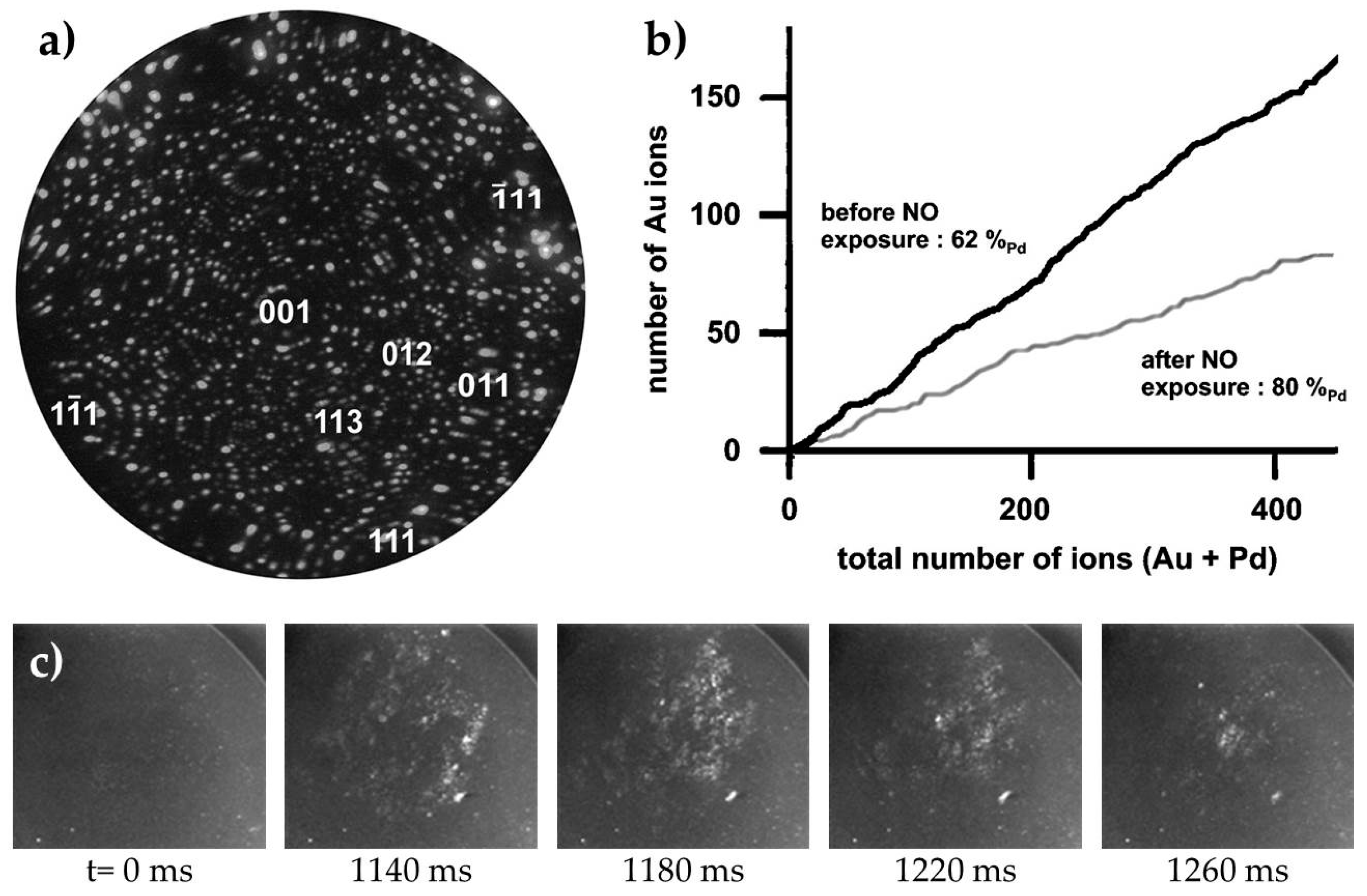
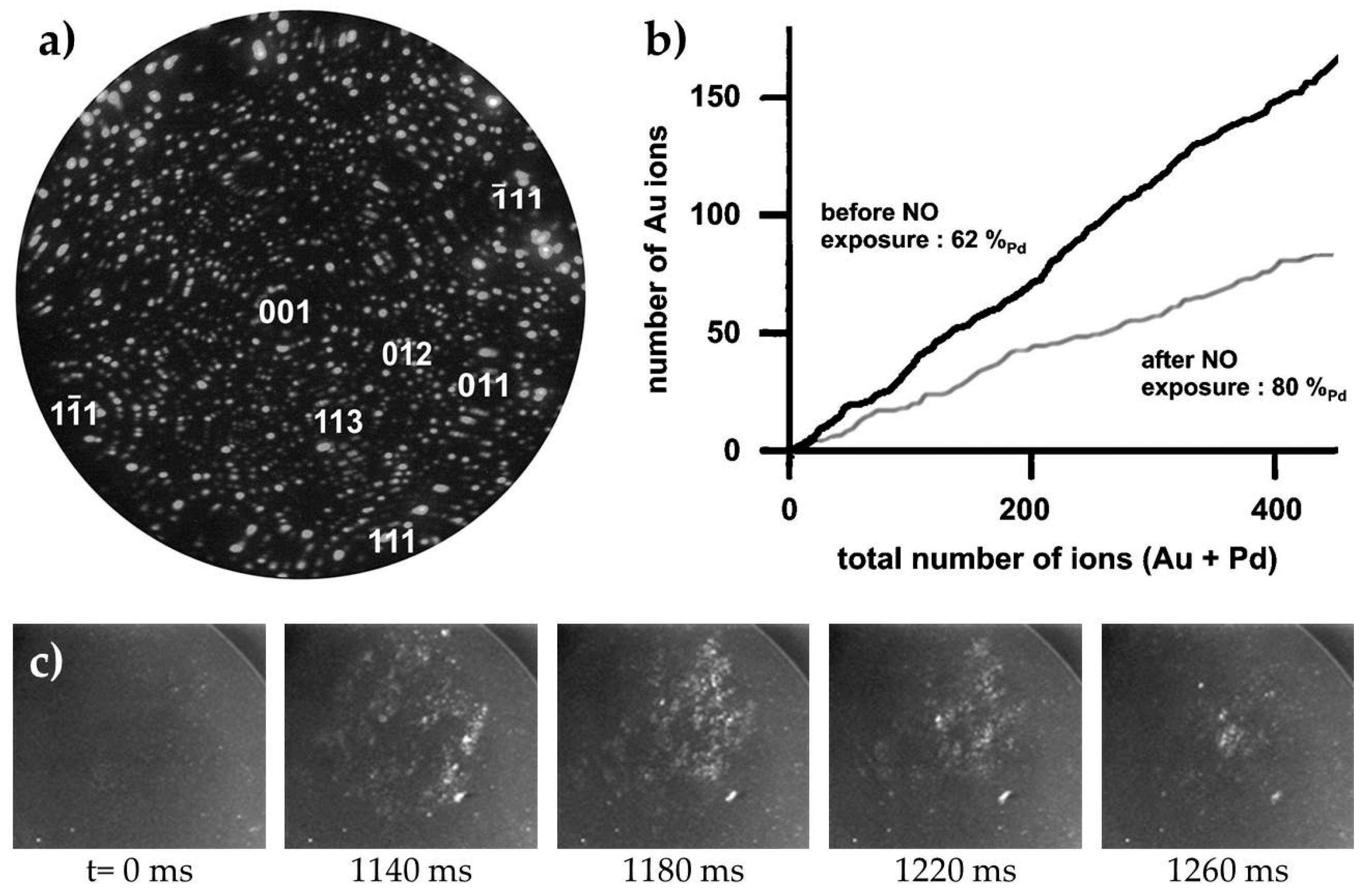
3.2.3. Processes on Au-Based Alloys
Oxygen Interaction with Au-Ag Alloys
Towards deNOx Reactions on Au-Based Catalysts
3.2.4. Perspectives
3.3. Study of Processes on Supported and Self-Supported Catalysts by E-TEM
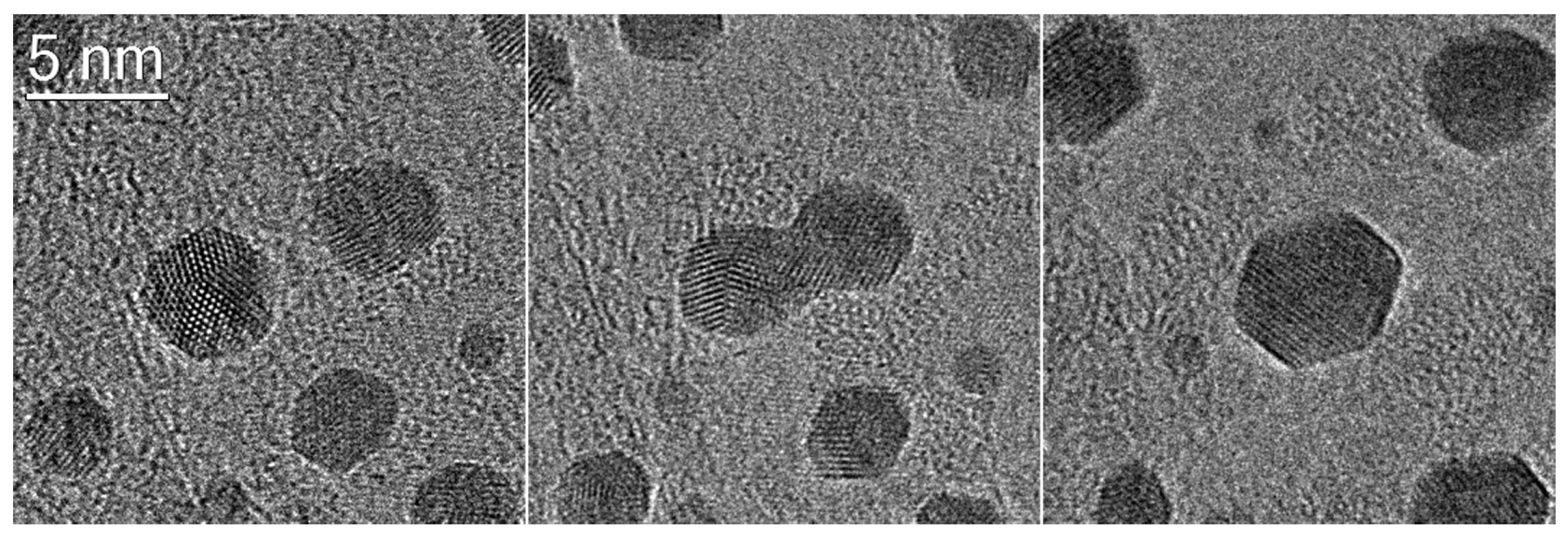
3.3.1. Processes on Supported Au Nanoparticles
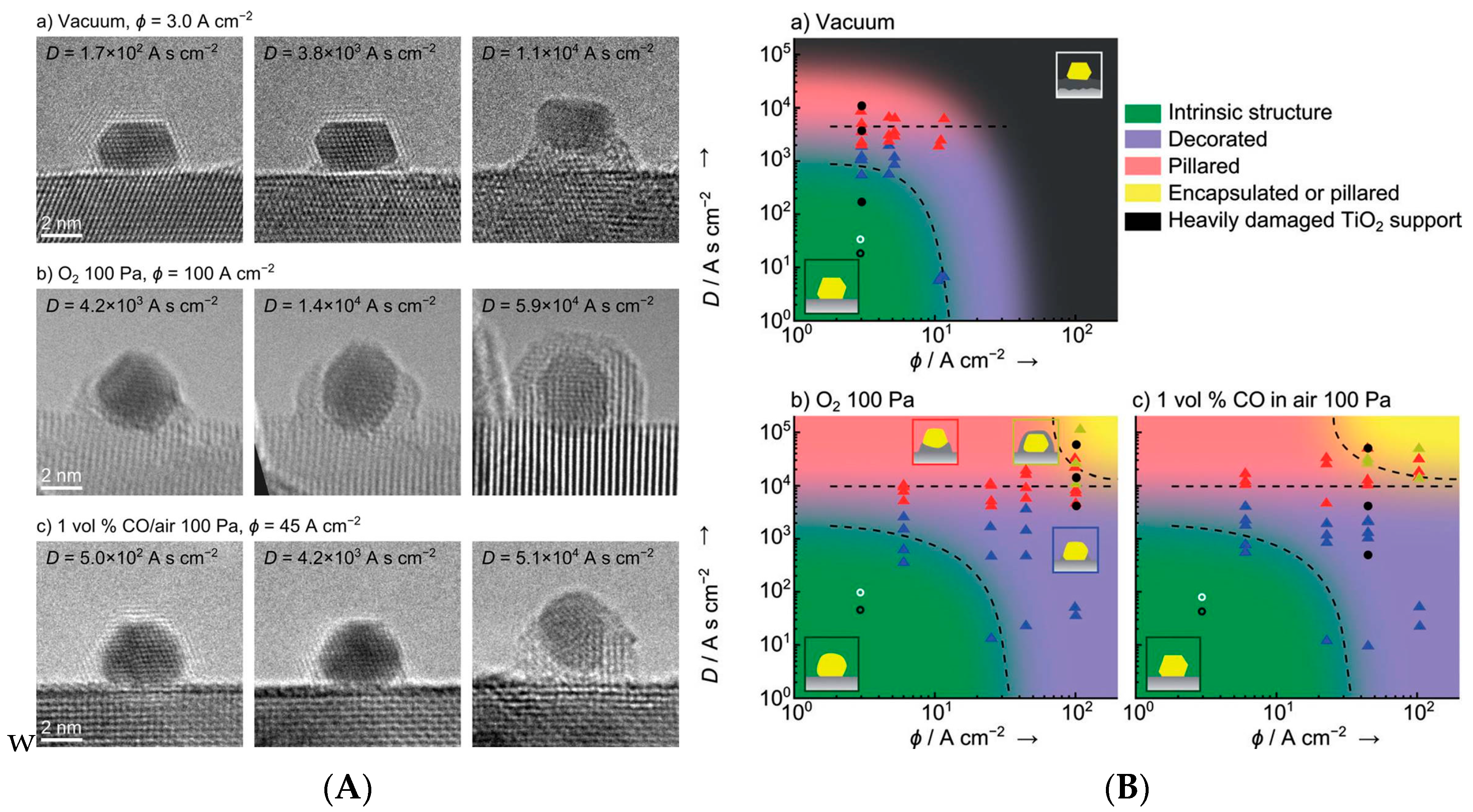
Coalescence of Au Particles
Importance of the Support for the Stability
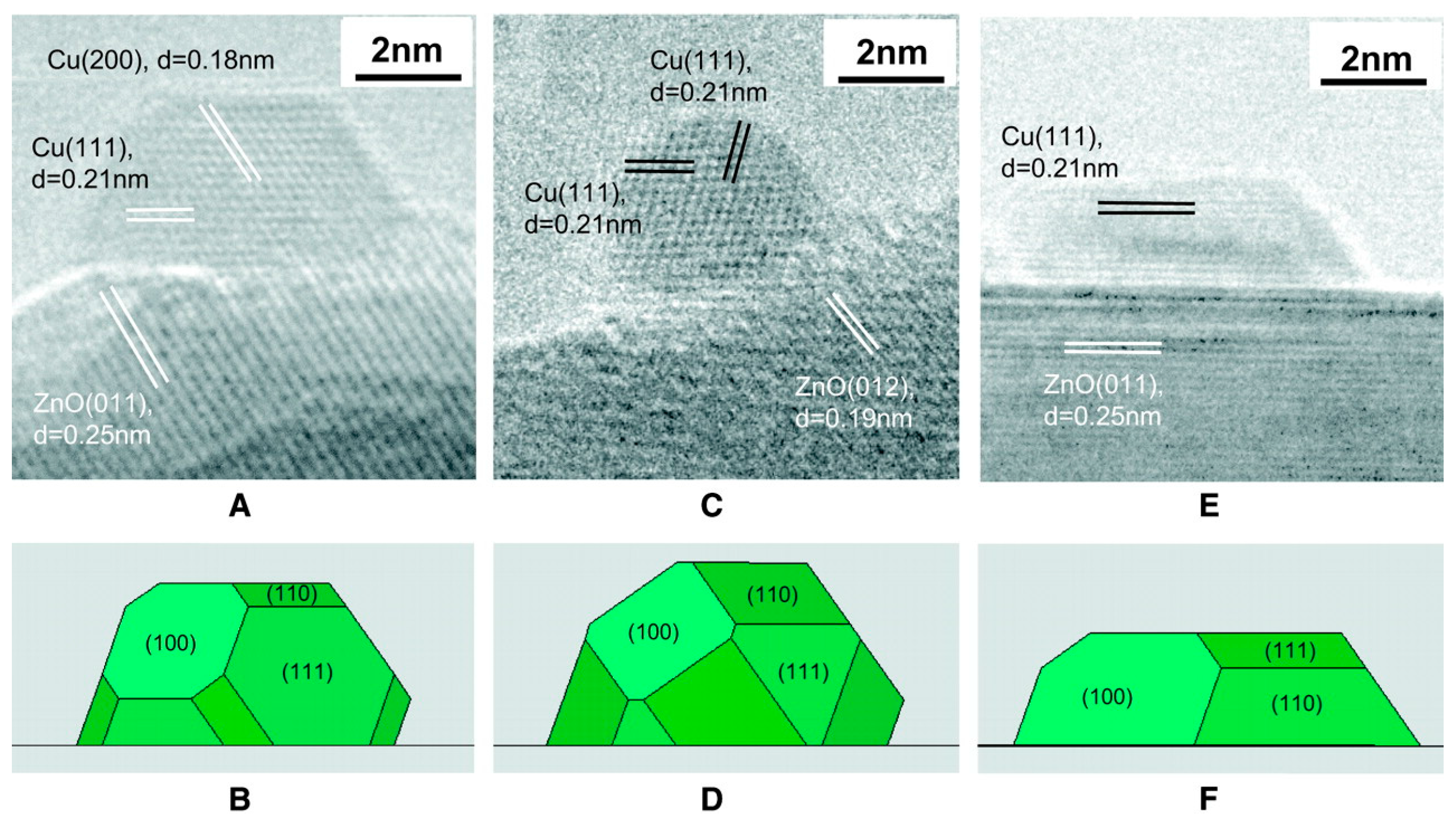
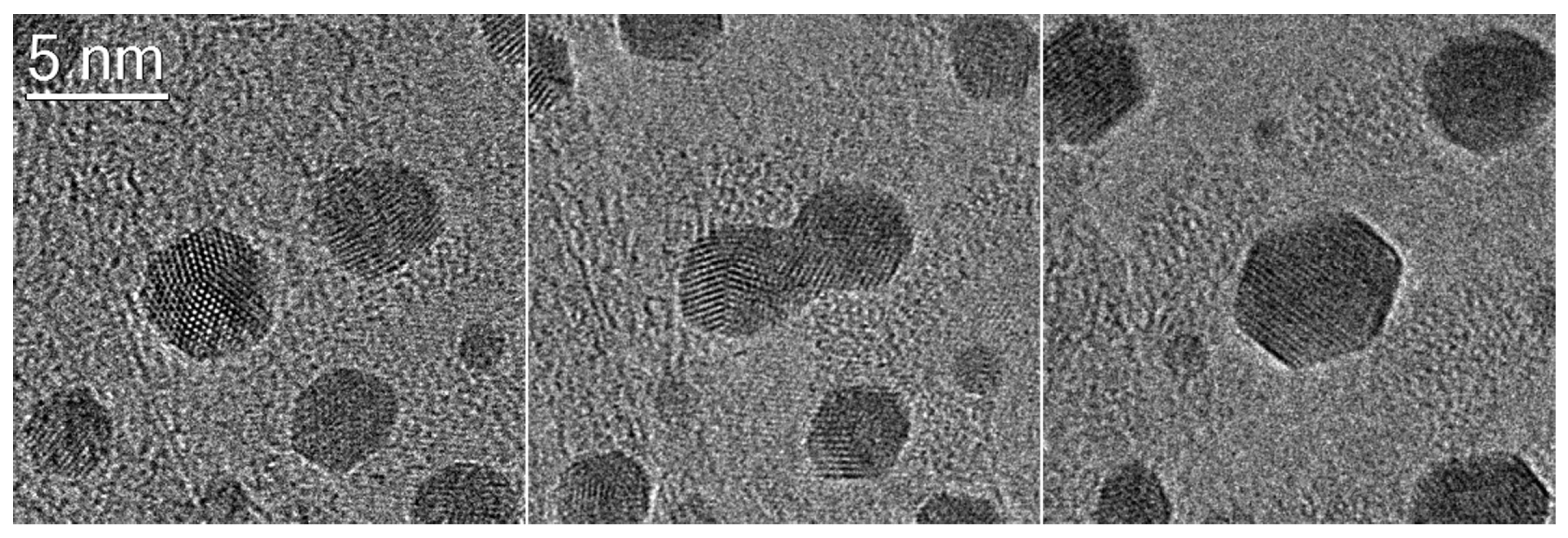
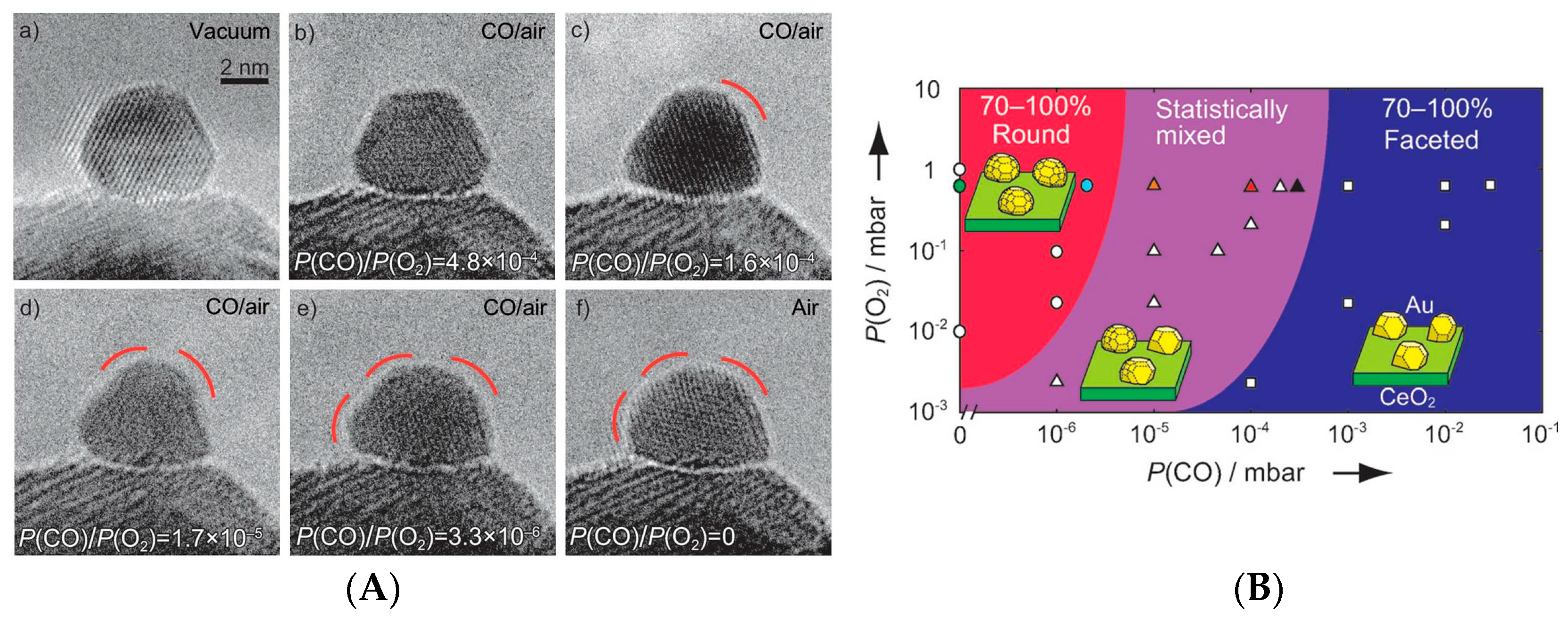
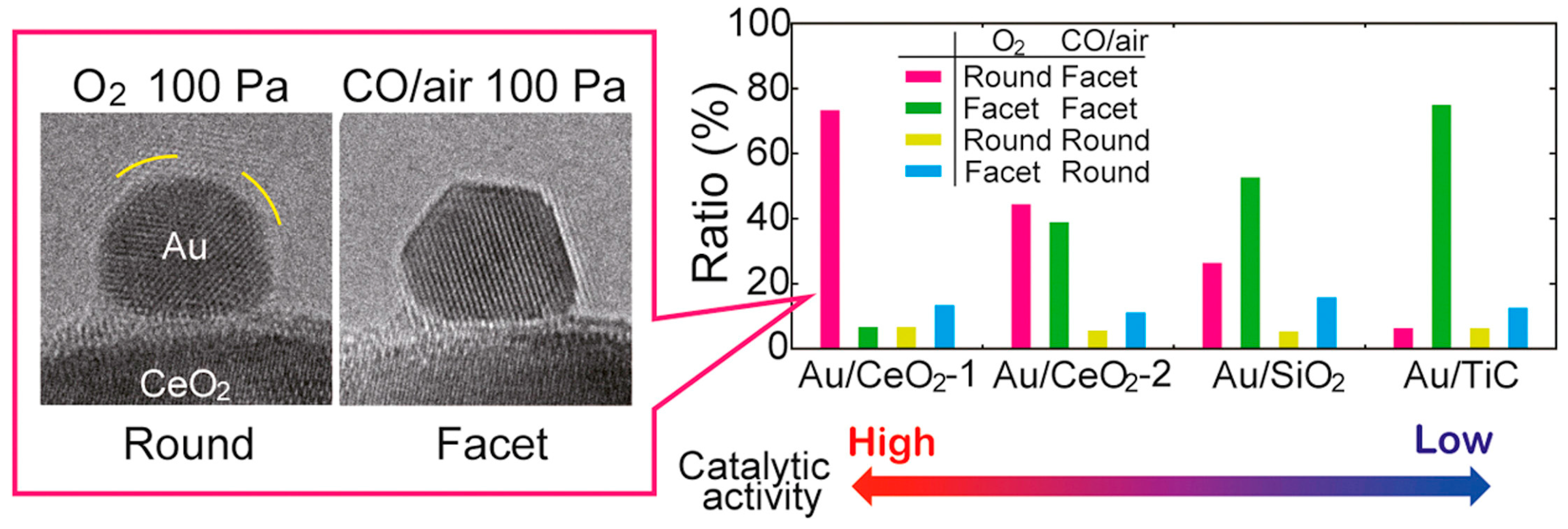
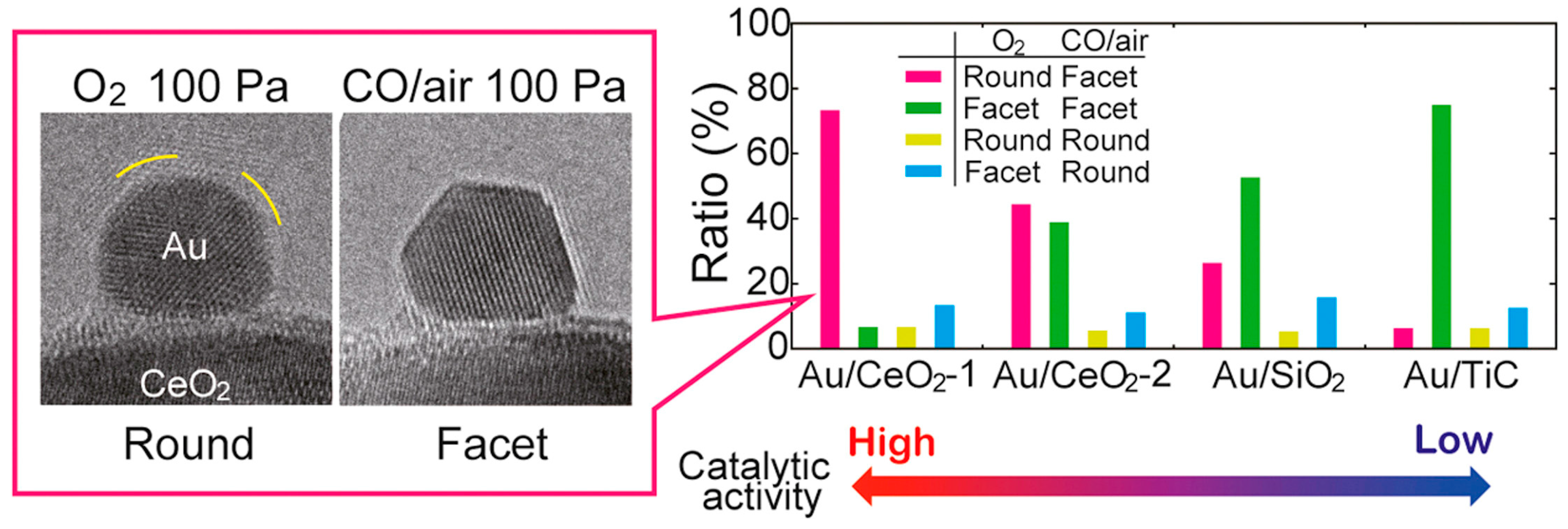
Morphology of the Active Nanoparticles
Importance of the Support for the Activity
3.3.2. Processes on Self-Supported Au
Nanoporous Au as Catalyst for CO Oxidation
Nanoporous Au as Catalyst for Selective Alcohol Oxidation
3.3.3. Perspectives
4. Conclusions and Perspectives
- -
- The addition of a minor amount of surface species may severely affect the global reactivity of the catalyst, as it was demonstrated on Au-covered Pt in PEEM and Au-Ag alloys in nanoporous gold by E-TEM;
- -
- Alloying induces synergistic effects, as shown on Au + Pd-covered Rh by LEEM and Au-Pd catalysts for NO hydrogenation by FEM, a reaction that was not possible on pure Au;
- -
- The size-effect in gold catalysis arises from the step and kinks density, as shown on nanoporous structures by E-TEM where the reactivity of the npAu depends on the size of the ligaments. These studies also prove the relevance of using a tip-sample of 20–50 nm by FIM to study fundamental processes;
- -
- The presence of silver can be used to stabilise the structure, but also to guide the selectivity of the reaction, as it was studied by FEM and E-TEM;
- -
- CO oxidation has been studied by PEEM, FIM and E-TEM; the water gas shift reaction by FIM and E-TEM, but current research focuses on more complex reactions over Au-based catalysts.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liotta, L.F. Catalytic oxidation of volatile organic compounds on supported noble metals. Appl. Catal. B Environ. 2010, 100, 403–412. [Google Scholar] [CrossRef]
- Wang, J.; Aguilar-Rios, G.; Wang, R. Inhibition of carbon monoxide on methanol oxidation over alumina supported Ag, Pd and Ag–Pd catalysts. Appl. Surf. Sci. 1999, 147, 44–51. [Google Scholar] [CrossRef]
- Brunet, J.; Genty, E.; Landkocz, Y.; Al Zallouha, M.; Billet, S.; Courcot, D.; Siffert, S.; Thomas, D.; De Weireld, G.; Cousin, R. Identification of by-products issued from the catalytic oxidation of toluene by chemical and biological methods. Comptes Rendus Chim. 2015, 18, 1084–1093. [Google Scholar] [CrossRef]
- Patterson, M.J.; Angove, D.E.; Cant, N.W. The effect of carbon monoxide on the oxidation of four C6 to C8 hydrocarbons over platinum, palladium and rhodium. Appl. Catal. B Environ. 2000, 26, 47–57. [Google Scholar] [CrossRef]
- Patterson, M.J.; Angove, D.E.; Cant, N.W. The effect of metal order on the oxidation of a hydrocarbon mixture over alumina-supported combined platinum/rhodium catalysts. Appl. Catal. B Environ. 2001, 35, 53–58. [Google Scholar] [CrossRef]
- Cant, N.W.; Angove, D.E.; Chambers, D.C. Nitrous oxide formation during the reaction of simulated exhaust streams over rhodium, platinum and palladium catalysts. Appl. Catal. B Environ. 1998, 17, 63–73. [Google Scholar] [CrossRef]
- Al Zallouha, M.; Landkocz, Y.; Brunet, J.; Cousin, R.; Genty, E.; Courcot, D.; Siffert, S.; Shirali, P.; Billet, S. Usefulness of toxicology validation of VOCs catalytic degradation by air-liquid interface exposure system. Environ. Res. 2017, 152, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Tidahy, H.L.; Siffert, S.; Wyrwalski, F.; Lamonier, J.-F.; Aboukaïs, A. Catalytic activity of copper and palladium based catalysts for toluene total oxidation. Catal. Today 2007, 119, 317–320. [Google Scholar] [CrossRef]
- Liotta, L.F.; Di Carlo, G.; Pantaleo, G.; Venezia, A.M.; Deganello, G. Co3O4/CeO2 composite oxides for methane emissions abatement: Relationship between Co3O4-CeO2 interaction and catalytic activity. Appl. Catal. B Environ. 2006, 66, 217–227. [Google Scholar] [CrossRef]
- Genty, E.; Cousin, R.; Capelle, S.; Gennequin, C.; Siffert, S. Catalytic oxidation of toluene and CO over nanocatalysts derived from hydrotalcite-like compounds (X62+Al23+): Effect of the bivalent cation. Eur. J. Inorg. Chem. 2012, 2012, 2802–2811. [Google Scholar] [CrossRef]
- Kim, S.C.; Shim, W.G. Catalytic combustion of VOCs over a series of manganese oxide catalysts. Appl. Catal. B Environ. 2010, 98, 180–185. [Google Scholar] [CrossRef]
- Cook, K.M.; Poudyal, S.; Miller, J.T.; Bartholomew, C.H.; Hecker, W.C. Reducibility of alumina-supported cobalt Fischer-Tropsch catalysts: Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size. Appl. Catal. A Gen. 2012, 449, 69–80. [Google Scholar] [CrossRef]
- Ataloglou, T.; Vakros, J.; Bourikas, K.; Fountzoula, C.; Kordulis, C.; Lycourghiotis, A. Influence of the preparation method on the structure–activity of cobalt oxide catalysts supported on alumina for complete benzene oxidation. Appl. Catal. B Environ. 2005, 57, 299–312. [Google Scholar] [CrossRef]
- Genty, E.; Brunet, J.; Pequeux, R.; Capelle, S.; Siffert, S.; Cousin, R. Effect of Ce substituted hydrotalcite-derived mixed oxides on total catalytic oxidation of air pollutant. Mater. Today Proc. 2016, 3, 277–281. [Google Scholar] [CrossRef]
- Fuentes, E.M.; da Costa Faro, A.; de Freitas Silva, T.; Assaf, J.M.; Rangel, M.D.C. A comparison between copper and nickel-based catalysts obtained from hydrotalcite-like precursors for WGSR. Catal. Today 2011, 171, 290–296. [Google Scholar] [CrossRef]
- Palomares, A.E.; Uzcategui, A.; Corma, A. NOx storage/reduction catalysts based in cobalt/copper hydrotalcites. Catal. Today 2008, 137, 261–266. [Google Scholar] [CrossRef]
- Casapu, M.; Bernhard, A.; Peitz, D.; Mehring, M.; Elsener, M.; Krocher, O. A Niobia-Ceria based multi-purpose catalyst for selective catalytic reduction of NOx urea hydrolysis and soot oxidation in diesel exhaust. Appl. Catal. B Environ. 2011, 103, 79–84. [Google Scholar] [CrossRef]
- Brunet, J.; Genty, E.; Bugnon, L.; De Weireld, G.; Thomas, D.; Decroly, A.; Siffert, S.; Cousin, R. Co-Al-Ce mixed oxide materials prepared by hydrotalcite way for VOCs total oxidation in micro- and semi-pilot scale. Mater. Today Proc. 2016, 3, 188–193. [Google Scholar] [CrossRef]
- Bera, P.; Hegde, M.S. Noble metal ions in CeO2 and TiO2: Synthesis, structure and catalytic properties. RSC Adv. 2015, 5, 94949–94979. [Google Scholar] [CrossRef]
- Zheng, T.; He, J.; Zhao, Y.; Xia, W.; He, J. Precious metal-support interaction in automotive exhaust catalysts. J. Rare Earths 2014, 32, 97–107. [Google Scholar] [CrossRef]
- Kinnunen, N.M.; Hirvi, J.T.; Kallinen, K.; Maunula, T.; Keenan, M.; Suvanto, M. Case study of a modern lean-burn methane combustion catalyst for automotive applications: What are the deactivation and regeneration mechanisms? Appl. Catal. B Environ. 2017, 207, 114–119. [Google Scholar] [CrossRef]
- Ordóñez, S.; Bello, L.; Sastre, H.; Rosal, R.; Fernando, V.D. Kinetics of the deep oxidation of benzene, toluene, n-hexane and their binary mixtures over a platinum on γ-alumina catalyst. Appl. Catal. B Environ. 2002, 38, 139–149. [Google Scholar] [CrossRef]
- Benard, S.; Ousmane, M.; Retailleau, L.; Boreave, A.; Vernoux, P.; Giroir-Fendler, A. Catalytic removal of propene and toluene in air over noble metal catalyst. Can. J. Civ. Eng. 2009, 36, 1935–1945. [Google Scholar] [CrossRef]
- Mergler, Y.J.; van Aalst, A.; van Delft, J.; Nieuwenhuys, B.E. CO oxidation over promoted Pt catalysts. Appl. Catal. B Environ. 1996, 10, 245–261. [Google Scholar] [CrossRef]
- Liotta, L.F.; Ousmane, M.; Di Carlo, G.; Pantaleo, G.; Deganello, G.; Boreave, A.; Giroir-Fendler, A. Catalytic removal of toluene over Co3O4–CeO2 mixed oxide catalysts: Comparison with Pt/Al2O3. Catal. Lett. 2008, 127, 270–276. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 405–408. [Google Scholar] [CrossRef]
