Microwave-Assisted Silver-Catalyzed Protodecarboxylation and Decarboxylative Iodination of Aromatic Carboxylic Acids

Department of Chemistry, Xi’an Jiaotong-Liverpool University, Suzhou 215123, China
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(11), 314; https://doi.org/10.3390/catal7110314
Submission received: 10 October 2017
/
Revised: 23 October 2017
/
Accepted: 24 October 2017
/
Published: 26 October 2017
Abstract
:Carboxylic acids and their derivatives are readily available from both natural and synthetic sources. Apart from being used as direct substrates in the functional transformation, aryl carboxylic acids have found more applications in aromatic functionalization, especially in decarboxylation coupling reactions. Microwave-assisted protodecarboxylation and decarboxylative iodination of aromatic carboxylic acids were achieved with excellent yields in the presence of Ag2CO3 catalyst and K2S2O8. These reactions will be helpful for better understanding of decarboxylation-related coupling reactions and also have the potential of being used as a practical labeling method to synthesize regioselective deuterium and iodine-labelled compounds for chemical, biological, and medicinal research.
1. Introduction
Carboxylic acids and their derivatives are common chemicals of relatively low price with diverse structures, readily available from both natural and synthetic sources. They have long functioned as valuable intermediates and precursors in the construction of carbon frameworks [1,2,3].
The decarboxylative cross-coupling reaction is one of the most powerful strategies developed over the decades to form a carbon–carbon bond or carbon–heteroatom bond and is advantageous over traditional cross-coupling or addition reactions, which generally require the use of preformed organometallic reagents [4,5,6]. Transition metals are essential for decarboxylation and coupling reaction, the two main steps in this process. Decarboxylative cross-coupling reaction starts with the formation of metal carboxylates from carboxylic acid substrate and metal catalyst, the pivotal step of CO2 extrusion from the metal salts of carboxylates typically proceeds under rather forcing reaction conditions. Over the years, various metal catalytic systems—both monometallic and bimetallic—have been discovered and developed to turn this strategy applicable for organic synthesis [4,7].
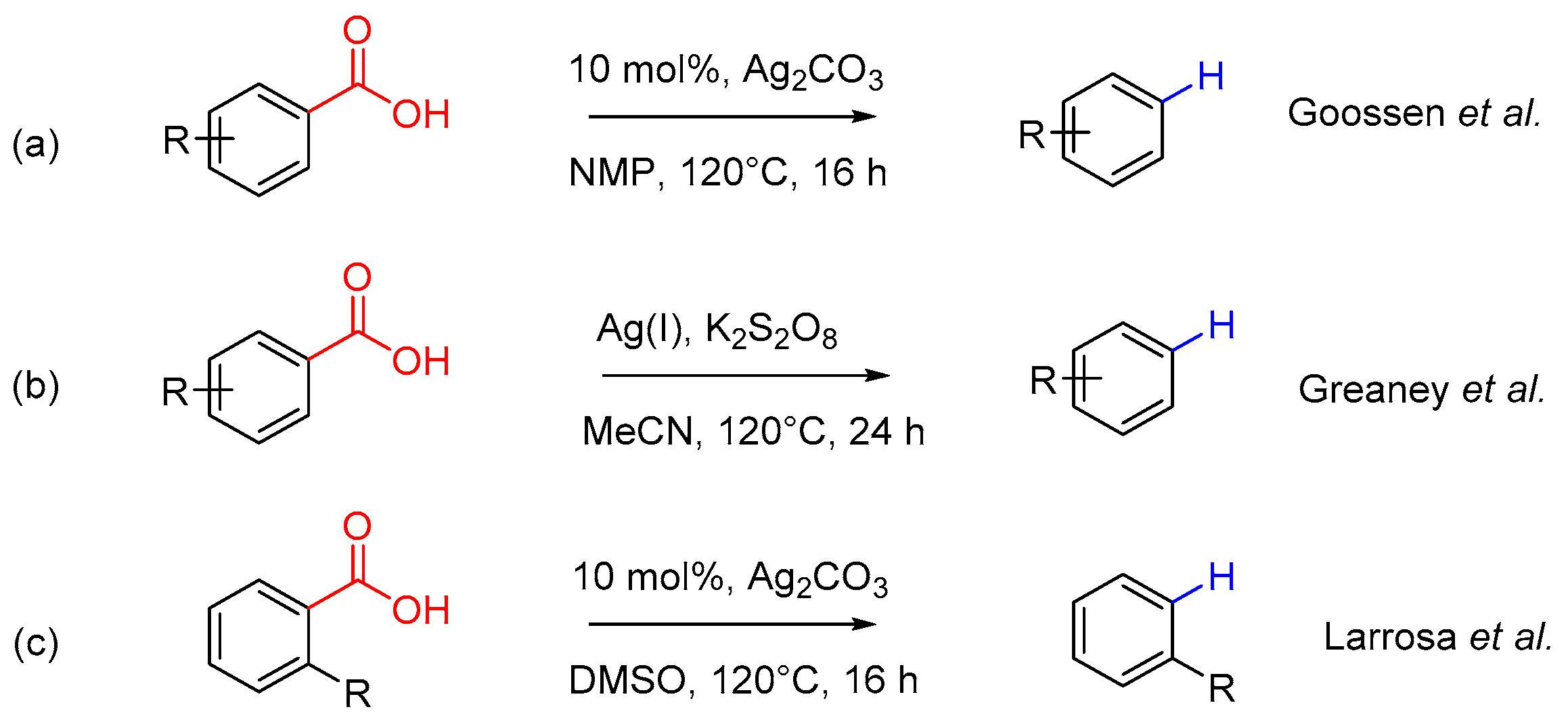
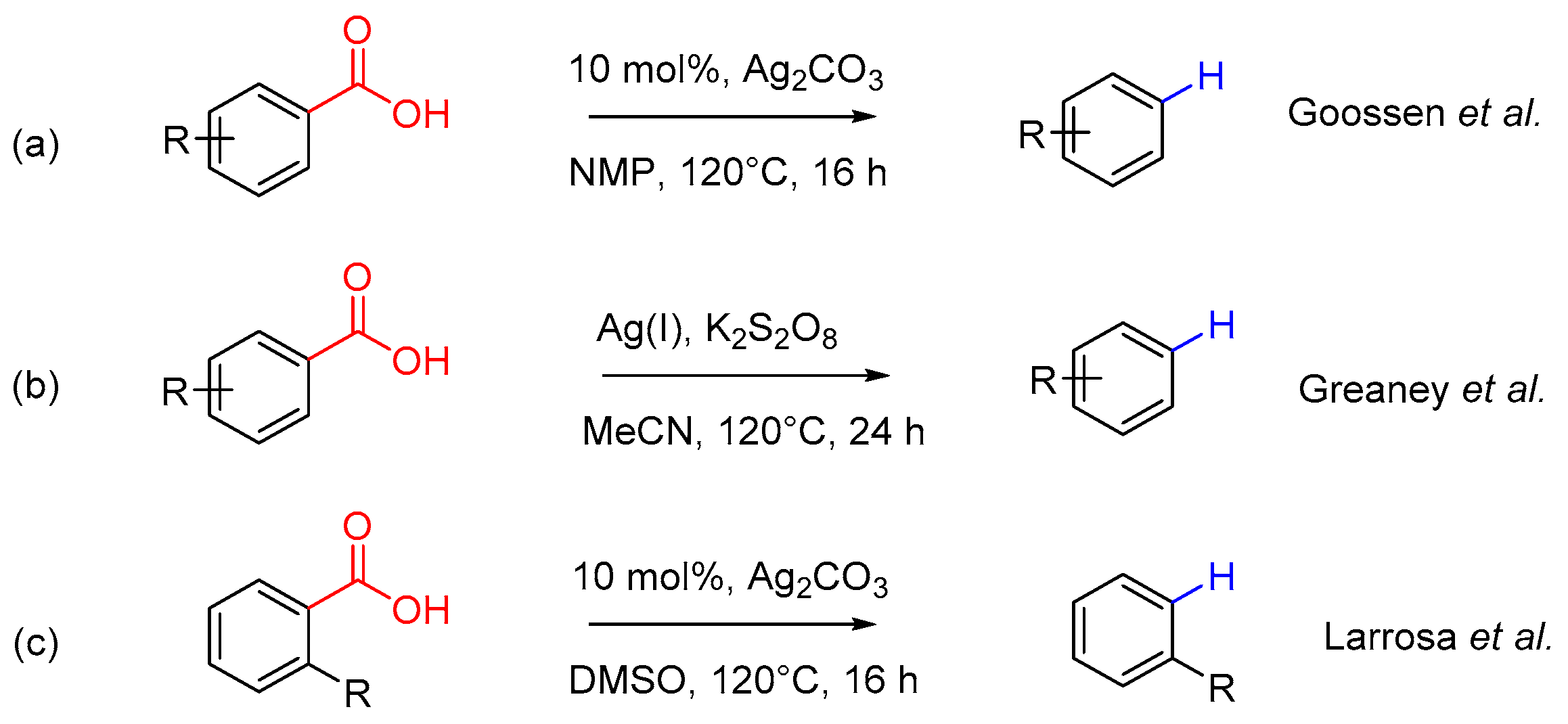
Protodecarboxylation reaction is of high preparative utility in this field, first as a precursor step of decarboxylative cross-coupling reaction and also as a constructive model method for further functionalization design [8,9,10]. The traditional copper-catalyzed decarboxylation is hampered by the extremely high temperature and costly ligands [11]. The recent research have found that silver in metallic catalyst systems can effectively promote this reaction (Scheme 1) [12,13,14]. However, these decarboxylative transformation reactions either require 20 h or an even longer time to obtain satisfied reaction yields under conventional heating conditions (Scheme 1a,b) or is only applicable for some specific substituted benzoic acids and heteroaromatic benzoic acids (Scheme 1c). Development of an effective and facile protodecarboxylation reaction is still highly desirable for useful organic transformations and their applications in the decarboxylative cross-coupling reaction.
