
C-Homoscorpionate Oxidation Catalysts—Electrochemical and Catalytic Activity


Abstract
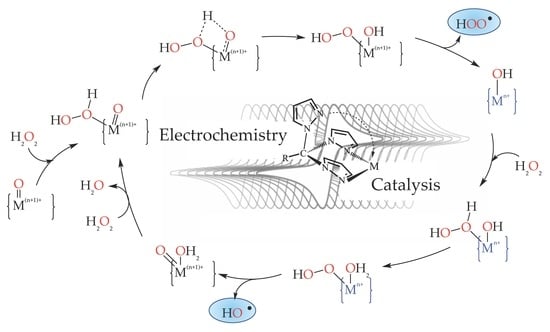
:
1. Introduction
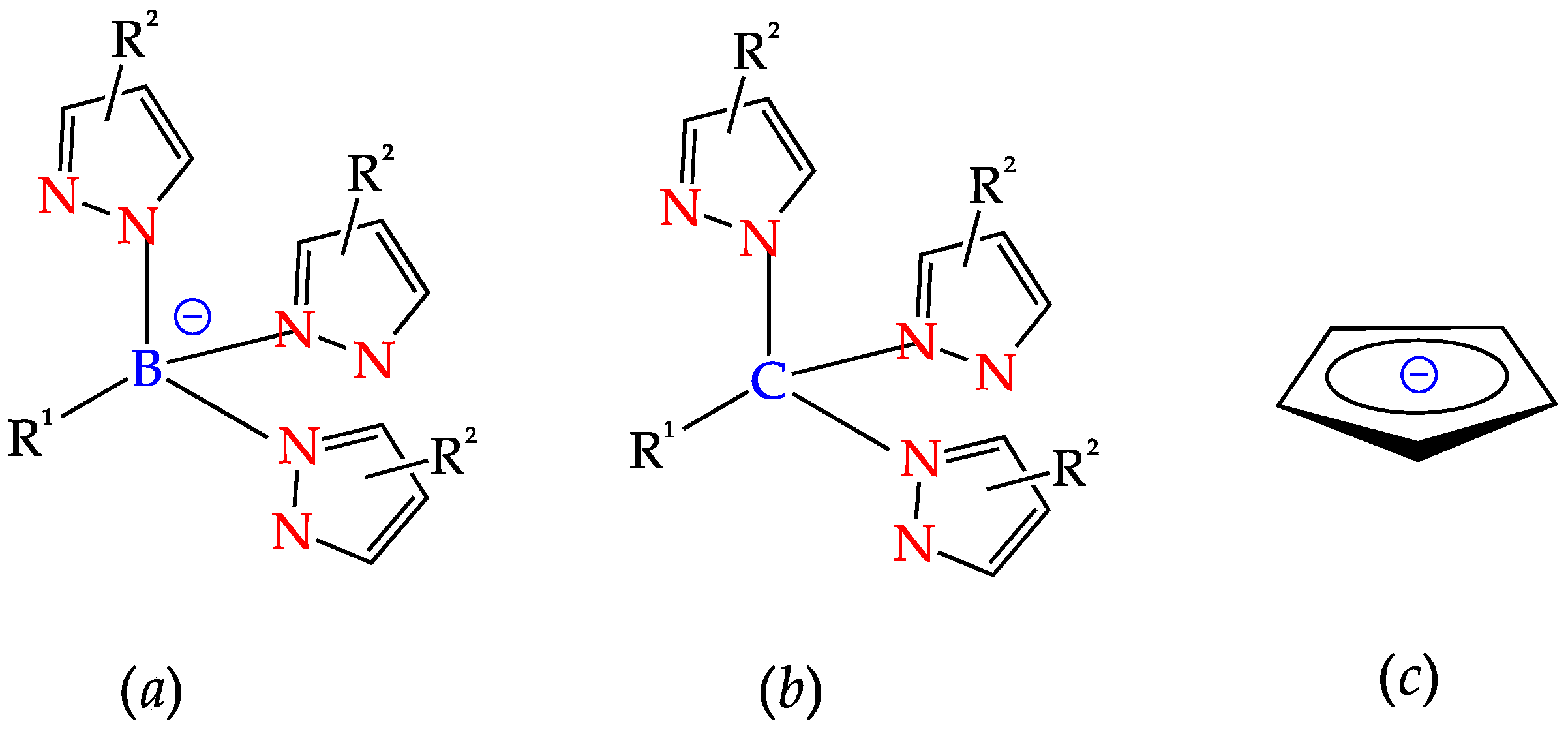
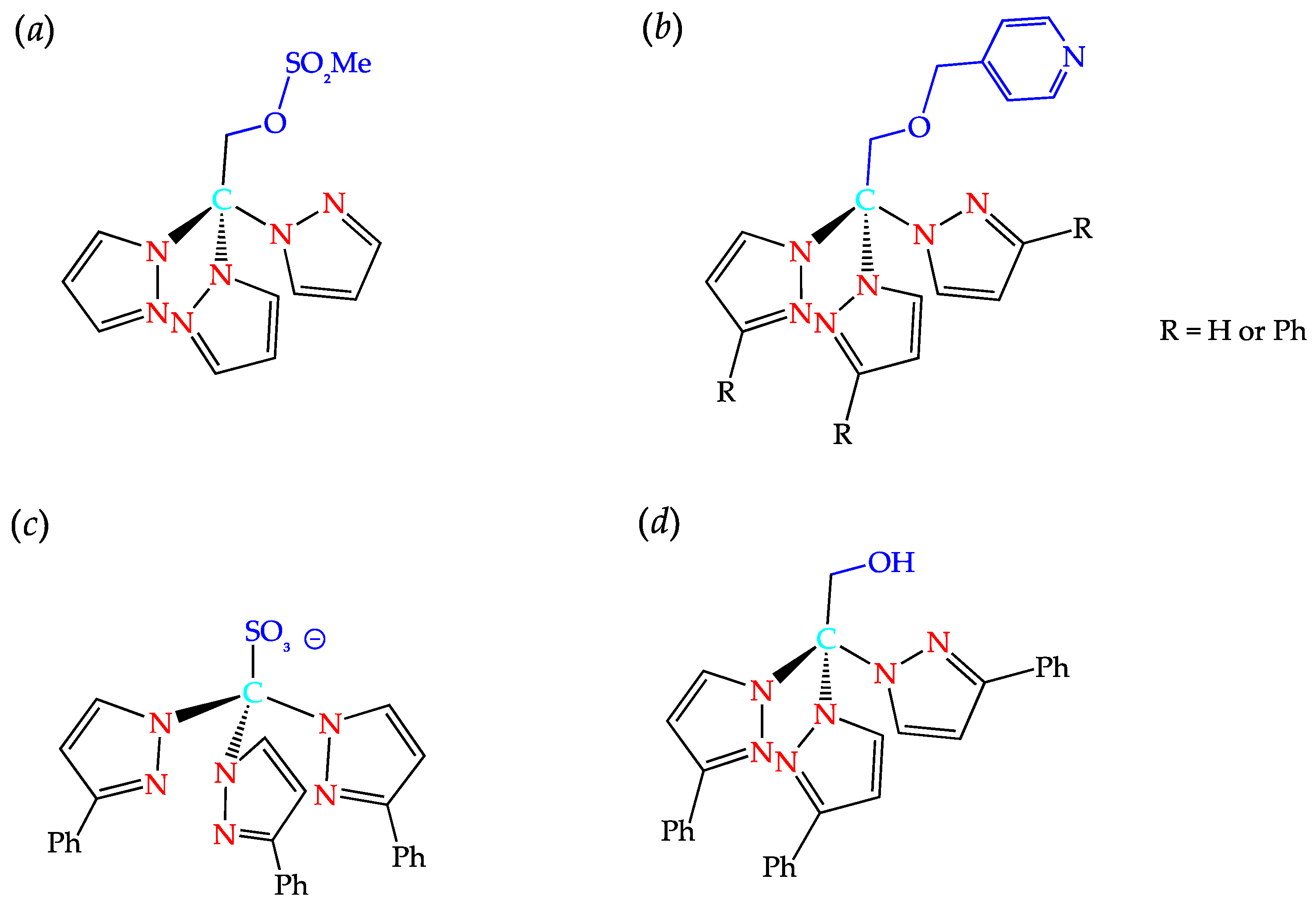
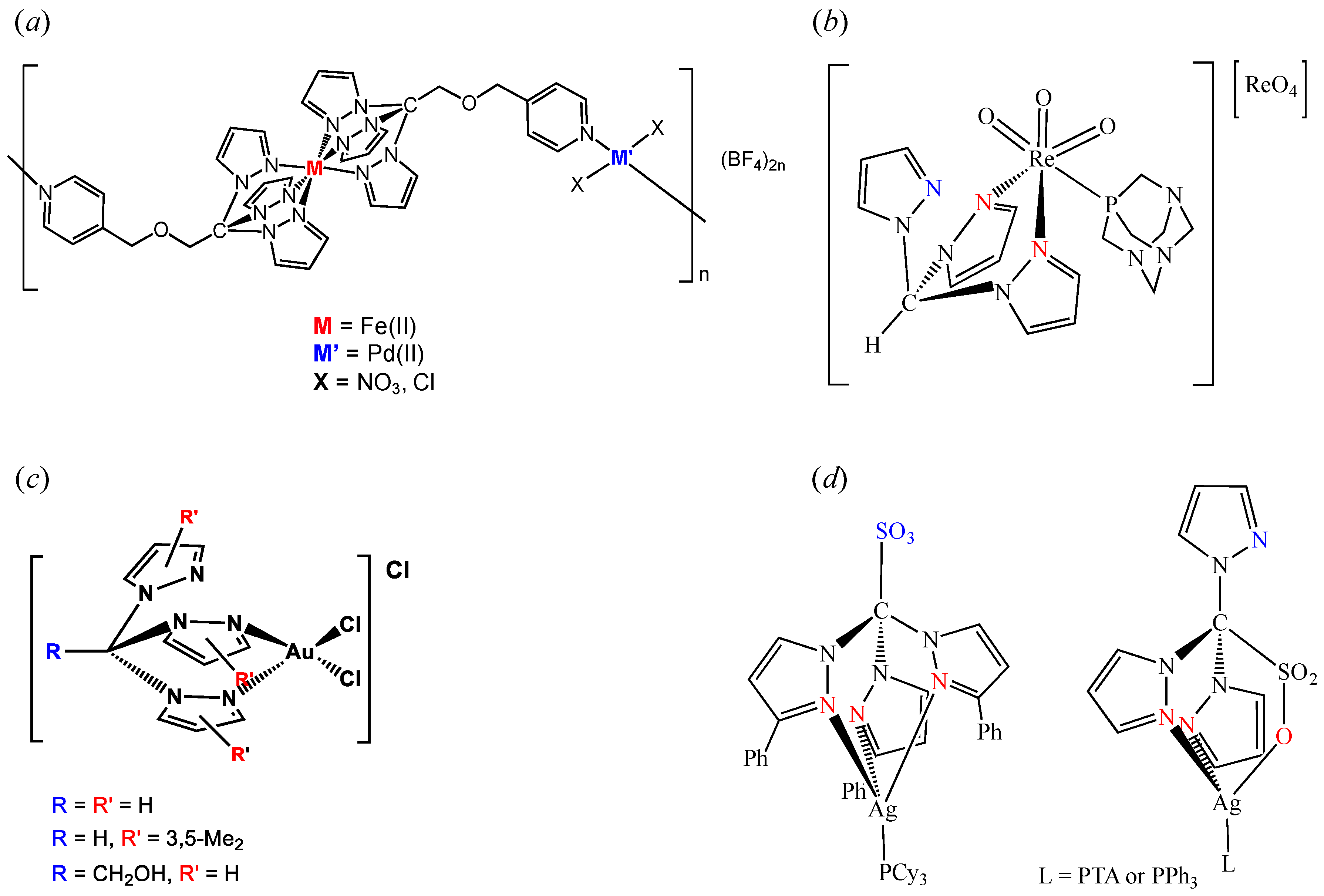
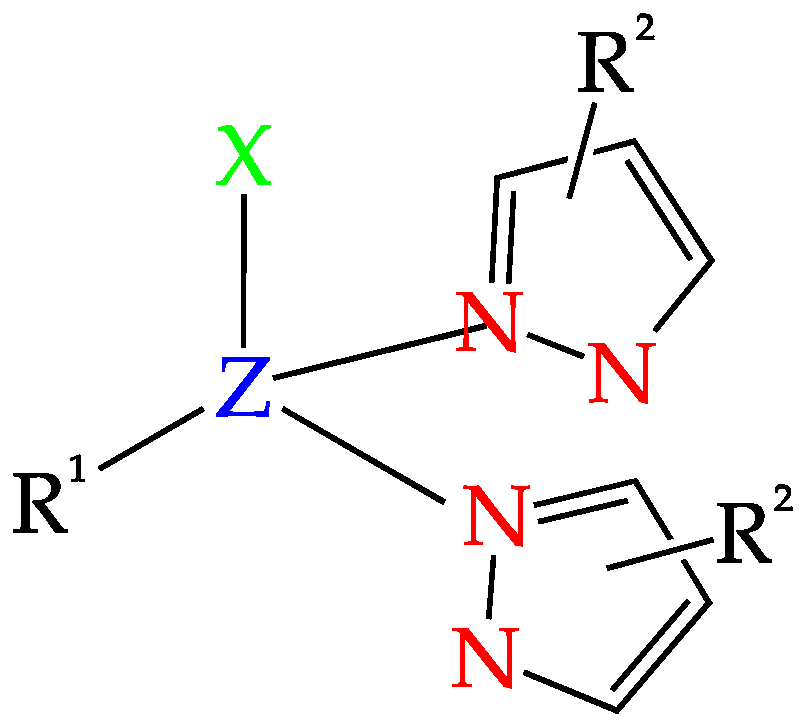
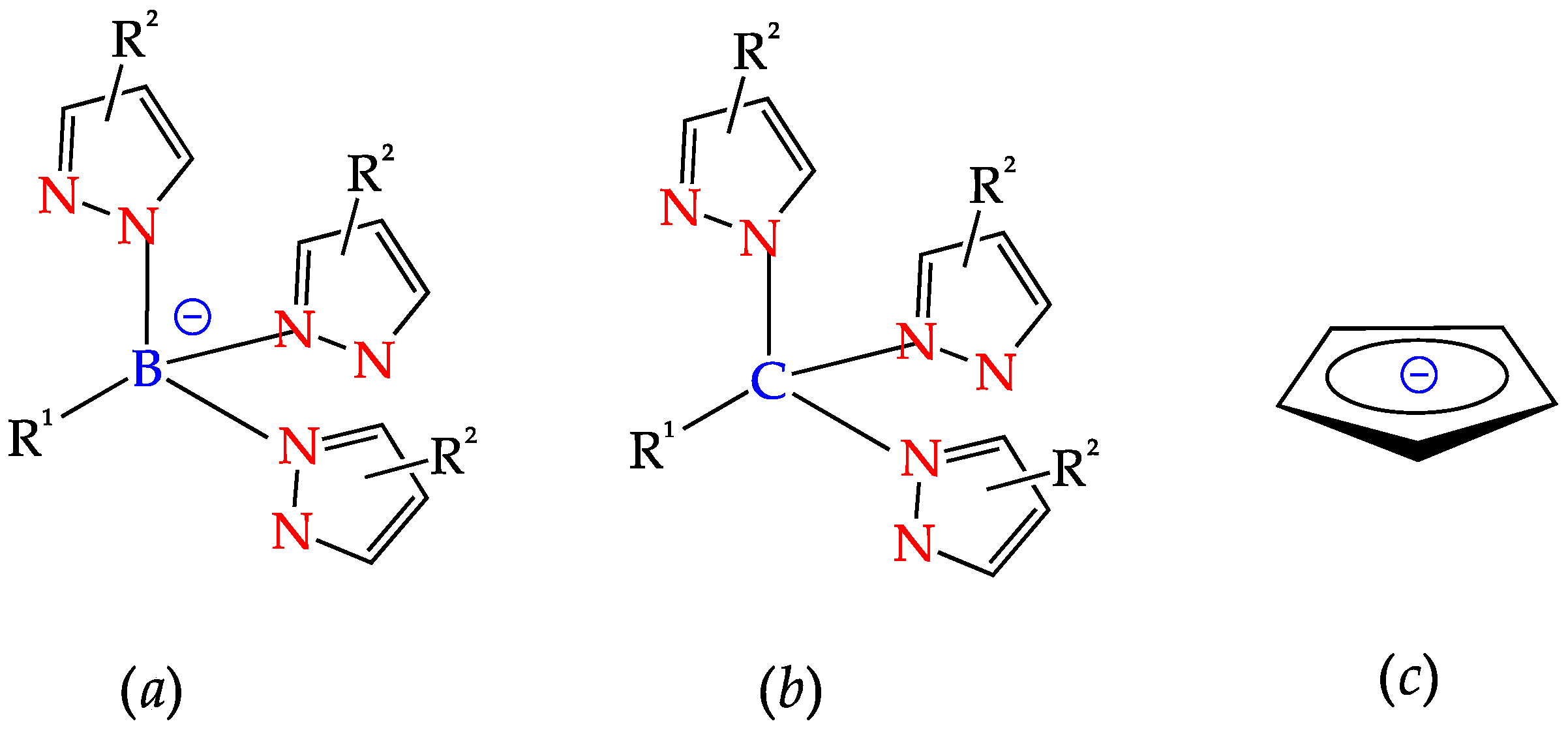
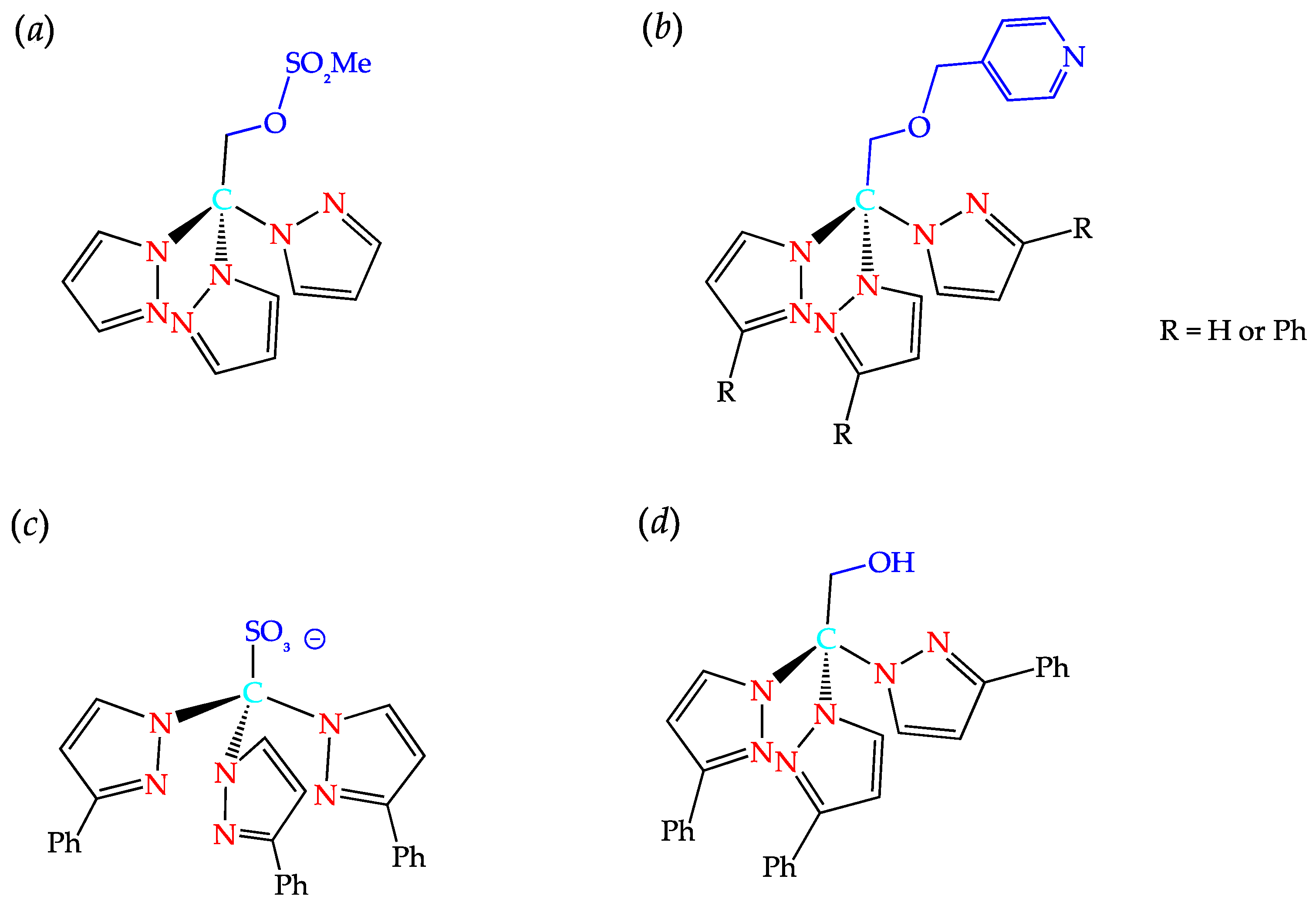
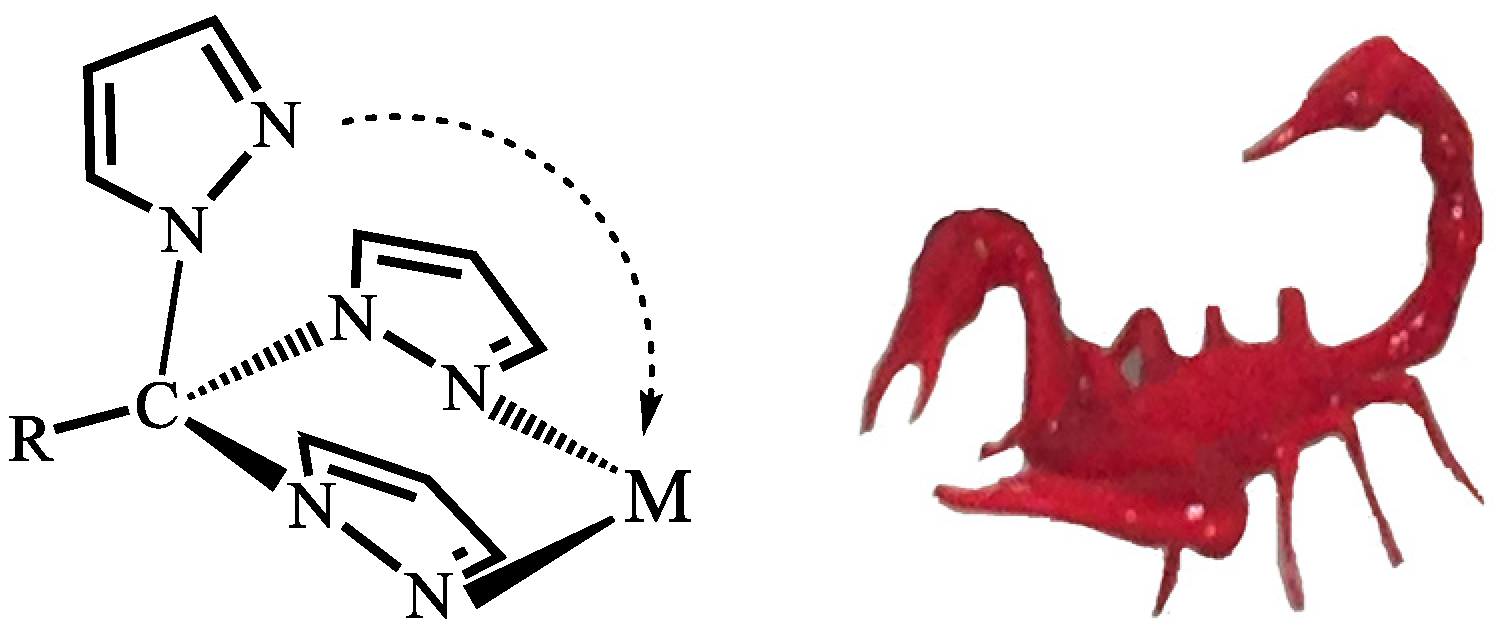
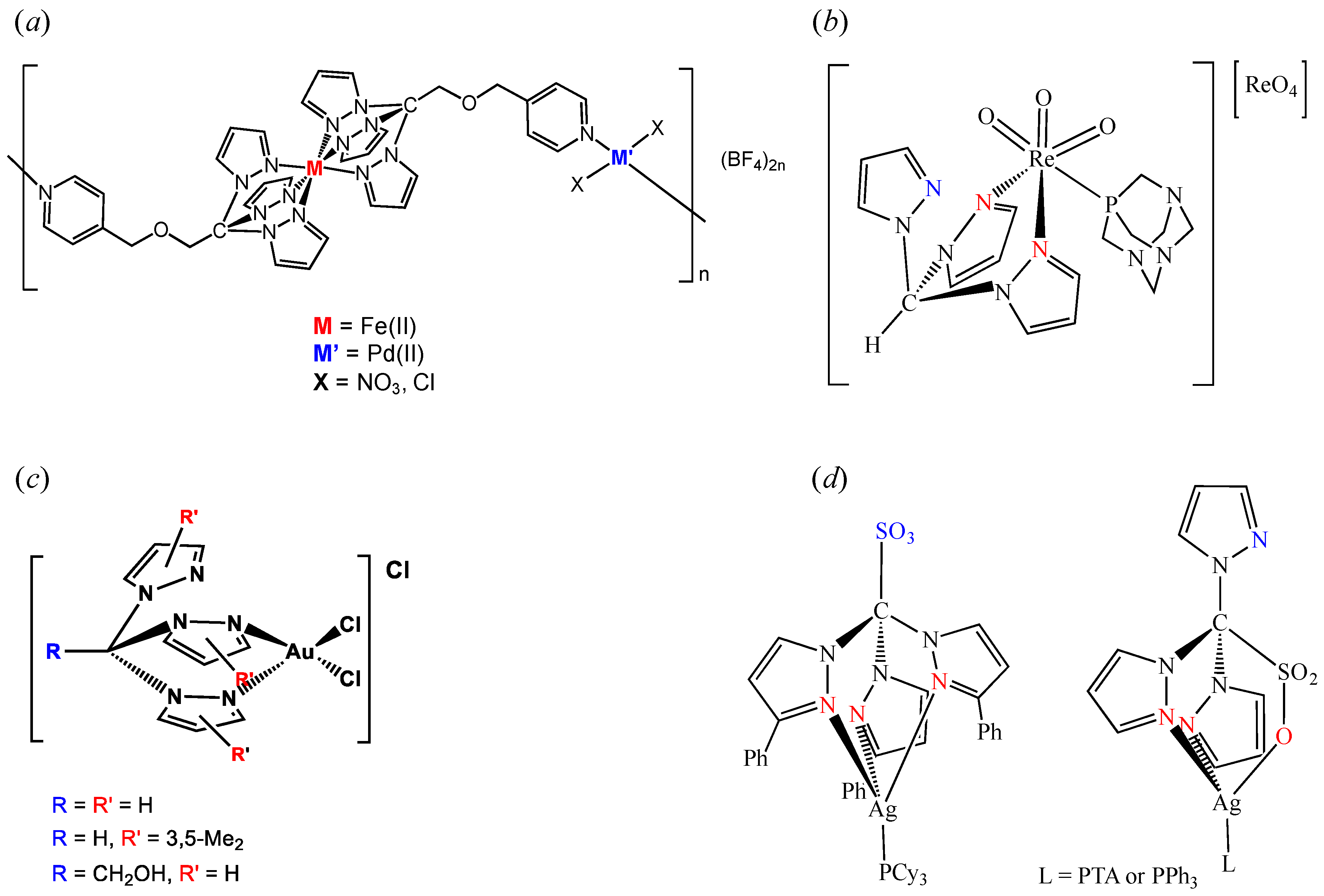
2. C-Homoscorpionates and Their Metal Catalysts
3. Electrochemical Properties of C-Scorpionate Metal Complexes
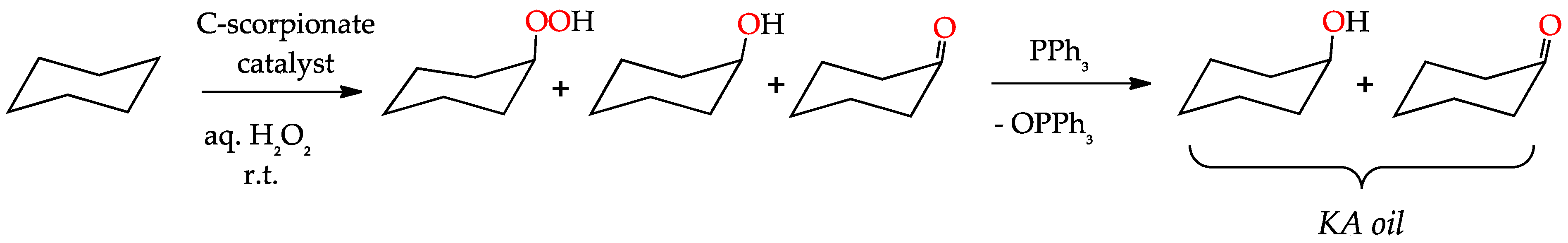
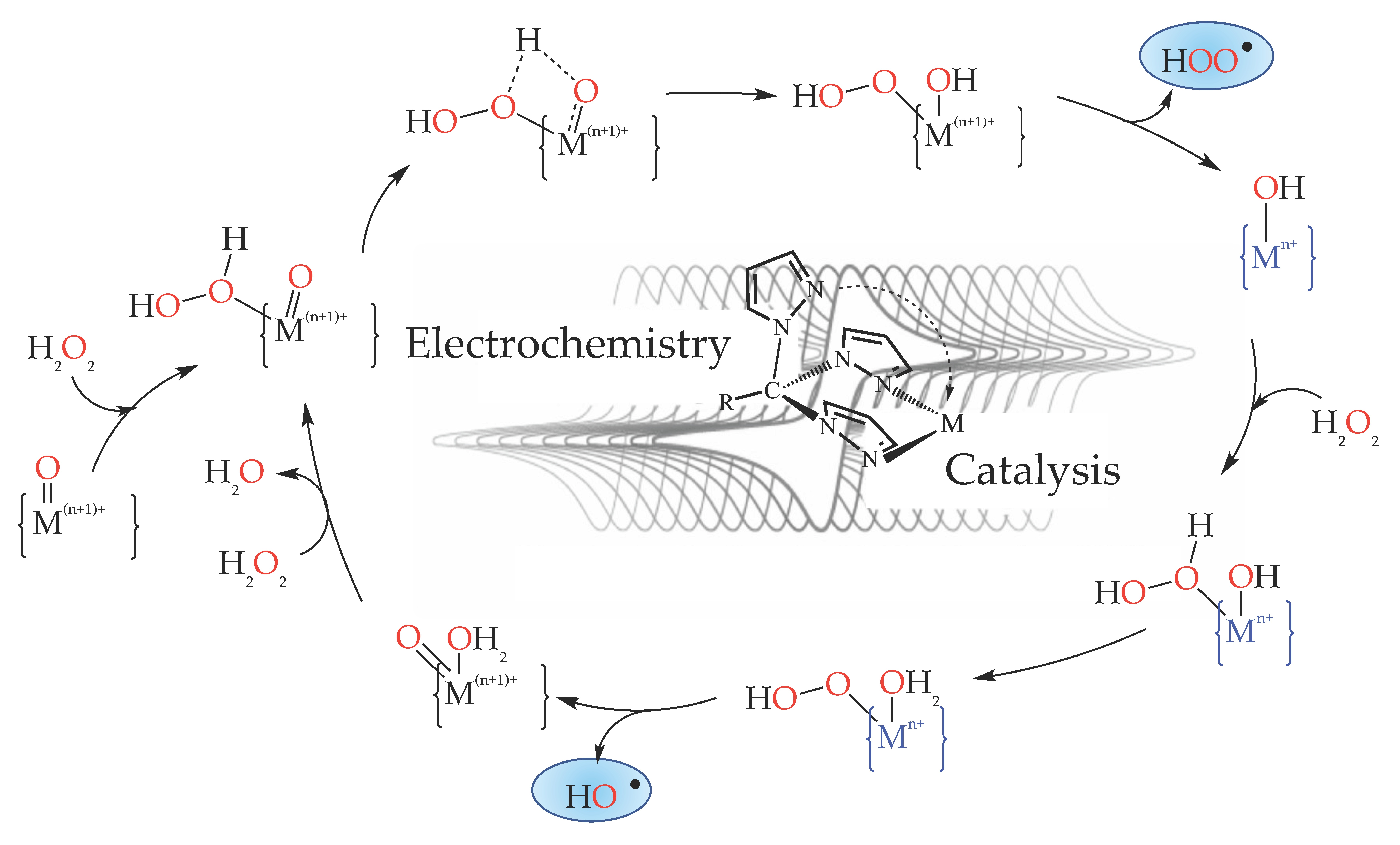
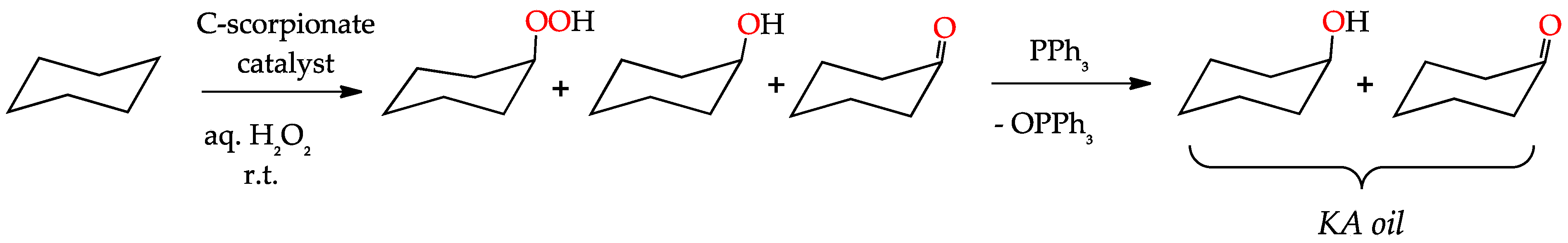
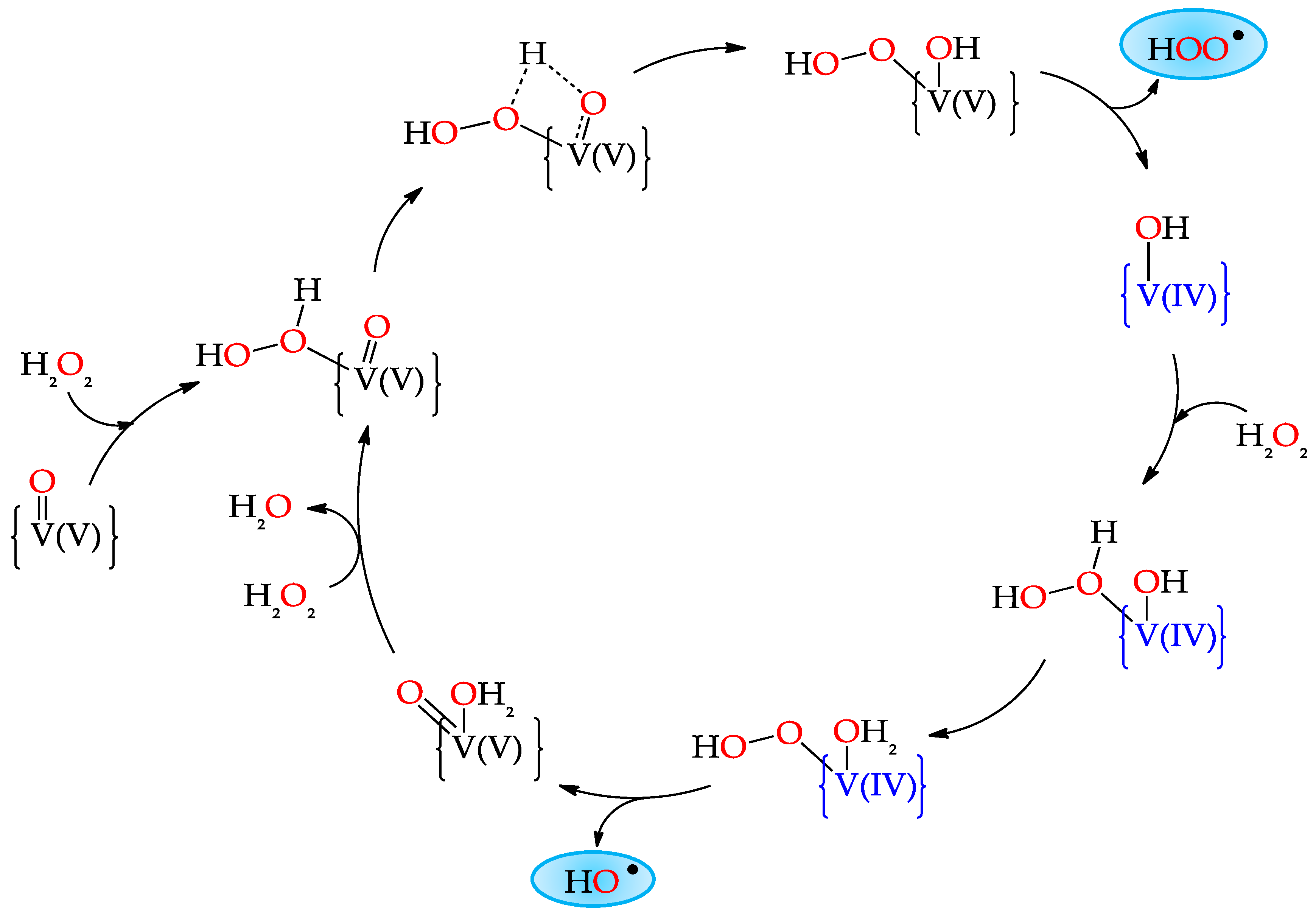
3.1. Oxidation of Alkanes to Alcohols and Ketones
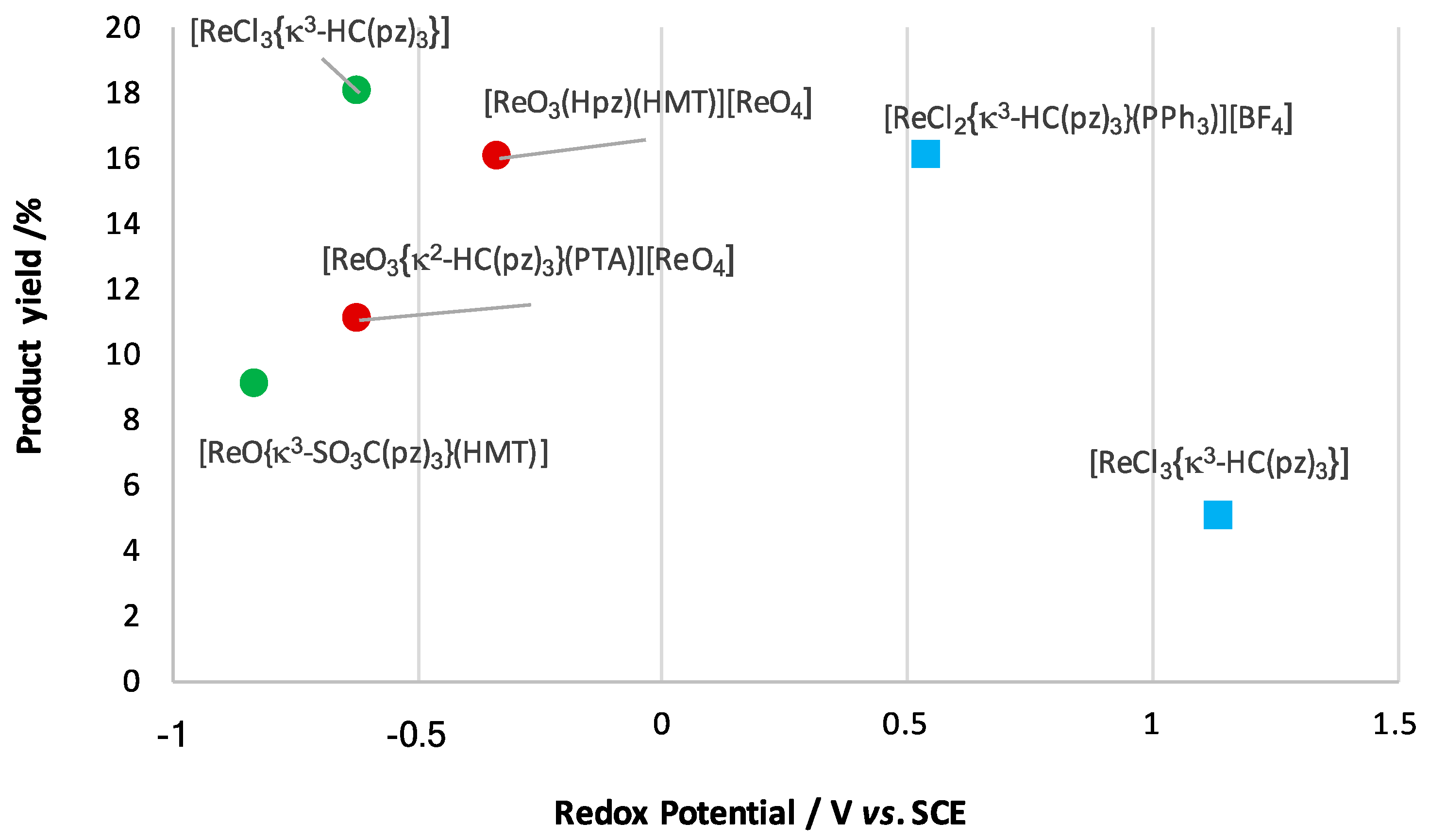
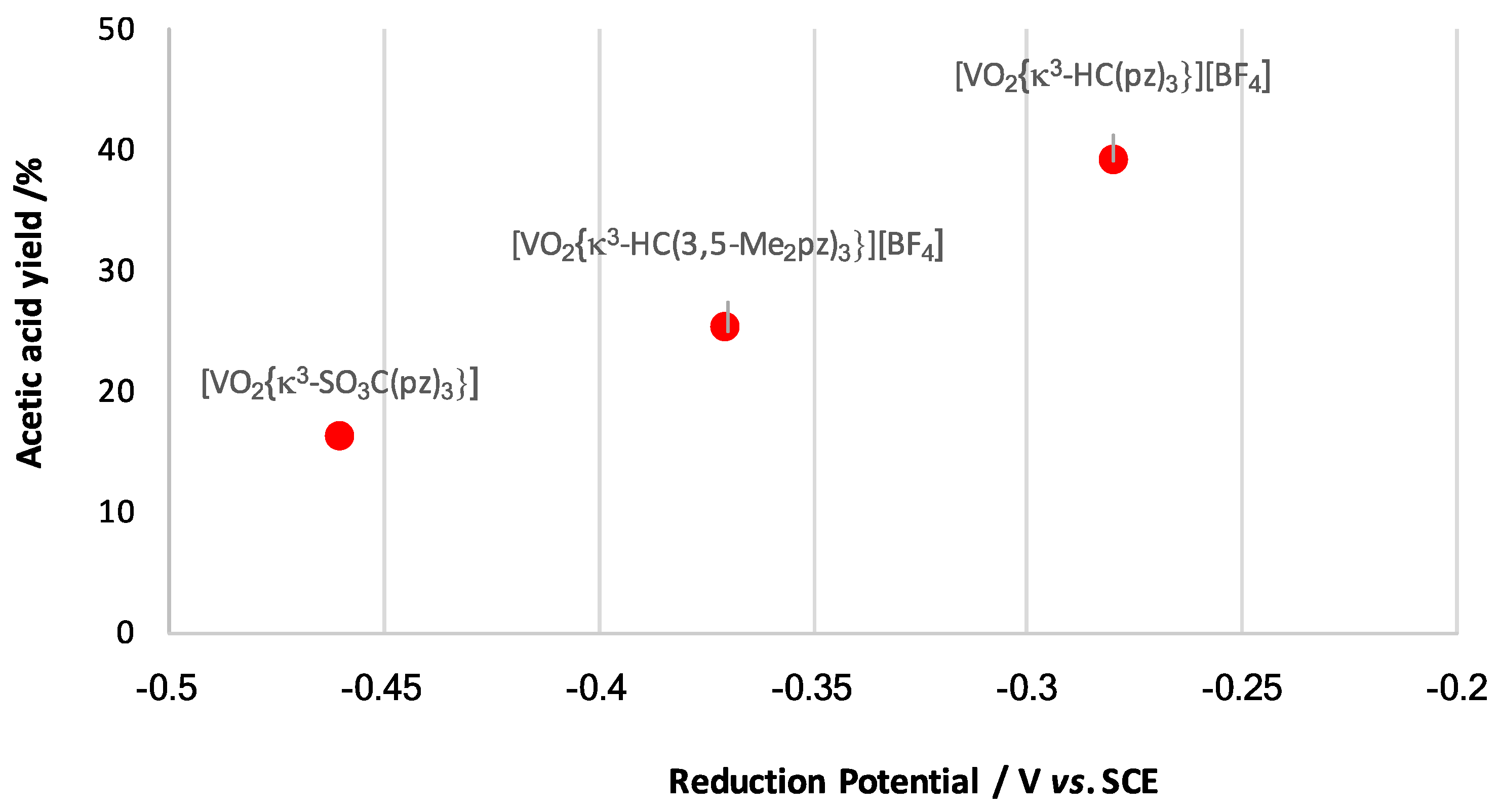
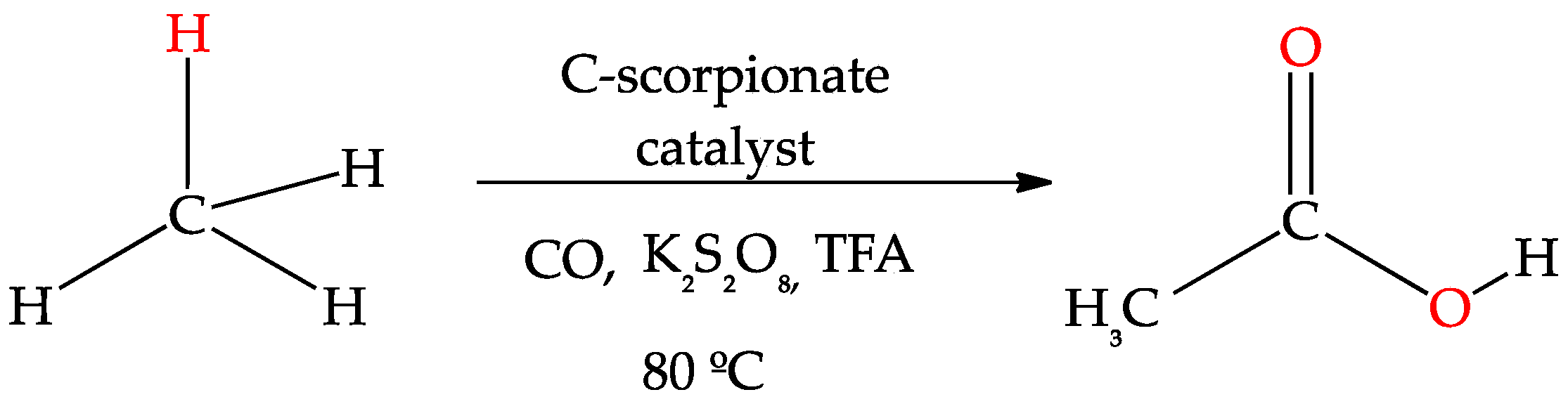
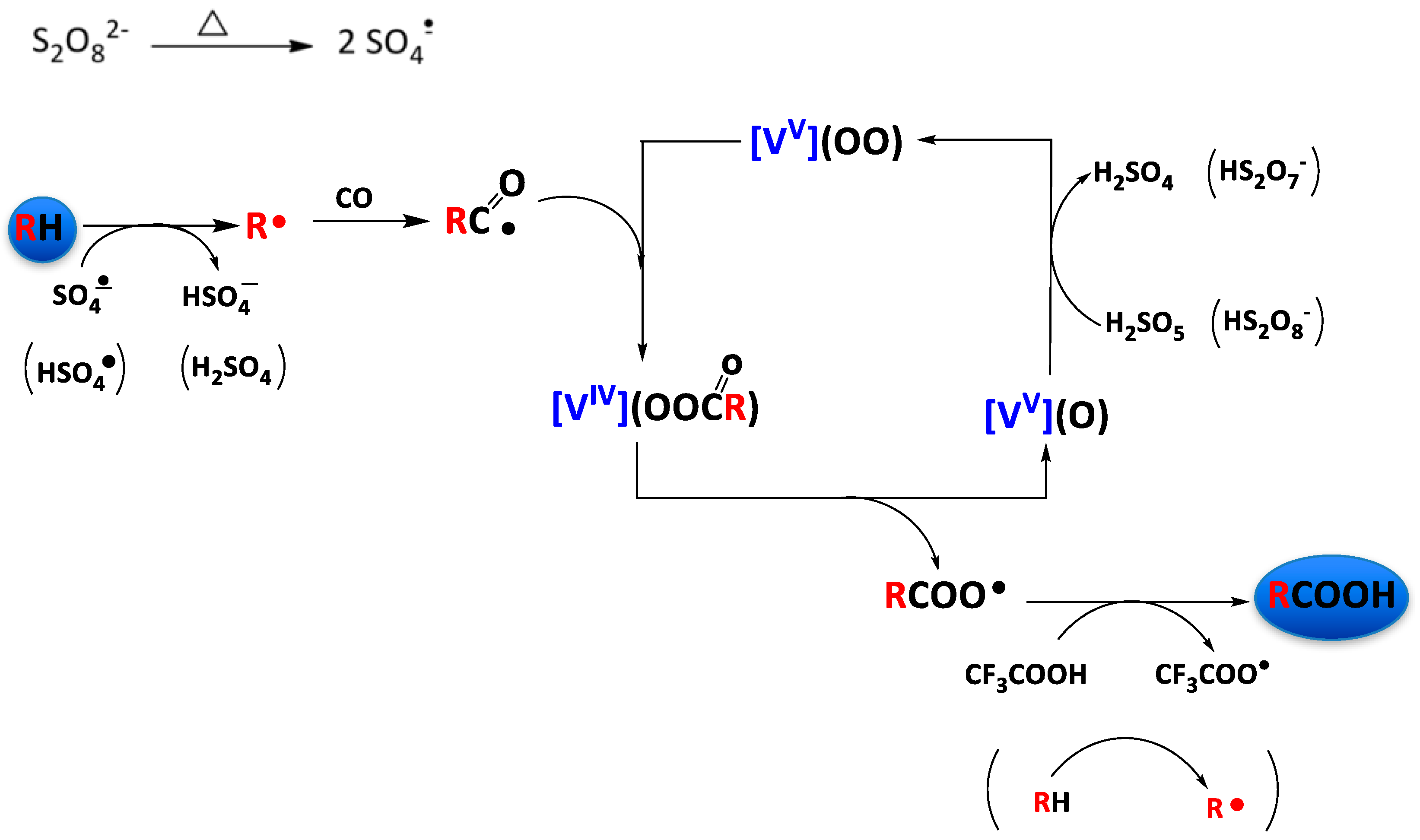
3.2. Oxidation of Alkanes to Carboxylic Acids
3.3. Baeyer-Villiger Oxidation of Ketones
3.4. Oxidation of 1,2-Diols
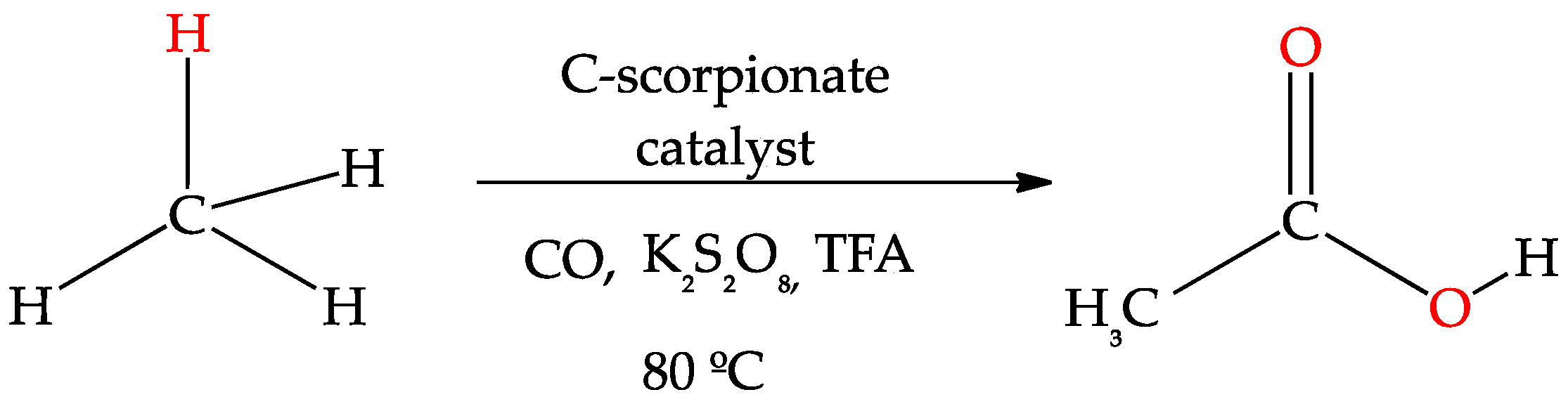
3.5. Carboxylation of Alkanes
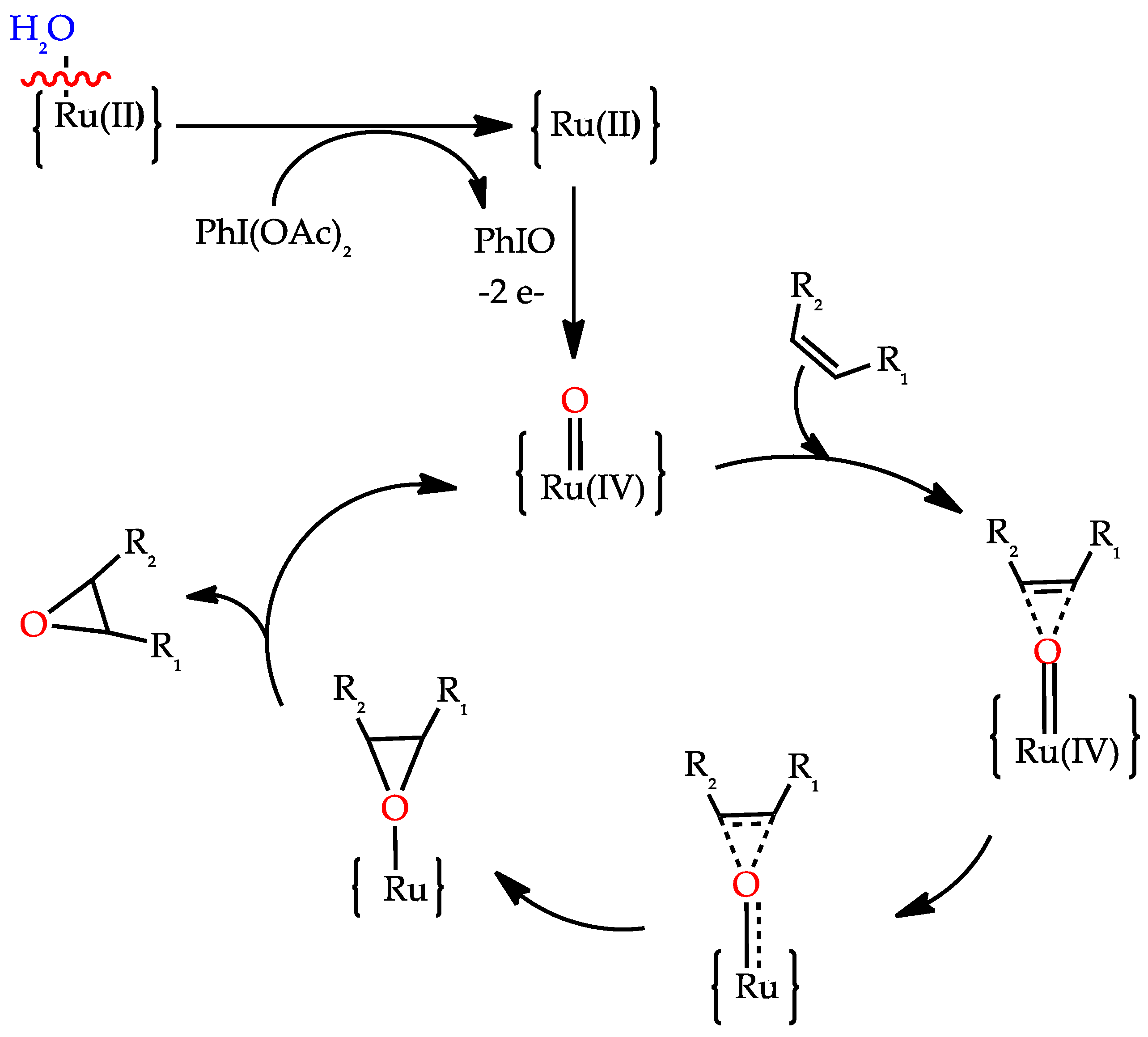
3.6. Epoxidation of Alkenes
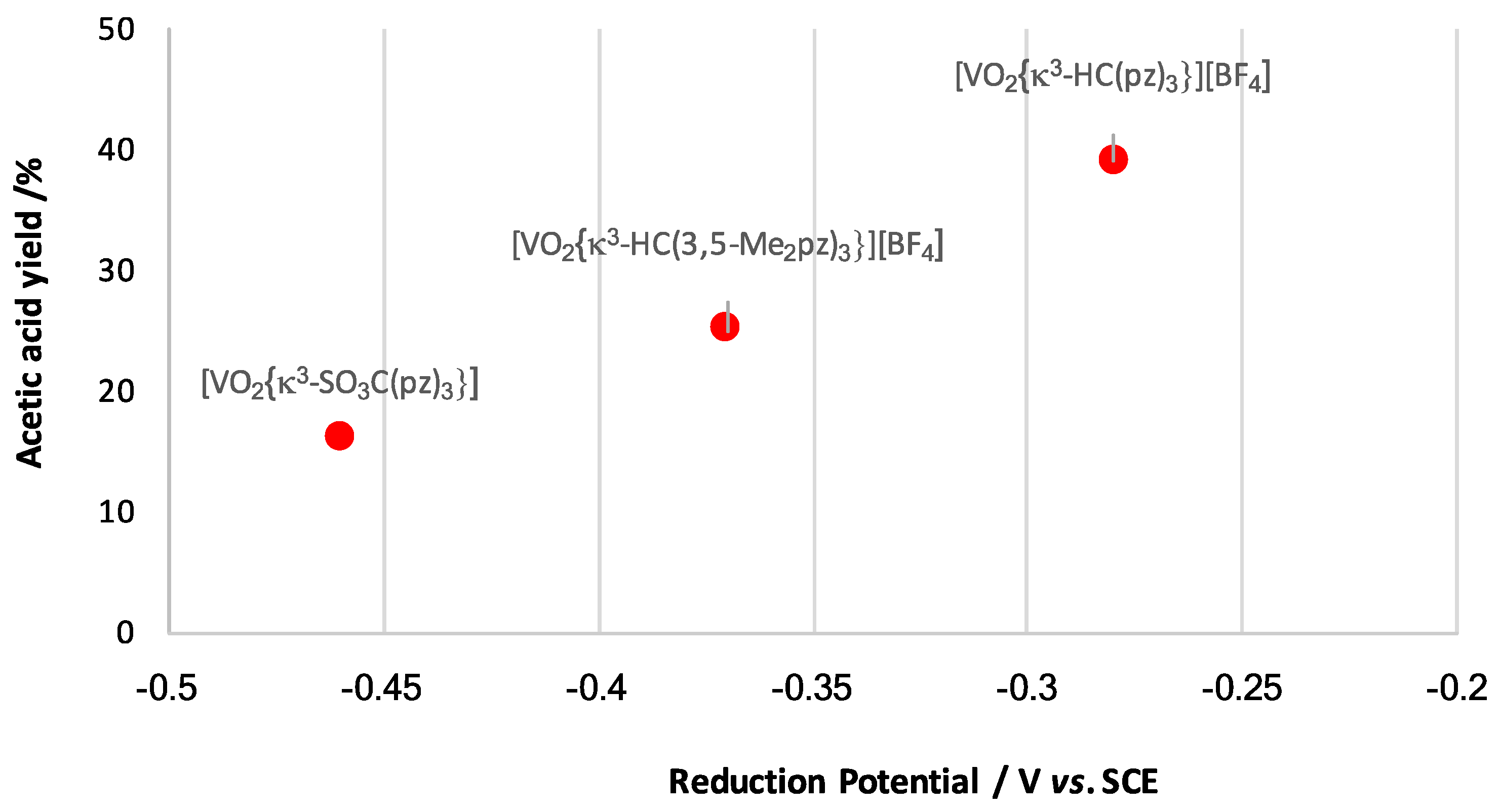
3.7. Redox Potential Parametrization
Acknowledgments
Conflicts of Interest
References
- Pettinari, C. Scorpionates II: Chelating Borate Ligands—Dedicated to Swiatoslaw Trofimenko; Imperial College Press, World Scientific Pub.: London, UK, 2008; ISBN 13-1-86094-876-3. [Google Scholar]
- Trofimenko, S. Scorpionates: The Coordination Chemistry of Polypyrazolylborates Ligands; Imperial College Press: London, UK, 1999; ISBN 1-86094-172-9. [Google Scholar]
- Pettinari, C.; Santini, C. Comprehensive Coordination Chemistry II: From Biology to Nanotechnology; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier Pergamon (APS): Amsterdam, The Netherlands, 2003; Volume 1, pp. 159–172. [Google Scholar]
- Pettinari, C.; Pettinari, R. Metal derivatives of poly(pyrazolyl)alkanes. I. Tris(pyrazolyl)alkanes and related systems. Coord. Chem. Rev. 2005, 249, 525–543. [Google Scholar] [CrossRef]
- Bigmore, H.R.; Lawrence, S.C.; Mountford, P.; Tredget, C.S. Coordination, organometallic and related chemistry of tris(pyrazolyl)methane ligands. Dalton Trans. 2005, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Sánchez-Barba, L.F. Metal complexes with heteroscorpionate ligands based on the bis(pyrazol-1-yl)methane moiety: Catalytic chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Tris(pyrazol-1yl)methane metal complexes for catalytic mild oxidative functionalizations of alkanes, alkenes and ketones. Coord. Chem. Rev. 2014, 265, 74–88. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Water-soluble C-scorpionate complexes: Catalytic and biological applications. Eur. J. Inorg. Chem. 2016, 2016, 2236–2252. [Google Scholar] [CrossRef]