- Haruta, M.; Yamada, N.; Kobayashi, T.; Iijima, S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide. J. Catal. 1989, 115, 301–309. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, Fe2O3 and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Haruta, M. Size- and support-dependency in the catalysis of gold. Catal. Today 1997, 36, 153–166. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Hutchings, G.J.; Joffe, R. A novel process for the co-synthesis of vinyl chloride monomer and sodium carbonate using a gold catalyst. Appl. Catal. 1986, 20, 215–218. [Google Scholar] [CrossRef]
- Nkosi, B.; Coville, N.J.; Hutchings, G.J. Vapour phase hydrochlorination of acetylene with group VIII and IB metal chloride catalysts. Appl. Catal. 1988, 43, 33–39. [Google Scholar] [CrossRef]
- Andreeva, D.; Ivanov, I.; Ilieva, L.; Abrashev, M.V.; Zanella, R.; Sobczak, J.W.; Lisowski, W.; Kantcheva, M.; Avdeev, G.; Petrov, K. Gold catalysts supported on ceria doped by rare earth metals for water gas shift reaction: Influence of the preparation method. Appl. Catal. A Gen. 2009, 357, 159–169. [Google Scholar] [CrossRef]
- Ivanova, S.; Petit, C.; Pitchon, V. Application of alumina supported gold-based catalysts in total oxidation of CO and light hydrocarbons mixture. Catal. Today 2006, 113, 182–186. [Google Scholar] [CrossRef]
- Vargas, J.C.; Ivanova, S.; Thomas, S.; Roger, A.-C.; Pitchon, V. Influence of gold on Ce-Zr-Co fluorite-type mixed oxide catalysts for ethanol steam reforming. Catalysts 2012, 2, 121–138. [Google Scholar] [CrossRef]
- Genty, E.; Cousin, R.; Gennequin, C.; Capelle, S.; Aboukaïs, A.; Siffert, S. Investigation of Au/hydrotalcite catalysts for toluene total oxidation. Catal. Today 2011, 176, 116–119. [Google Scholar] [CrossRef]
- Ivanova, S.; Petit, C.; Pitchon, V. A new preparation method for the formation of gold nanoparticles on an oxide support. Appl. Catal. A Gen. 2004, 267, 191–201. [Google Scholar] [CrossRef]
- Delannoy, L.; Fajerwerg, K.; Lakshmanan, P.; Potvin, C.; Méthivier, C.; Louis, C. Supported gold catalysts for the decomposition of VOC: Total oxidation of propene in low concentration as model reaction. Appl. Catal. B Environ. 2010, 94, 117–124. [Google Scholar] [CrossRef]
- Zanella, R.; Louis, C.; Giorgio, S.; Touroude, R. Crotonaldehyde hydrogenation by gold supported on TiO2: Structure sensitivity and mechanism. J. Catal. 2004, 223, 328–339. [Google Scholar] [CrossRef]
- Zanella, R.; Giorgio, S.; Shin, C.-H.; Henry, C.R.; Louis, C. Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J. Catal. 2004, 222, 357–367. [Google Scholar] [CrossRef]
- Zanella, R.; Delannoy, L.; Louis, C. Mechanism of deposition of gold precursors onto TiO2 during the preparation by cation adsorption and deposition–precipitation with NaOH and urea. Appl. Catal. A Gen. 2005, 291, 62–72. [Google Scholar] [CrossRef]
- Park, E.D.; Lee, J.S. Effects of Pretreatment Conditions on CO oxidation over supported Au catalysts. J. Catal. 1999, 186, 1–11. [Google Scholar] [CrossRef]
- Ueda, A.; Haruta, M. Nitric oxide reduction with hydrogen, carbon monoxide, and hydrocarbons over gold catalysts. Gold Bull. 1999, 32, 3–11. [Google Scholar] [CrossRef]
- Golunski, S.; Rajaram, R.; Hodge, N.; Hutchings, G.J.; Kiely, C.J. Low-temperature redox activity in co-precipitated catalysts: A comparison between gold and platinum-group metals. Catal. Today 2002, 72, 107–113. [Google Scholar] [CrossRef]
- Barakat, T.; Rooke, J.C.; Genty, E.; Cousin, R.; Siffert, S.; Su, B.-L. Gold catalysts in environmental remediation and water-gas shift technologies. Energy Environ. Sci. 2013, 6, 371. [Google Scholar] [CrossRef]
- Genty, E.; Cousin, R.; Capelle, S.; Siffert, S. Influence of gold on hydrotalcite-like compound catalysts for toluene and CO total oxidation. Catalysts 2013, 3, 966–977. [Google Scholar] [CrossRef]
- Centeno, M.; Ramírez Reina, T.; Ivanova, S.; Laguna, O.; Odriozola, J. Au/CeO2 catalysts: Structure and CO oxidation activity. Catalysts 2016, 6, 158. [Google Scholar] [CrossRef]
- Alshammari, A.; Kalevaru, V.N.; Martin, A. Bimetallic catalysts containing gold and palladium for environmentally important reactions. Catalysts 2016, 6, 1–24. [Google Scholar] [CrossRef]
- Luo, Y.; Seo, H.O.; Kim, K.D.; Kim, M.J.; Tai, W.S.; Burkhart, M.; Kim, Y.D. CO oxidation of Au-Pt nanostructures: Enhancement of catalytic activity of Pt nanoparticles by Au. Catal. Lett. 2010, 134, 45–50. [Google Scholar] [CrossRef]
- Destro, P.; Marras, S.; Manna, L.; Colombo, M.; Zanchet, D. AuCu alloy nanoparticles supported on SiO2: Impact of redox pretreatments in the catalyst performance in CO oxidation. Catal. Today 2016, 282, 105–110. [Google Scholar] [CrossRef]
- Hosseini, M.; Barakat, T.; Cousin, R.; Aboukaïs, A.; Su, B.-L.; De Weireld, G.; Siffert, S. Catalytic performance of core–shell and alloy Pd–Au nanoparticles for total oxidation of VOC: The effect of metal deposition. Appl. Catal. B Environ. 2012, 111, 218–224. [Google Scholar] [CrossRef]
- Peneau, V.; He, Q.; Shaw, G.; Kondrat, S.A.; Davies, T.E.; Miedziak, P.; Forde, M.; Dimitratos, N.; Kiely, C.J.; Hutchings, G.J. Selective catalytic oxidation using supported gold–platinum and palladium–platinum nanoalloys prepared by sol-immobilisation. Phys. Chem. Chem. Phys. 2013, 15, 10636–10644. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Siffert, S.; Cousin, R.; Aboukaïs, A.; Hadj-Sadok, Z.; Su, B.-L. Total oxidation of VOCs on Pd and/or Au supported on TiO2/ZrO2 followed by “operando” DRIFT. Comptes Rendus Chim. 2009, 12, 654–659. [Google Scholar] [CrossRef]
- Qian, L.; Sha, Y.; Yang, X. Simple and convenient preparation of Au-Pt core-shell nanoparticles on surface via a seed growth method. Thin Solid Films 2006, 515, 1349–1353. [Google Scholar] [CrossRef]
- Zhang, L.; Xie, Z.; Gong, J. Shape-controlled synthesis of Au-Pd bimetallic nanocrystals for catalytic applications. Chem. Soc. Rev. 2016, 45, 3916–3934. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Chen, B.; Ji, N.; Chen, F.; Zhang, Y.; Zhang, Z. Nanocomposites of size-controlled gold nanoparticles and graphene oxide: Formation and applications in SERS and catalysis. Nanoscale 2010, 2, 2733–2738. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Juárez, R.; Marino, T.; Molinari, R.; García, H. Influence of excitation wavelength (UV or visible light) on the photocatalytic activity of titania containing gold nanoparticles for the generation of hydrogen or oxygen from water. J. Am. Chem. Soc. 2011, 133, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Fenger, R.; Fertitta, E.; Kirmse, H.; Thünemann, A.F.; Rademann, K. Size dependent catalysis with CTAB-stabilized gold nanoparticles. Phys. Chem. Chem. Phys. 2012, 14, 9343–9349. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Ma, H.; Xiao, S.; Shen, M.; Guo, R.; Cao, X.; Shi, X. Facile immobilization of gold nanoparticles into electrospun polyethyleneimine/polyvinyl alcohol nanofibers for catalytic applications. J. Mater. Chem. 2011, 21, 4493–4501. [Google Scholar] [CrossRef]
- Shang, C.; Liu, Z.P. Origin and activity of gold nanoparticles as aerobic oxidation catalysts in aqueous solution. J. Am. Chem. Soc. 2011, 133, 9938–9947. [Google Scholar] [CrossRef] [PubMed]