One of the interesting, synthetically valuable functionalizations from the controlled protodecarboxylation of carboxylic acids is halogenation on the aromatic ring where the carboxylate groups are removed. Aryl iodides have played essential roles in organic synthesis as substrates for many organic reactions, and also found increasing applications in medical research and drug discovery [15,16,17]. Specifically, radioactive organic iodides has been widely used in nuclear medicine and radiotherapy science, such as hypothyroidism treatment, single-electron-emission computed tomography, and preclinical X-ray imaging [18,19,20]. Several radioactive iodine isotopes, for example, 125I, 131I, and 123I, act successfully as labeling agents in the pharmacokinetic studies [21]. However, aryl and heteroaryl iodides are much more difficult to acquire compared to their corresponding bromides and chlorides, and the facile iodination of aromatic compounds with high yield and broad substrate scope are still out of reach in organic chemistry [17,22,23].
Great efforts have been devoted and several useful strategies were developed to synthesize functionalized aromatic, hetero-aromatic iodides, but mainly based on Finkelstein reaction [24,25,26], the traditional aromatic halogen exchange reaction. The practical application of this approach has been limited by its high reaction temperature, polar solvents, costly ligands, and long reaction time, using aromatic halides as starting materials, which itself needs multistep synthesis, even if it is achievable under a mild reaction conditions, such as photo-induced metal-catalyst-free aromatic halogen exchange reaction, as discovered by Li (Scheme 2) [27]. As aromatic carboxylic acids are more widely available and cheaper, the successful decarboxylative iodination reaction will provide a reliable route for aryl iodides and has the potential of general application in organic synthesis. Very recently, an interesting transition metal-free decarboxylative iodination was reported by Larrosa [28], demonstrating that there is still room for improvement in this area.
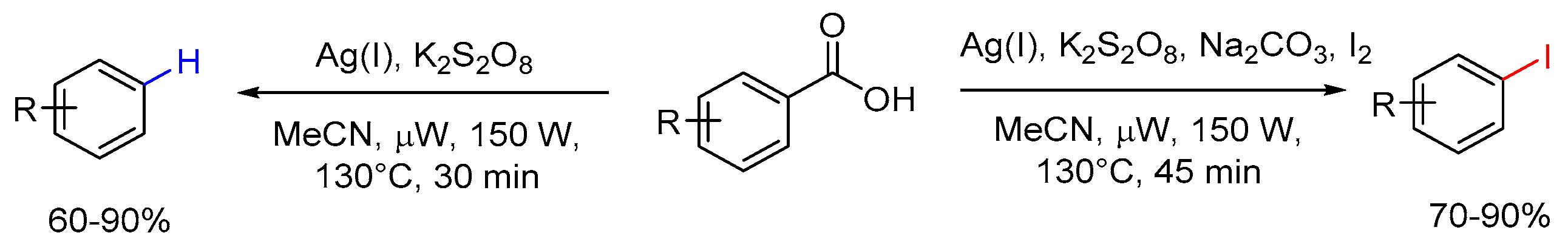
Our ongoing efforts in the development of synthetic methodologies and their applications in the synthesis of natural products and the active pharmaceutical ingredients prompted us to explore the possibilities of using microwave technology to promote protodecarboxylation reaction and decarboxylative iodination reaction, taking advantage of the efficiency generally rendered by this approach [29,30,31,32]. Herein, we report the outcomes of this work, in which the silver-catalyzed protodecarboxylation occurred smoothly under microwave-assisted heating and the decarboxylative iodination reaction afforded aryl iodides in high yields and great regioselectivity (Scheme 3).
2. Results and Discussion
2.1. Microwave-Assisted Protodecarboxylation Reaction of Aromatic Carboxylic Acids
Our investigation started with ortho-bromo benzoic acid which has a moderate acid dissociation constant (pKa 2.88, 25 °C). Potassium peroxydisulfate, reported as an efficient oxidant by Greaney in their works [14], was employed as the oxidant. We were delighted to find that, under microwave irradiation (150 W, 2.45 GHz magnetic frequency) at 130 °C with acetonitrile as solvent in the presence of Ag2CO3 (10 mol %), the reaction completed in one hour to afford the expected protodecarboxylation product at a 64% yield. Encouraged by the initial results, we investigated the influence of the loading of silver catalyst and several different silver salts as candidate catalysts to optimize the reaction. Increasing the loading of silver catalyst from 10% to 40% did improve reaction yields to some extent (entries 1–5, Table 1). Of catalyst loading, 15% was selected for further experimentation on the basis of its good efficiency and lower cost, when two equivalents of potassium peroxydisulfate was used. Although all the silver salts tested could promoted the protodecarboxylation effectively, Ag2CO3 gave the highest yield, followed by silver oxide, silver acetate, and silver triflate (entries 6–8). Potassium peroxydisulfate proved to be the oxidant of choice, when it was replaced by Oxone® (Sigma-Aldrich, St. Louis, MO, USA), the yield dropped to 30% (entry 9). Further optimization showed that the solvent was important for the protodecarboxylation, acetonitrile, which is a polar solvent with adequate loss factor (tanδ = 0.062), provided the suitable substrate solubility and the energy absorbing efficiency, was the better solvent for the reaction, compared to DMF and DMSO. Also, the protodecarboxylation reaction occurred efficiently at 130 °C (entries 10–12), completed in one hour at the initial trials, but the reaction time could be further reduced to half hour after optimization of the other factors (entries 13 and 14).