- Reger, D.L.; Grattan, T.C.; Brown, K.J.; Little, C.A.; Lamba, J.J.S.; Rheingold, A.L.; Sommer, R.D. Syntheses of tris(pyrazolyl)methane ligands and {[tris(pyrazolyl)methane]Mn(CO)3}SO3CF3 complexes: Comparison of ligand donor properties. J. Organomet. Chem. 2000, 607, 120–128. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Alegria, E.C.B.A. Use of Microwaves for the Synthesis of Substituted Tris(pyrazolyl)methanes. Patent 103,681, 4 December 2009. [Google Scholar]
- Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Pombeiro, A.J.L. Ligands: Synthesis, Characterization and Role in Biotechnology; Smolenski, P., Gawryszewska, P., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2014; Chapter 4; pp. 117–140. [Google Scholar]
- Silva, T.F.S.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Fernandes, A.R.; Silva, A.; Borralho, P.M.; Santos, S.; Rodrigues, C.M.P.; Pombeiro, A.J.L. Cobalt complexes bearing scorpionate ligands: Synthesis, characterization, cytotoxicity and DNA cleavage. Dalton Trans. 2012, 41, 12888–12897. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.F.S.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Kuznetsov, M.L.; Fernandes, A.R.; Silva, A.; Santos, S.; Pan, C.-J.; Lee, J.-F.; Hwang, B.-J.; et al. Cobalt complexes with pyrazole ligands as catalysts for the peroxidative oxidation of cyclohexane. XAS studies and biological applications. Chem. Asian J. 2014, 9, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Pettinari, C.; Marchetti, F.; Lupidi, G.; Quassinti, L.; Bramucci, M.; Petrelli, D.; Vitali, L.A.; Guedes da Silva, M.F.C.; Martins, L.M.D.R.S.; Smoleński, P.; et al. Synthesis, Antimicrobial and antiproliferative activity of novel silver(I) tris(pyrazolyl)methanesulfonate and 1,3,5-triaza-7-phosphadamantane complexes. Inorg. Chem. 2011, 50, 11173–11183. [Google Scholar] [CrossRef] [PubMed]
- Niesel, J.; Pinto, A.; N’Dongo, H.W.P.; Merz, K.; Ott, I.; Gust, R.; Schatzschneider, U. Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex. Chem. Commun. 2008, 1798–1800. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. Carbon-scorpionate Complexes in Oxidation Catalysis. In Advances in Organometallic Chemistry and Catalysis, The Silver/Gold Jubilee ICOMC Celebratory Book; Pombeiro, A.J.L., Ed.; J. Wiley & Sons: Hoboken, NJ, USA, 2014; Chapter 22; pp. 285–294. [Google Scholar]
- Martins, L.M.D.R.S.; Ribeiro, A.P.C.; Carabineiro, S.A.C.; Figueiredo, J.L.; Pombeiro, A.J.L. Highly efficient and reusable CNT supported iron(II) catalyst for microwave assisted alcohol oxidation. Dalton Trans. 2016, 45, 6816–6819. [Google Scholar] [CrossRef] [PubMed]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Ribeiro, A.P.C.; Carabineiro, S.A.C.; Figueiredo, J.L. Production Process of Ketones from Secondary Alcohols. Patent 109,062, 29 December 2015. [Google Scholar]
- Santos, A.M.; Kuhn, F.E.; Bruus-Jensen, K.; Lucas, I.; Romão, C.C.; Herdtweck, E. Molybdenum(VI) cis-dioxo complexes bearing (poly)pyrazolylmethane and -borate ligands: Syntheses, characterization and catalytic applications. J. Chem. Soc. Dalton Trans. 2001, 1332–1337. [Google Scholar] [CrossRef]
