
Enantiopure C1-symmetric N-Heterocyclic Carbene Ligands from Desymmetrized meso-1,2-Diphenylethylenediamine: Application in Ruthenium-Catalyzed Olefin Metathesis

 ,
,  ,
,  and
and
Abstract
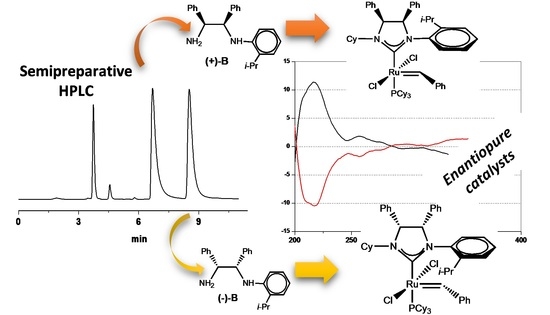
:
1. Introduction
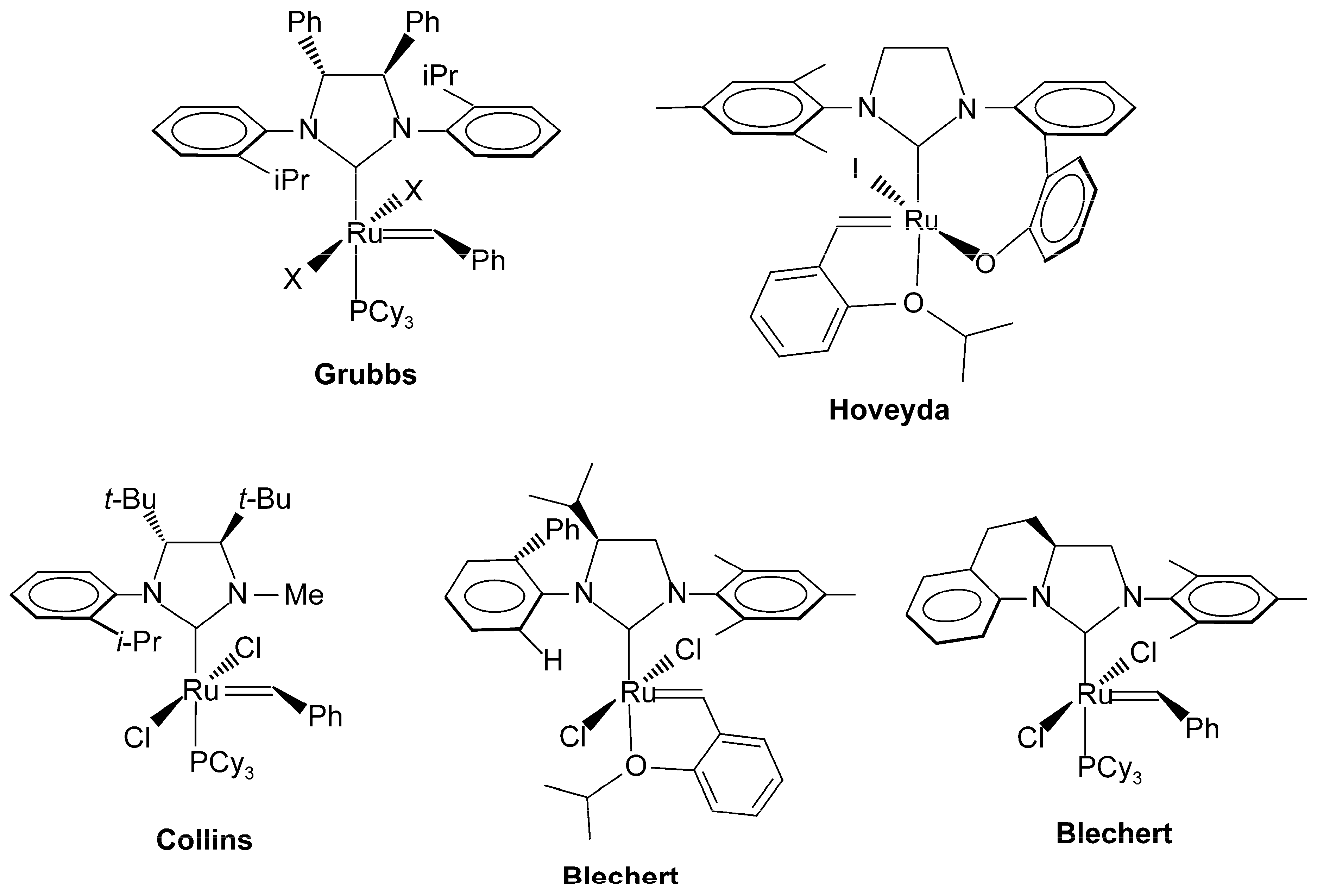
2. Results and Discussion
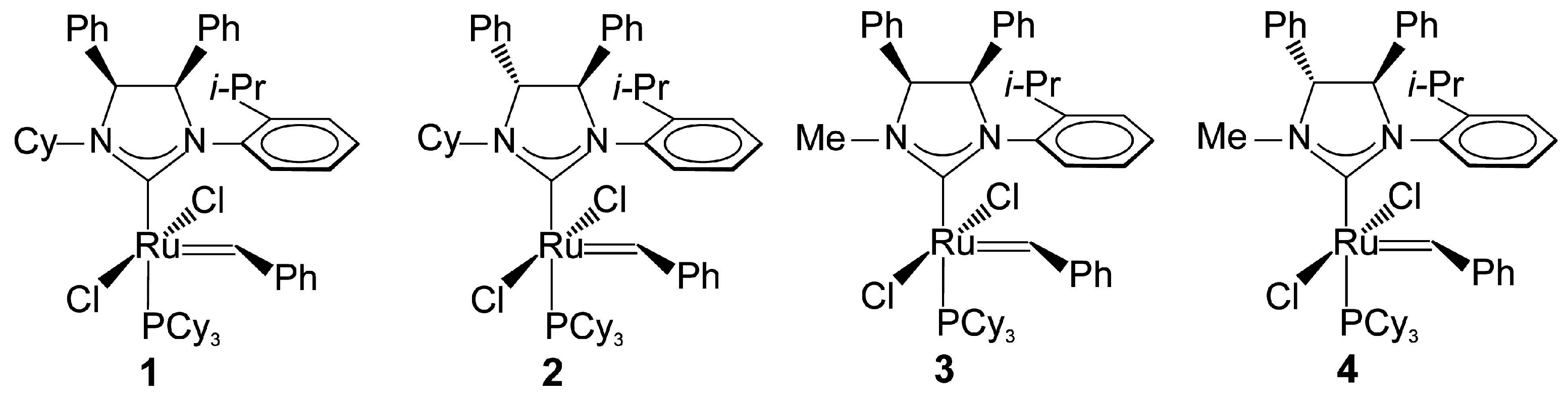
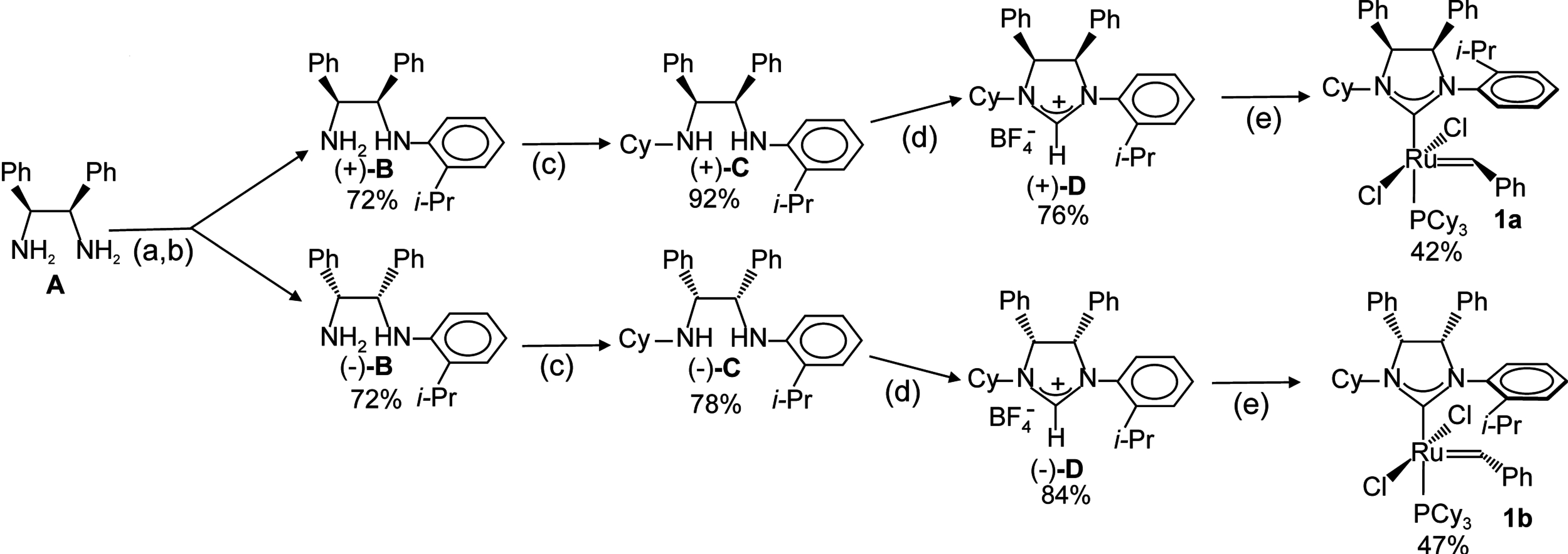
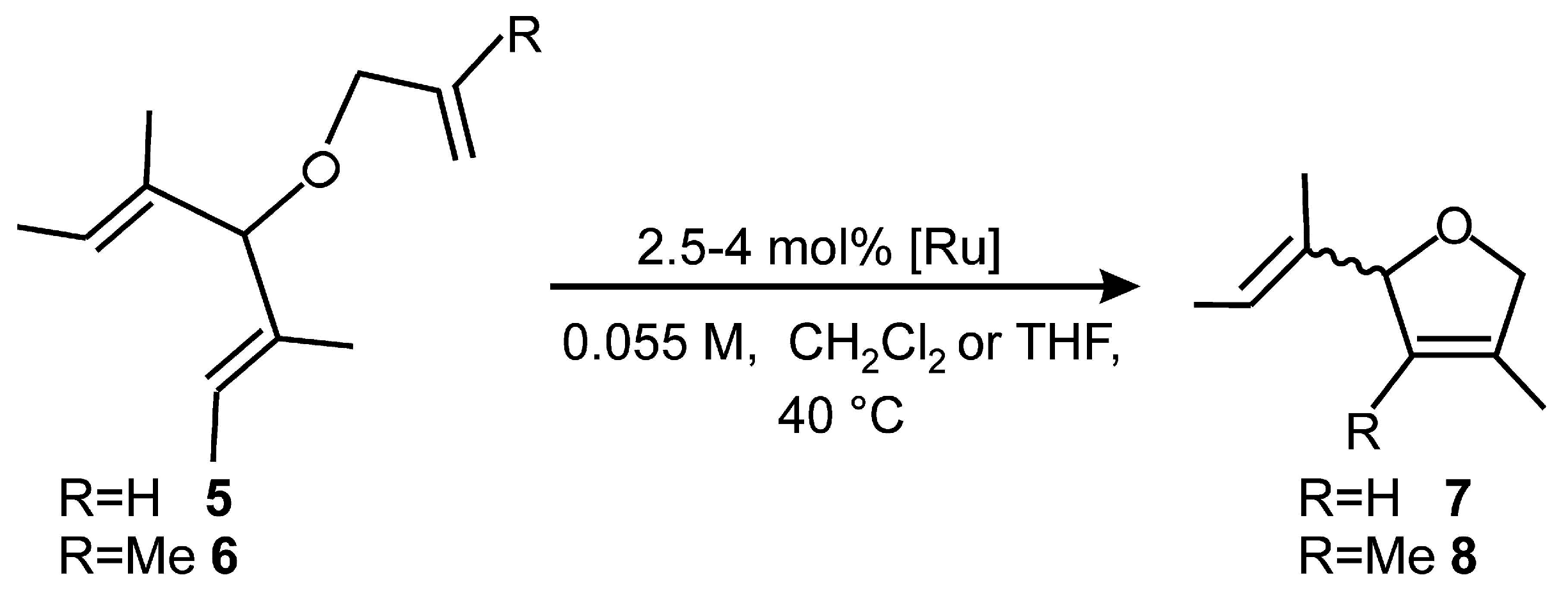
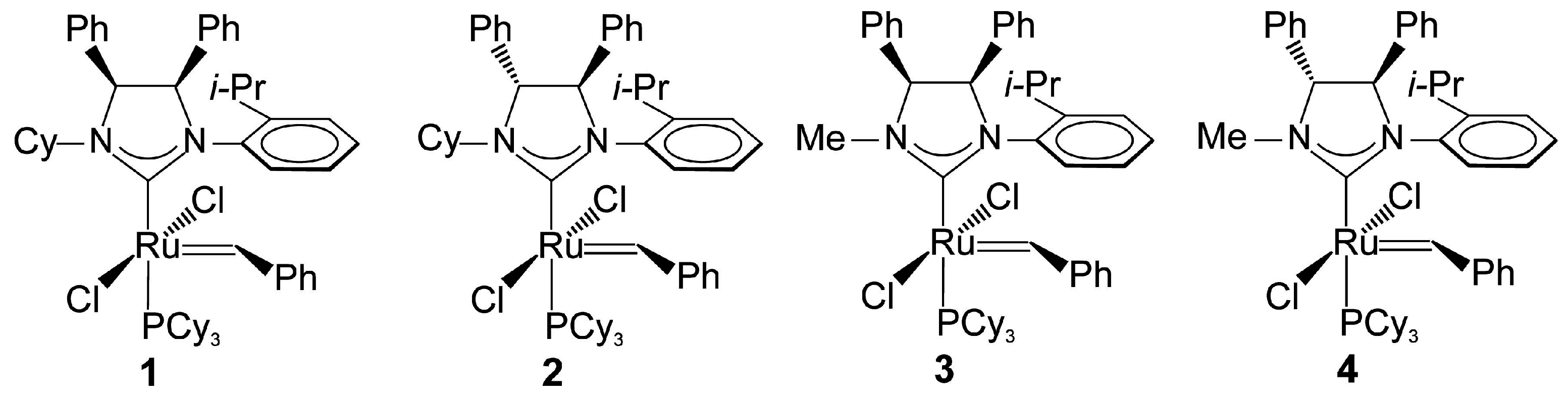
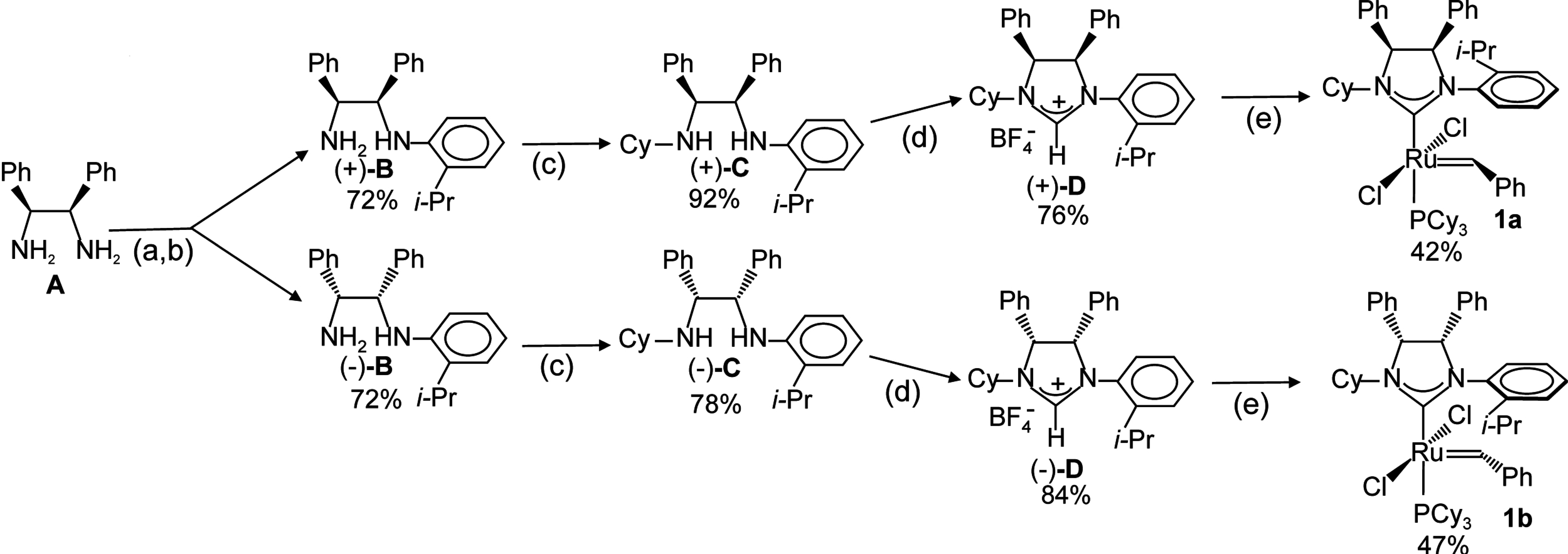
2.1. Synthesis of Catalyst 1a and 1b
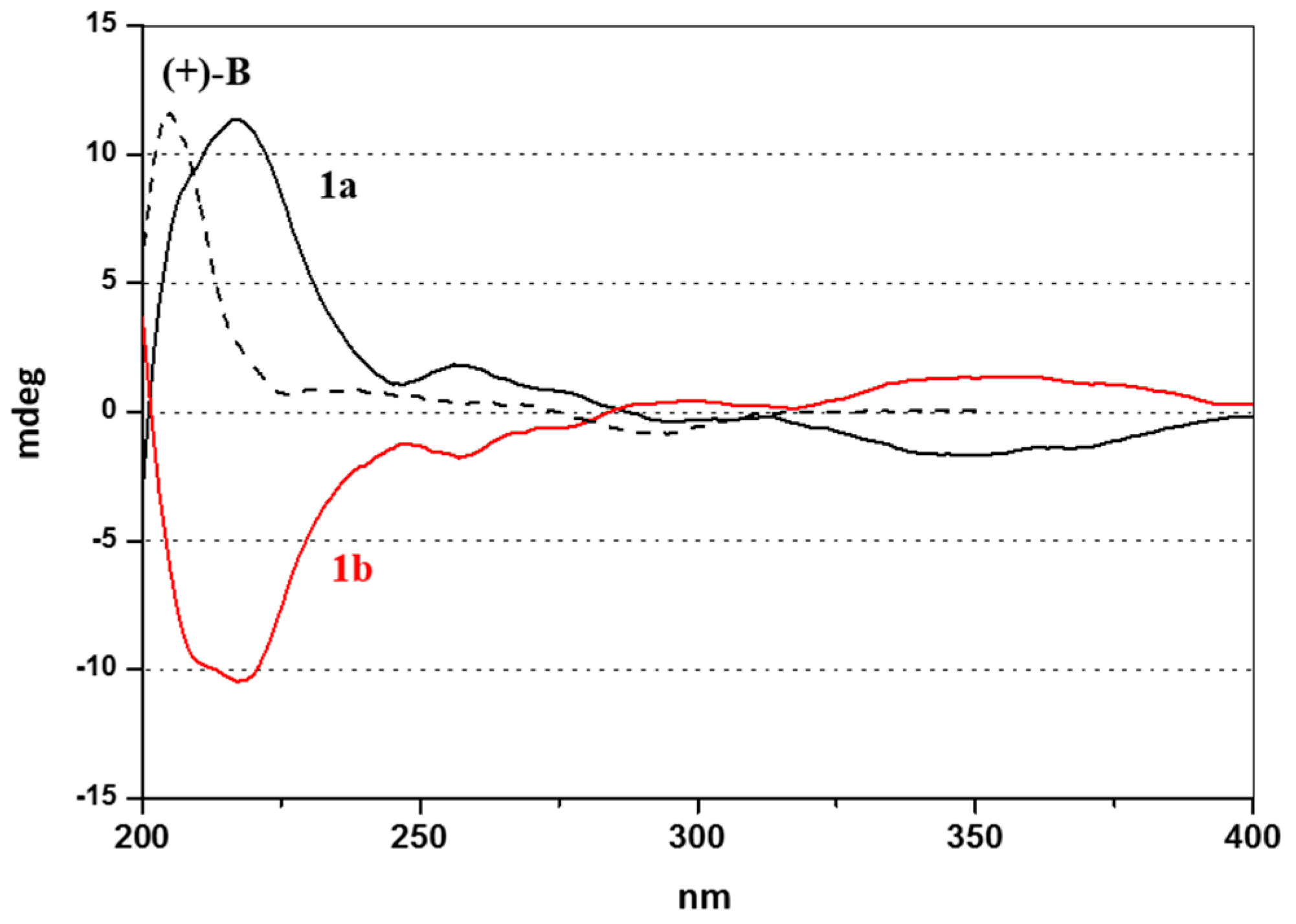
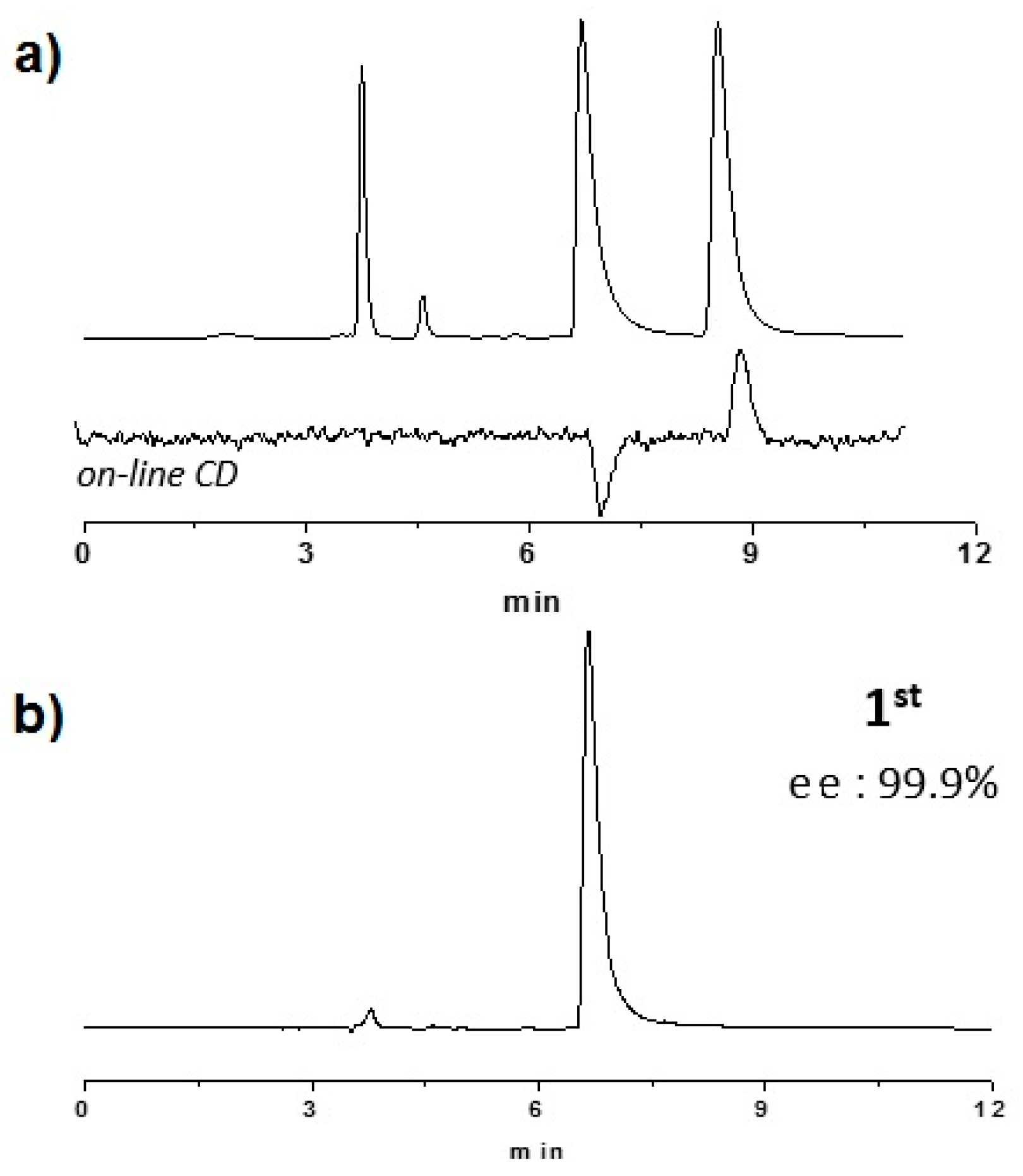
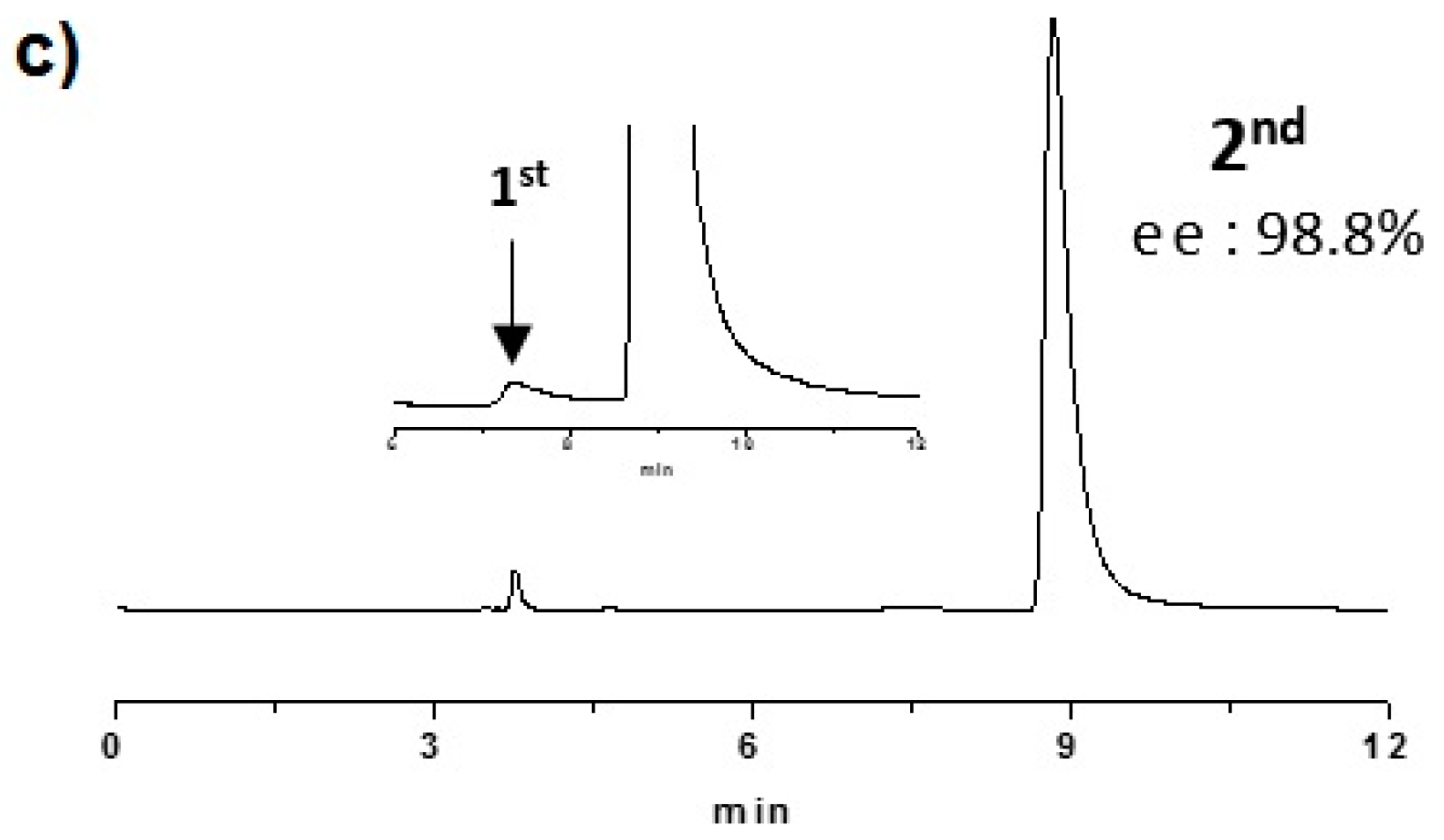
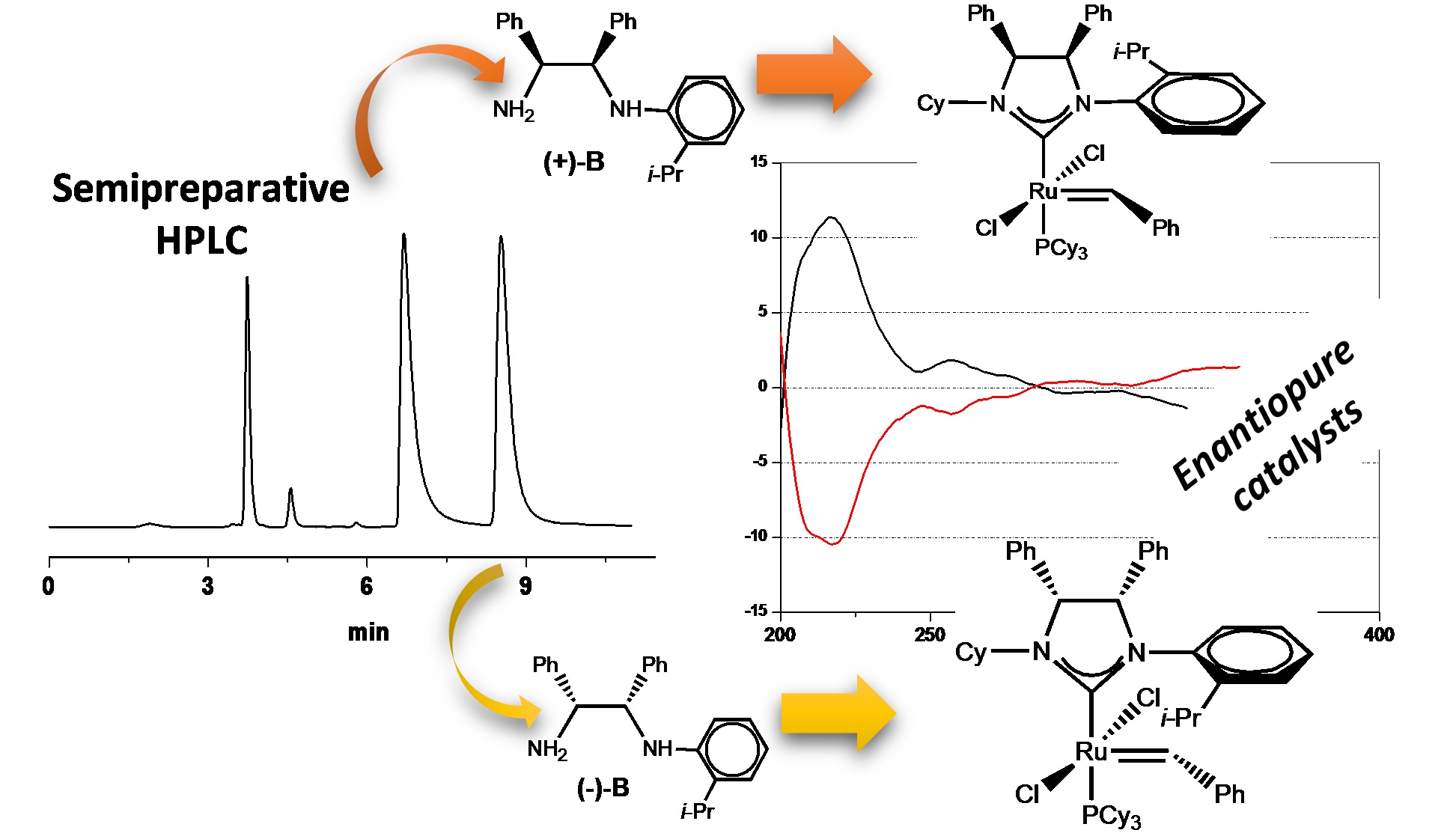
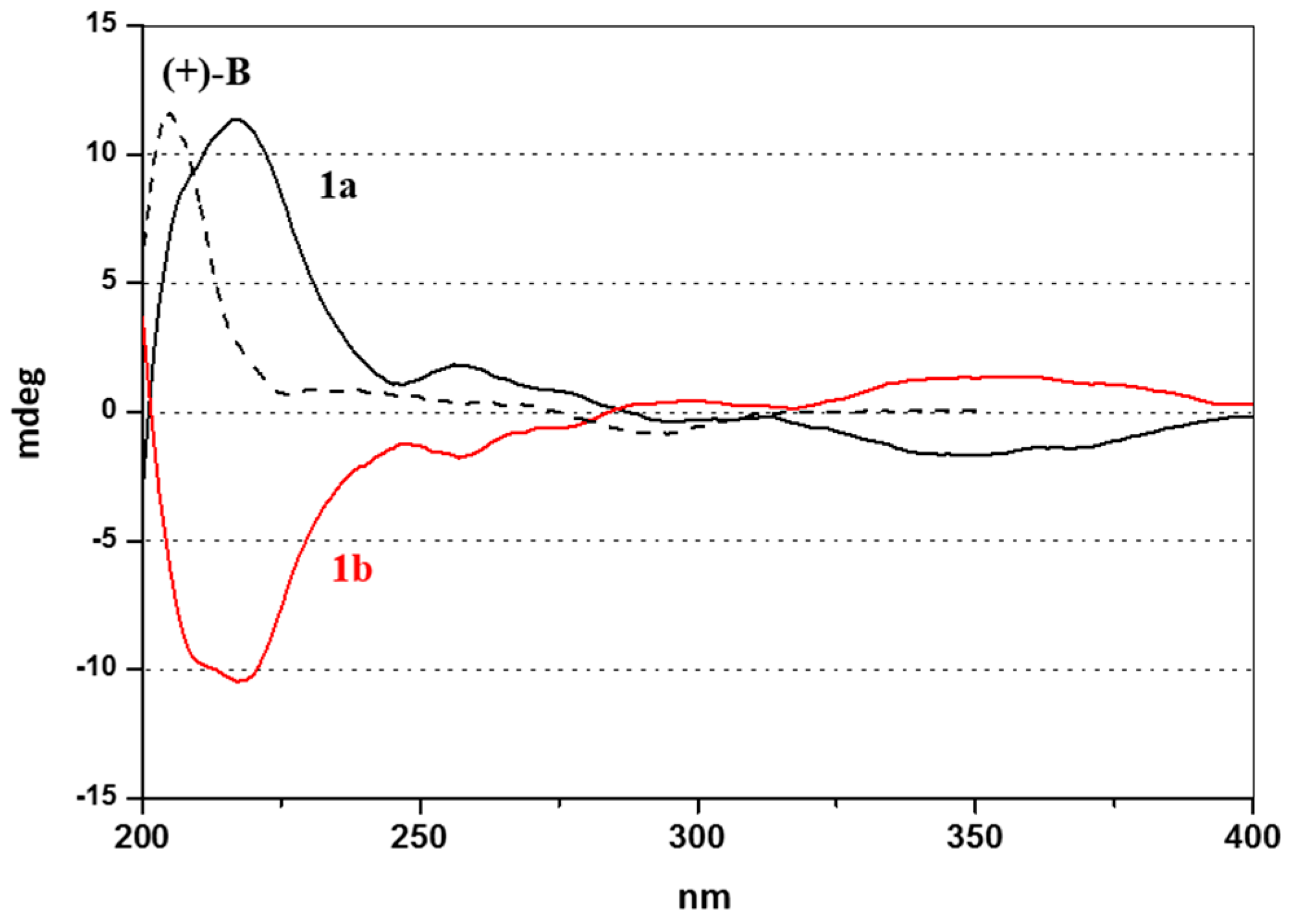
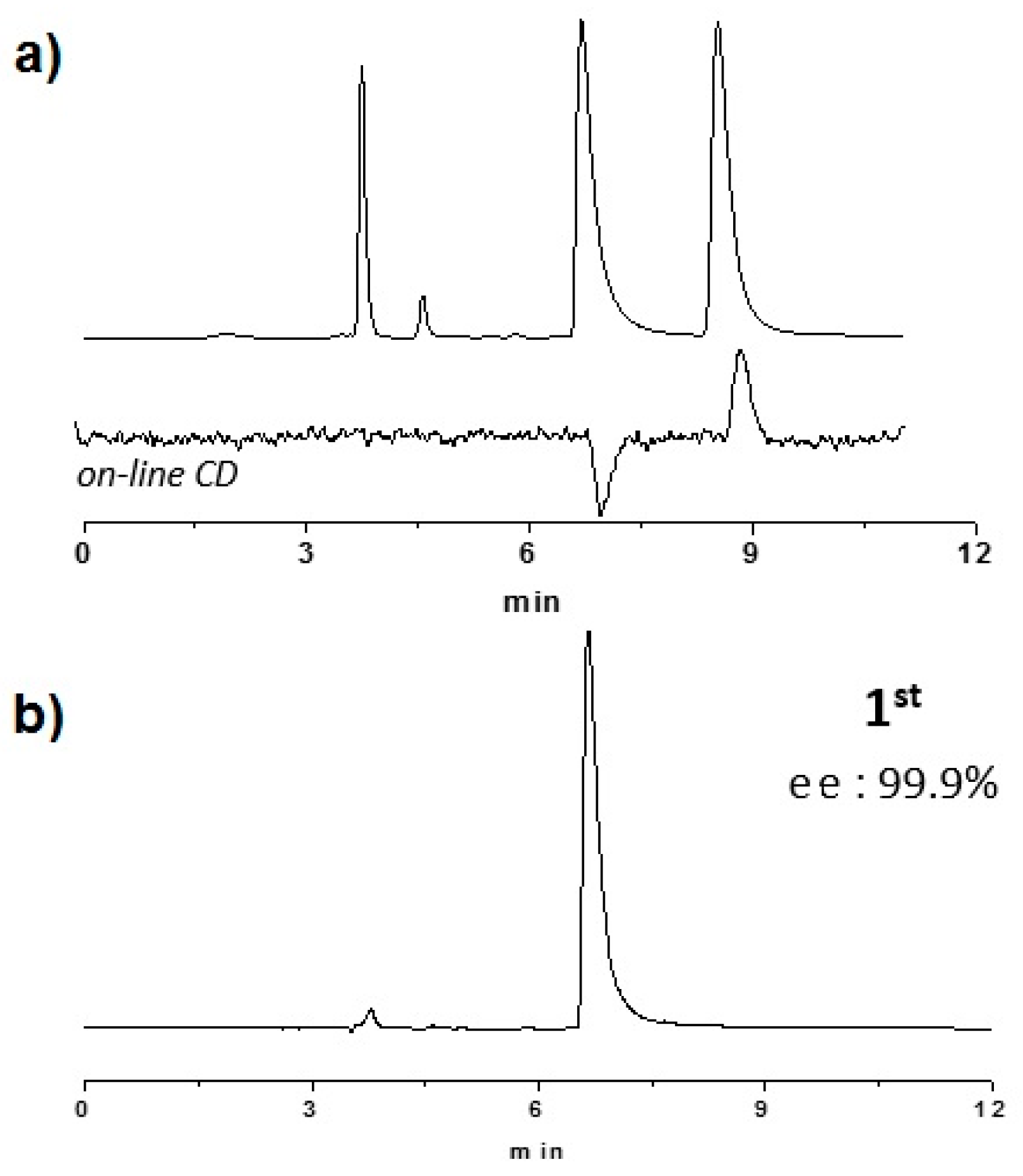
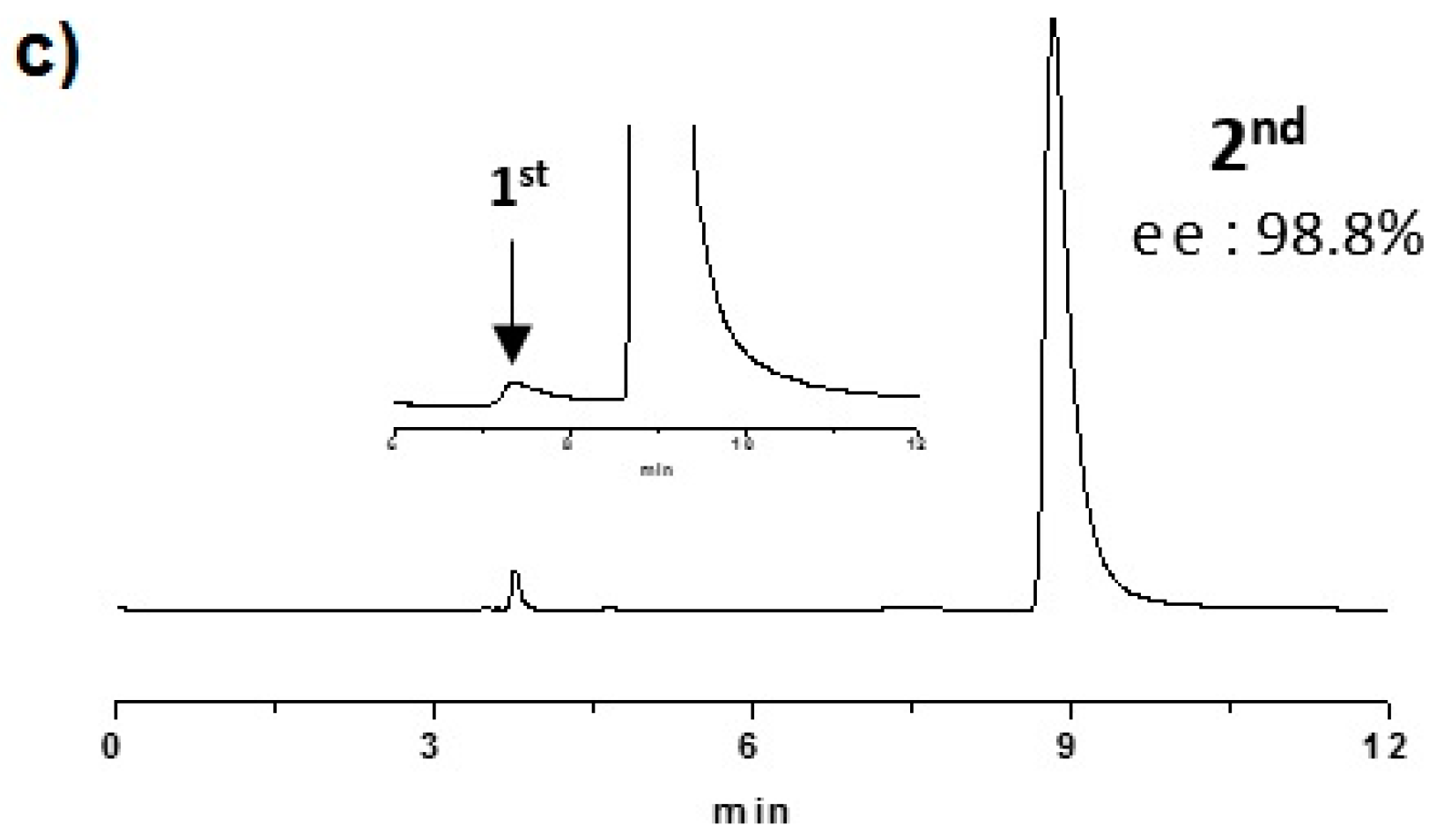
2.2. Enantioselective Chromatography and Chiro-Optical Characterization
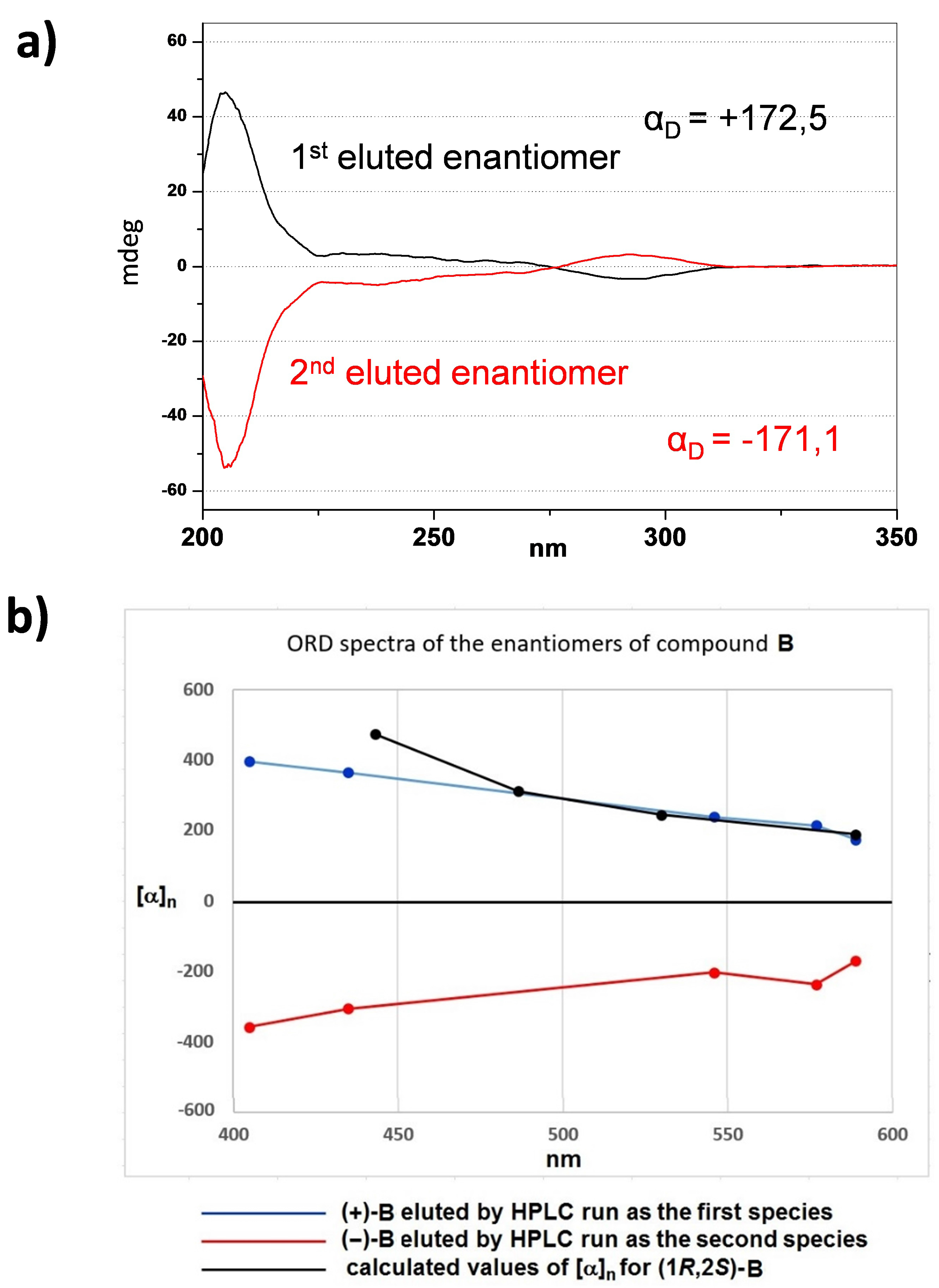
2.3. Determination of Absolute Configuration by Density Functional Theory (DFT)/ORD Calculations
(1R, 2S)-B = N-((1R, 2S)-2-amino-1,2-diphenylethyl)-2-isopropylbenzenamine
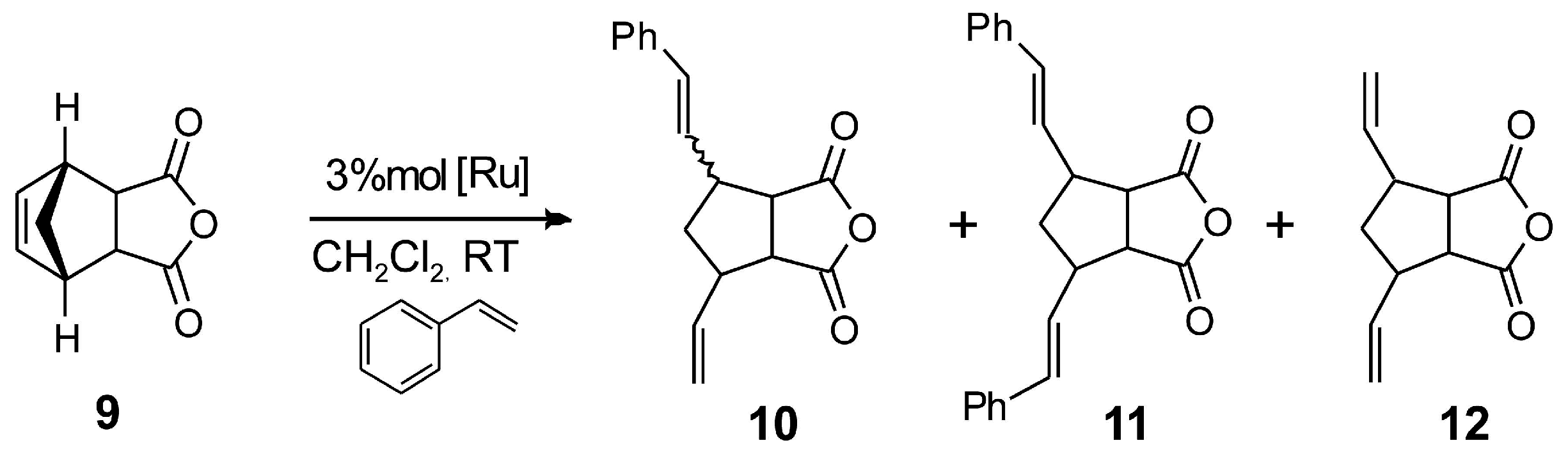
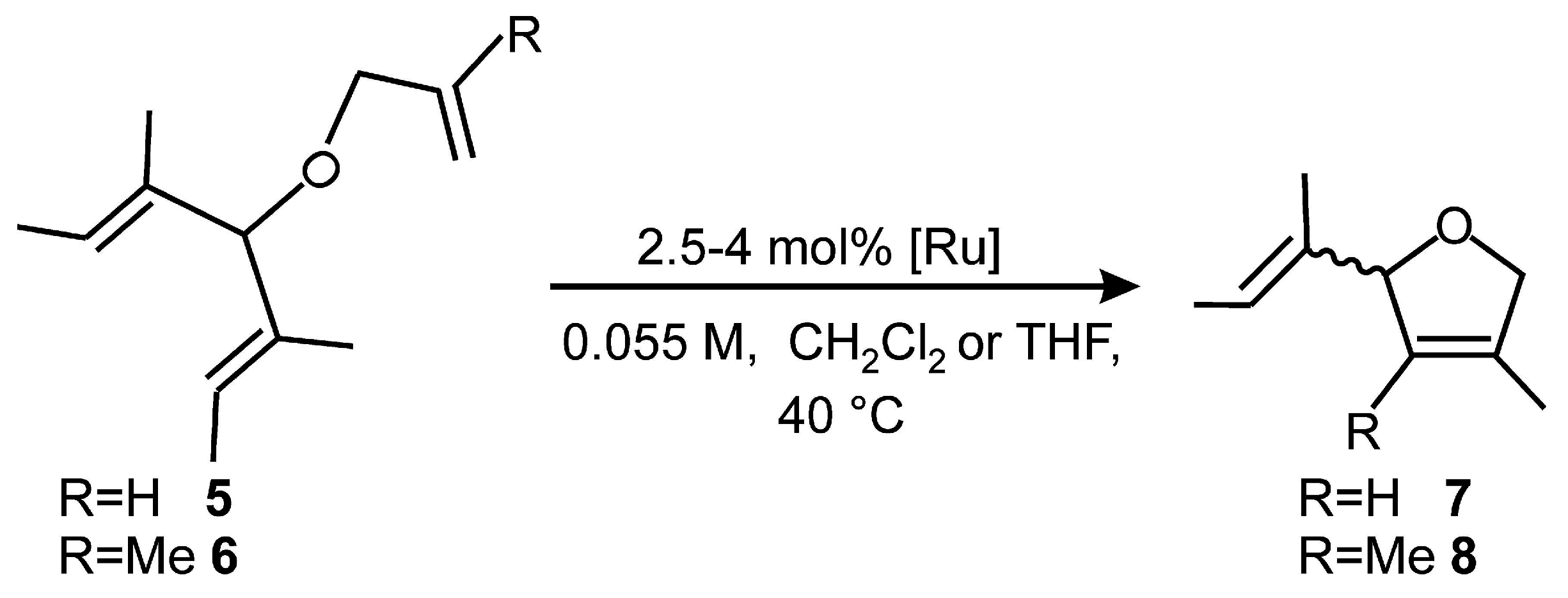
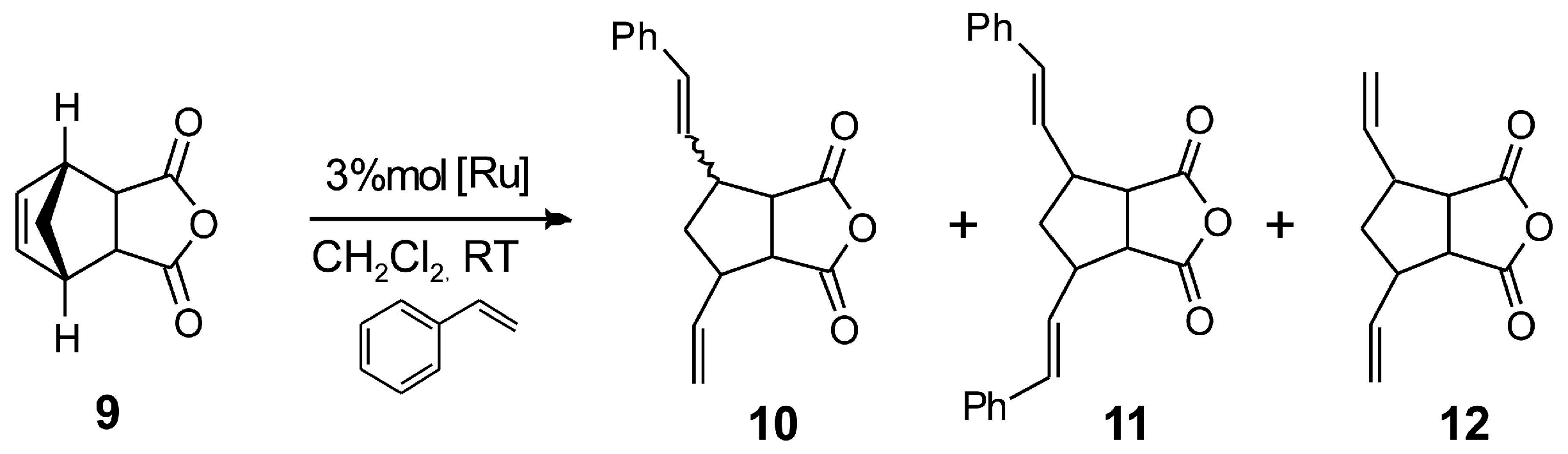
2.4. Asymmetric Metathesis Transformations
3. Materials and Methods
3.1. Enantioselective Chromatography and Chiro-Optical Characterization
3.2. Simulation of the ORD Spectrum of the (1R, 2S)-B Enantiomer.