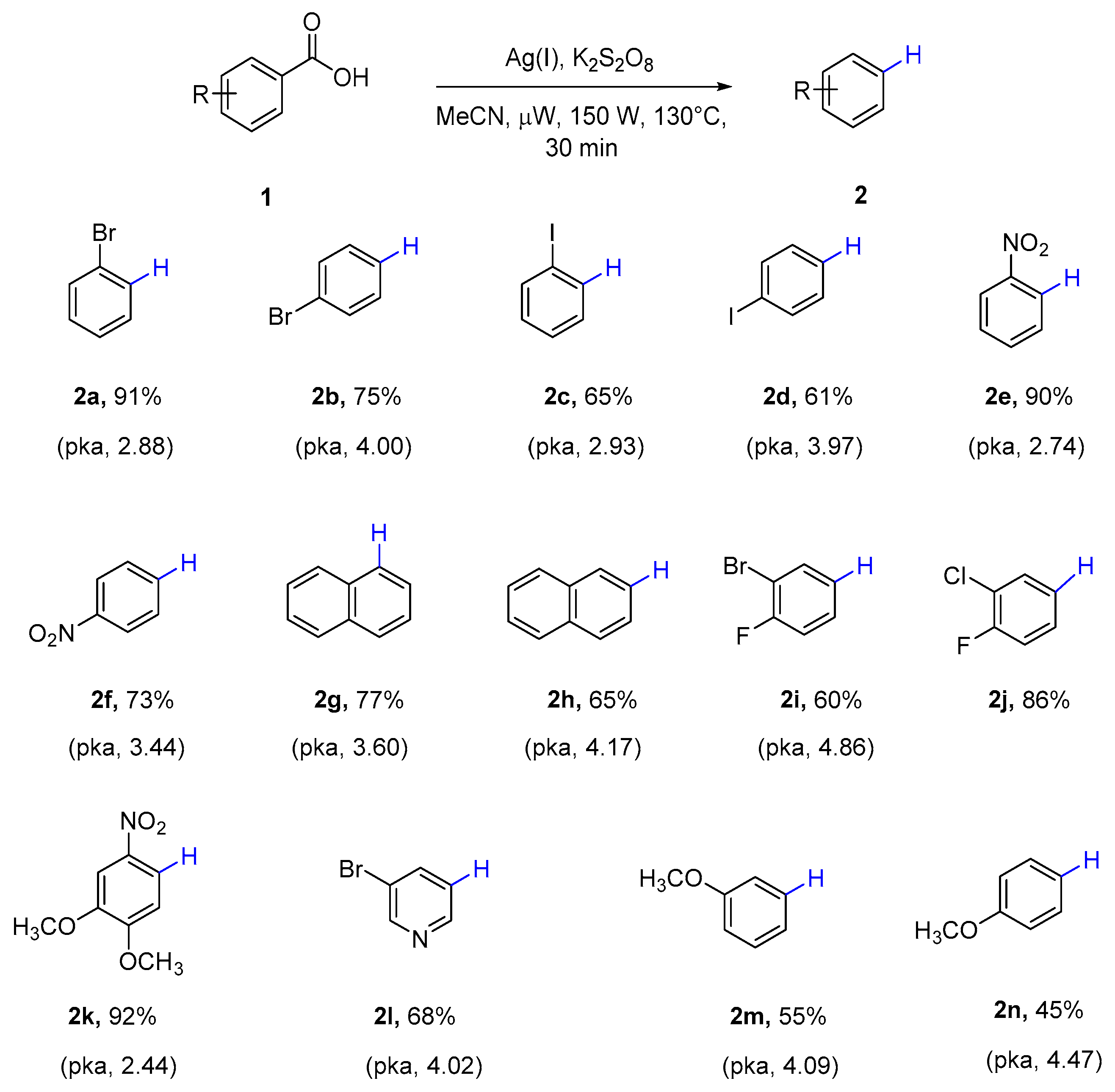
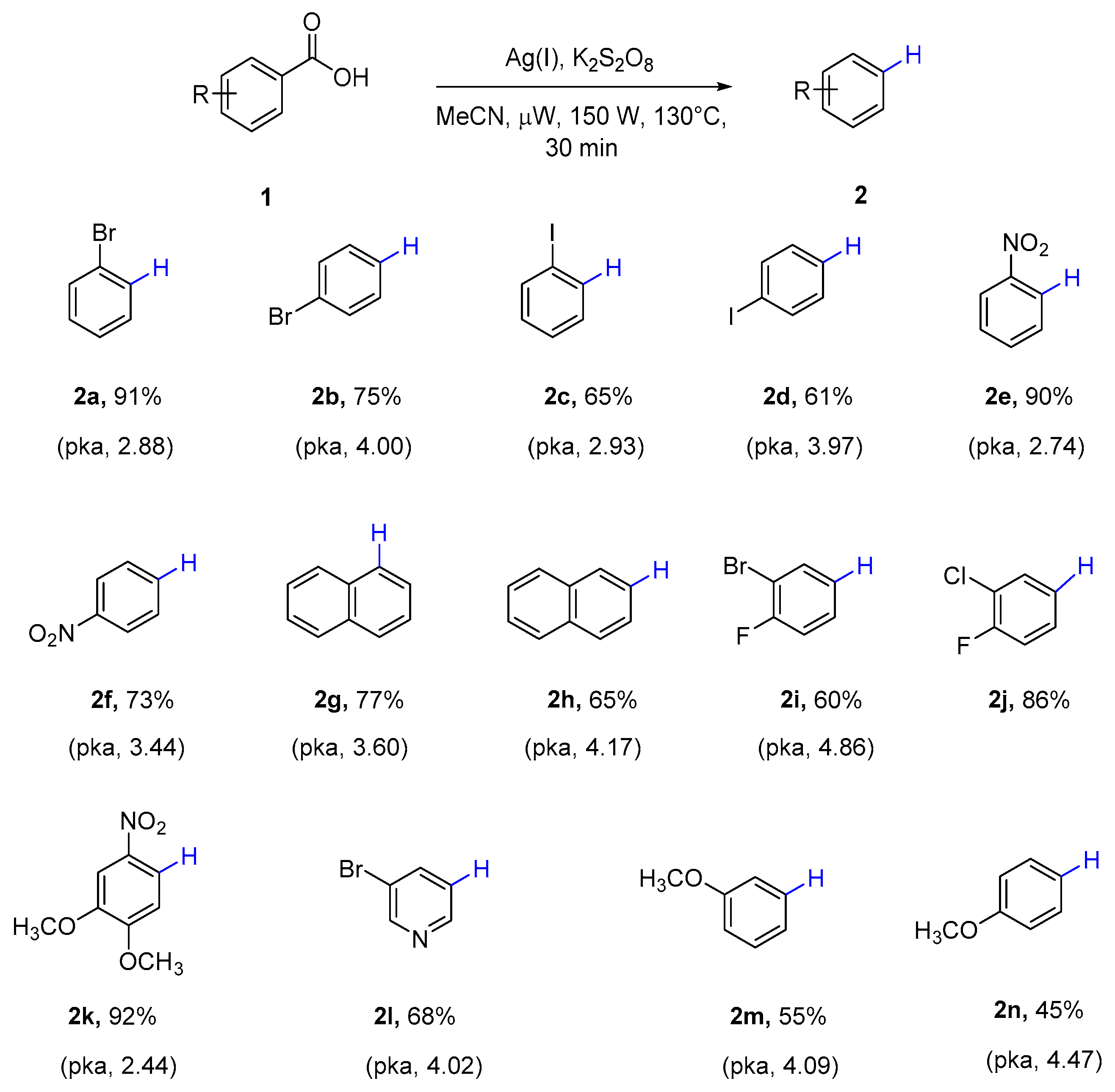
With the optimized conditions in hand, the feasibility of this protocol was investigated. A variety of substituted aromatic carboxylic acids (Scheme 4) could proceed smoothly to afford the corresponding protodecarboxylation products. A relationship was revealed between the dissociation constants of carboxylic acids with the productivities of protodecarboxylation processes. Better yields were obtained for the carboxylic acids with relatively low pKa values, such as 2-bromo benzoic acid (2a), 2-nitro benzoic acid (2e), and 2-nitro-4, 5-dimethoxy benzoic acid (2k). In contrast, 4-bromo benzoic acid (2b), 4-nitro benzoic acid (2f), 3-bromo-4-fluoro benzoic acid (2i), and para- and meta-methoxy benzoic acid (2m, 2n)—which have higher acid dissociation constants—showed relatively poor yields. Interestingly, polycyclic carboxylic acid 1-naphthoic acid (2g, pKa 3.6) showed more favorable yield than 2-naphthoic acid (2h, pKa 4.17) and 5-bromonicotinic acid (2l, pKa 4.02).