- Neves, P.; Gago, S.; Balula, S.S.; Lopes, A.D.; Valente, A.A.; Cunha-Silva, L.; Paz, F.A.A.; Pillinger, M.; Rocha, J.; Silva, C.M.; et al. Synthesis and catalytic properties of molybdenum(VI) complexes with tris(3,5-dimethyl-1-pyrazolyl)methane. Inorg. Chem. 2011, 50, 3490–3500. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Smoleński, P.; Kuznetsov, M.L.; Pombeiro, A.J.L. Oxorhenium complexes bearing the water-soluble tris(pyrazol-1-yl)methanesulfonate, 1,3,5-triaza-7-phosphaadamantane or related ligands, as catalysts for the Baeyer-Villiger oxidation of ketones. Inorg. Chem. 2013, 52, 4534–4546. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.D.R.S.; Pombeiro, A.J.L. C-scorpionate rhenium complexes and their application as catalysts in Baeyer-Villiger oxidation of ketones. Inorg. Chim. Acta 2016. [Google Scholar] [CrossRef]
- Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Redox potential parameterization in coordination compounds with polydentate scorpionate and benzene ligands. Electrochim. Acta 2012, 82, 478–483. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L. Characterization of coordination compounds by electrochemical parameters. Eur. J. Inorg. Chem. 2007, 2007, 1473–1482. [Google Scholar] [CrossRef]
- Guedes da Silva, M.F.C.; Martins, L.M.D.R.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Redox potential-structure relationships in 18-and 17-electron mononitrile (or monocarbonyl) diphosphine complexes of Re and Fe. Collect. Czech. Chem. Commun. 2001, 66, 139–154. [Google Scholar] [CrossRef]
- Bursten, B.E. Ligand additivity: Applications to the electrochemistry and photoelectron spectroscopy of D6 octahedral complexes. J. Am. Chem. Soc. 1982, 104, 1299–1304. [Google Scholar] [CrossRef]
- Lever, A.B.P. Electrochemical parametrization of metal complex redox potentials, using the ruthenium(III)/ruthenium(II) couple to generate a ligand electrochemical series. Inorg. Chem. 1990, 29, 1271–1285. [Google Scholar] [CrossRef]
- Lever, A.B.P.; Dodsworth, E.S. Inorganic Electronic Structure and Spectroscopy; Solomon, E.I., Lever, A.B.P., Eds.; Wiley: New York, NY, USA, 1999; Volume 2, p. 227. [Google Scholar]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Boron-pyrazole chemistry. II. Poly(1-pyrazolyl)-borates. J. Am. Chem. Soc. 1967, 89, 3170–3177. [Google Scholar] [CrossRef]
- Hückel, W.; Bretschneider, H. N-tripyrazolyl-methane. Ber. Dtsch. Chem. Ges. 1937, 70, 2024–2026. [Google Scholar] [CrossRef]
- Jesson, J.P. Isotropic nuclear resonance shifts in some trigonal Co(II) and Ni(II) chelate systems. J. Chem. Phys. 1966, 45, 1049–1056. [Google Scholar] [CrossRef]
- Juliá, S.; del Mazo, J.M.; Avila, L.; Elguero, J. Improved synthesis of polyazolylmethanes under solid-liquid phase-transfer catalysis. Org. Prep. Proced. Int. 1984, 16, 299–307. [Google Scholar] [CrossRef]
- Titze, C.; Hermann, J.; Vahrenkamp, H. Highly substituted tris(pyrazolyl)methane ligands and some zinc-complexes thereof. Chem. Ber. 1995, 128, 1095–1103. [Google Scholar] [CrossRef]