3.3. Catalytic Tests
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-Heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Cazin, C.S.J. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis; Springer: Dordrecht, The Netherlands, 2011. [Google Scholar]
- Fevre, M.; Pinaud, J.; Gnanou, Y.; Vignolle, J.; Taton, D. N-Heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis. Chem. Soc. Rev. 2013, 42, 2142–2172. [Google Scholar] [CrossRef] [PubMed]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, L.-J.; Wang, W.; Li, S.; Shi, M. Chiral NHC–metal-based asymmetric catalysis. Coord. Chem. Rev. 2012, 256, 804–853. [Google Scholar] [CrossRef]
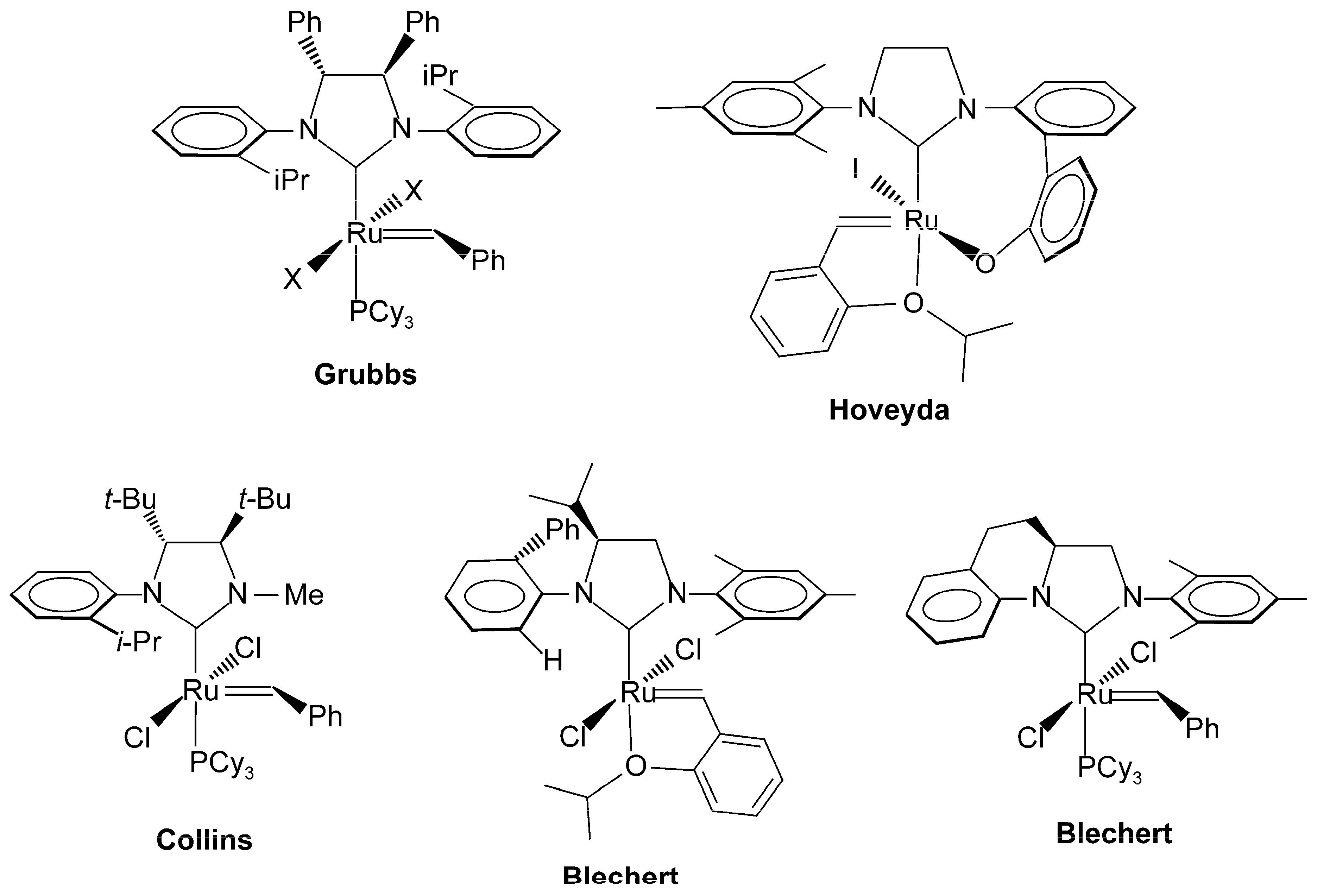
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Samojlowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Tornatzky, J.; Kannenberg, A.; Blechert, S. New catalysts with unsymmetrical N-heterocyclic carbene ligands. Dalton Trans. 2012, 41, 8215–8225. [Google Scholar] [CrossRef] [PubMed]
- Kress, S.; Blechert, S. Asymmetric catalysts for stereocontrolled olefin metathesis reactions. Chem. Soc. Rev. 2012, 41, 4389–4408. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Costabile, C.; Grisi, F. NHC backbone configuration in ruthenium-catalyzed olefin metathesis. Molecules 2016, 21, E117. [Google Scholar] [CrossRef] [PubMed]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective Ruthenium-Catalyzed Ring-Closing Metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.W.; Berlin, J.M.