- Matassa, R.; Familiari, G.; Battaglione, E.; Sibilia, C.; Leahu, G.; Belardini, A.; Venditti, I.; Fontana, L.; Fratoddi, I. Electron microscopy reveals a soluble hybrid network of individual nanocrystals self-anchored by bifunctional thiol fluorescent bridges. Nanoscale 2016, 8, 18161–18169. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Sanchez, J.A.; Dimitratos, N.; Hammond, C.; Brett, G.L.; Kesavan, L.; White, S.; Miedziak, P.; Tiruvalam, R.; Jenkins, R.L.; Carley, A.F.; et al. Facile removal of stabilizer-ligands from supported gold nanoparticles. Nat. Chem. 2011, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Venditti, I.; Fontana, L.; Fratoddi, I.; Battocchio, C.; Cametti, C.; Sennato, S.; Mura, F.; Sciubba, F.; Delfini, M.; Russo, M.V. Direct interaction of hydrophilic gold nanoparticles with dexamethasone drug: Loading and release study. J. Colloid Interface Sci. 2014, 418, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Venditti, I.; Palocci, C.; Chronopoulou, L.; Fratoddi, I.; Fontana, L.; Diociaiuti, M.; Russo, M.V. Candida rugosa lipase immobilization on hydrophilic charged gold nanoparticles as promising biocatalysts: Activity and stability investigations. Colloids Surf. B 2015, 131, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Porcaro, F.; Battocchio, C.; Antoccia, A.; Fratoddi, I.; Venditti, I.; Fracassi, A.; Luisetto, I.; Russo, M.V.; Polzonetti, G. Synthesis of functionalized gold nanoparticles capped with 3-mercapto-1-propansulfonate and 1-thioglucose mixed thiols and “in vitro” bioresponse. Colloids Surf. B 2016, 142, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Venditti, I.; Hassanein, T.F.; Fratoddi, I.; Fontana, L.; Battocchio, C.; Rinaldi, F.; Carafa, M.; Marianecci, C.; Diociaiuti, M.; Agostinelli, E.; et al. Bioconjugation of gold-polymer core-shell nanoparticles with bovine serum amine oxidase for biomedical applications. Colloids Surf. B 2015, 134, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Fratoddi, I.; Venditti, I.; Ksenzov, D.; Russo, M.V.; Grigorian, S. Structural studies on drop-cast film based on functionalized gold nanoparticles network: The effect of thermal treatment. Appl. Surf. Sci. 2016, 369, 115–119. [Google Scholar] [CrossRef]
- Ertl, G.; Freund, H.-J. Catalysis and surface science. Phys. Today 1999, 32–38. [Google Scholar] [CrossRef]
- Rotermund, H.H. Imaging pattern formation in surface reactions from ultra-high vacuum up to atmospheric pressures. Surf. Sci. 1997, 386, 10–23. [Google Scholar] [CrossRef]
- Osterlund, L.; Rasmussen, P.B.; Thostrup, P.; Laegsgaard, E.; Stensgaard, I.; Besenbacher, F. Bridging the pressure gap in surface science at the atomic level: H/Cu(110). Phys. Rev. Lett. 2001, 86, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Haas, G.; Pletcher, T.D.; Bonilla, G.; Jachimowski, T.A.; Rotermund, H.H.; Lauterbach, J. Ellipsomicroscopy for surface imaging: A novel tool to investigate surface dynamics. J. Vac. Sci. Technol. 1998, 16, 1117–1121. [Google Scholar] [CrossRef]
- Besenbacher, F.; Lauritsen, J.V.; Wendt, S. STM studies of model catalysts. Nano Today 2007, 2, 30–39. [Google Scholar] [CrossRef]
- Besenbacher, F.; Lauritsen, J.V.; Linderoth, T.R.; Lægsgaard, E.; Vang, R.T.; Wendt, S. Atomic-scale surface science phenomena studied by scanning tunneling microscopy. Surf. Sci. 2009, 603, 1315–1327. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2003. [Google Scholar]
- Imbihl, R.; Behm, R.J.; Schlogl, R. Bridging the pressure and material gap in heterogeneous catalysis. Phys. Chem. Chem. Phys. 2007, 9, 3459. [Google Scholar] [PubMed]
- Genty, E.; Brunet, J.; Poupin, C.; Casale, S.; Capelle, S.; Massiani, P.; Siffert, S.; Cousin, R. Co-Al mixed oxides prepared via LDH route using microwaves or ultrasound: Application for catalytic toluene total oxidation. Catalysts 2015, 5, 851–867. [Google Scholar] [CrossRef]
- Iablokov, V.; Xiang, Y.; Meffre, A.; Fazzini, P.F.; Chaudret, B.; Kruse, N. Size-dependent activity and selectivity of Fe/MCF-17 in the catalytic hydrogenation of carbon monoxide using Fe(0) nanoparticles as precursors. ACS Catal. 2016, 6, 2496–2500. [Google Scholar] [CrossRef]
- De Decker, Y.; Bullara, D.; Barroo, C.; Visart de Bocarmé, T. Nonlinear dynamics of reactive nanosystems: Theory and experiments. In Bottom-Up Self-Organization in Supramolecular Soft Matter; Springer: Berlin/Heidelberg, Germany, 2015; pp. 127–150. [Google Scholar]
- Vogel, D.; Spiel, C.; Schmid, M.; Stoger-Pollach, M.; Schlogl, R.; Suchorski, Y.; Rupprechter, G. The role of defects in the local reaction kinetics of CO oxidation on low- index Pd surfaces. J. Phys. Chem. C 2012, 117, 12054–12060. [Google Scholar] [CrossRef] [PubMed]
- Hiebel, F.; Shong, B.; Chen, W.; Madix, R.J.; Kaxiras, E.; Friend, C.M. Self-assembly of acetate adsorbates drives atomic rearrangement on the Au(110) surface. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, P.W.; Spence, J.C.H. Science of Microscopy; Springer: Berlin/Heidelberg, Germany, 2007; Volume 53. [Google Scholar]
- Gai, P.L. Environmental high resolution electron microscopy of gas-catalyst reactions. Top. Catal. 1999, 8, 97–113. [Google Scholar] [CrossRef]
- Block, J.H.; Ehsasi, M.; Gorodetskii, V. Dynamic studies of surface reactions with microscopic techniques. Prog. Surf. Sci. 1993, 42, 143–168. [Google Scholar] [CrossRef]
- Chenna, S.; Crozier, P.A. In situ environmental transmission electron microscopy to determine transformation pathways in supported Ni nanoparticles. Micron 2012, 43, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Bagot, P.A.J.; Kreuzer, H.J.; Cerezo, A.; Smith, G.D.W. A model for oxidation-driven surface segregation and transport on Pt-alloys studied by atom probe tomography. Surf. Sci. 2011, 605, 1541–1546. [Google Scholar] [CrossRef]
- Crozier, P.A.; Hansen, T.W. In situ and operando transmission electron microscopy of catalytic materials. MRS Bull. 2015, 40, 38–45. [Google Scholar] [CrossRef]
- Miller, M.K.M.K.; Cerezo, A.; Hetherington, M.G.G.; Smith, G.D.W.D.W. Atom Probe Field Ion Microscopy; Clarendon Press: Wotton-under-Edge, UK, 1996. [Google Scholar]
- Barroo, C.; Bagot, P.A.J.; Smith, G.D.W.; Visart, T. Investigating nano-structured catalysts at the atomic scale by field ion microscopy and atom probe tomography. In Atomically-Precise Methods for Synthesis of Solid Catalysts; Royal Society of Chemistry: London, UK, 2015; pp. 248–295. [Google Scholar]
- Gorodetskii, V.; Lauterbach, J.; Rotermund, H.H.; Block, J.H.; Ertl, G. Coupling between adjacent crystal planes in heterogeneous catalysis by propagating reaction-diffusion waves. Nature 1994, 370, 276–279. [Google Scholar] [CrossRef]
- Müller, E. Field Ion Microscopy Principles and Applications; American Elsevier Pub.: Amsterdam, The Netherlands, 1969. [Google Scholar]
- Gorodetskii, V.V.; Elokhin, V.I.; Bakker, J.W.; Nieuwenhuys, B.E. Field electron and field ion microscopy studies of chemical wave propagation in oscillatory reactions on platinum group metals. Catal. Today 2005, 105, 183–205. [Google Scholar] [CrossRef]
- Barroo, C.; De Decker, Y.; Visart de Bocarmé, T.; Kruse, N. Complex oscillation patterns during the catalytic hydrogenation of NO2 over platinum nanosized crystals. J. Phys. Chem. C 2014, 118, 6839–6846. [Google Scholar] [CrossRef]
- Barroo, C.; De Decker, Y.; Visart de Bocarmé, T.; Kruse, N. Emergence of chemical oscillations from nanosized target patterns. Phys. Rev. Lett. 2016, 144501, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Suchorski, Y.; Imbihl, R.; Medvedev, V.K. Compatibility of field emitter studies of oscillating surface reactions with single crystal measurements: Catalytic CO oxidation on Pt. Surf. Sci. 1998, 401, 392–399. [Google Scholar] [CrossRef]
- Suchorski, Y.; Beben, J.; James, E.; Evans, J.; Imbihl, R. Fluctuation-induced transitions in a bistable surface reaction: Catalytic CO oxidation on a Pt field emitter tip. Phys. Rev. Lett. 1999, 82, 1907–1910. [Google Scholar] [CrossRef]
- Suchorski, Y.; Beben, J.; Imbihl, R. Spatiotemporal dynamics of fluctuations in a surface reaction by Karhunen-Loeve decomposition of field emission images. Surf. Sci. 2000, 454, 331–336. [Google Scholar] [CrossRef]
- Cobden, P.D.; Gorodetskii, V.V.; Nieuwenhuys, B.E. Field emission microscope study of the initial behaviour of the palladium-hydrogen system at low temperatures. Surf. Sci. 1999, 432, 61–68. [Google Scholar] [CrossRef]
- Barroo, C.; Lambeets, S.V.; Devred, F.; Chau, T.D.; Kruse, N.; De Decker, Y.; Visart de Bocarmé, T. Hydrogenation of NO and NO2 over palladium and platinum nanocrystallites: Case studies using field emission techniques. New J. Chem. 2014, 38, 2090–2097. [Google Scholar] [CrossRef]
- Ertl, G.; Rotermund, H.-H. Spatiotemporal pattern formation in reactions at surfaces. Curr. Opin. Solid State Mater. Sci. 1996, 1, 617–621. [Google Scholar] [CrossRef]
- Engel, W.; Kordesch, M.E.; Rotermund, H.H.; Kubala, S.; von Oertzen, A. A UHV-compatible photoelectron emission microscope for applications in surface science. Ultramicroscopy 1991, 36, 148–153. [Google Scholar] [CrossRef]
- Rotermund, H.H.; Engel, W.; Jakubith, S.; von Oertzen, A.; Ertl, G. Methods and application of UV photoelectron in heterogeneous catalysis microscopy. Ultramicroscopy 1991, 36, 164–172. [Google Scholar] [CrossRef]
