
Novel Mesoporous Carbon Supports for PEMFC Catalysts

Abstract
:1. Introduction
1.1. Carbon Nanotubes (CNTs)
1.2. Ordered Mesoporous Carbons (OMCs)
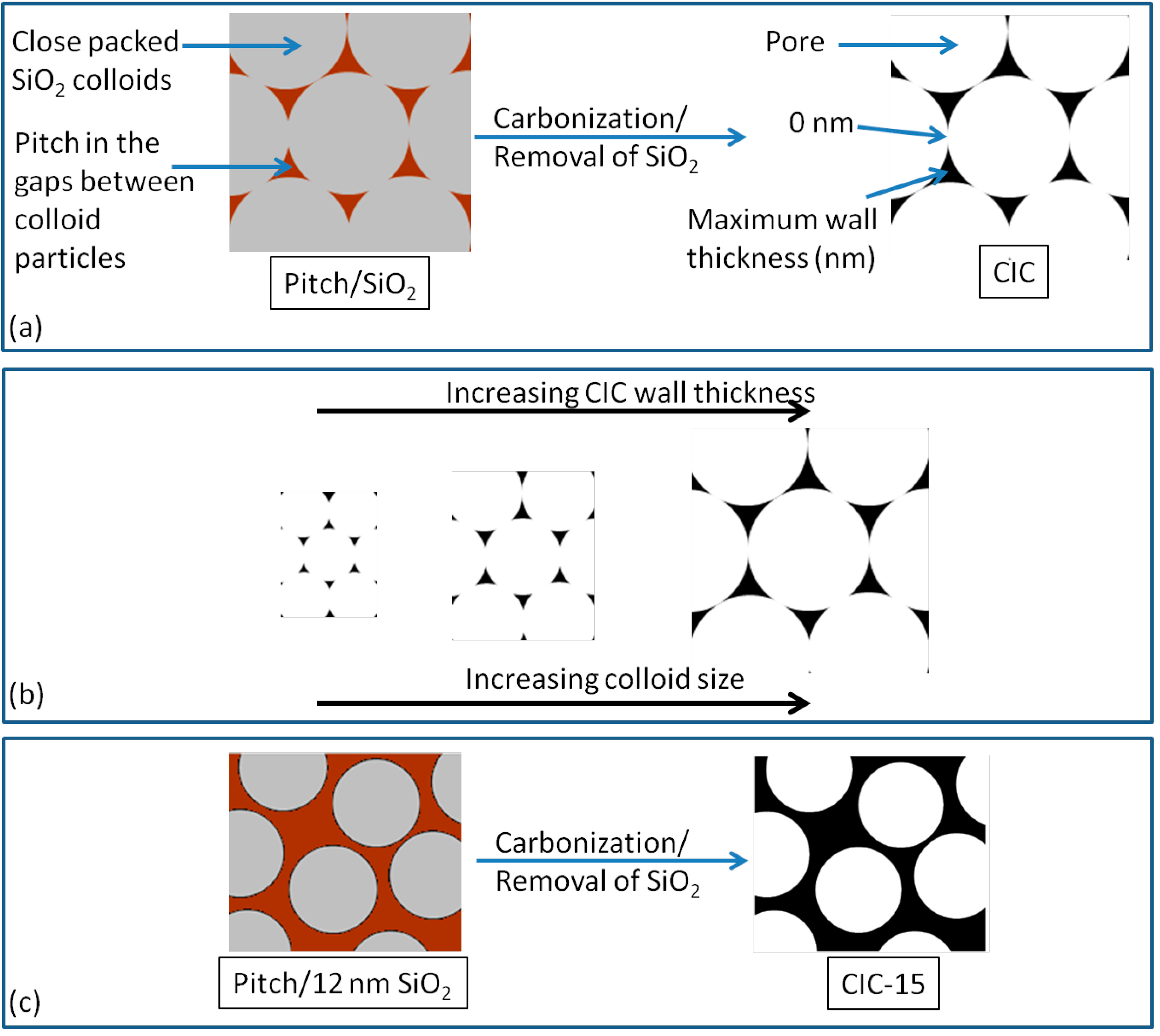