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Ring-Closing Olefin Metathesis. J. Am. Chem. Soc. 2006, 128, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.M.; Goldberg, S.D.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Cross- and Ring-Opening Cross-Metathesis. Angew. Chem. Int. Ed. 2006, 45, 7591–7595. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Collins, S.K. A Highly Active Chiral Ruthenium-Based Catalyst for Enantioselective Olefin Metathesis. Organometallics 2007, 26, 2945–2949. [Google Scholar] [CrossRef]
- Fournier, P.; Savoie, J.; Stenne, B.; Bédard, M.; Grandbois, A.; Collins, S.K. Mechanistically Inspired Catalysts for Enantioselective Desymmetrizations by Olefin Metathesis. Chem. Eur. J. 2008, 14, 8690–8695. [Google Scholar] [CrossRef] [PubMed]
- Grandbois, A.; Collins, S.K. Enantioselective Synthesis of [7] Helicene: Dramatic Effects of Olefin Additives and Aromatic Solvents in Asymmetric Olefin Metathesis. Chem. Eur. J. 2008, 14, 9323–9329. [Google Scholar] [CrossRef] [PubMed]
- Savoie, T.; Stenne, B.; Collins, S.K. Improved Chiral Olefin Metathesis Catalysts: Increasing the Thermal and Solution Stability via Modification of a C1-Symmetrical N-Heterocyclic Carbene Ligand. Adv. Synth. Catal. 2009, 351, 1826–1832. [Google Scholar] [CrossRef]