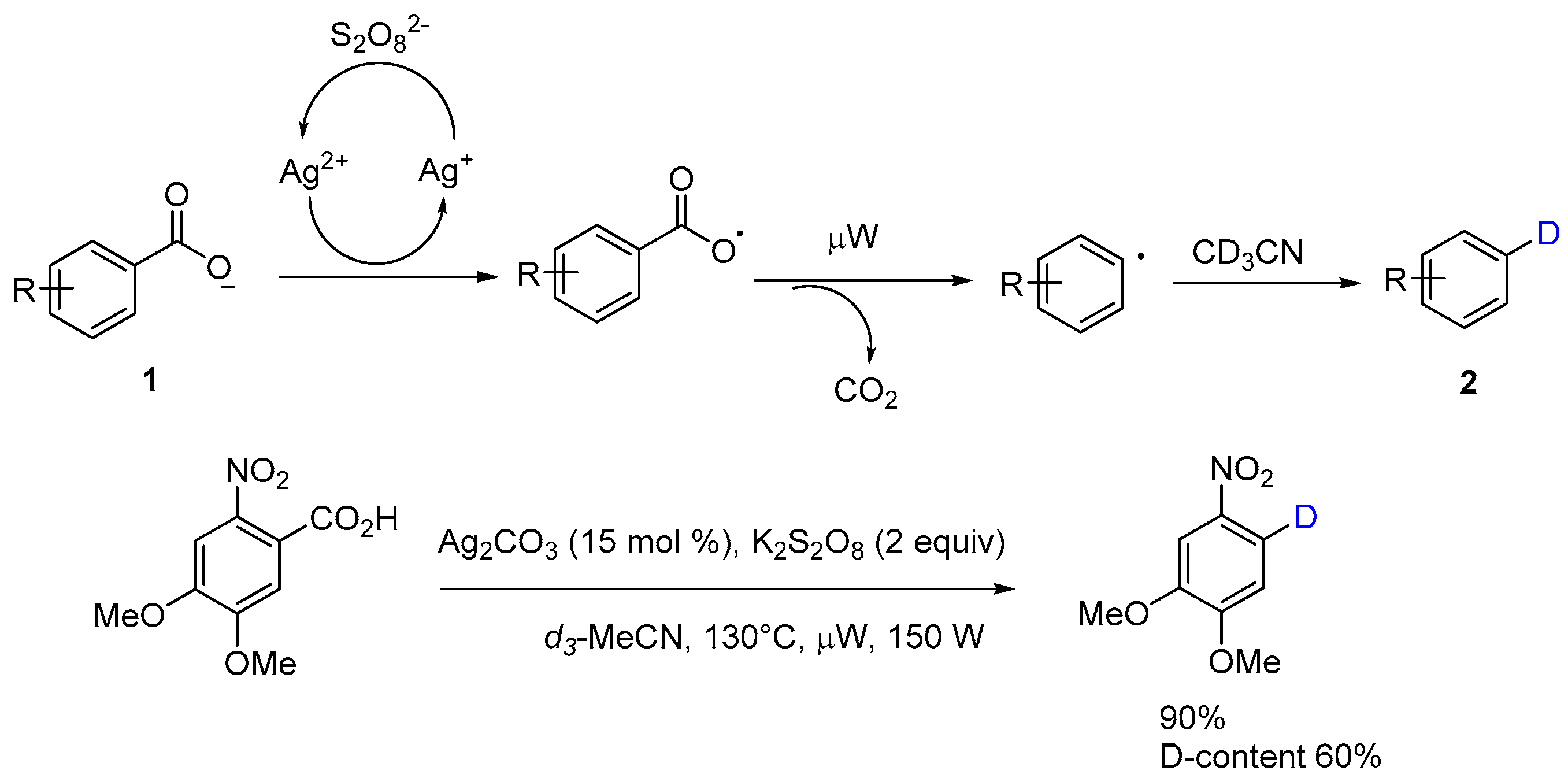
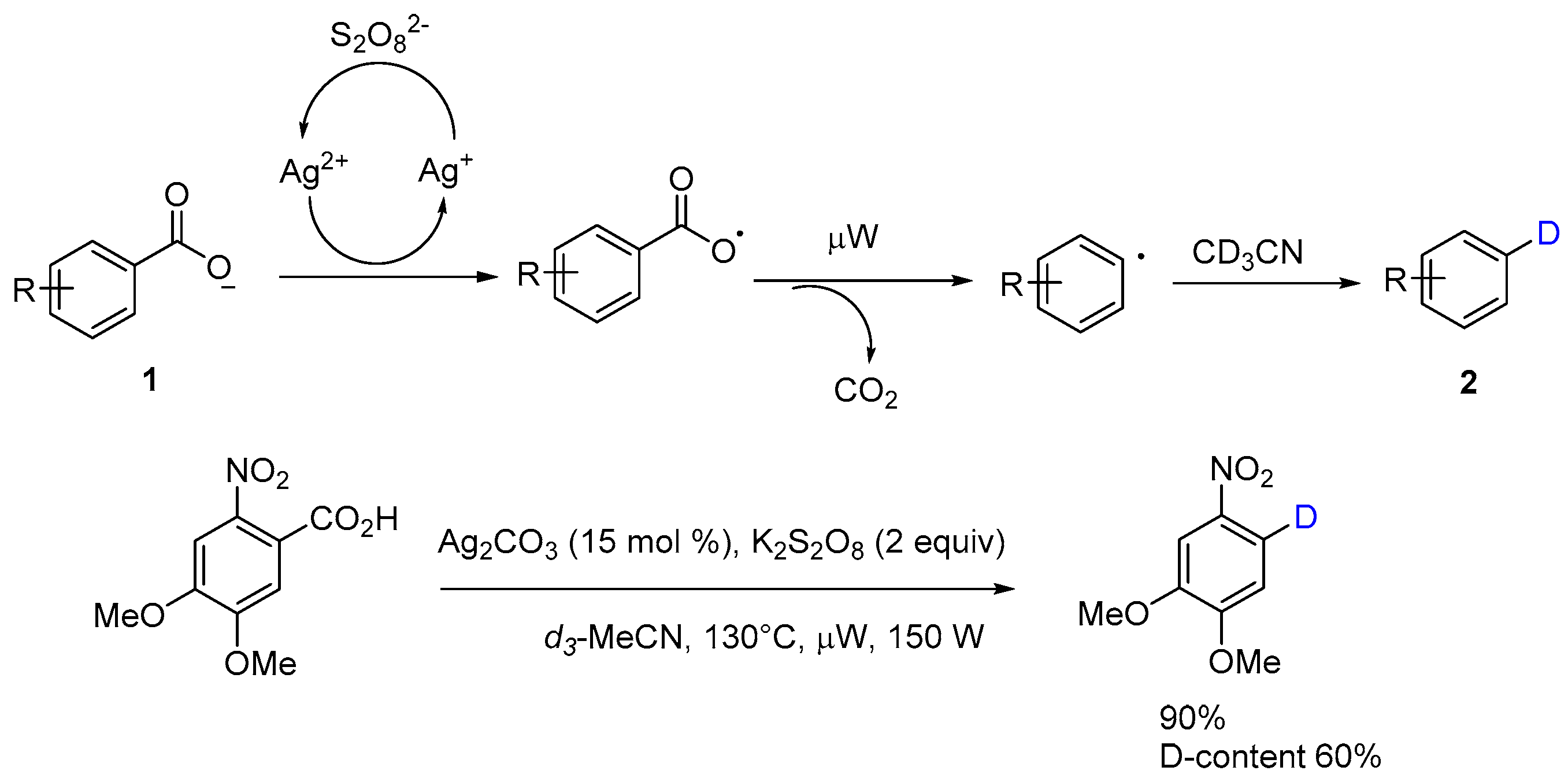
The mechanism of oxidative decarboxylation with Ag(I) and K2S2O8 has been investigated and proposed in the literature [12,14]. Potassium peroxydisulfate can oxidize the silver(I) salts to silver(II), which could covert carboxylate anion into aryl carboxylate radical. Microwaving facilitates and accelerates the elimination of carbon dioxide from carboxylate radical to generate the aryl radical, which can pick up a hydrogen from acetonitrile to form the product 2 (Scheme 5). Protodecarboxylation of 4,5-dimethoxy-2-nitrobenzoic acid in deuterated acetonitrile afforded the corresponding 1,2-dimethoxy-4-nitrobenzene-5-d in the yield of 90% with D-content of 60%, which properly vindicates the mechanism previously proposed. As this example showed, the deuterium exchange with free carboxylic acids in this process might provide an alternative route for the synthesis of deuterium-labeled compounds, useful for molecular structure analysis investigations of the reaction mechanism, and be employed as intermediates or metabolites for chemical biology research.