- Bauer, E. A brief history of PEEM. J. Electron. Spectrosc. Relat. Phenom. 2012, 185, 314–322. [Google Scholar] [CrossRef]
- Rotermund, H.H. Real time imaging of catalytic reactions on surfaces: Past, present and future. Surf. Sci. 2009, 603, 1662–1670. [Google Scholar] [CrossRef]
- Cobden, P.D.; De Wolf, C.A.; Smirnov, M.Y.; Makeev, A.; Nieuwenhuys, B.E. Non-linear processes on Pt, Rh, Pd, Ir and Ru surfaces during the NO-hydrogen reactions. J. Mol. Catal. A Chem. 2000, 158, 115–128. [Google Scholar] [CrossRef]
- Bauer, E. Low energy electron microscopy. Rep. Prog. Phys. 1994, 57, 895–938. [Google Scholar] [CrossRef]
- Świȩch, W.; Rausenberger, B.; Engel, W.; Bradshaw, A.M.; Zeitler, E. In-situ studies of heterogeneous reactions using mirror electron microscopy. Surf. Sci. 1993, 294, 297–307. [Google Scholar] [CrossRef]
- Rausenberger, B.; Swiech, W.; Rastomjee, C.S.; Mundschau, M.; Engel, W.; Zeitler, E.; Bradshaw, A.M. Imaging reaction-diffusion fronts with low-energy electron microscopy. Chem. Phys. Lett. 1993, 215, 109–113. [Google Scholar] [CrossRef]
- Rausenberger, B.; Świȩch, W.; Engel, W.; Bradshaw, A.M.; Zeitler, E. LEEM and selected-area LEED studies of reaction front propagation. Surf. Sci. 1993, 287, 235–240. [Google Scholar] [CrossRef]
- Tromp, R.M.; Hannon, J.B.; Ellis, A.W.; Wan, W.; Berghaus, A.; Schaff, O. A new aberration-corrected, energy-filtered LEEM/PEEM instrument. I. Principles and design. Ultramicroscopy 2010, 110, 852–861. [Google Scholar] [CrossRef] [PubMed]
- McCarty, K.F.; Bartelt, N.C. Spatially resolved dynamics of the TiO2(110) surface reconstruction. Surf. Sci. 2003, 540, 157–171. [Google Scholar] [CrossRef]
- Kodambaka, S.; Israeli, N.; Bareño, J.; Święch, W.; Ohmori, K.; Petrov, I.; Greene, J.E. Low-energy electron microscopy studies of interlayer mass transport kinetics on TiN(111). Surf. Sci. 2004, 560, 53–62. [Google Scholar] [CrossRef]
- Donald, A.M. The use of environmental scanning electron microscopy for imaging wet and insulating materials. Nat. Mater. 2003, 2, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Meng, Y.S.; Zhu, Y. Frontiers of in situ electron microscopy. MRS Bull. 2015, 40, 12–18. [Google Scholar] [CrossRef]
- Yuan, W.; Yu, J.; Li, H.; Zhang, Z.; Sun, C.; Wang, Y. In situ TEM observation of dissolution and regrowth dynamics of MoO2 nanowires under oxygen. Nano Res. 2017, 10, 397–404. [Google Scholar] [CrossRef]
- Gai, P.L.; Boyes, E.D. In situ TEM measurement methods. In Characterization of Materials; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 1–11. [Google Scholar]
- Alayoglu, S.; Beaumont, S.K.; Zheng, F.; Pushkarev, V.V.; Zheng, H.; Iablokov, V.; Liu, Z.; Guo, J.; Kruse, N.; Somorjai, G.A. CO2 hydrogenation studies on Co and CoPt bimetallic nanoparticles under reaction conditions using TEM, XPS and NEXAFS. Top. Catal. 2011, 54, 778–785. [Google Scholar] [CrossRef]
- Zhang, X.; Kamino, T. Imaging gas-solid interactions in an atomic resolution environmental TEM. Micros. Today 2006, 16–18. [Google Scholar]
- Boyes, E.D.; Gai, P.L. Environmental high resolution electron microscopy and applications to chemical science. Ultramicroscopy 1997, 67, 219–232. [Google Scholar] [CrossRef]
- Gai, P.L.; Boyes, E.D.; Yoshida, K.; Hansen, T.W. Development of the atomic-resolution environmental transmission electron microscope. In Controlled Atmosphere Transmission Electron Microscopy; Springer: Berlin/Heidelberg, Germany, 2016; pp. 45–62. [Google Scholar]
- Xin, H.L.; Niu, K.; Alsem, D.H.; Zheng, H. In situ TEM study of catalytic nanoparticle reactions in atmospheric pressure gas environment. Microsc. Microanal. 2013, 19, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, H.H. Imaging of dynamic processes on surfaces by light. Surf. Sci. Rep. 1997, 29, 265–364. [Google Scholar] [CrossRef]
- Punckt, C.; Rotermund, H.H. Optical imaging of pattern formation: Reflection anisotropy microscopy applied to globally coupled oscillatory CO-oxidation. Phys. Chem. Chem. Phys. 2007, 9, 3635–3640. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, T.; Wintterlin, J.; Trost, J.; Ertl, G. Identification of the “active sites” of a surface-catalyzed reaction. Science 1996, 273, 1688–1690. [Google Scholar] [CrossRef]
- Wintterlin, J.; Trost, J.; Renisch, S.; Schuster, R.; Zambelli, T.; Ertl, G. Real-time STM observations of atomic equilibrium fluctuations in an adsorbate system: O/Ru(0001). Surf. Sci. 1997, 394, 159–169. [Google Scholar] [CrossRef]
- Hendriksen, B.L.M.; Frenken, J.W.M. CO oxidation on Pt(110): Scanning tunneling microscopy inside a high-pressure flow reactor. Phys. Rev. Lett. 2002, 89, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Helveg, S.; Lauritsen, J.; Laegsgaard, E.; Stensgaard, I.; Norskov, J.; Clausen, B.; Topsoe, H.; Besenbacher, F. Atomic-scale structure of single-layer MoS2 nanoclusters. Phys. Rev. Lett. 2000, 84, 951–954. [Google Scholar] [CrossRef] [PubMed]
- Wintterlin, J. Scanning tunneling microscopy studies of catalytic reactions. Adv. Catal. 2000, 45, 131–206. [Google Scholar]
- Zhu, Z.; Barroo, C.; Lichtenstein, L.; Eren, B.; Wu, C.H.; Mao, B.; Visart de Bocarmé, T.; Liu, Z.; Kruse, N.; Salmeron, M.; et al. Influence of step geometry on the reconstruction of stepped platinum surfaces under coadsorption of ethylene and CO. J. Phys. Chem. Lett. 2014, 5, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Barroo, C.; Gilis, N.; Lambeets, S.V.; Devred, F.; Visart de Bocarmé, T. Oxygen assisted reconstructions of rhodium and platinum nanocrystals and their effects on local catalytic activity of hydrogenation reactions. Appl. Surf. Sci. 2014, 304, 2–10. [Google Scholar] [CrossRef]
- Ast, D.G.; Seidman, D.N. The field ion microscopy of gold. Appl. Phys. Lett. 1968, 13, 348–350. [Google Scholar] [CrossRef]
- Schmid, T.E.; Balluffi, R.W. Formation and migration of artifact vacancies induced on gold surfaces by neon field ion microscopy. Surf. Sci. 1971, 28, 574–580. [Google Scholar] [CrossRef]
- Nam, A.J.; Teren, A.; Lusby, T.A.; Melmed, A.J. Benign making of sharp tips for STM and FIM-Pt, Ir, Au, Pd, and Rh. J. Vac. Sci. Technol. B 1995, 13, 1556–1559. [Google Scholar] [CrossRef]
- Eisele, M.; Krger, M.; Schenk, M.; Ziegler, A.; Hommelhoff, P. Note: Production of sharp gold tips with high surface quality. Rev. Sci. Instrum. 2011, 82, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Fujioka, H.; Sakai, A. Confinement of surface state electrons on Cu and Au field emitters. Ultramicroscopy 2001, 89, 89–94. [Google Scholar] [CrossRef]
- Nomura, K.; Rokuta, E.; Itagaki, T.; Oshima, C.; Kuo, H.; Tsong, T.T. Electron emission characteristics of Au-covered tungsten <111> nanotips. e-Journal Surf. Sci. Nanotechnol. 2008, 6, 25–28. [Google Scholar] [CrossRef]
- Plsek, J. Field emission study of pulsed laser deposition of gold on clean and oxidized tungsten tip. Appl. Surf. Sci. 2014, 292, 717–725. [Google Scholar] [CrossRef]
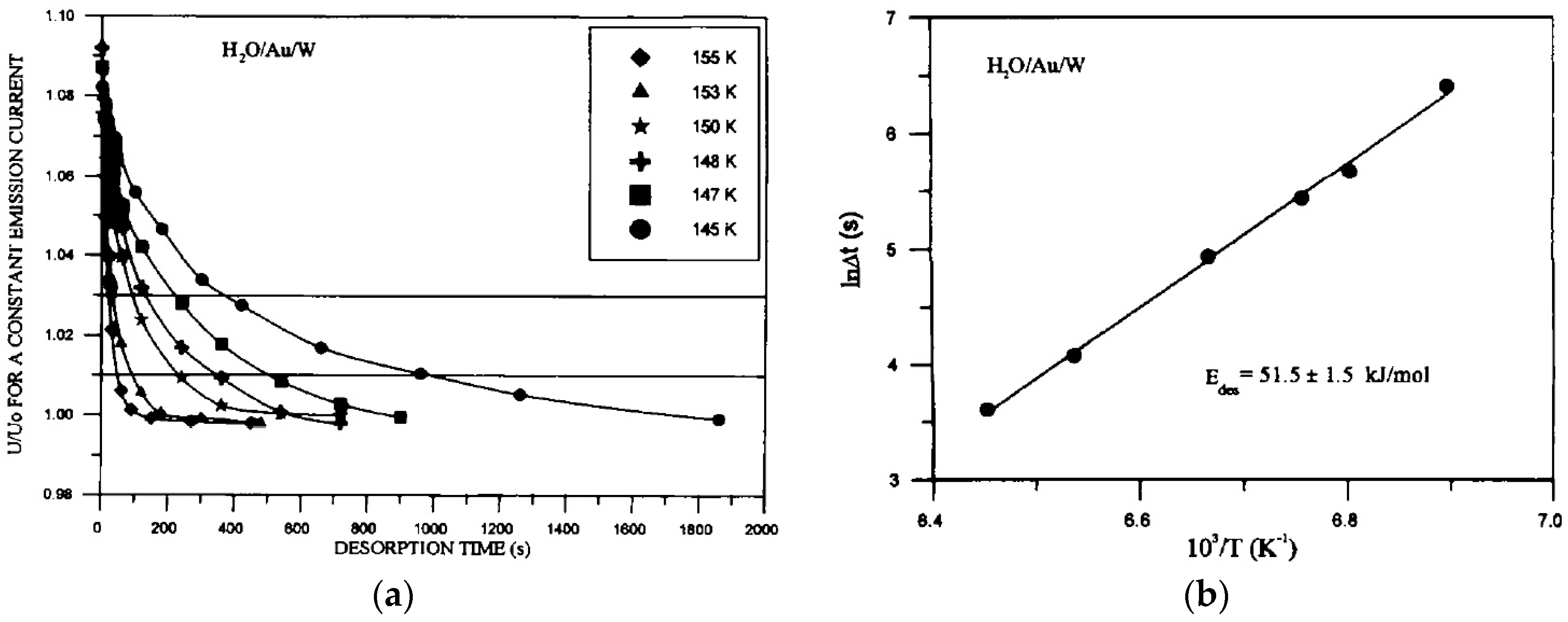
- Bryl, R.; Błaszczyszyn, R.; Galewska, E. Interaction of water with clean and gold-precovered tungsten field emitters: Adsorption and desorption. Vacuum 1997, 48, 329–332. [Google Scholar] [CrossRef]
- Visart de Bocarmé, T.; Moors, M.; Kruse, N.; Atanasov, I.S.; Hou, M.; Cerezo, A.; Smith, G.D.W. Surface segregation of Au-Pd alloys in UHV and reactive environments: Quantification by a catalytic atom probe. Ultramicroscopy 2009, 109, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Sobyanin, V.; Gorodetskii, V.; Bulgakov, N. Oxygen adsorption on silver-gold alloy. React. Kinet. Catal. Lett. 1977, 7, 285–290. [Google Scholar] [CrossRef]
- Moors, M.; Amara, H.; Visart de Bocarmé, T.; Bichara, C.; Ducastelle, F.; Kruse, N.; Charlier, J.C. Early stages in the nucleation process of carbon nanotubes. ACS Nano 2009, 3, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, M.F.H.; Gielbert, A.; Nieuwenhuys, B.E. Oscillatory behaviour of the reduction of NO by H2 over Rh. Catal. Lett. 1992, 16, 297–309. [Google Scholar] [CrossRef]
- Visart de Bocarmé, T.; Kruse, N. Kinetic instabilities during the NOx reduction with hydrogen on Pt crystals studied with field emission on the nanoscale. Chaos 2002, 12, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Imbihl, R.; Ertl, G. Oscillatory kinetics in heterogeneous catalysis. Chem. Rev. 1995, 95, 697–733. [Google Scholar] [CrossRef]