- Reger, D.L.; Collins, J.E.; Rheingold, A.L.; Liable-Sands, L.M. Synthesis and characterization of cationic [tris(pyrazolyl)methane]copper(I) carbonyl and acetonitrile complexes. Organometallics 1996, 15, 2029–2032. [Google Scholar] [CrossRef]
- Trofimenko, S. Recent advances in poly(pyrazolyl)borate (scorpionate) chemistry. Chem. Rev. 1993, 93, 943–980. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Antiñolo, A.; Tejeda, J.; Lara-Sánchez, A. Heteroscorpionate ligands based on bis(pyrazol-1-yl)methane: Design and coordination chemistry. Dalton Trans. 2004, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Brunker, T.J.; Cowley, A.R.; O’Hare, D. Synthesis, structures, and redox properties of mixed-sandwich complexes of cyclopentadienyl and hydrotris(pyrazolyl)borate ligands with first-row transition metals. Organometallics 2002, 21, 3123–3138. [Google Scholar] [CrossRef]
- McKeown, B.A.; Lee, J.P.; Mei, J.; Cundari, T.R.; Gunnoe, T.B. Transition metal mediated C–H activation and functionalization: The role of poly(pyrazolyl)borate and poly(pyrazolyl)alkane ligands. Eur. J. Inorg. Chem. 2016, 2016, 2296–2311. [Google Scholar] [CrossRef]
- Tellers, D.M.; Skoog, S.J.; Bergman, R.G.; Gunnoe, T.B.; Harman, W.D. Comparison of the relative electron-donating abilities of hydridotris(pyrazolyl)borate and cyclopentadienyl ligands: Different interactions with different transition metals. Organometallics 2000, 19, 2428–2432. [Google Scholar] [CrossRef]
- Jones, W.D.; Feher, F.J. Comparative reactivities of hydrocarbon C–H bonds with a transition-metal complex. Acc. Chem. Res. 1989, 22, 91–100. [Google Scholar] [CrossRef]
- Vahrenkamp, H. Transitions, transition states, transition state analogues: Zinc pyrazolylborate chemistry related to zinc enzymes. Acc. Chem. Res. 1999, 32, 589–596. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Mishra, G.S.; Silva, M.F.G.; Wanke, R.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Cu(II) complexes bearing the 2,2,2-tris(1-pyrazolyl)ethanol or 2,2,2-tris(1-pyrazolyl)ethyl methanesulfonate scorpionates. X-ray structural characterization and application in the mild catalytic peroxidative oxidation of cyclohexane. Dalton Trans. 2009, 42, 9207–9215. [Google Scholar] [CrossRef] [PubMed]
- Wanke, R.; Silva, M.F.C.G.; Lancianesi, S.; Silva, T.F.S.; Martins, L.M.D.R.S.; Pettinari, C.; Pombeiro, A.J.L. Synthesis and coordination chemistry of a new N4-Polydentate class of pyridyl-functionalized scorpionate ligands: Complexes of FeII, ZnII, NiII, VIV, PdII and use for heterobimetallic systems. Inorg. Chem. 2010, 49, 7941–7952. [Google Scholar] [CrossRef] [PubMed]
- Wanke, R.; Smoleński, P.; Guedes da Silva, M.F.C.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Cu(I) complexes bearing the new sterically hindered and coordination flexible tris(3-phenyl-1-pyrazolyl)methanesulfonate (TpmsPh) ligand and the water-soluble phosphine 1,3,5-triaza-7-phosphaadamantane (PTA) or related ligands. Inorg. Chem. 2008, 47, 10158–10168. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.F.S.; Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Scorpionate vanadium, iron and copper complexes as selective catalysts for the peroxidative oxidation of cyclohexane under mild conditions. Adv. Synth. Catal. 