2.2. Microwave-Assisted Decarboxylative Iodination of Aromatic Carboxylic Acid
With the optimized reaction condition established for protodecarboxylation, we were in a good position to investigate microwave-assisted decarboxylative iodination of aromatic carboxylic acid. We reckon that the aryl radical generated from decarboxylation of aromatic carboxylic acid will readily pick up iodine from a suitable source. Treatment of ortho-bromo benzoic acid with silver(I) carbonate (15 mol %), potassium persulfate (2 equiv.) and iodine (2 equiv.) in acetonitrile under heating in microwave reactor (150 W) at 130 °C for 30 min afforded the corresponding 1-iodo-2-bromobenzene in the yield of 76% (Table 2, entry 1), no obvious formation of protodecarboxylation product had been observed. It clearly showed that the decarboxylative iodination was a faster reaction in the competition during the process. Additionally, an improved yield was observed when the loading of silver catalyst was increased from 15% to 30% (entries 2–4). Meanwhile, sodium iodide and potassium iodide were examined as the iodine source, although the reactions employing iodine generally gave good yields, no reaction occurred when NaI or KI was used (entries 5 and 6). Further optimization showed that the addition of sodium carbonate (2.0 equiv.) promoted the progress of the reaction (entry 7), supposedly facilitating the formation of carboxylate anion for decarboxylation and removal of hydrogen iodide from iodination reaction. The optimal temperature was 130 °C, the reaction yield deceased when the temperature was further raised (entries 8 and 9). Also this iodination required slightly longer time (45 min) to complete (entries 10 and 11), compared to half an hour for protodecarboxylation reaction.
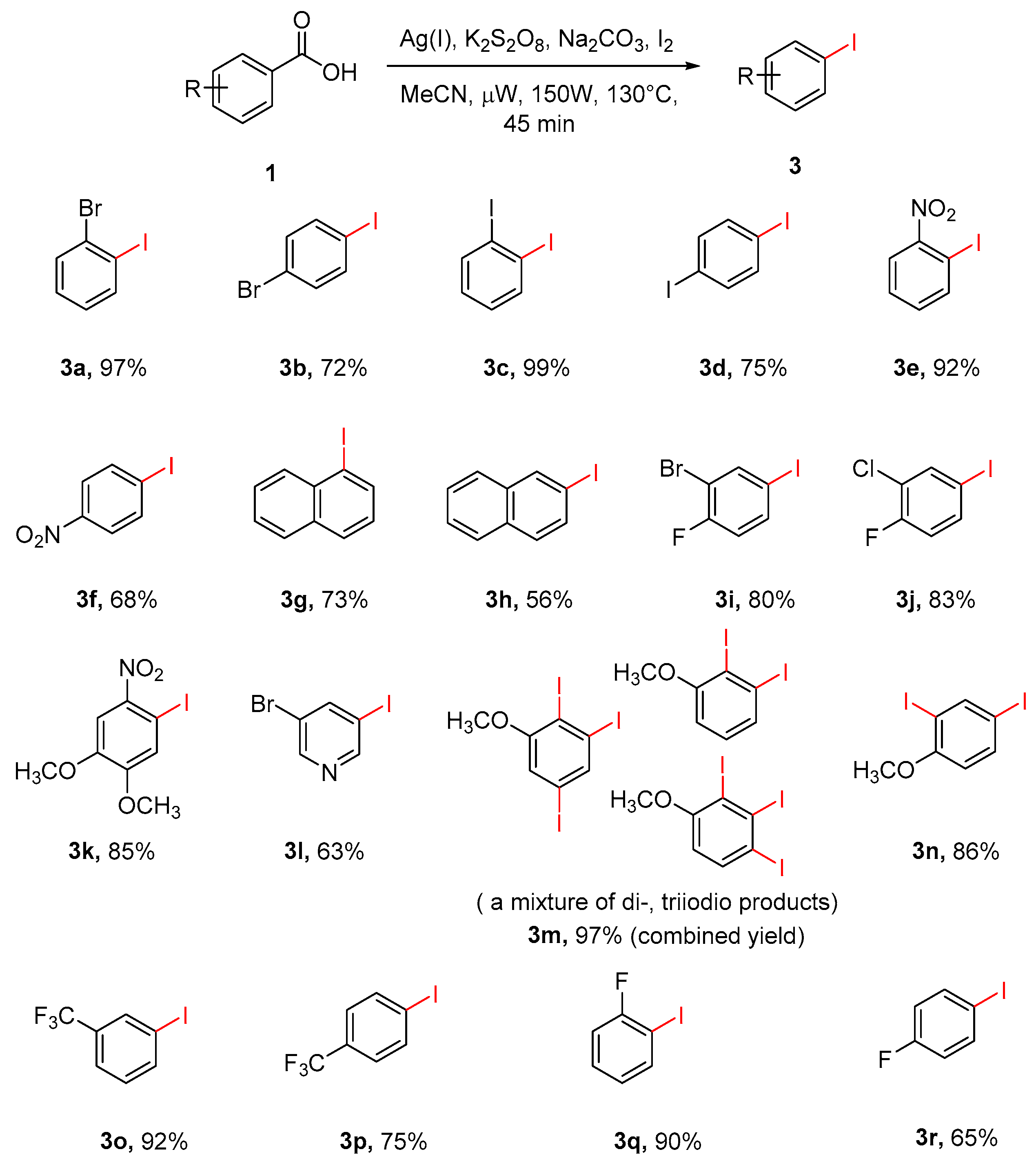
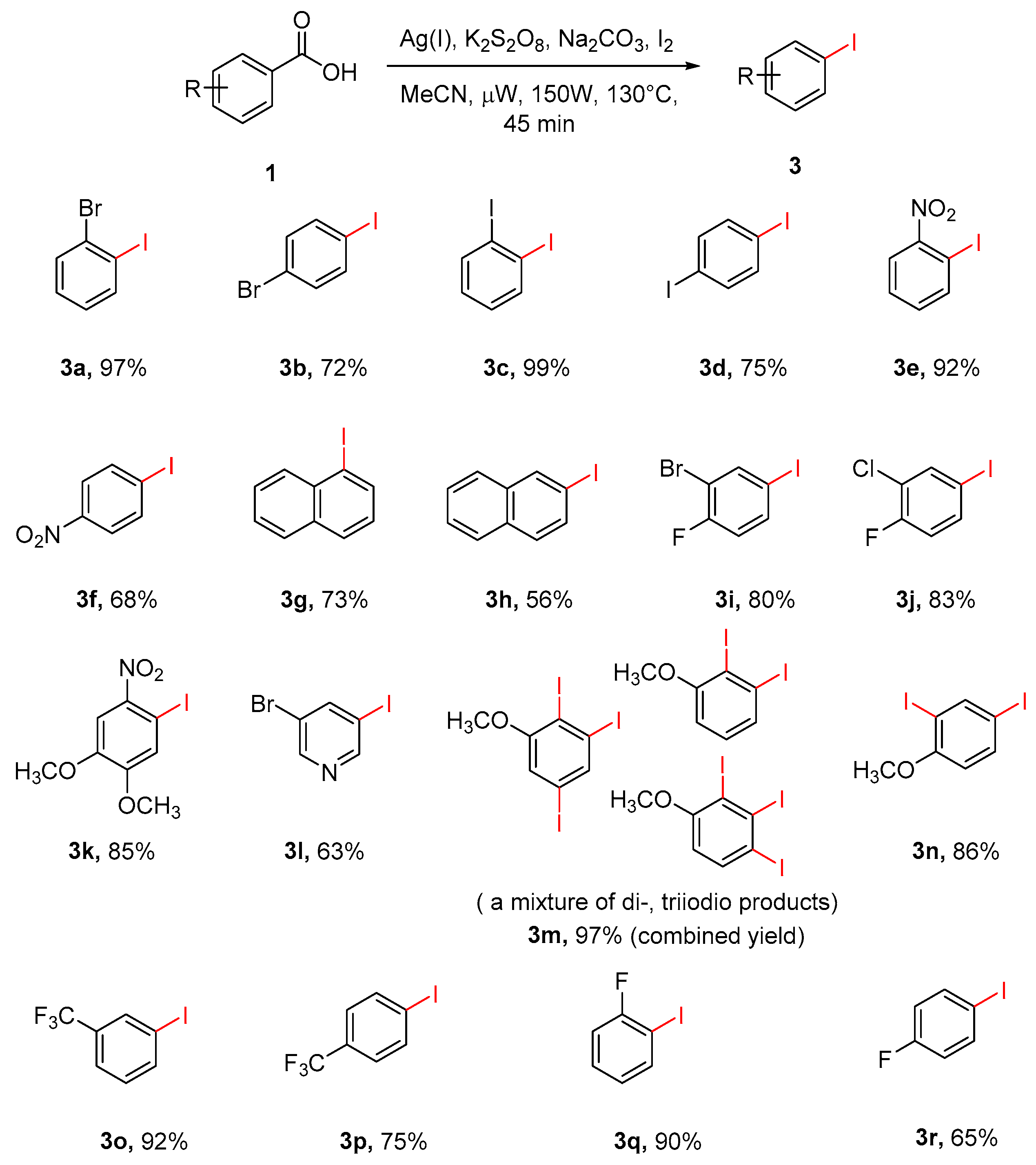
To explore the scope of this transformation, we investigated the decarboxylative iodination reaction of various aryl and heteroaryl carboxylic acids under the optimized reaction condition. In general, the procedure typically delivered the clean, corresponding aryl or heteroaryl iodides in the yield ranging from 56% to 99% (Scheme 6). The ortho-substituted aryl carboxylic acids generally gave very good yields, comparable to the protodecarboxylation abovementioned. Polysubstituted benzoic acids, such as 3-bromo-4-fluorobenzoic acid (3i) and 4, 5-dimethoxy-2-nitrobenzoic acid (3k) smoothly underwent the reaction to produce aryl iodides in good yields.
In terms of nitro-substituted benzoic acids (3e, 3f) and monofluorine-substituted benzoic acids (3q, 3r), ortho-substituted acids showed better yields than their para-substituted isomers. Iodination occurred smoothly on trifluoromethyl-substituted benzoic acids as well with satisfactory yields (3o, 3p). Diverse results were obtained for 1 and 2-substituted naphthoic acids (3g, 3h) and heterocyclic carboxylic acids, such as 5-Bromonicotinic acid (3l). Interestingly, aryl carboxylic acids with strong electron-donating methoxy group, produced a mixture of diiodo- and triiodo-substituted products for 3-methoxybenzoic acid (3m) and a single disubstituted product for 4-methoxybenzoic acid (3n). Although the exact rational for the formation of these compounds is not clear, it was tentatively suggested that the presence of electron-donating group activates the aromatic ring for a direct electrophilic iodination under the adopted reaction condition.