- Van Veldhuizen, J.J.; Garber, S.B.; Kingsbury, J.S.; Hoveyda, A.H. A Recyclable Chiral Ru Catalyst for Enantioselective Olefin Metathesis. Efficient Catalytic Asymmetric Ring-Opening/Cross Metathesis in Air. J. Am. Chem. Soc. 2002, 124, 4954–4955. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, D.G.; Kataoka, K.; Garber, S.B.; Hoveyda, A.H. Efficient Enantioselective Synthesis of Functionalized Tetrahydropyrans by Ru-Catalyzed Asymmetric Ring-Opening Metathesis/Cross-Metathesis (AROM/CM). J. Am. Chem. Soc. 2004, 126, 12288–12290. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, J.J.; Campbell, J.E.; Giudici, R.E.; Hoveyda, A.H. A Readily Available Chiral Ag-Based N-Heterocyclic Carbene Complex for Use in Efficient and Highly Enantioselective Ru-Catalyzed Olefin Metathesis and Cu-Catalyzed Allylic Alkylation Reactions. J. Am. Chem. Soc. 2005, 127, 6877–6882. [Google Scholar] [CrossRef] [PubMed]
- Giudici, R.E.; Hoveyda, A.H. Directed Catalytic Asymmetric Olefin Metathesis. Selectivity Control by Enoate and Ynoate Groups in Ru-Catalyzed Asymmetric Ring-Opening/Cross-Metathesis. J. Am. Chem. Soc. 2007, 129, 3824–3825. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Berger, A.; Schlesiger, D.; Rost, D.; Lühl, A.; Blechert, S. Highly Active Chiral Ruthenium-Based Metathesis Catalysts through a Monosubstitution in the N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2010, 49, 3972–3975. [Google Scholar] [CrossRef] [PubMed]