- Mc Ewen, J.-S.; Gaspard, P.; De Decker, Y.; Barroo, C.; Visart de Bocarmé, T.; Kruse, N. Catalytic reduction of NO2 with hydrogen on Pt field emitter tips: Kinetic instabilities on the nanoscale. Langmuir 2010, 26, 16381–16391. [Google Scholar] [CrossRef] [PubMed]
- Barroo, C.; De Decker, Y.; Visart de Bocarmé, T.; Gaspard, P. Fluctuating dynamics of nanoscale chemical oscillations: Theory and experiments. J. Phys. Chem. Lett. 2015, 6, 2189–2193. [Google Scholar] [CrossRef] [PubMed]
- Barroo, C.; De Decker, Y.; Jacobs, L.; Visart de Bocarmé, T. Nonlinear behavior during NO2 hydrogenation on a nanosized Pt-Rh catalyst sample. Appl. Surf. Sci. 2017, 412, 564–570. [Google Scholar] [CrossRef]
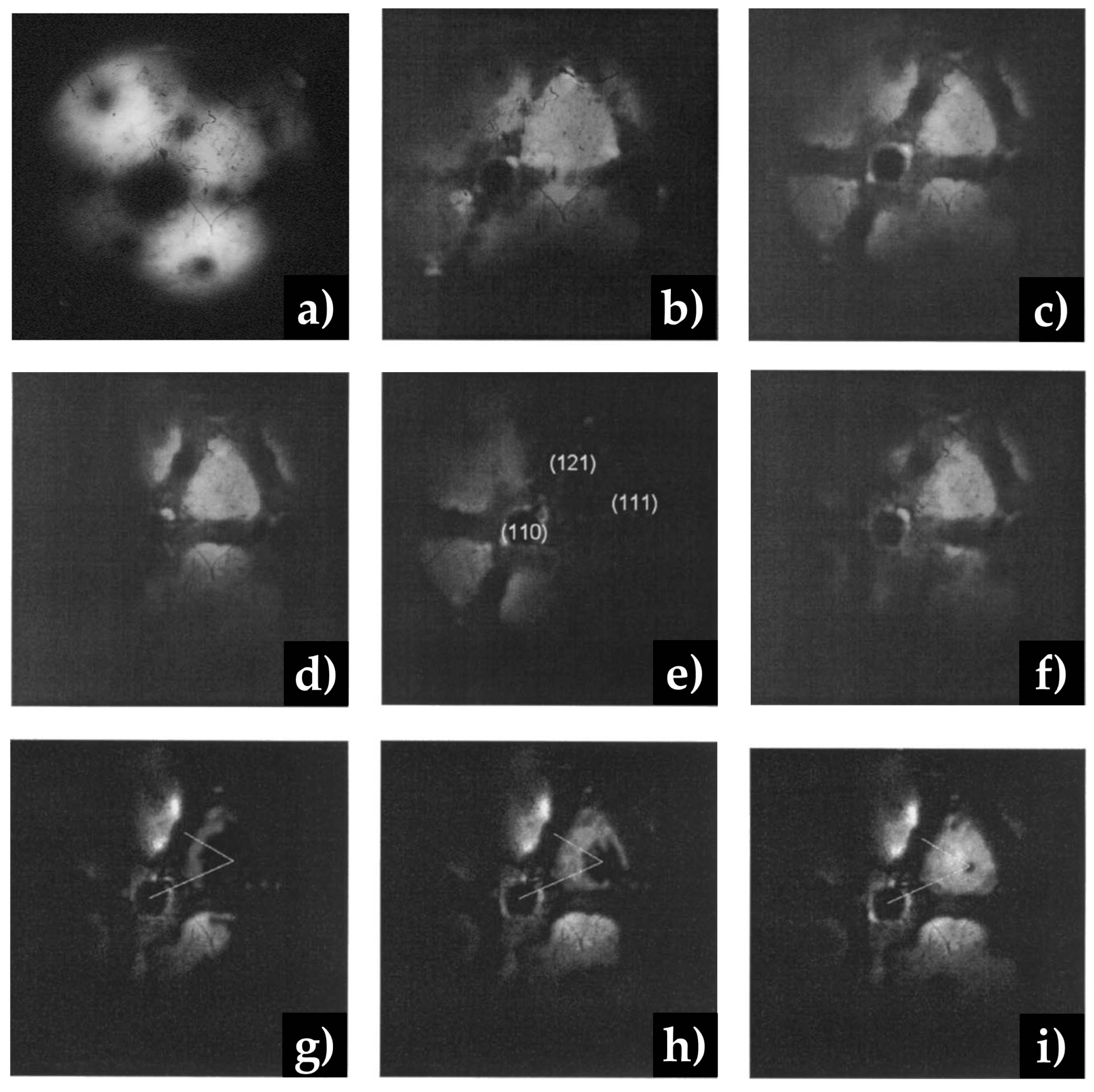
- Kruse, N.; Gaussmann, A. Changes in the morphology of Rh field emitter tips due to the reaction with carbon monoxide. Surf. Sci. 1992, 266, 51–55. [Google Scholar] [CrossRef]
- Moors, M.; Visart de Bocarmé, T.; Kruse, N. Surface reaction kinetics studied with nanoscale lateral resolution. Catal. Today 2007, 124, 61–70. [Google Scholar] [CrossRef]
- Miller, M.K. Development of atom probe field-ion microscopy. Mater. Charact. 2000, 44, 11–27. [Google Scholar] [CrossRef]
- Moody, M.P.; Vella, A.; Gerstl, S.S.A.; Bagot, P.A.J. Advances in atom probe tomography instrumentation: Implications for materials research. MRS Bull. 2016, 41, 40–45. [Google Scholar] [CrossRef]
- Bagot, P.A.J.; Visart de Bocarmé, T.; Cerezo, A.; Smith, G.D.W. 3D atom probe study of gas adsorption and reaction on alloy catalyst surfaces I: Instrumentation. Surf. Sci. 2006, 600, 3028–3035. [Google Scholar] [CrossRef]
- Ernst, N.; Bozdech, G.; Block, J.H. Filed Ion appearance spectroscopy: Investigations on ion generating processes at fiels emitter surfaces. Surf. Sci. 1979, 80, 645–655. [Google Scholar] [CrossRef]
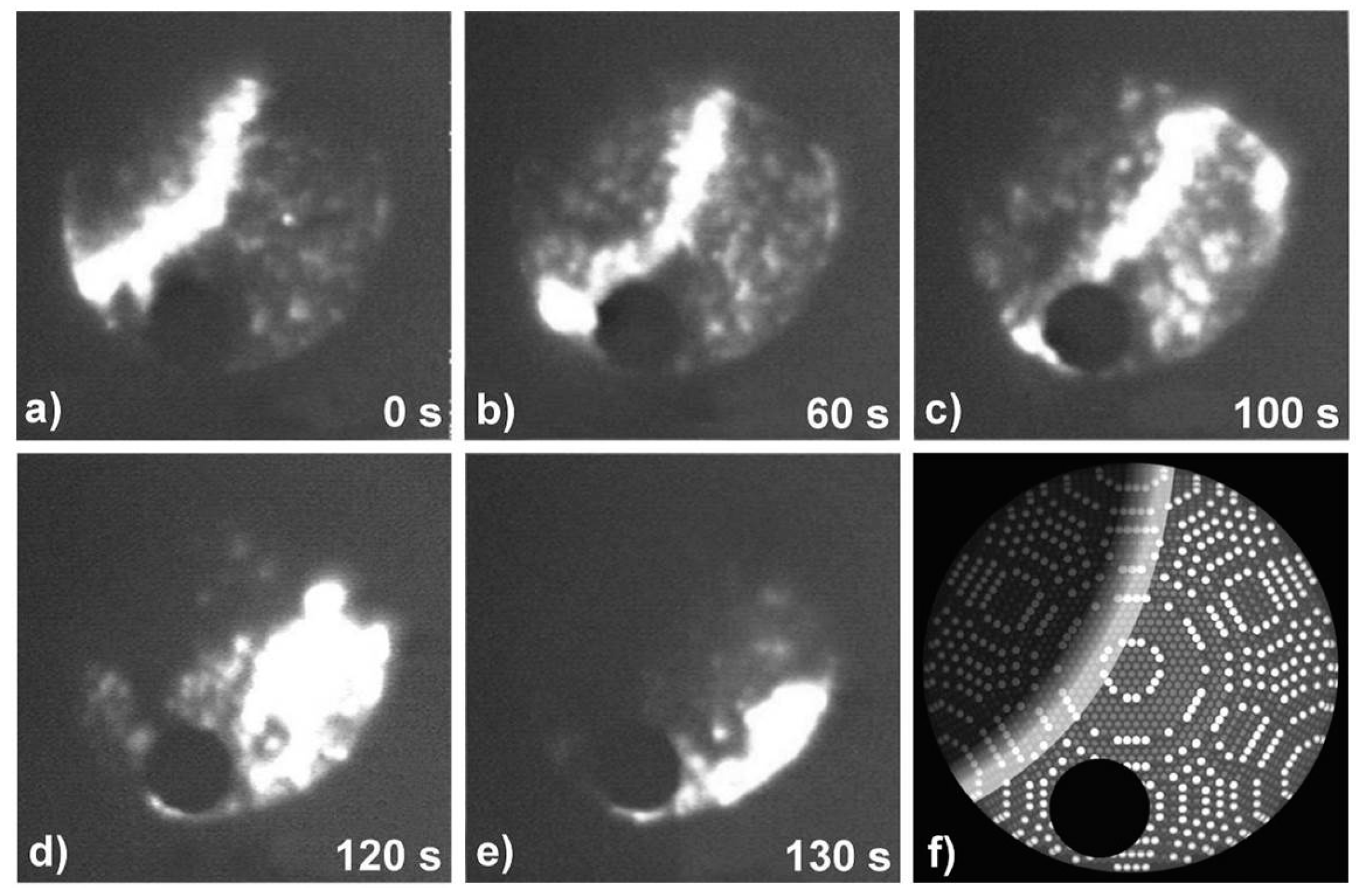
- Locatelli, A.; Heun, S.; Kiskinova, M. Direct observation of reaction-induced lateral redistribution of sub-monolayers of Au deposited on a Rh(110) surface. Surf. Sci. 2004, 566, 1130–1136. [Google Scholar] [CrossRef]
- Bauer, E. LEEM basics. Surf. Rev. Lett. 1998, 5, 1275–1286. [Google Scholar] [CrossRef]
- Schmidt, T.; Heun, S.; Slezak, J.; Diaz, J.; Prince, K.C.; Lilienkamp, G.; Bauer, E. SPELEEM: Combining LEEM and spectroscopic imaging. Surf. Rev. Lett. 1998, 5, 1287–1296. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Takakusagi, S.; Sakai, Y.; Kato, M.; Asakura, K.; Iwasawa, Y. X-ray photoemission electron microscopy (XPEEM) as a new promising tool for the real-time chemical imaging of active surfaces. J. Mol. Catal. A Chem. 1999, 141, 129–137. [Google Scholar] [CrossRef]
- Bauer, E.; Mundschau, M.; Swiech, W.; Telieps, W. Surface studies by low-energy electron microscopy (LEEM) and conventional UV photoemission electron microscopy (PEEM). Ultramicroscopy 1989, 31, 49–57. [Google Scholar] [CrossRef]
- Nettesheim, S.; von Oertzen, A.; Rotermund, H.H.; Ertl, G. Reaction diffusion patterns in the catalytic CO-oxidation on Pt(110): Front propagation and spiral waves. J. Chem. Phys. 1993, 98, 9977–9985. [Google Scholar] [CrossRef]
- Günther, S.; Kolmakov, A.; Kovac, J.; Marsi, M.; Kiskinova, M. Au on Ag/Si(111)-(3 × 3)R30°: A spectromicroscopy study of a bimetal-silicon interface. Phys. Rev. B 1997, 56, 5003–5013. [Google Scholar] [CrossRef]
- Suchorski, Y.; Spiel, C.; Vogel, D.; Drachsel, W.; Schlögl, R.; Rupprechter, G. Local reaction kinetics by imaging: CO oxidation on polycrystalline platinum. ChemPhysChem 2010, 11, 3231–3235. [Google Scholar] [CrossRef] [PubMed]
- Suchorski, Y.; Rupprechter, G. Local reaction kinetics by imaging. Surf. Sci. 2016, 643, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, A.; Kiskinova, M. Imaging with chemical analysis: Adsorbed structures formed during surface chemical reactions. Chem. A Eur. J. 2006, 12, 8890–8896. [Google Scholar] [CrossRef] [PubMed]
- Aballe, L.; Barinov, A.; Locatelli, A.; Heun, S.; Kiskinova, M. Tuning surface reactivity via electron quantum confinement. Phys. Rev. Lett. 2004, 93, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, A.; Bauer, E. Recent advances in chemical and magnetic imaging of surfaces and interfaces by XPEEM. J. Phys. Condens. Matter 2008, 20, 1–22. [Google Scholar] [CrossRef]
- Podor, R.; Ravaux, J.; Brau, H.-P. In situ experiments in the scanning electron microscope chamber. In Scanning Electron Microscopy; inTech: Rijeka, Croatia, 2012; pp. 31–35. [Google Scholar]
- Kamino, T.; Yaguchi, T.; Watabe, A.; Saka, H.; Kishita, K. Environmental transmission electron microscopy using a conventional TEM and a gas injection-specimen heating holder. Microsc. Microanal. 2006, 12, 766–767. [Google Scholar] [CrossRef]
- Saka, H.; Mima, T.; Takeuchi, Y.; Marukawa, T.; Arai, S.; Kuroda, K.; Kishita, K.; Kamino, T. Observation of gas-solid and gas-liquid Reactions by In-situ environmental holder in TEM. Microsc. Microanal. 2006, 12, 764–766. [Google Scholar] [CrossRef]
- Sharma, R.; Crozier, P.A. Environmental transmission electron microscopy in nanotechnology. Handb. Microsc. Nanotechnol. 2005, 531–565. [Google Scholar]
- Williams, D.B.; Barry Carter, C. Transmission Electron Microscopy. Diffraction, Imaging, Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Crozier, P.A.; Chenna, S. In situ analysis of gas composition by electron energy-loss spectroscopy for environmental transmission electron microscopy. Ultramicroscopy 2011, 111, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Tao, F.; Crozier, P.A. Atomic-scale observations of catalyst structures under reaction conditions and during catalysis. Chem. Rev. 2016, 116, 3487–3539. [Google Scholar] [CrossRef] [PubMed]
- Chenna, S.; Banerjee, R.; Crozier, P.A. Atomic-scale observation of the Ni activation process for partial oxidation of methane using in situ environmental TEM. ChemCatChem 2011, 3, 1051–1059. [Google Scholar] [CrossRef]
- Hansen, P.L. Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals. Science 2002, 295, 2053–2055. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, H.H.; Engel, W.; Kordesch, M.; Ertl, G. Imaging of spatio-temporal pattern evolution during carbon monoxide oxidation on platinum. Nature 1990, 343, 355–357. [Google Scholar] [CrossRef]
- Rotermund, H.H.; Jakubith, S.; Von Oertzen, A.; Ertl, G. Solitons in a surface reaction. Phys. Rev. Lett. 1991, 66, 3083–3086. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, H.H. Imaging of dynamic patterns on surfaces. Curr. Opin. Solid State Mater. Sci. 1998, 3, 354–360. [Google Scholar] [CrossRef]
- Ertl, G. Reactions at surfaces: From atoms to complexity. Angew. Chem. Int. Ed. 2008, 47, 3524–3535. [Google Scholar] [CrossRef] [PubMed]
- Asakura, K.; Lauterbach, J.; Rotermund, H.H.; Ertl, G. Modification of spatiotemporal pattern formation in an excitable medium by continuous variation of its intrinsic parameters: CO oxidation on Pt(110). Phys. Rev. B 1994, 50, 8043–8046. [Google Scholar] [CrossRef]
- Asakura, K.; Lauterbach, J.; Rotermund, H.H.; Ertl, G. Spatiotemporal concentration patterns associated with the catalytic oxidation of CO and Au covered Pt(110) surfaces. J. Chem. Phys. 1995, 102, 8175–8184. [Google Scholar] [CrossRef]
- Ertl, G. Dynamics of catalytic processes on atomic and mesoscopic scale. Appl. Surf. Sci. 1997, 121, 20–25. [Google Scholar] [CrossRef]
- Asakura, K.; Lauterbach, J.; Rotermund, H.H.; Ertl, G. Spatio-temporal pattern formation during catalytic CO oxidation on a Pt(100) surface modified with submonolayers of Au. Surf. Sci. 1997, 374, 125–141. [Google Scholar] [CrossRef]
- Locatelli, A.; Sbraccia, C.; Heun, S.; Baroni, S.; Kiskinova, M. Energetically driven reorganization of a modified catalytic surface under reaction conditions. J. Am. Chem. Soc. 2005, 127, 2351–2357. [Google Scholar] [CrossRef] [PubMed]
- Mertens, F.; Imbihl, R. Parameter-dependent anisotropy of front propagation in the H2 + O2 reaction on Rh(110). Chem. Phys. Lett. 1995, 242, 221–227. [Google Scholar] [CrossRef]