2008, 350, 706–716. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Luzyanin, K.V.; Kirilova, M.V.; Silva, M.F.C.G.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Novel scorpionate and pyrazole dioxovanadium complexes, catalysts for carboxylation and peroxidative oxidation of alkanes. Adv. Synth. Catal. 2010, 352, 171–187. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Mac Leod, T.C.O.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.; Pombeiro, A.J.L. Pyrazole or tris(pyrazolyl)ethanol oxo-vanadium(IV) complexes as homogeneous or supported catalysts for oxidation of cyclohexane under mild conditions. J. Mol. Catal. A Chem. 2013, 367, 52–60. [Google Scholar] [CrossRef]
- Silva, T.F.S.; Rocha, B.G.M.; Guedes da Silva, M.F.C.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. VIV, FeII, NiII and CuII complexes bearing 2,2,2-tris(pyrazol-1-yl)ethyl methanesulfonate: Application as catalysts for the cyclooctane oxidation. New J. Chem. 2016, 40, 528–537. [Google Scholar] [CrossRef]
- Dinoi, C.; Silva, M.F.C.G.; Alegria, E.C.B.A.; Smoleński, P.; Martins, L.M.D.R.S.; Poli, R.; Pombeiro, A.J.L. Molybdenum complexes bearing the tris(1-pyrazolyl)methanesulfonate ligand: Synthesis, characterization and electrochemical behavior. Eur. J. Inorg. Chem. 2010, 16, 2415–2424. [Google Scholar] [CrossRef]
- Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Syntheses and properties of Re(III) complexes derived from hydrotris(1-pyrazolyl)methanes. Molecular structure of [ReCl2(HCpz3)(PPh3)][BF4]. J. Organomet. Chem. 2005, 690, 1947–1958. [Google Scholar] [CrossRef]
- Alegria, E.C.B.A.; Martins, L.M.D.R.S.; Haukka, M.; Pombeiro, A.J.L. Rhenium complexes of tris(pyrazolyl)methanes and sulfonate derivative. Dalton Trans. 2006, 4954–4961. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.F.S.; Silva, M.F.C.G.; Mishra, G.S.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Synthesis and structural characterization of iron complexes with 2,2,2-tris(1-pyrazolyl)ethanol ligands: Application in the peroxidative oxidation of cyclohexane under mild conditions. J. Organomet. Chem. 2011, 696, 1310–1318. [Google Scholar] [CrossRef]
- Marchetti, F.; Pettinari, C.; Pettinari, R.; Cerquetella, A.; Martins, L.M.D.R.S.; Guedes da Silva, M.F.; Silva, T.F.S.; Pombeiro, A.J.L. Ru(II) arene complexes bearing tris(pyrazolyl)methanesulfonate capping ligands. Electrochemistry, spectroscopic and X-ray structural characterization. Organometallics 2011, 30, 6180–6188. [Google Scholar] [CrossRef]
- Rocha, B.G.M.; Mac Leod, T.C.O.; Guedes da Silva, M.F.C.; Luzyanin, K.V.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. NiII, CuII and ZnII complexes with a sterically hindered scorpionate ligand (TpmsPh) and catalytic application in the diasteroselective nitroaldol (Henry) reaction. Dalton Trans. 2014, 43, 15192–15200. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.G.M.; Wanke, R.; Guedes da Silva, M.F.C.; Luzyanin, K.V.; Martins, L.M.D.R.S.; Smolénski, P.; Pombeiro, A.J.L. Reactivity of bulky tris(phenylpyrazolyl)methanesulfonate copper(I) complexes towards small unsaturated molecules. J. Organomet. Chem. 2012, 714, 47–52. [Google Scholar] [CrossRef]