1.3. Colloid Imprinted Carbons (CICs)
2. Results and Discussion
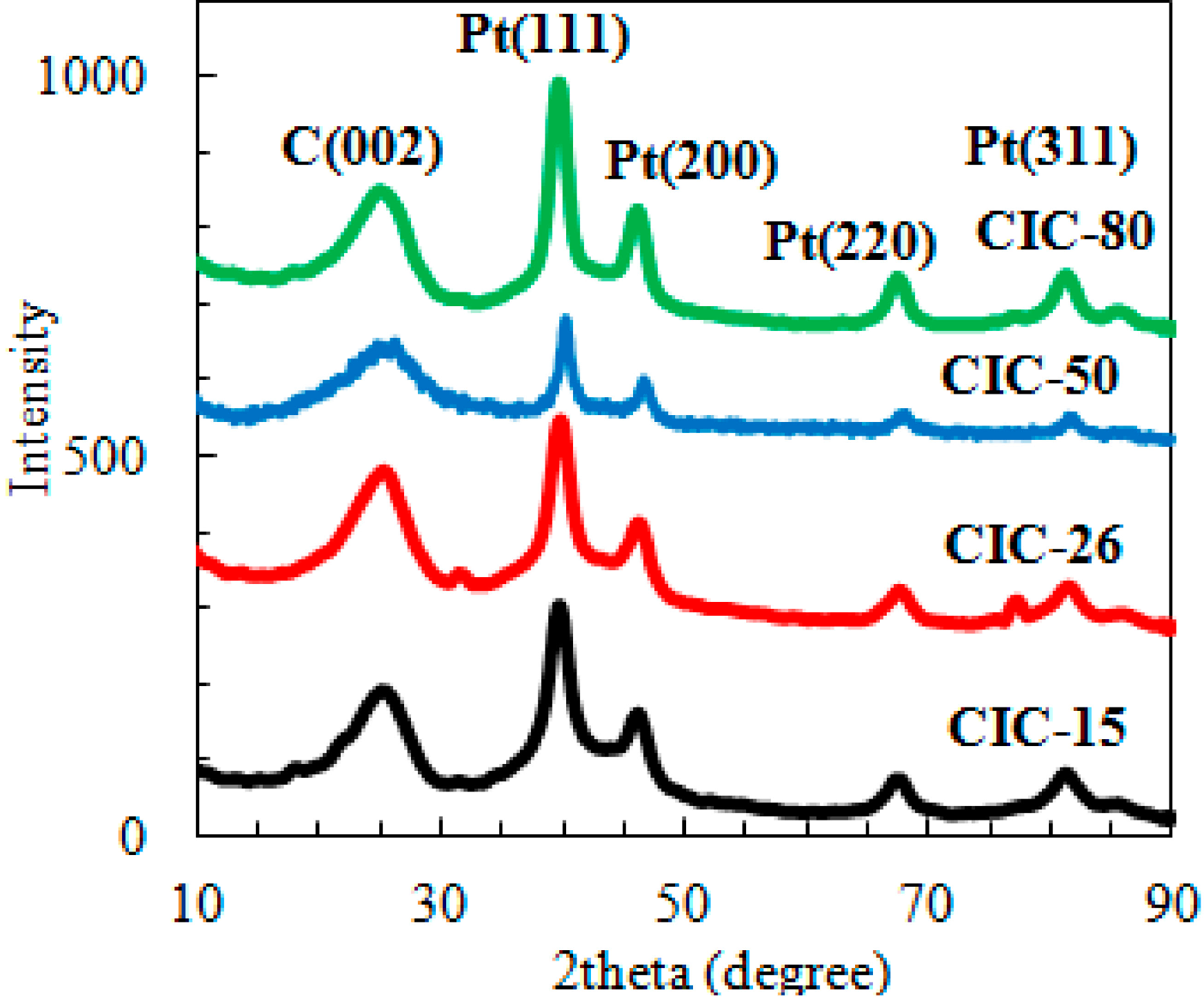
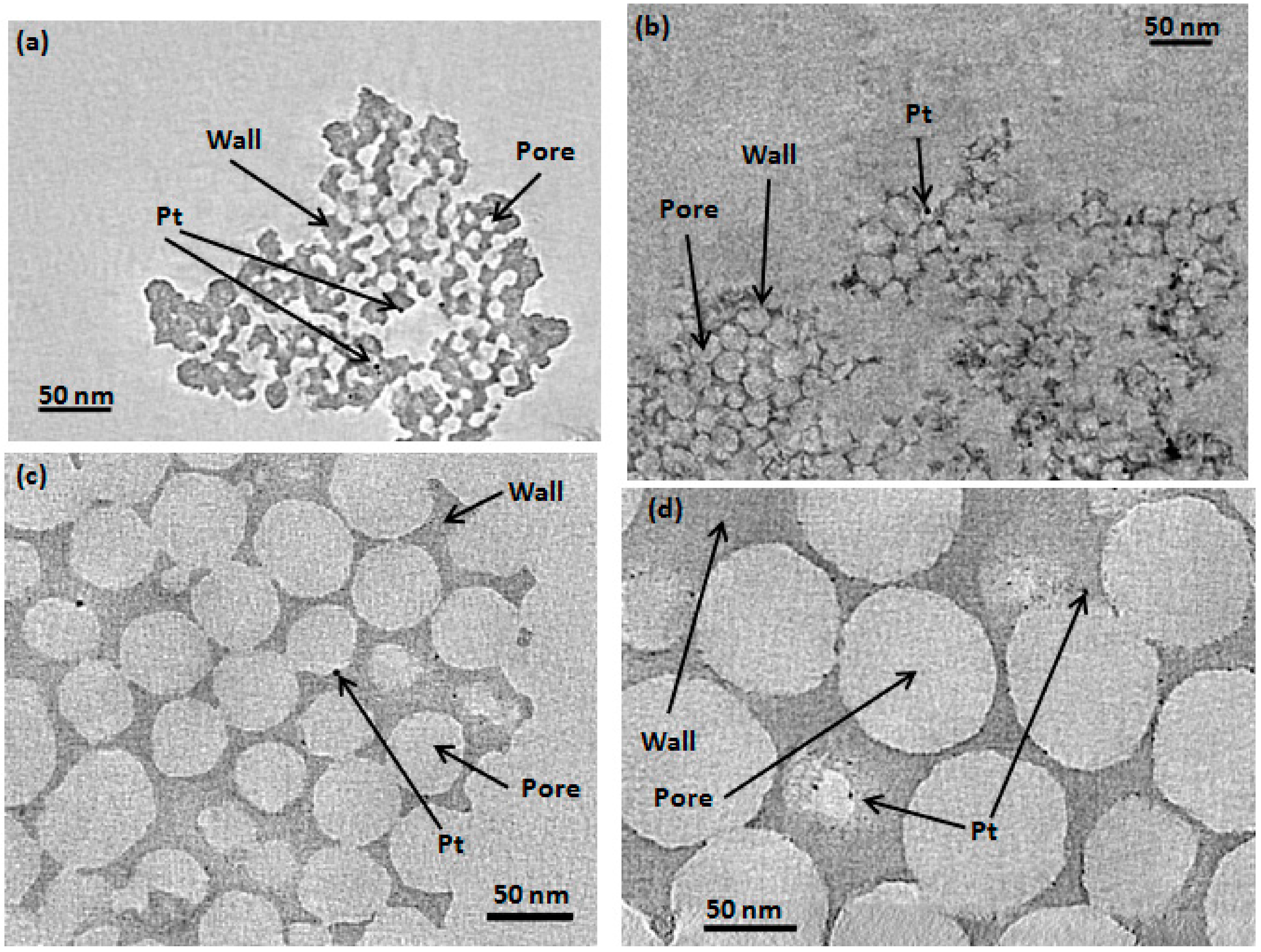