- Makeev, A.; Imbihl, R. Simulations of anisotropic front propagation in the H2 + O2 reaction on a Rh(110) surface. J. Chem. Phys. 2000, 113, 3854–3863. [Google Scholar] [CrossRef]
- Locatelli, A.; Mentes, T.O.; Aballe, L.; Mikhailov, A.; Kiskinova, M. Formation of regular surface-supported mesostructures with periodicity controlled by chemical reaction rate. J. Phys. Chem. B 2006, 110, 19108–19111. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, A.; Aballe, L.; Mentes, T.O.; Guo, F.Z.; Kiskinova, M. A spectro-microscopic study of the reactive phase separation of Au + Pd and O on Rh(110). Surf. Sci. 2007, 601, 4663–4668. [Google Scholar] [CrossRef]
- Johannesson, G.H.; Bligaard, T.; Ruban, A.V.; Skriver, H.L.; Jacobsen, K.W.; Norskov, J.K. Combined electronic structure and evolutionary search approach to materials design. Phys. Rev. Lett. 2002, 88, 2555061–2555065. [Google Scholar] [CrossRef] [PubMed]
- De Decker, Y.; Marbach, H.; Hinz, M.; Gunther, S.; Kiskinova, M.; Mikhailov, A.S.; Imbihl, R. Promoter-induced reactive phase separation in surface reactions. Phys. Rev. Lett. 2004, 92, 198305. [Google Scholar] [CrossRef] [PubMed]
- De Decker, Y.; Mikhailov, A.S. Promoter-induced nonlinear pattern formation in surface chemical reactions. J. Phys. Chem. B 2004, 108, 14759–14765. [Google Scholar] [CrossRef]
- Grinter, D.C.; Muryn, C.; Santos, B.; Shaw, B.J.; Menteş, T.O.; Locatelli, A.; Thornton, G. Spectromicroscopy of a model water-gas shift catalyst: Gold nanoparticles supported on ceria. J. Phys. Chem. C 2014, 118, 19194–19204. [Google Scholar] [CrossRef]
- Lamallem, M.; El Ayadi, H.; Gennequin, C.; Cousin, R.; Siffert, S.; Aïssi, F.; Aboukaïs, A. Effect of the preparation method on Au/Ce-Ti-O catalysts activity for VOCs oxidation. Catal. Today 2008, 137, 367–372. [Google Scholar] [CrossRef]
- Nolan, M.; Parker, S.C.; Watson, G.W. Reduction of NO2 on ceria surfaces. J. Phys. Chem. B 2006, 110, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Carrettin, S.; Fierro-Gonzalez, J.C.; Hao, Y.; Gates, B.C.; Corma, A. CO oxidation catalyzed by supported gold: Cooperation between gold and nanocrystalline rare-earth supports forms reactive surface superoxide and peroxide species. Angew. Chem. Int. Ed. 2005, 44, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Carrettin, S.; Guzman, J.; Corma, A. Supported gold catalyzes the homocoupling of phenylboronic acid with high conversion and selectivity. Angew. Chem. Int. Ed. 2005, 44, 2242–2245. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, H.; Inoue, Y. PEEM study of work function changes in Cu, Au and Pd metal surfaces with surface acoustic wave propagation. Surf. Sci. 2006, 600, 2644–2649. [Google Scholar] [CrossRef]
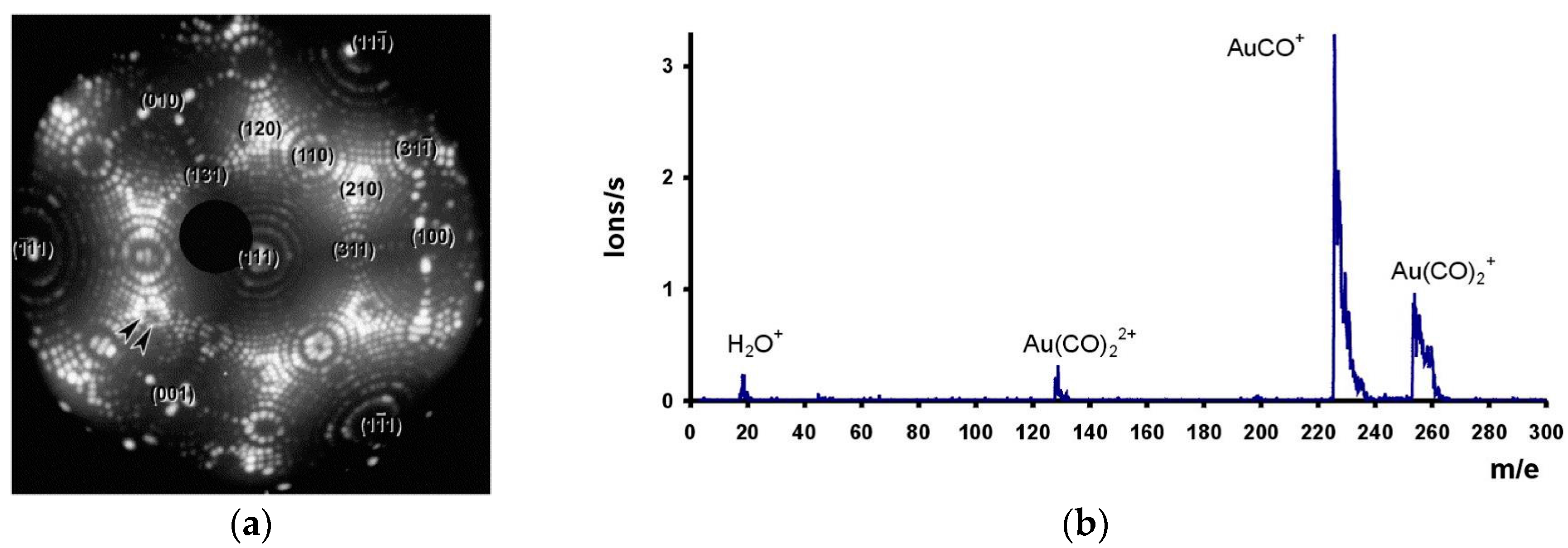
- Visart de Bocarmé, T.; Chau, T.-D.; Kruse, N. The interaction of CO–O2 gas mixtures with Au tips: In situ imaging and local chemical probing. Surf. Interface Anal. 2007, 39, 166–171. [Google Scholar] [CrossRef]
- Bär, T.; Visart de Bocarmé, T.; Nieuwenhuys, B.E.; Kruse, N. CO oxidation on gold surfaces studied on the atomic scale. Catal. Lett. 2001, 74, 127–131. [Google Scholar] [CrossRef]
- Gorodetskii, V.; Drachsel, W.; Block, J.H. The surface specificity of the oscillating CO oxidation on platinum investigated by field ion microscopy. Appl. Surf. Sci. 1994, 76, 122–128. [Google Scholar] [CrossRef]
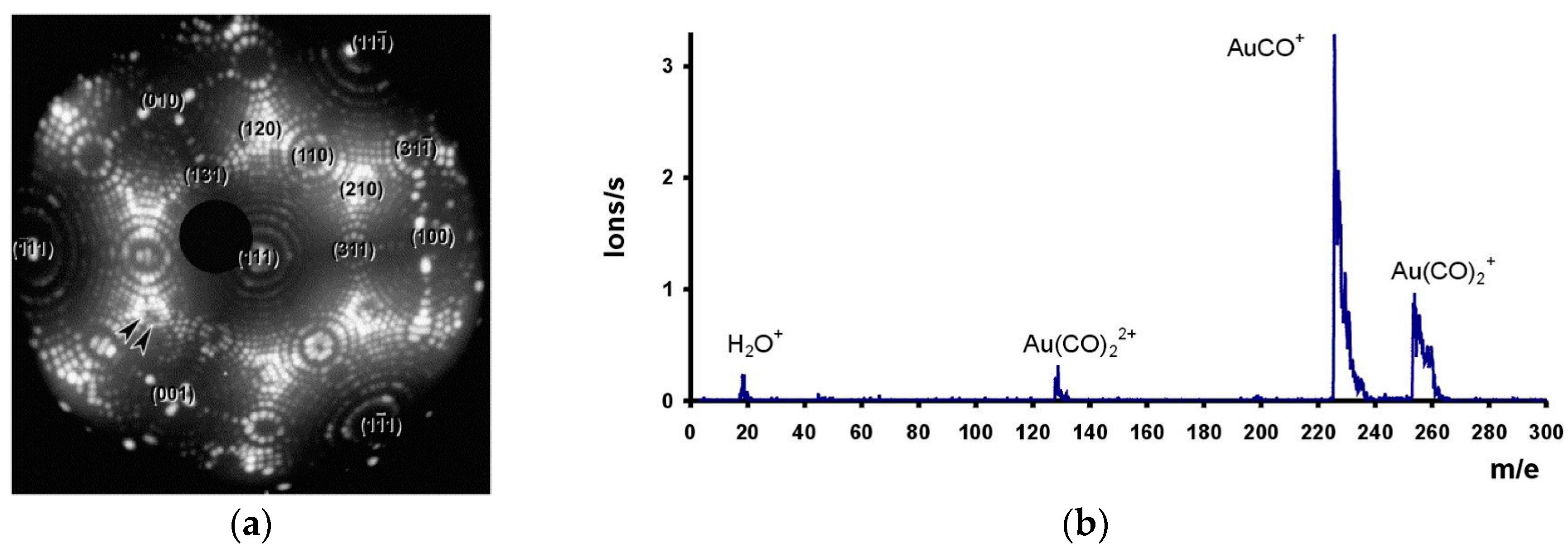
- Chau, T.D.; Visart de Bocarmé, T.; Kruse, N.; Wang, R.L.C.; Kreuzer, H.J. Formation of neutral and charged gold carbonyls on highly facetted gold nanostructures. J. Chem. Phys. 2003, 119, 12605–12610. [Google Scholar] [CrossRef]
- Visart de Bocarmé, T.; Chau, T.D.; Tielens, F.; Andrés, J.; Gaspard, P.; Wang, R.L.C.; Kreuzer, H.J.; Kruse, N. Oxygen adsorption on gold nanofacets and model clusters. J. Chem. Phys. 2006, 125, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Visart de Bocarmé, T.; Kruse, N.; Gaspard, P.; Kreuzer, H.J. Field-induced CO adsorption and formation of carbonyl waves on gold nanotips. J. Chem. Phys. 2006, 125, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.A.; Suchorski, Y.; Block, J.H.; Kreuzer, H.J.; Wang, R.L.C. Field ion appearance energy spectroscopy of CO+ originated from Rh(111) and Au(111) surface step sites. Surf. Sci. 1995, 326, 243–251. [Google Scholar] [CrossRef]
- Taketoshi, A.; Haruta, M. Size- and structure-specificity in catalysis by gold clusters. Chem. Lett. 2014, 43, 380–387. [Google Scholar] [CrossRef]
- Visart de Bocarmé, T.; Chau, T.D.; Kruse, N. Dynamic interaction of CO/H2O mixtures with gold nanocrystals: Real-time imaging and local chemical probing. Surf. Sci. 2006, 600, 4205–4210. [Google Scholar] [CrossRef]
- Chesters, M.A.; Somorjai, G.A. The chemisorption of oxygen, water and selected hydrocarbons on the (111) and stepped gold surfaces. Surf. Sci. 1975, 52, 21–28. [Google Scholar] [CrossRef]
- Schrader, M.E. Chemisorption of oxygen to gold: AES study of catalytic effect of calcium. Surf. Sci. 1978, 78, 227–232. [Google Scholar] [CrossRef]
- Pireaux, J.J.; Chtaib, M.; Delrue, J.P.; Thiry, P.A.; Liehr, M.; Caudano, R. Electron spectroscopic characterization of oxygen adsorption on gold surfaces. Surf. Sci. 1984, 141, 211–220. [Google Scholar] [CrossRef]
- Tielens, F.; Andrés, J.; Chau, T.D.; Visart de Bocarmé, T.; Kruse, N.; Geerlings, P. Molecular oxygen adsorption on electropositive nano gold tips. Chem. Phys. Lett. 2006, 421, 433–438. [Google Scholar] [CrossRef]
- McEwen, J.S.; Gaspard, P. CO oxidation on electrically charged gold nanotips. J. Chem. Phys. 2006, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Andreev, A.; Andreeva, D.; Idakiev, V.; Tabakova, T. Low-temperature water–gas shift reaction over Au/α-Fe2O3. J. Catal. 1996, 158, 354–355. [Google Scholar] [CrossRef]
- Andreeva, D. Low temperature water gas shift over gold catalysts. Gold Bull. 2002, 35, 82–88. [Google Scholar] [CrossRef]
- Costello, C.K.; Yang, J.H.; Law, H.Y.; Wang, Y.; Lin, J.; Marks, L.D.; Kung, M.C.; Kung, H.H. On the potential role of hydroxyl groups in CO oxidation over Au/Al2O3. Appl. Catal. A Gen. 2003, 243, 15–24. [Google Scholar] [CrossRef]
- Daniells, S.; Overweg, A.; Makkee, M.; Moulijn, J. The mechanism of low-temperature CO oxidation with Au/Fe2O3 catalysts: A combined Mössbauer, FT-IR, and TAP reactor study. J. Catal. 2005, 230, 52–65. [Google Scholar] [CrossRef]
- Bryl, R. Diffusion of H2O on surfaces of clean and Au covered tungsten field emitters. Vacuum 1997, 48, 333–335. [Google Scholar] [CrossRef]
- Bryl, R.; Błaszczyszyn, R. Surface diffusion of water on clean and Au-covered tungsten field emitter tips. Vacuum 1999, 54, 103–112. [Google Scholar] [CrossRef]
- Cetronio, A.; Jones, J.P. A study by high field microscopy of the effect of substrate surface structure on the work function of layers of group 1b metals adsorbed on tungsten. Surf. Sci. 1974, 44, 109–128. [Google Scholar] [CrossRef]
- Zugic, B.; Wang, L.; Heine, C.; Zakharov, D.N.; Lechner, B.A.J.; Stach, E.A.; Biener, J.; Salmeron, M.; Madix, R.J.; Friend, C.M. Dynamic restructuring drives catalytic activity on nanoporous gold–silver alloy catalysts. Nat. Mater. 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Barroo, C.; Montemore, M.M.; Janvelyan, N.; Zugic, B.; Akey, A.J.; Magyar, A.P.; Ye, J.; Kaxiras, E.; Biener, J.; Bell, D.C. Macroscopic 3D nanoporosity formation by dry oxidation of AgAu alloys. J. Phys. Chem. C 2017, 121, 5115–5122. [Google Scholar] [CrossRef]
- Nga, N.L.T.; Potvin, C.; Djéga-Mariadassou, G.; Delannoy, L.; Louis, C. Catalytic reduction of nitrogen monoxide by propene in the presence of excess oxygen over gold based ceria catalyst. Top. Catal. 2007, 42–43, 91–94. [Google Scholar] [CrossRef]
- More, P.M.; Nguyen, D.L.; Granger, P.; Dujardin, C.; Dongare, M.K.; Umbarkar, S.B. Activation by pretreatment of Ag–Au/Al2O3 bimetallic catalyst to improve low temperature HC-SCR of NOx for lean burn engine exhaust. Appl. Catal. B Environ. 2015, 174, 145–156. [Google Scholar] [CrossRef]
- Chau, T.-D.; Visart de Bocarmé, T.; Kruse, N. Formation of N2O and (NO)2 during NO adsorption on Au 3D crystals. Catal. Lett. 2004, 98, 85–87. [Google Scholar] [CrossRef]
- Burch, R.; Shestov, A.A.; Sullivan, J.A. A steady-state isotopic transient kinetic analysis of the NO/O2/H2 reaction over Pt/SiO2 catalysts. J. Catal. 1999, 188, 69–82. [Google Scholar] [CrossRef]