- Peixoto de Almeida, M.; Martins, L.M.D.R.S.; Carabineiro, S.A.C.; Lauterbach, T.; Rominger, F.; Hashmi, A.S.K.; Pombeiro, A.J.L.; Figueiredo, J.L. Homogeneous and heterogenised new gold C-scorpionate complexes as catalysts for cyclohexane oxidation. Catal. Sci. Technol. 2013, 3, 3056–3069. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Ribeiro, A.P. Process for the Microwave-Assisted Conversion of Cycloalkanes to the Corresponding Alcohol-Ketone Mixtures, with Hydrogen Peroxide, and Using a Scorpionate Chloro-Complex of Iron(II) as Catalyst. Patent 107,797, 25 July 2014. [Google Scholar]
- Vitze, H.; Bolte, M.; Lerner, H.-W.; Wagner, M. Third-generation scorpionates [RBpz3]−—How influential is the nondonor substituent R? Eur. J. Inorg. Chem. 2016, 2016, 2443–2454. [Google Scholar] [CrossRef]
- Trofimenko, S. Geminal poly(1-pyrazolyl)alkanes and their coordination chemistry. J. Am. Chem. Soc. 1970, 92, 5118–5126. [Google Scholar] [CrossRef]
- Astley, T.; Gulbis, J.M.; Hitchman, M.A.; Tiekink, E.R.T. Syntheses and characterisation of tris(3-(pyridin-2-yl)-1H-pyrazol-1-yl)methane and its bis(µ-hydroxo) dicobalt(II) complex. J. Chem. Soc. Dalton Trans. 1993, 509–515. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Silva, T.F.S.; Silva, M.F.C.G.; Luzyanin, K.V.; Kirillova, M.V. Oxocomplexes of Vanadium(IV-V) with Scorpionate or Pyrazole Ligands and Their Application as Catalysts for the Peroxidative Oxidation of Cycloalkanes and the Carboxylation of Gaseous Alkanes. Patent 104,887, 15 September 2011. [Google Scholar]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Silva, M.F.C.G.; Mishra, G.S.; Silva, T.F.S.; Wanke, R. Copper(II) Complexes with Hydrophilic C-Functionalized Scorpionate Ligands and Their Application as Catalysts for the Peroxidative Oxidation of Cyclohexane under Environmentally Tolerable Conditions, in Particular in Aqueous Medium. Patent 104713, 8 September 2010. [Google Scholar]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Silva, T.F.S.; Mishra, G.S. Process for conversion of cyclohexane to cyclohexanol and cyclohexanone using scorpionate chloro-complexes of vanadium(III or IV) as catalysts, with oxygen in the absence of solvents. Patent 104,447, 30 December 2009. [Google Scholar]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Mishra, G.S.; Fraústo da Silva, J.J.R. Complexes of Rhenium and Pyrazole Supported on Functionalized Silica as Catalysts for the Partial Oxidation of n-hexane and Cyclohexane with Dioxygen and under Environmentally Acceptable Conditions. Patent 104,197, 27 July 2009. [Google Scholar]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Silva, T.F.S. Scorpionate Chloro-Complexes of Iron and Vanadium and Their Application as Catalysts for the Partial Oxidation, under Mild and Environmentally Tolerable Conditions, of Cyclohexane to Cyclohexanol and Cyclohexanone. Patent 104153, 1 July 2009. [Google Scholar]