2.1. Determination of CIC Pore Diameter and Pt Nanoparticle Size
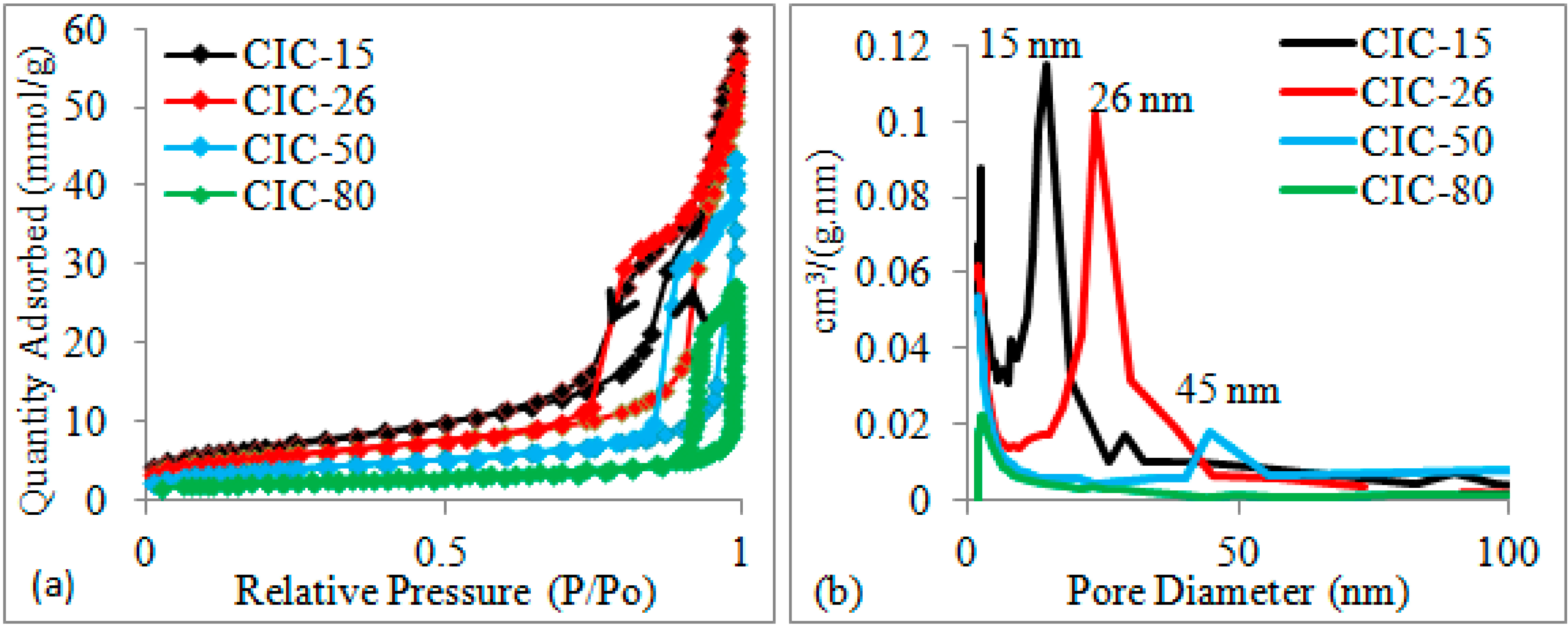


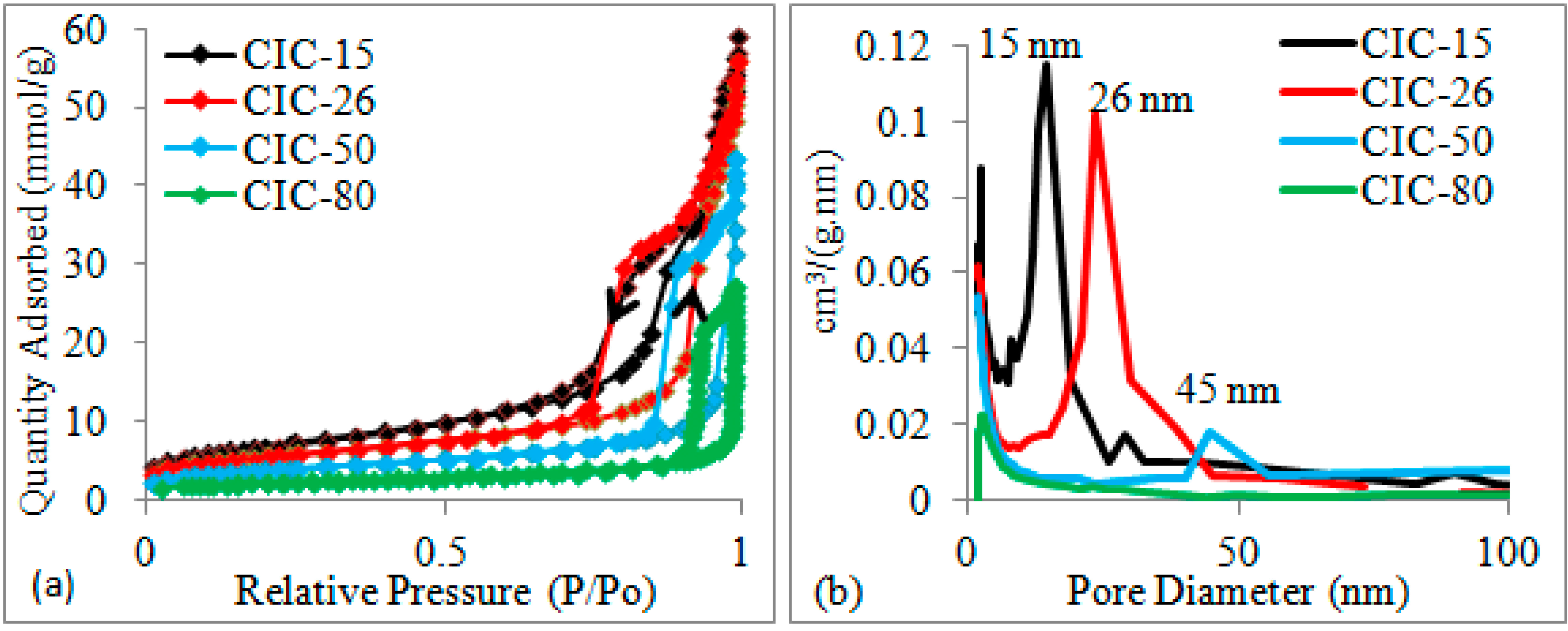{kind=link}
{kind=link}
{kind=link}