- Burch, R.; Shestov, A.A.; Sullivan, J.A. A steady-state isotopic transient kinetic analysis of the NO/O2/H2 reaction over Pt/SiO2 catalysts 1. Isotopic transient kKinetics and temperature programmed analysis. J. Catal. 1999, 186, 353–361. [Google Scholar] [CrossRef]
- Wei, C.; Seidman, D.N. The stage II recovery behavior of a series of ion-irradiated platinum (gold) alloys as studied by field-ion microscopy. Radiat. Eff. 1977, 32, 229–249. [Google Scholar] [CrossRef]
- Ivchenko, V.A. Field ion microscopy of phase transformations in a Cu2Au (Pt, Pd, Ag) alloy. Surf. Sci. 1992, 276, 273–280. [Google Scholar] [CrossRef]
- Miller, M.K.; Russell, K.F.; Jostsons, A.; Blake, R.G. Characterization of neutron-irradiated FeAu alloys. Appl. Surf. Sci. 1995, 87, 216–222. [Google Scholar] [CrossRef]
- Bernatskii, D.P.; Pavlov, V.G. Field desorption of a potassium-gold film on tungsten. Phys. Solid State 2004, 46, 1538–1541. [Google Scholar] [CrossRef]
- Nomura, K.; Nagao, T.; Cho, B.L.; Katsuda, H.; Matsumura, T.; Oshima, C. Thermodynamically stable nanotips of Au–Mo alloy. J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 2009, 27, 2432–2434. [Google Scholar] [CrossRef]
- Carrasquillo-Flores, R.; Ro, I.; Kumbhalkar, M.D.; Burt, S.; Carrero, C.A.; Alba-Rubio, A.C.; Miller, J.T.; Hermans, I.; Huber, G.W.; Dumesic, J.A. Reverse water–gas shift on interfacial sites formed by deposition of oxidized molybdenum moieties onto gold nanoparticles. J. Am. Chem. Soc. 2015, 137, 10317–10325. [Google Scholar] [CrossRef] [PubMed]
- Ro, I.; Carrasquillo-Flores, R.; Dumesic, J.A.; Huber, G.W. Intrinsic kinetics of plasmon-enhanced reverse water gas shift on Au and Au-Mo interfacial sites supported on silica. Appl. Catal. A Gen. 2016, 521, 182–189. [Google Scholar] [CrossRef]
- Martirez, J.M.P.; Carter, E.A. Thermodynamic constraints in using AuM (M = Fe, Co, Ni, and Mo) alloys as N2 dissociation catalysts: Functionalizing a plasmon-active metal. ACS Nano 2016, 10, 2940–2949. [Google Scholar] [CrossRef] [PubMed]
- Jahn, S.; Lechner, S.J.; Freichels, H.; Möller, M.; Spatz, J.P. Precise AuxPt1−xAlloy Nanoparticle Array of Tunable Composition for Catalytic Applications. Nature 2016, 6, 1–8. [Google Scholar]
- Cui, H.-Z.; Guo, Y.; Wang, X.; Jia, C.-J.; Si, R. Gold-iron oxide catalyst for CO oxidation: Effect of support structure. Catalysts 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Son, U.T.; Hren, J.J. Field-ion microscopy of ordered Cu–Au alloy. J. Appl. Phys. 1971, 42, 5895–5896. [Google Scholar] [CrossRef]
- Cao, Z.; Guo, L.; Liu, N. A theoretical study of the water–gas-shift reaction on Cu6TM (TM = Co, Ni, Cu, Rh, Pd, Ag, Ir, Pt, Au) clusters. J. Clust. Sci. 2016, 27, 523–535. [Google Scholar] [CrossRef]
- Saqlain, M.A.; Hussain, A.; Siddiq, D.M.; Leenaerts, O.; Leitao, A.A. DFT study of synergistic catalysis of the water-gas-shift reaction on Cu-Au bimetallic surfaces. ChemCatChem 2016, 8, 1208–1217. [Google Scholar] [CrossRef]
- Klesper, G.; Röllgen, F.W. Field ionization behavior of cyclohexane on Au tips. Int. J. Mass Spectrom. 1999, 185–187, 189–194. [Google Scholar] [CrossRef]
- Castro, T. Studies of individual nanometer-sized metallic clusters using scanning tunneling microscopy, field emission, and field ion microscopy. J. Vac. Sci. Technol. A 1989, 7, 2845. [Google Scholar] [CrossRef]
- Lovall, D.; Buss, M.; Andres, R.; Reifenberger, R. Resolving the atomic structure of supported nanometer-size Au clusters. Phys. Rev. B 1998, 58, 889–896. [Google Scholar] [CrossRef]
- Akita, T.; Okumura, M.; Tanaka, K.; Kohyama, M.; Haruta, M. TEM observation of gold nanoparticles deposited on cerium oxide. J. Mater. Sci. 2005, 40, 3101–3106. [Google Scholar] [CrossRef]
- Akita, T.; Kohyama, M.; Haruta, M. Electron microscopy study of gold nanoparticles deposited on transition metal oxides. Acc. Chem. Res. 2013, 46, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, Y. Electron beam induced evolution in Au, Ag, and interfaced heterogeneous Au/Ag nanoparticles. Nanoscale 2015, 7, 13687–13693. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.J.; Yoshida, K.; Pay, M.L.; Gai, P.L.; Boyes, E.D. On the effect of atomic structure on the activity and deactivation of catalytic gold nanoparticles. ChemCatChem 2012, 4, 1638–1644. [Google Scholar] [CrossRef]
- Hansen, T.W.; Wagner, J.B. Environmental transmission electron microscopy in an aberration-corrected environment. Microsc. Microanal. 2012, 18, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.W.; Wagner, J.B. Catalysts under controlled atmospheres in the transmission electron microscope. ACS Catal. 2014, 4, 1673–1685. [Google Scholar] [CrossRef]
- Gai, P.L.; Yoshida, K.; Ward, M.R.; Walsh, M.; Baker, R.T.; van de Water, L.; Watson, M.J.; Boyes, E.D. Visualisation of single atom dynamics in water gas shift reaction for hydrogen generation. Catal. Sci. Technol. 2016, 6, 2214–2227. [Google Scholar] [CrossRef]
- Chen, M.S.; Goodman, D.W. The structure of catalytically active Au on titania. Science 2004, 306, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Landman, U.; Yoon, B.; Zhang, C.; Heiz, U.; Arenz, M. Factors in gold nanocatalysis: Oxidation of CO in the non-scalable size regime. Top. Catal. 2007, 44, 145–158. [Google Scholar] [CrossRef]
- Aguilar-Guerrero, V.; Gates, B.C. Kinetics of CO oxidation catalyzed by highly dispersed CeO2-supported gold. J. Catal. 2008, 260, 351–357. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I. Mechanism and active sites of the oxidation of CO over Au/TiO2. Angew. Chem. Int. Ed. 2011, 50, 10144–10147. [Google Scholar] [CrossRef] [PubMed]
- Kuwauchi, Y.; Takeda, S.; Yoshida, H.; Sun, K.; Haruta, M.; Kohno, H. Stepwise displacement of catalytically active gold nanoparticles on cerium oxide. Nano Lett. 2013, 13, 3073–3077. [Google Scholar] [CrossRef] [PubMed]
- Ta, N.; Liu, J.; Chenna, S.; Crozier, P.A.; Li, Y.; Chen, A.; Shen, W. Stabilized gold nanoparticles on ceria nanorods by strong interfacial anchoring. J. Am. Chem. Soc. 2012, 134, 20585–20588. [Google Scholar] [CrossRef] [PubMed]
- Kuwauchi, Y.; Yoshida, H.; Akita, T.; Haruta, M.; Takeda, S. Intrinsic catalytic structure of gold nanoparticles supported on TiO2. Angew. Chem. Int. Ed. 2012, 51, 7729–7733. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Kawasaki, T.; Hasegawa, H.; Tanji, T.; Ichihashi, M. First observation of dynamic shape changes of a gold nanoparticle catalyst under reaction gas environment by transmission electron microscopy. Surf. Interface Anal. 2008, 40, 1725–1727. [Google Scholar] [CrossRef]
- Giorgio, S.; Sao Joao, S.; Nitsche, S.; Chaudanson, D.; Sitja, G.; Henry, C.R. Environmental electron microscopy (ETEM) for catalysts with a closed E-cell with carbon windows. Ultramicroscopy 2006, 106, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, S.; Cabié, M.; Henry, C.R. Dynamic observations of Au catalysts by environmental electron microscopy. Gold Bull. 2008, 41, 167–173. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yoshida, H.; Kuwauchi, Y.; Ichikawa, S.; Shimada, S.; Haruta, M.; Takeda, S. Systematic morphology changes of gold nanoparticles supported on CeO2 during CO oxidation. Angew. Chem. Int. Ed. 2011, 50, 10157–10160. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Kuwauchi, Y.; Jinschek, J.R.; Sun, K.; Tanaka, S.; Kohyama, M.; Shimada, S.; Haruta, M.; Takeda, S. Visualizing gas molecules interacting with supported nanoparticulate catalysts at reaction conditions. Science 2011, 628, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Yoshida, H.; Kuwauchi, Y.; Sun, K.; Kohyama, M. Operand structural study of supported gold nanoparticulate catalysts by quantitative environmental transmission electron microscopy. Microsc. Microanal. 2012, 18, 1112–1113. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yoshida, H.; Kamiuchi, N. Correlation of catalytic activity with the morphology change of supported Au nanoparticles in gas. Surf. Sci. 2017, 659, 16–19. [Google Scholar] [CrossRef]
- Biener, J.; Wittstock, A.; Baumann, T.F.; Weissmüller, J.; Bäumer, M.; Hamza, A.V. Surface chemistry in nanoscale materials. Materials 2009, 2, 2404–2428. [Google Scholar] [CrossRef]