- Alegria, E.C.B.A.; Kirillova, M.V.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Pyrazole and tris(pyrazolyl)methane rhenium complexes as catalysts for ethane and cyclohexane oxidations. Appl. Catal. A Gen. 2007, 317, 43–52. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L.; Martins, L.M.D.R.S.; Alegria, E.C.B.A.; Kirillova, M.V. New Complexes of Rhenium with Pyrazole or Tris(1-pyrazolyl)methanes and Their Application as Catalysts for the Partial Oxidation, under Mild Conditions, of Ethane to Acetic and Acetaldehyde and of Cyclohexane to Cyclohexanol and Cyclohexanone. Patent 103,735, 31 March 2008. [Google Scholar]
- Labinger, J.A. Catalysis by Metal Complexes; Pérez, P.J., Ed.; Springer Science + Business Media: Dordrecht, The Netherlands, 2012; Chapter 2. [Google Scholar]
- Shilov, A.E.; Shul’pin, G.B. Activation of Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes; Kluwer Academic Press: New York, NY, USA, 2000. [Google Scholar]
- Derouane, E.D.; Haber, J.; Lemos, F.; Ramôa Ribeiro, F.; Guinet, M. (Eds.) Catalytic Activation and Functionalisation of Light Alkanes; NATO ASI Series; Kluwer Academic Publ.: Dordrecht, The Netherlands, 1998; Volume 44.
- Shul’pin, G.B. New trends in oxidative functionalization of carbon–hydrogen bonds: A review. Catalysts 2016, 6, 50. [Google Scholar] [CrossRef]
- Shul’pin, G.B. C–H functionalization: Thoroughly tuning ligands at a metal ion, a chemist can greatly enhance catalyst’s activity and selectivity. Dalton Trans. 2013, 42, 12794–12818. [Google Scholar] [CrossRef] [PubMed]
- Shul’pin, G.B. Metal-catalyzed hydrocarbon oxidations. C. R. Chim. 2003, 6, 163–178. [Google Scholar] [CrossRef]
- Shul’pin, G.B. Hydrocarbon oxygenations with peroxides catalyzed by metal compounds. Mini-Rev. Org. Chem. 2009, 6, 95–104. [Google Scholar] [CrossRef]
- Kirillova, M.V.; Kuznetsov, M.L.; Reis, P.M.; Silva, J.A.L.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Direct and remarkably efficient conversion of methane into acetic acid catalyzed by amavadine and related vanadium complexes. A synthetic and a theoretical DFT mechanistic study. J. Am. Chem. Soc. 2007, 129, 10531–10545. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, M.V.; Kuznetsov, M.L.; Silva, J.A.L.; Guedes da Silva, M.F.C.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Amavadin and other vanadium complexes as remarkably efficient catalysts for one-pot conversion of ethane to propionic and acetic acids. Chem. Eur. J. 2008, 14, 1828–1842. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, A.M.; Kopylovich, M.N.; Kirillova, M.V.; Haukka, M.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Multinuclear copper triethanolamine complexes as selective catalysts for the peroxidative oxidation of alkanes under mild conditions. Angew. Chem. Int. Ed. 2005, 44, 4345–4349. [Google Scholar] [CrossRef] [PubMed]
- Kirk-Othmer Encyclopedia of Chemical Technology; Seidel, A.; Bickford, M. (Eds.) J. Wiley & Sons: New York, NY, USA, 2014.