- Kannenberg, A.; Rost, D.; Eibauer, S.; Tiede, S.; Blechert, S. A Novel Ligand for the Enantioselective Ruthenium-Catalyzed Olefin Metathesis. Angew. Chem. Int. Ed. 2011, 50, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Bertolasi, V.; Grisi, F. Novel Olefin Metathesis Ruthenium Catalysts Bearing Backbone-Substituted Unsymmetrical NHC Ligands. Organometallics 2014, 33, 5932–5935. [Google Scholar] [CrossRef]
- Paradiso, V.; Bertolasi, V.; Costabile, C.; Grisi, F. Ruthenium Olefin Metathesis Catalysts Featuring Unsymmetrical N-Heterocyclic Carbenes. Dalton Trans. 2016, 45, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Gasparrini, F.; Ciogli, A.; Spinelli, D.; Cosimelli, B. Determination of the absolute configuration of a chiral oxadiazol-3-one calcium channel blocker, resolved using chiral chromatography, via concerted density functional theory calculations of its vibrational circular dichroism, electronic circular dichroism, and optical rotation. J. Org. Chem. 2007, 72, 4707–4715. [Google Scholar] [PubMed]
- Ioan, P.; Ciogli, A.; Sirci, F.; Budriesi, R.; Cosimelli, B.; Pierini, M.; Severi, E.; Chiarini, A.; Cruciani, G.; Gasparrini, F.; Spinelli, D.; Carosati, E. Absolute configuration and biological profile of two thiazinooxadiazol-3-ones with L-type calcium channel activity: A study of the structural effects. Org. Biomol. Chem. 2012, 10, 8994–9003. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, E.; Viglione, R.G.; Zanasi, R.; Rosini, C. Ab Initio Calculation of Optical Rotatory Dispersion (ORD) Curves: A Simple and Reliable Approach to the Assignment of the Molecular Absolute Configuration. J. Am. Chem. Soc. 2004, 126, 12968–12976. [Google Scholar] [CrossRef] [PubMed]
- Rotili, D.; Samuele, A.; Tarantino, D.; Ragno, R.; Musmuca, I.; Ballante, F.; Botta, G.; Morera, L.; Pierini, M.; Cirilli, R.; et al. 2-(Alkyl/aryl)amino-6-benzylpyrimidin-4(3H)-ones as inhibitors of wild-type and mutant HIV-1: Enantioselectivity studies. J. Med. Chem. 2012, 55, 3558–3562. [Google Scholar] [CrossRef] [PubMed]
- Vaghi, L.; Benincori, T.; Cirilli, R.; Alberico, E.; Mussini, P.R.; Pierini, M.; Pilati, T.; Rizzo, S.; Sannicolò, F. Ph-tetraMe-bithienine, the first member of the class of chiral heterophosphepines: Synthesis, electronic and steric properties, metal complexes and catalytic activity. Eur. J. Org. Chem. 2013, 2013, 8174–8184. [Google Scholar] [CrossRef]
- Stenne, B.; Timperio, J.; Savoie, J.; Dudding, T.; Collins, S.K. Desymmetrizations Forming Tetrasubstituted Olefins Using Enantioselective Olefin Metathesis. Org. Lett. 2010, 12, 2032–2035. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Grubbs, R.H. Mechanistic Studies of Enantioselective N-aryl, N-alkyl NHC Ruthenium Metathesis Catalysts in Asymmetric Ring-Opening Cross-Metathesis. Chem. N. Z. 2011, 75, 65–71. [Google Scholar]