We also explored the probabilities of use this methodology for decarboxylative bromination and chlorination reaction, using Br2, NBS (N-bromosuccinimide) as a bromine source, and NCS (N-chlorosuccinimide) as a chlorine source. These reactions occurred to give the corresponding bromine and chlorine substituted aromatic compounds but with poor yields of 20–40%. Transformation of carboxylic acid group into trifluoromethyl functional group attracts great interests from drug discovery and industry. Our work to trap trifluoromethyl group with various trifluoromethyl reagents, such as CF3I, TMSCF3, Umemoto reagent, Togni reagent, and radical reagents (Na2SO3CF3, trifluoroacetic acid) is still ongoing, and will be reported in due course.
3. Experimental Details
3.1. General Procedure for Microwave-Assisted Protodecarboxylation Reaction of Aromatic Carboxylic Acid
An oven-dried microwave reaction vial (35 mL) was charged with benzoic acid (0.50 mmol), silver(I) carbonate (15 mol %), potassium persulfate (2 equiv.) in 4 mL acetonitrile. The mixture was heated in microwave reactor (CEM Discover-SP, Buckingham, UK) at 130 °C for 30 min, then allowed to cool down. For the measurement of volatile products, chlorobenzene (0.50 mmol) was added into the reaction mixture, and analysis was carried out on GC-MS (Agilent 5975C Triple Axis GC-MS, Santa Clara, CA, USA). Otherwise, the reaction mixture was diluted with Et2O (10 mL) and washed with saturated aqueous NaHCO3 (10 mL). The aqueous layer was extracted with Et2O (2 × 10 mL) and the combined organic layers were dried with anhydrous MgSO4, then filtered and concentrated under reduced pressure. The crude product was purified by the short silica column and eluted with n-hexane to give the product.
3.2. General Procedure for Microwave-Assisted Decarboxylative Iodination Reaction of Aromatic Carboxylic Acid
An oven-dried microwave reaction vial (35 mL) was charged with 2-nitro benzoic acid (0.50 mmol), silver(I) carbonate (25 mol %), potassium persulfate (2 equiv.), sodium carbonate (2.0 equiv.), and iodine (2 equiv.) in 4 mL acetonitrile. The mixture was heated in microwave reactor at 130 °C for 45 min, then allowed to cool down, diluted with EtOAc (10 mL), and washed with saturated aqueous Na2S2O3 (10 mL). The resulting mixture was extracted with EtOAc (2 × 10 mL) and the combined organic layers were dried with anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by the short silica column and eluted with n-hexane (see Supplementary Materials).
4. Conclusions
In summary, we have demonstrated that microwave-assisted protodecarboxylation and decarboxylative iodination could proceed smoothly on aromatic and heterocyclic carboxylic acids to produce the corresponding aromatic compounds and regioselective aromatic iodide. Significantly, employing microwave technology greatly facilitates the process, the reactions could be completed in less than one hour. This work provides a practical and general protocol for the synthesis of deuterated and iodine-labelling intermediate or compounds, which could be used for the radio medicine and biological research.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/7/11/314/s1, Experimental procedure and spectral data for the iodides.