- Weissermel, W.; Horpe, H.J. Industrial Organic Chemistry, 2nd ed.; VCH Press: Weinheim, Germany, 1993. [Google Scholar]
- Schuchardt, U.; Cardoso, D.; Sercheli, R.; Pereira, R.; da Cruz, R.S.; Guerreiro, M.C.; Mandelli, D.; Spinace, E.V.; Pires, E.L. Cyclohexane oxidation continues to be a challenge. Appl. Catal. A Gen. 2001, 211, 1–17. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Peixoto de Almeida, M.; Carabineiro, S.A.C.; Figueiredo, J.L.; Pombeiro, A.J.L. Heterogenisation of a C-scorpionate Fe(II) complex in carbon materials for cyclohexane oxidation with hydrogen peroxide. ChemCatChem 2013, 5, 3847–3856. [Google Scholar] [CrossRef]
- Martins, L.M.D.R.S.; Martins, A.; Alegria, E.C.B.A.; Carvalho, A.P.; Pombeiro, A.J.L. Efficient cyclohexane oxidation with hydrogen peroxide catalyzed by a C-scorpionate iron(II) complex immobilized on desilicated MOR zeolite. Appl. Catal. A Gen. 2013, 464–465, 43–50. [Google Scholar] [CrossRef]
- Pombeiro, A.J.L. Vanadium-catalyzed alkane functionalization reactions under mild conditions. In Vanadium: The Versatile Metal; Kustin, K., Pessoa, J.C., Crans, D.C., Eds.; ACS Symposium Series, Nº 974; American Chemical Society; Oxford University Press: Oxford, UK, 2007; Chapter 4; p. 51. [Google Scholar]
- Kirillova, M.V.; Kuznetsov, M.L.; Romakh, V.B.; Shul’pina, L.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L.; Shul’pin, G.B. Mechanism of H2O2 oxidations catalyzed by vanadate anion or oxovanadium(V) triethanolaminate (vanadatrane) in combination with pyrazine-2-carboxylic acid (PCA): Kinetic and DFT studies. J. Catal. 2009, 267, 140–157. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Kirillova, M.V.; Pombeiro, A.J.L. Multicopper complexes and coordination polymers for mild oxidative functionalization of alkanes. Coord. Chem. Rev. 2012, 256, 2741–2759. [Google Scholar] [CrossRef]
- Rachmilovich-Calis, S.; Masarwa, A.; Meyerstein, N.; Meyerstein, D.; van Eldik, R. New Mechanistic Aspects of the Fenton Reaction. Chem. Eur. J. 2009, 15, 8303–8309. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.C.; Neves, P.; Figueiredo, S.; Fernandes, J.A.; Valente, A.A.; Paz, F.A.A.; Pillinger, M.; Lopes, A.D.; Gonçalves, I.S. Tris(pyrazolyl)methane molybdenum tricarbonyl complexes as catalyst precursors for olefin epoxidation. J. Mol. Catal. A Chem. 2013, 370, 64–74. [Google Scholar] [CrossRef]
- Agarwala, H.; Ehret, F.; Chowdhury, A.D.; Maji, S.; Mobin, S.M.; Kaim, W.; Lahiri, G.K. Electronic structure and catalytic aspects of [Ru(tpm)(bqdi)(Cl/H2O)]n, tpm = tris(1-pyrazolyl)methane and bqdi = o-benzoquinonediimine. Dalton Trans. 2013, 42, 3721–3734. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2474. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.F.S.; Martins, L.M.D.R.S.; Pombeiro, A.J.L. Redox behaviour of a tris(pyrazolyl) methanesulfonate vanadium complex, a preliminary study. Port. Electrochim. Acta 2006, 24, 257–259. [Google Scholar] [CrossRef]
- Dhanalakshmi, T.; Sureshb, E.; Palaniandavar, M. Synthesis, structure, spectra and reactivity of iron(III) complexes of imidazole and pyrazole containing ligands as functional models for catechol dioxygenases. Dalton Trans. 2009, 8317–8328. [Google Scholar] [CrossRef] [PubMed]
- Underwood, C.C.; Stadelman, B.S.; Sleeper, M.L.; Brumaghim, J.L. Synthesis and electrochemical characterization of [Ru(NCCH3)6]2+, tris(acetonitrile) tris(pyrazolyl)borate, and tris(acetonitrile) tris(pyrazolyl)methane ruthenium(II) complexes. Inorg. Chim. Acta 2013, 405, 470–476. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods–Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Fettinger, J.C.; Keogh, D.W.; Poli, R. Stable mononuclear, 17-electron molybdenum(III) carbonyl complexes. Synthesis, structure, thermal decomposition, and Cl− addition reactions. J. Am. Chem. Soc. 1996, 118, 3617–3625. [Google Scholar] [CrossRef]
- Quadrelli, E.A.; Kraatz, H.-B.; Poli, R. Oxidation and protonation of transition metal hydrides: Role of an added base as proton shuttle and nature of protonated water in acetonitrile. Inorg. Chem. 1996, 35, 5154–5162. [Google Scholar] [CrossRef]
- Venâncio, A.I.F.; Kuznetsov, M.L.; Guedes da Silva, M.F.C.; Martins, L.M.D.R.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Metal-hydride bond activation and metal-metal interaction in dinuclear iron complexes with linking dinitriles: A synthetic, electrochemical and theoretical study. Inorg. Chem. 2002, 41, 6456–6467. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.D.R.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L.; Henderson, R.A.; Evans, D.J.; Benetollo, F.; Bombieri, G.; Michelin, R.A. Syntheses, properties and mössbauer studies of cyanamide and cyanoguanidine complexes of iron(II). Crystal structures of trans-[FeH(NCNH2)(Ph2PCH2CH2PPh2)2] [BF4] and trans-[Fe(NCNEt2)2(Et2PCH2CH2PEt2)2][BF4]2. Inorg. Chim. Acta 1999, 291, 39–48. [Google Scholar] [CrossRef]
- Marchetti, F.; Pettinari, C.; Pettinari, R.; Cerquetella, A.; Cingolani, A.; Chan, E.J.; Kozawa, K.; Skelton, B.W.; White, A.H.; Wanke, R.; et al. Areneruthenium(II) 4-acyl-5-pyrazolonate derivatives: Coordination chemistry and reactivity. Inorg. Chem. 2007, 46, 8245–8257. [Google Scholar] [CrossRef] [PubMed]
- Lever, A.B.P. Electrochemical parametrization of rhenium redox couples. Inorg. Chem. 1991, 30, 1980–1985. [Google Scholar] [CrossRef]
- Lever, A.B.P. (Ed.) Comprehensive Coordination Chemistry II; Elsevier: Oxford, UK, 2004; Chapter 2.19; Volume 2, p. 251.
- Lu, S.; Strelets, V.V.; Ryan, M.F.; Pietro, W.J.; Lever, A.B.P. Electrochemical Parametrization in Sandwich Complexes of the First Row Transition Metals. Inorg. Chem. 1996, 35, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Kopylovich, M.N.; Mahmudov, K.T.; Silva, M.F.C.G.; Martins, L.M.D.R.S.; Kuznetsov, M.L.; Silva, T.F.S.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Trends in properties of para-substituted 3-(phenylhydrazo)pentane-2,4-diones. J. Phys. Org. Chem. 2011, 24, 764–773. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C-Scorpionate Compound | Redox Potential/V vs. SCE | Ref. | ||
|---|---|---|---|---|
| IEpox (IE½ox) | IEpred (IE½red) | IIEpred (IIE½red) | ||
| [VCl3{κ3-SO3C(pz)3}] | (1.14) | - | - | [91] |
| [VO2{κ3-SO3C(pz)3}] b | - | −0.46 | −1.82 | [47] |
| [VOCl2{κ3-CH3SO2OCH2C(pz)3}] c | (1.35) | −0.78 | - | [49] |
| [VO2{κ3-HC(pz)3}][BF4] b | - | −0.28 | −1.70 | [47] |
| [VO2{κ3-HC(3,5-Me2pz)3}][BF4] | - | −0.37 | −1.75 | [47] |
| Li[Mo{κ3-SO3C(pz)3}(CO)3] | (0.18) | - | - | [50] |
| [Mo{κ3-SO3C(pz)3}I(CO)3] | 0.44 | - | - | [50] |
| [Mo{κ3-SO3C(pz)3}H(CO)3] | 0.09 | - | - | [50] |
| [ReCl2{κ3-HC(pz)3}(PPh3)][BF4] d | (0.54) | (−0.74) | - | [51] |
| [ReCl3{κ3-HC(pz)3}] | 1.14 | −0.62 | −1.70 | [52] |
| [ReCl3{κ3-HC(3,5-Me2pz)3}] | (1.25) | (−0.13) | (−0.72) | [52] |
| [ReCl4{κ2-HC(pz)3}] | 1.79 | (−0.06) | −1.50 | [52] |
| [ReO3{κ3-SO3C(pz)3}] | - | −0.83 | - | [52] |
| [ReO{κ3-SO3C(pz)3}(HMT)] b | (0.86) | −0.83 | - | [21] |
| [ReOCl{κ3-SO3C(pz)3}(PPh3)]Cl | 1.45 | (−0.94) | (−1.41) | [52] |
| [ReO3{κ2-HC(pz)3}(PTA)][ReO4] b | - | (−0.62) | - | [21] |
| [ReO3(Hpz)(HMT)][ReO4] b | - | (−0.33) | - | [21] |
| [FeCl2{κ3-CH3SO2OCH2C(pz)3}] c | (1.06) | −0.38 | - | [49] |
| [FeCl3{κ3-HC(pz)3}] d | (−0.11) | - | - | [92] |
| [FeCl3{κ3-HC(3,5-Me2pz)3}] d | (−0.20) | - | - | [92] |
| [FeCl3{κ3-HC(3-iPrpz)3}] d | (−0.04) | - | - | [92] |
| [Ru(p-cymene){κ3-SO3C(pz)3}]Cl | (0.95) | (−0.97) | - | [54] |
| [Ru(p-cymene){κ3-SO3C(pz)3}][BF4] | (0.96) | (−0.97) | - | [54] |
| [Ru(p-cymene){κ3-SO3C(3-Phpz)3}]Cl | 1.02 | (−1.00) | - | [54] |
| [Ru(benzene){κ3-SO3C(pz)3}]Cl | (1.07) | (−0.87) | - | [54] |
| [Ru(benzene){κ3-SO3C(3-Phpz)3}]Cl | (1.37) | (−0.92) | - | [54] |
| [Ru(HMB){κ3-SO3C(pz)3}]Cl | 0.95 | (−1.11) | - | [54] |
| [Ru(cod)Cl{κ3-SO3C(pz)3}] | (0.96) | (−1.10) | - | [54] |
| [Ru(cod)Cl{κ3-SO3C(3-Phpz)3}] | (0.99) | (−1.27) | - | [54] |
| [RuCl{κ3-HC(pz)3}(bqdi)][ClO4] c | (0.82) | (−0.79) | −1.39 | [89] |
| [Ru(H2O){κ3-HC(pz)3}(bqdi)][ClO4]2 c | (0.44) | - | - | [89] |
| [Ru{κ3-HC(3,5-Me2pz)3}(NCCH3)3][BF4]2 c | (0.42) | - | - | [93] |
| [Ru{κ3-HC(3,5-Ph2pz)3}(NCCH3)3][BF4]2 c | (0.71) | - | - | [93] |
| [Co(OSO3H)(OCH3)(HOCH3){κ3-HC(pz)3}] b | 1.03 | −0.40 | [12] | |
| [Co{κ3-HOCH2C(pz)3}2](NO3)2 | (0.58) | −0.68 | [12] | |
| [Co{κ3-HOCH2C(pz)3}2]·[Co{κ3-HOCH2C(pz)3} (H2O)3]2(Cl)6·6H2O | (0.60) | −0.67 | −1.21 | [12] |
| [CoCl2(H2O){κ3-PyCH2OCH2C(pz)3}] | 1.28 | −0.60 | - | [12] |
| [CoCl2(H2O){κ3-CH3SO2OCH2C(pz)3}] c | 1.10 | −0.64 | - | [12] |
| [CuCl2{κ3-CH3SO2OCH2C(pz)3}] c | - | −0.70 | - | [49] |
| [AuCl2{κ2-HC(pz)3}]Cl c | - | −0.02 | −0.60 | [57] |
| [AuCl2{κ2-HOCH2C(pz)3}]Cl c | - | −0.01 | −0.58 | [57] |
| [AuCl2{κ2-HC(3,5-Me2pz)3}]Cl c | - | −0.11 | −0.69 | [57] |
| Tris(pyrazol-1-yl)methane | EL/V vs. SHE a |
|---|---|
| HC(pz)3 | 0.14 |
| {SO3C(pz)3}− | −0.09 |
| {SO3C(3-Phpz)3}− | −0.05 |
© 2017 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, L.M.D.R.S. C-Homoscorpionate Oxidation Catalysts—Electrochemical and Catalytic Activity. Catalysts 2017, 7, 12. https://doi.org/10.3390/catal7010012
Martins LMDRS. C-Homoscorpionate Oxidation Catalysts—Electrochemical and Catalytic Activity. Catalysts. 2017; 7(1):12. https://doi.org/10.3390/catal7010012
Chicago/Turabian StyleMartins, Luísa M. D. R. S. 2017. "C-Homoscorpionate Oxidation Catalysts—Electrochemical and Catalytic Activity" Catalysts 7, no. 1: 12. https://doi.org/10.3390/catal7010012
APA StyleMartins, L. M. D. R. S. (2017). C-Homoscorpionate Oxidation Catalysts—Electrochemical and Catalytic Activity. Catalysts, 7(1), 12. https://doi.org/10.3390/catal7010012