- Stewart, I.C.; Keitz, B.K.; Kuhn, K.M.; Thomas, R.M.; Grubbs, R.H. Nonproductive events in ring-closing metathesis using ruthenium catalysts. J. Am. Chem. Soc. 2010, 132, 8534–8535. [Google Scholar] [CrossRef] [PubMed]
- Keitz, B.K.; Grubbs, R.H. Probing the Origin of Degenerate Metathesis Selectivity via Characterization and Dynamics of Ruthenacyclobutanes Containing Variable NHCs. J. Am. Chem. Soc. 2011, 133, 16277–16284. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavelength of [α]n (nm) | [α]n of (+)-B | [α]n of (−)-B | [α]n of (1R, 2S)-B |
|---|---|---|---|
| 589 | 172 | −171 | 186 |
| 577 | 212 | −238 | - |
| 546 | 237 | −204 | - |
| 530 | - | - | 242 |
| 487 | - | - | 309 |
| 443 | - | - | 473 |
| 435 | 363 | −307 | - |
| 405 | 395 | −359 | - |
| Entry a | Substrate | Catalyst (mol %) | Additive | Time (h) | Yield (%) b | ee % c |
|---|---|---|---|---|---|---|
| 1 | 5 | 1b (2.5) | none | 2 | >98 | 37 (R) [1a:39(S)] d |
| 2 | 5 | 1b (4.0) | NaI | 2 | 46 | 44 (R) |
| 3 | 5 | 1b (4.0) | NaI | 16 | 57 | 44 (R) |
| 4 | 5 | 1b (2.5) | none | 2 | >98 | 39 (R) (22 °C) |
| 5 | 5 | 1b (2.5) | NaI | 2 | 56 | 39 (R) (0 °C) |
| 6 e | 5 | 2 (2.5) | none | 2 | >98 | 18 (S) |
| 7 e | 5 | 2 (4.0) | NaI | 2 | >95 | 53 (S) |
| 8 e | 5 | 4 (2.5) | none | 2 | >98 | 33 (S) |
| 9 e | 5 | 4 (4.0) | NaI | 2 | >95 | 50 (S) |
| 10 f | 5 | Collins (2.5) | none | 2 | >95 | 82 (S) |
| 11 f | 5 | Collins (4.0) | NaI | 2 | >95 | 48 (S) |
| 12 | 6 | 1b (2.5) | none | 2 | >98 | 14 (S) |
| 13 | 6 | 1b (4.0) | NaI | 3 | - | - |