Acknowledgments
The authors thank the Jiangsu Science and Technology Program (Basic Research Program) (Grant No. BK20131180) for the funding.
Author Contributions
Y.L. conceived and designed the experiments; K.Z. performed the experiments and analyzed the data; Y.L. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goossen, L.J.; Rodriguez, N.; Goossen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. Engl. 2008, 47, 3100–3120. [Google Scholar] [CrossRef] [PubMed]
- Goossen, L.J.; Goossen, K.; Rodriguez, N.; Blanchot, M.; Linder, C.; Zimmermann, B. New catalytic transformations of carboxylic acids. Pure Appl. Chem. 2008, 80, 1725–1733. [Google Scholar] [CrossRef]
- Font, M.; Quibell, J.M.; Perry, G.J.P.; Larrosa, I. The use of carboxylic acids as traceless directing groups for regioselective C-H bond functionalisation. Chem. Commun. 2017, 53, 5584–5597. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C-C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A.K.; Lei, A. Recent advances in radical C-H activation/radical cross-coupling. Chem. Rev. 2017, 117, 9016–9085. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Arcos, U.A.; Rojas-Ocampo, J.; Porcel, S. Oxidative cyclization of alkenoic acids promoted by AgOAc. Dalton Trans. 2016, 45, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Jafarpour, F.; Zarei, S.; Olia, M.B.; Jalalimanesh, N.; Rahiminejadan, S. Palladium-catalyzed decarboxylative cross-coupling reactions: A route for regioselective functionalization of coumarins. J. Org. Chem. 2013, 78, 2957–2964. [Google Scholar] [CrossRef] [PubMed]
- Arroniz, C.; Ironmonger, A.; Rassias, G.; Larrosa, I. Direct ortho-arylation of ortho-substituted benzoic acids: Overriding Pd-catalyzed protodecarboxylation. Org. Lett. 2013, 15, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.; Dzik, W.I.; Goossen, L.J. Synthesis of aryl ethers from benzoates through carboxylate-directed C-H-activating alkoxylation with concomitant protodecarboxylation. Angew. Chem. Int. Ed. Engl. 2013, 52, 2959–2962. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, S.; Nolan, S.P. Gold(I)-catalyzed protodecarboxylation of (hetero)aromatic carboxylic acids. Chemistry 2013, 19, 14034–14038. [Google Scholar] [CrossRef] [PubMed]
- Goossen, L.J.; Thiel, W.R.; Rodriguez, N.; Linder, C.; Melzer, B. Copper-catalyzed protodecarboxylation of aromatic carboxylic acids. Adv. Synth. Catal. 2007, 349, 2241–2246. [Google Scholar] [CrossRef]
- Cornella, J.; Sanchez, C.; Banawa, D.; Larrosa, I. Silver-catalysed protodecarboxylation of ortho-substituted benzoic acids. Chem. Commun. 2009, 7176–7178. [Google Scholar] [CrossRef] [PubMed]
- Grainger, R.; Cornella, J.; Blakemore, D.C.; Larrosa, I.; Campanera, J.M. The ortho-substituent effect on the Ag-catalysed decarboxylation of benzoic acids. Chemistry 2014, 20, 16680–16687. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Taylor, J.B.; Greaney, M.F. Protodecarboxylation of benzoic acids under radical conditions. Chem. Commun. 2012, 48, 8270–8272. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Cai, Q. Copper/amino acid catalyzed cross-couplings of aryl and vinyl halides with nucleophiles. Acc. Chem. Res. 2008, 41, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Blanchard, N.; Toumi, M. Copper-mediated coupling reactions and their applications in natural products and designed biomolecules synthesis. Chem. Rev. 2008, 108, 3054–3131. [Google Scholar] [CrossRef] [PubMed]
- Barluenga, J.; Gonzalez, J.M.; Garciamartin, M.A.; Campos, P.J.; Asensio, G. Acid-mediated reaction of bis(pyridine)iodonium(I) tetrafluoroborate with aromatic-compounds—A selective and general iodination method. J. Org. Chem. 1993, 58, 2058–2060. [Google Scholar] [CrossRef]
- Bunevicius, R.; Kazanavicius, G.; Zalinkevicius, R.; Prange, A.J., Jr. Effects of thyroxine as compared with thyroxine plus triiodothyronine in patients with hypothyroidism. N. Engl. J. Med. 1999, 340, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Hallouard, F.; Anton, N.; Choquet, P.; Constantinesco, A.; Vandamme, T. Iodinated blood pool contrast media for preclinical X-ray imaging applications—A review. Biomaterials 2010, 31, 6249–6268. [Google Scholar] [CrossRef] [PubMed]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and spect imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Seevers, R.H.; Counsell, R.E. Radioiodination techniques for small organic-molecules. Chem. Rev. 1982, 82, 575–590. [Google Scholar] [CrossRef]
- Baird, W.C.; Surridge, J.H. Halogenation with copper(II) halides—Synthesis of aryl iodides. J. Org. Chem. 1970, 35, 3436–3442. [Google Scholar] [CrossRef]
- Naumann, D.; Ruther, G. Synthesis of aryl iodine(III) difluorides by direct fluorination of aryl iodides. J. Fluorine Chem. 1980, 15, 213–222. [Google Scholar] [CrossRef]
- Cant, A.A.; Bhalla, R.; Pimlott, S.L.; Sutherland, A. Nickel-catalysed aromatic Finkelstein reaction of aryl and heteroaryl bromides. Chem. Commun. 2012, 48, 3993–3995. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ichikawa, S.; Buchwald, S.L. Rapid and efficient copper-catalyzed Finkelstein reaction of (hetero)aromatics under continuous-flow conditions. Angew. Chem. Int. Ed. Engl. 2015, 54, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Buchwald, S.L. Copper-catalyzed halogen exchange in aryl halides: An aromatic Finkelstein reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, W.; Zeng, H.; Mu, X.; Cosa, G.; Mi, Z.; Li, C.J. Photo-induced metal-catalyst-free aromatic Finkelstein reaction. J. Am. Chem. Soc. 2015, 137, 8328–8331. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.J.P.; Quibell, J.M.; Panigrahi, A.; Larrosa, I. Transition-metal-free decarboxylative iodination: New routes for decarboxylative oxidative cross-couplings. J. Am. Chem. Soc. 2017, 139, 11527–11536. [Google Scholar] [CrossRef] [PubMed]
- Mavandadi, F.; Pilotti, A. The impact of microwave-assisted organic synthesis in drug discovery. Drug Discov. Today 2006, 11, 165–174. [Google Scholar] [CrossRef]
- Lidstrom, P.; Westman, J.; Lewis, A. Enhancement of combinatorial chemistry by microwave-assisted organic synthesis. Comb. Chem. High Throughput Screen. 2002, 5, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Goossen, L.J.; Manjolinho, F.; Khan, B.A.; Rodriguez, N. Microwave-assisted Cu-catalyzed protodecarboxylation of aromatic carboxylic acids. J. Org. Chem. 2009, 74, 2620–2623. [Google Scholar] [CrossRef] [PubMed]
- Ferro, S.; Grazia, S.D.; De Luca, L.; Gitto, R.; Faliti, C.E.; Debyzer, Z.; Chimirri, A. Microwave assisted organic synthesis (MAOS) of small molecules as potential HIV-1 integrase inhibitors. Molecules 2011, 16, 6858–6870. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Silver-catalyzed protodecarboxylation reactions.
Scheme 2.
Photo-induced aromatic halogen exchange reaction [27].
Scheme 2.