{kind=link}
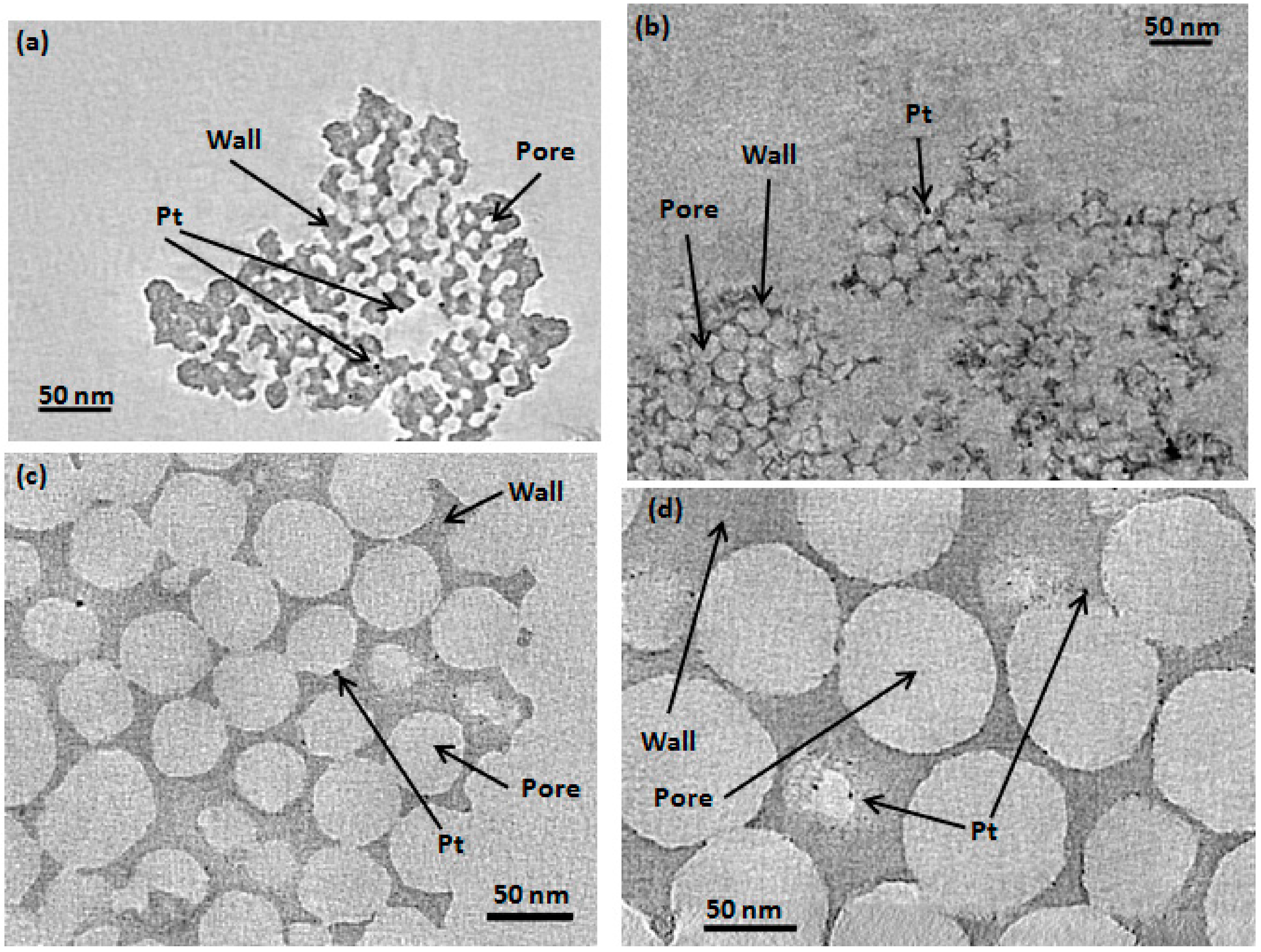{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Pore Diameter a (nm) | Pt Particle Size b (nm) | Graphite