| 14 e | 6 | 2 (2.5) | none | 2 | >95 | 42 (S) |
| 15 e | 6 | 2 (4.0) | NaI | 3 | - | - |
| 16 e | 6 | 4 (2.5) | none | 2 | >95 | 25 (R) |
| 17 e | 6 | 4 (4.0) | NaI | 3 | - | - |
| 18 g | 6 | Collins (2.5) | none | 3 | 95 | 8 (S) |
| Entry a | Catalyst | 10 Yield b | 11 Yield b | 12 Yield b | ee % (10) c |
|---|---|---|---|---|---|
| 1 | 1b | 70 | 1 | 29 | 15 |
| 2 d | 2 | 45 | 10 | 10 | 32 |
| 3 d | 4 | 20 | 5 | 11 | 29 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paradiso, V.; Menta, S.; Pierini, M.; Della Sala, G.; Ciogli, A.; Grisi, F. Enantiopure C1-symmetric N-Heterocyclic Carbene Ligands from Desymmetrized meso-1,2-Diphenylethylenediamine: Application in Ruthenium-Catalyzed Olefin Metathesis. Catalysts 2016, 6, 177. https://doi.org/10.3390/catal6110177
Paradiso V, Menta S, Pierini M, Della Sala G, Ciogli A, Grisi F. Enantiopure C1-symmetric N-Heterocyclic Carbene Ligands from Desymmetrized meso-1,2-Diphenylethylenediamine: Application in Ruthenium-Catalyzed Olefin Metathesis. Catalysts. 2016; 6(11):177. https://doi.org/10.3390/catal6110177
Chicago/Turabian StyleParadiso, Veronica, Sergio Menta, Marco Pierini, Giorgio Della Sala, Alessia Ciogli, and Fabia Grisi. 2016. "Enantiopure C1-symmetric N-Heterocyclic Carbene Ligands from Desymmetrized meso-1,2-Diphenylethylenediamine: Application in Ruthenium-Catalyzed Olefin Metathesis" Catalysts 6, no. 11: 177. https://doi.org/10.3390/catal6110177