Photo-induced aromatic halogen exchange reaction [27].

Scheme 3.
Microwave-assisted silver-catalyzed protodecarboxylation and decarboxylative iodination of aromatic carboxylic acids (this work).
Scheme 3.
Microwave-assisted silver-catalyzed protodecarboxylation and decarboxylative iodination of aromatic carboxylic acids (this work).

Scheme 4.
Scope of microwave assisted protodecarboxylation of aromatic carboxylic acids. a Reaction conditions: Aryl carboxylic acid (0.50 mmol), silver(I) carbonate (15 mol %), potassium persulfate (2 equivalents) in CH3CN (4 mL) was heated in microwave reactor at 130 °C for 30 min. b Yields were determined by gas chromatography-mass spectrometry (GC-MS) analysis of the crude reaction mixture using chlorobenzene as an internal standard. Isolated yields for 2c, 2g, 2i, 2k. c pKa data (25 °C in water), sources: U.S. National Library of Medicine and Reaxys.
Scheme 4.
Scope of microwave assisted protodecarboxylation of aromatic carboxylic acids. a Reaction conditions: Aryl carboxylic acid (0.50 mmol), silver(I) carbonate (15 mol %), potassium persulfate (2 equivalents) in CH3CN (4 mL) was heated in microwave reactor at 130 °C for 30 min. b Yields were determined by gas chromatography-mass spectrometry (GC-MS) analysis of the crude reaction mixture using chlorobenzene as an internal standard. Isolated yields for 2c, 2g, 2i, 2k. c pKa data (25 °C in water), sources: U.S. National Library of Medicine and Reaxys.
Scheme 5.
Proposed mechanism of protodecarboxylation in d3-acetonitrile.
Scheme 6.
Scope of microwave assisted decarboxylative iodination of aromatic carboxylic acids. a Reaction conditions: Aryl carboxylic acid (0.50 mmol), silver(I) carbonate (25 mol %), sodium carbonate (2.0 equiv.), potassium persulfate (2 equiv.), and iodine (2 equiv.) in CH3CN (4 mL) was heated in microwave reactor at 130 °C for 45 min. b Isolated yields.
Scheme 6.
Scope of microwave assisted decarboxylative iodination of aromatic carboxylic acids. a Reaction conditions: Aryl carboxylic acid (0.50 mmol), silver(I) carbonate (25 mol %), sodium carbonate (2.0 equiv.), potassium persulfate (2 equiv.), and iodine (2 equiv.) in CH3CN (4 mL) was heated in microwave reactor at 130 °C for 45 min. b Isolated yields.

{kind=link}
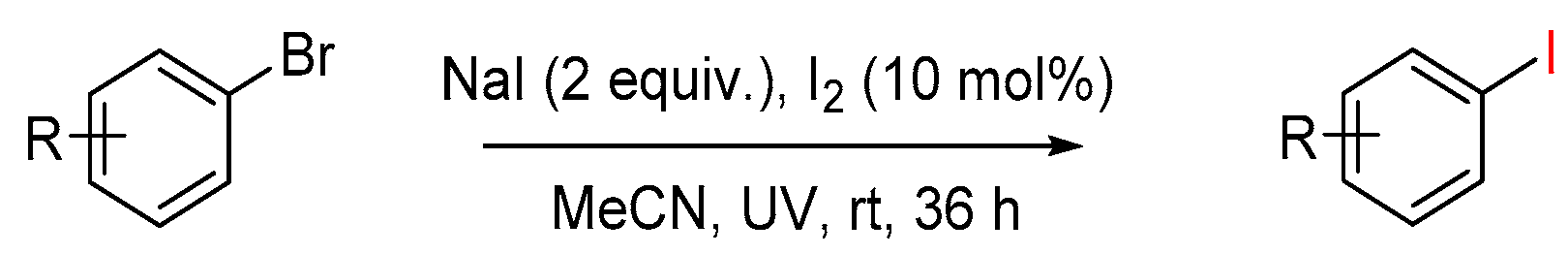{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected results for the optimization of protodecarboxylation reaction conditions.

| Entry | Catalyst Loading (mol %) | Ag(I) Catalyst (mol %) | K2S2O8 (equiv.) | Temperature (°C) | Time (h) | Yield (%) 1 |
|---|---|---|---|---|---|---|
| 1 | 10 | Ag2CO3 | 2 | 130 | 1 | 64 |
| 2 | 15 | Ag2CO3 | 2 | 130 | 1 | 87 |
| 3 | 20 | Ag2CO3 | 2 | 130 | 1 | 90 |
| 4 | 30 | Ag2CO3 | 2 | 130 | 1 | 89 |
| 5 | 40 | Ag2CO3 | 2 | 130 | 1 | 91 |
| 6 | 15 | Ag2O | 2 | 130 | 1 | 68 |
| 7 | 15 | AgOAc | 2 | 130 | 1 | 51 |
| 8 | 15 | AgSO3CF3 | 2 | 130 | 1 | 43 |
| 9 | 15 | Ag2CO3 | 2 (Oxone®) | 130 | 1 | 30 |
| 10 | 15 | Ag2CO3 | 2 | 100 | 1 | 45 |
| 11 | 15 | Ag2CO3 | 2 | 110 | 1 | 63 |
| 12 | 15 | Ag2CO3 | 2 | 140 | 1 | 80 |
| 13 | 15 | Ag2CO3 | 2 | 130 | 0.5 | 90 |
| 14 | 15 | Ag2CO3 | 2 | 130 | 0.3 | 63 |
1 Yields were determined by gas chromatography-mass spectrometry (GC-MS) analysis of the crude reaction mixture using chlorobenzene as an internal standard.
Table 2.
Selected results for the optimization of decarboxylative iodination conditions.

| Entry | Loading of Ag2CO3 (mol %) | Iodine Source (equiv.) | Temperature (°C) | Time (h) | Yield (%) 1 |
|---|---|---|---|---|---|
| 1 | 15 | I2 (2) | 130 | 0.5 | 76 |
| 2 | 20 | I2 (2) | 130 | 0.5 | 80 |
| 3 | 25 | I2 (2) | 130 | 0.5 | 86 |
| 4 | 30 | I2 (2) | 130 | 0.5 | 88 |
| 5 | 25 | KI (2) | 130 | 0.5 | trace |
| 6 | 25 | NaI (2) | 130 | 0.5 | trace |
| 7 2 | 25 | I2 (2) | 130 | 0.5 | 93 |
| 8 2 | 25 | I2 (2) | 140 | 0.5 | 80 |
| 9 2 | 25 | I2 (2) | 150 | 0.5 | 75 |
| 10 2 | 25 | I2 (2) | 130 | 0.75 | 97 |
| 11 2 | 25 | I2 (2) | 130 | 1 | 91 |
1 Yields were determined by gas chromatography-mass spectrometry (GC-MS) analysis of the crude reaction mixture using chlorobenzene as an internal standard. 2 Sodium carbonate (2.0 equiv.) was added.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhan, K.; Li, Y. Microwave-Assisted Silver-Catalyzed Protodecarboxylation and Decarboxylative Iodination of Aromatic Carboxylic Acids. Catalysts 2017, 7, 314. https://doi.org/10.3390/catal7110314
AMA Style
Zhan K, Li Y. Microwave-Assisted Silver-Catalyzed Protodecarboxylation and Decarboxylative Iodination of Aromatic Carboxylic Acids. Catalysts. 2017; 7(11):314. https://doi.org/10.3390/catal7110314
Chicago/Turabian StyleZhan, Kun, and Yi Li. 2017. "Microwave-Assisted Silver-Catalyzed Protodecarboxylation and Decarboxylative Iodination of Aromatic Carboxylic Acids" Catalysts 7, no. 11: 314. https://doi.org/10.3390/catal7110314
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.