- Biener, J.; Biener, M.M.; Madix, R.J.; Friend, C.M. Nanoporous gold: Understanding the origin of the reactivity of a 21st century catalyst made by pre-columbian technology. ACS Catal. 2015, 5, 6263–6270. [Google Scholar] [CrossRef]
- Personick, M.L.; Zugic, B.; Biener, M.M.; Biener, J.; Madix, R.J.; Friend, C.M. Ozone-activated nanoporous gold: A stable and storable material for catalytic oxidation. ACS Catal. 2015, 5, 4237–4241. [Google Scholar] [CrossRef]
- Stowers, K.J.; Madix, R.J.; Biener, M.M.; Biener, J.; Friend, C.M. Facile ester synthesis on Ag-modified nanoporous Au: Oxidative coupling of ethanol and 1-Butanol under UHV conditions. Catal. Lett. 2015, 145, 1217–1223. [Google Scholar] [CrossRef]
- Wang, L.-C.; Personick, M.L.; Karakalos, S.; Fushimi, R.; Friend, C.M.; Madix, R.J. Active sites for methanol partial oxidation on nanoporous gold catalysts. J. Catal. 2016, 344, 778–783. [Google Scholar] [CrossRef]
- Falcucci, G.; Succi, S.; Montessori, A.; Melchionna, S.; Prestininzi, P.; Barroo, C.; Bell, D.C.; Biener, M.M.; Biener, J.; Zugic, B.; Kaxiras, E. Mapping reactive flow patterns in monolithic nanoporous catalysts. Microfluid. Nanofluidics 2016, 20, 105. [Google Scholar] [CrossRef]
- Erlebacher, J.; Aziz, M.J.; Karma, A.; Dimitrov, N.; Sieradzki, K. Evolution of nanoporosity in dealloying. Nature 2001, 410, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Erlebacher, J. Nanoporous metals with controlled multimodal pore size distribution. J. Am. Chem. Soc. 2003, 125, 7772–7773. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.A.R.; Chen, H.; Stamou, M.; Biener, J.; Biener, M.M.; Lein, P.J.; Seker, E. Nanoporous gold as a neutral interface coating: Effects of topography, surface chemistry, and feature size. ACS Appl. Mater. Interfaces 2015, 7, 7093–7100. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Su, J.; Xu, X.; Liu, P.; Zhao, H.; Tian, F.; Ding, Y. Low temperature CO oxidation over unsupported nanoporous gold. J. Am. Chem. Soc. 2007, 129, 42–43. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Biener, J.; Bäumer, M. Nanoporous gold: A new material for catalytic and sensor applications. Phys. Chem. Chem. Phys. 2010, 12, 12919–12930. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Zielasek, V.; Biener, J.; Friend, C.M.; Baumer, M. Nanoporous gold catalysts for selective methanol at low temperature. Science 2010, 327, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Bäumer, M. Catalysis by unsupported skeletal gold catalysts. Acc. Chem. Res. 2014, 47, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Tokunaga, T.; Zhang, L.; Li, D.; Chen, L.; Arai, S.; Yamamoto, Y.; Hirata, A.; Tanaka, N.; Ding, Y.; Chen, M. Atomic observation of catalysis-induced nanopore coarsening of nanoporous gold. Nano Lett. 2014, 14, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.Q.; Feng, Q.; Liu, D.Z.; Nie, A.M.; Liu, J.B.; Zhang, X.B.; Geng, L.M. In situ high resolution transmission electron microscopy investigation of deformation mechanism in sub-10-nm Au crystals. Mater. Sci. Technol. 2014, 30, 774–781. [Google Scholar] [CrossRef]
- Baier, S.; Wittstock, A.; Damsgaard, C.D.; Diaz, A.; Reinhardt, J.; Benzi, F.; Shi, J.; Scherer, T.; Wang, D.; Kübel, C.; et al. Influence of gas atmospheres and ceria on the stability of nanoporous gold studied by environmental electron microscopy and in situ ptychography. RSC Adv. 2016, 6, 83031–83043. [Google Scholar] [CrossRef]
- Fujita, T.; Guan, P.; McKenna, K.; Lang, X.; Hirata, A.; Zhang, L.; Tokunaga, T.; Arai, S.; Yamamoto, Y.; Tanaka, N.; et al. Atomic origins of the high catalytic activity of nanoporous gold. Nat. Mater. 2012, 11, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.-M.; Liu, C.; Yu, W.-J.; Zhang, L.-L.; Hou, P.-X.; Li, J.-C.; Li, F.; Bando, Y.; Golberg, D.; Cheng, H.-M. Structural changes in iron oxide and gold catalysts during nucleation of carbon nanotubes studied by in situ transmission electron microscopy. ACS Nano 2014, 8, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Juluri, R.R.; Satyam, P.V. Real time nanoscale structural evaluation of gold structures on Si(100) surface using in-situ transmission electron microscopy. J. Appl. Phys. 2014, 115, 1–5. [Google Scholar] [CrossRef]
- Duchstein, L.D.L.; Damsgaard, C.D.; Hansen, T.W.; Wagner, J.B. Low-pressure ETEM studies of Au assisted MgO nanorod growth. J. Phys. Conf. Ser. 2014, 522, 1–4. [Google Scholar] [CrossRef]
- Gamalski, A.D.; Tersoff, J.; Sharma, R.; Ducati, C.; Hofmann, S. Metastable crystalline AuGe catalysts formed during isothermal germanium nanowire growth. Phys. Rev. Lett. 2012, 108, 255702. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Chee, S.W.; Herzing, A.; Miranda, R.; Rez, P. Evaluation of the role of Au in improving catalytic activity of Ni nanoparticles for the formation of one-dimensional carbon nanostructures. Nano Lett. 2011, 11, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Kang, Y.; Liu, Z.; Yang, J.C.; Zhou, G. Effect of oxygen pressure on the initial oxidation behavior of Cu and Cu-Au alloys. Mater. Res. Soc. Symp. Proc. 2011, 1318, 31–36. [Google Scholar] [CrossRef]
- Luo, L.; Kang, Y.; Yang, J.C.; Zhou, G. Effect of gold composition on the orientations of oxide nuclei during the early stage oxidation of Cu-Au alloys. J. Appl. Phys. 2012, 111, 1–9. [Google Scholar] [CrossRef]
- Zhou, G.W.; Eastman, J.A.; Birtcher, R.C.; Baldo, P.M.; Pearson, J.E.; Thompson, L.J.; Wang, L.; Yang, J.C. Composition effects on the early-stage oxidation kinetics of (001) Cu-Au alloys. J. Appl. Phys. 2007, 101, 1–6. [Google Scholar] [CrossRef]
- Albrecht, W.; Van der Hoeven, J.E.; Deng, T.-S.; de Jongh, P.E.; van Blaaderen, A. Fully alloyed metal nanorods with highly tunable properties. Nanoscale 2017, 9, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Shan, H.; Chen, W.; Gu, X.; Tao, P.; Song, C.; Shang, W.; Deng, T. In situ environmental TEM in imaging gas and liquid phase chemical reactions for materials research. Adv. Mater. 2016, 28, 9686–9712. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, W.; Wen, J.; Miller, D.J.; Lu, P.; Zuo, J.M.; Yang, H. Growth of Au on Pt icosahedral nanoparticles revealed by low-dose in situ TEM. Nano Lett. 2015, 15, 2711–2715. [Google Scholar] [CrossRef] [PubMed]
- Jungjohann, K.L.; Bliznakov, S.; Sutter, P.W.; Stach, E.A.; Sutter, E.A. In situ liquid cell electron microscopy of the solution growth of Au-Pd core-shell nanostructures. Nano Lett. 2013, 13, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
- Ring, E.A.; de Jonge, N. Microfluidic system for transmission electron microscopy. Microsc. Microanal. 2010, 16, 622–629. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Techniques | Description | Advantage | Inconvenient | Applications in Catalysis | References |
|---|---|---|---|---|---|
| Field Ion Microscopy (FIM) |
|
|
|
| [87,88,89,90,91,92,93] |
| Field Emission Microscopy (FEM) |
|
|
|
| [94,95,96,97,98] |
| Photoemission electron microscopy (PEEM) |
|
|
|
| [89,99,100,101,102,103,104] |
| Low energy electron microscopy (LEEM) |
|
|
|
| [102,105,106,107,108,109,110,111] |
| Environmental scanning electron microscopy (E-SEM) |
|
|
|
| [74,112] |
| Environmental transmission electron microscopy (E-TEM) |
|
|
|
| [113,114,115,116,117,118,119,120] |
| Ellipsomicroscopy for surface imaging (EMSI) |
|
|
|
| [69,71,103,121] |
| Reflection anisotropy microscopy (RAM) |
|
|
|
| [69,103,121,122] |
| Scanning tunnelling microscopy (STM) |
|
|
|
| [70,73,123,124,125,126,127,128] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genty, E.; Jacobs, L.; Bocarmé, T.V.d.; Barroo, C. Dynamic Processes on Gold-Based Catalysts Followed by Environmental Microscopies. Catalysts 2017, 7, 134. https://doi.org/10.3390/catal7050134
Genty E, Jacobs L, Bocarmé TVd, Barroo C. Dynamic Processes on Gold-Based Catalysts Followed by Environmental Microscopies. Catalysts. 2017; 7(5):134. https://doi.org/10.3390/catal7050134
Chicago/Turabian StyleGenty, Eric, Luc Jacobs, Thierry Visart de Bocarmé, and Cédric Barroo. 2017. "Dynamic Processes on Gold-Based Catalysts Followed by Environmental Microscopies" Catalysts 7, no. 5: 134. https://doi.org/10.3390/catal7050134