Crystallite Size c (nm) | BET Surface Area (m2·g−1 ± 10%) | Pore Volume (mL·g−1) ± 0.1 d | Wall Thickness (nm) e |
|---|---|---|---|---|---|---|
| CIC-15 | 15 (10–15) | - | 1.5 | 330 | 1.2 | 10 ± 5 |
| CIC-26 | 26 (20–25) | - | 1.5 | 380 | 1.6 | 0–3 |
| CIC-50 | 45 (50–60) | - | 1.5 | 240 | 1.4 | 0–20 |
| CIC-80 | N/A (80–90) | - | 1.5 | 150 | 1.4 | 0–50 |
| Pt/CIC-15 | 9 | 4.0 (4.3) | - | 280 | 0.9 | - |
| Pt/CIC-26 | 24 | 4.0 (4.7) | - | 360 | 1.6 | - |
| Pt/CIC-50 | 45 | 6.0 (4.4) | - | 260 | 1.6 | - |
| Pt/CIC-80 | N/A | 4.5 (4.5) | - | 140 | 1.3 | - |
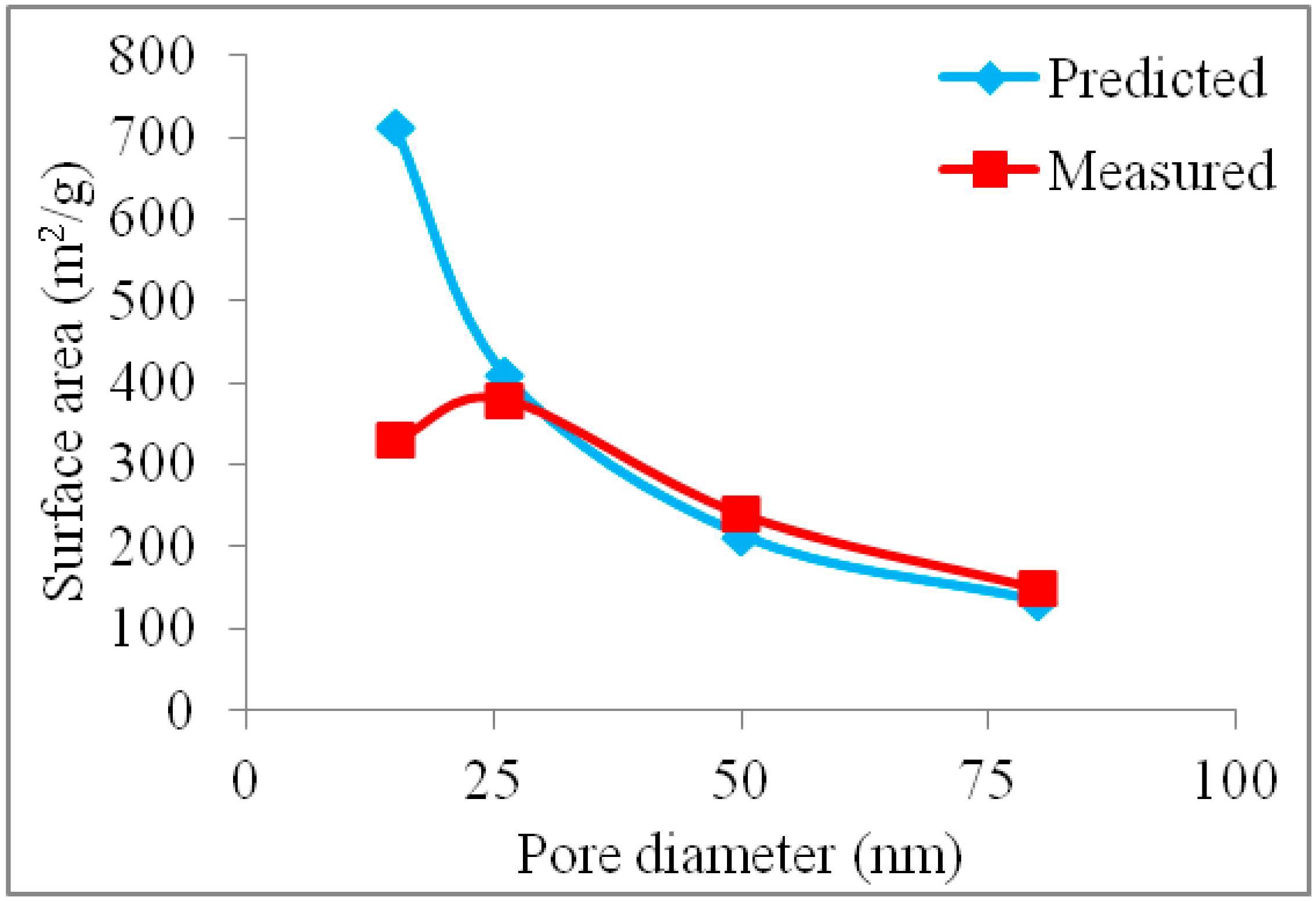
2.2. Surface Area of Pt-Free and Pt-Loaded CICs
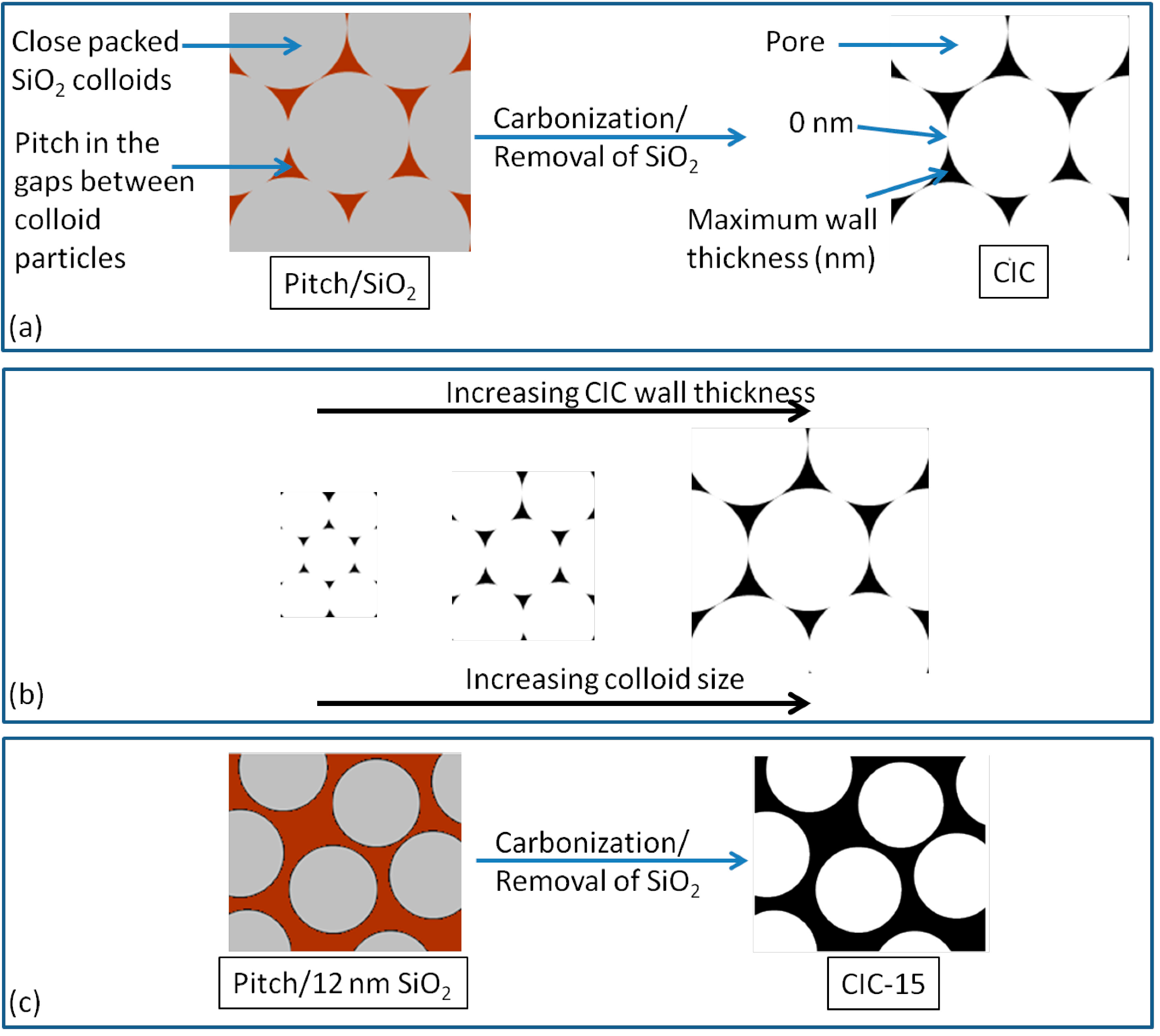
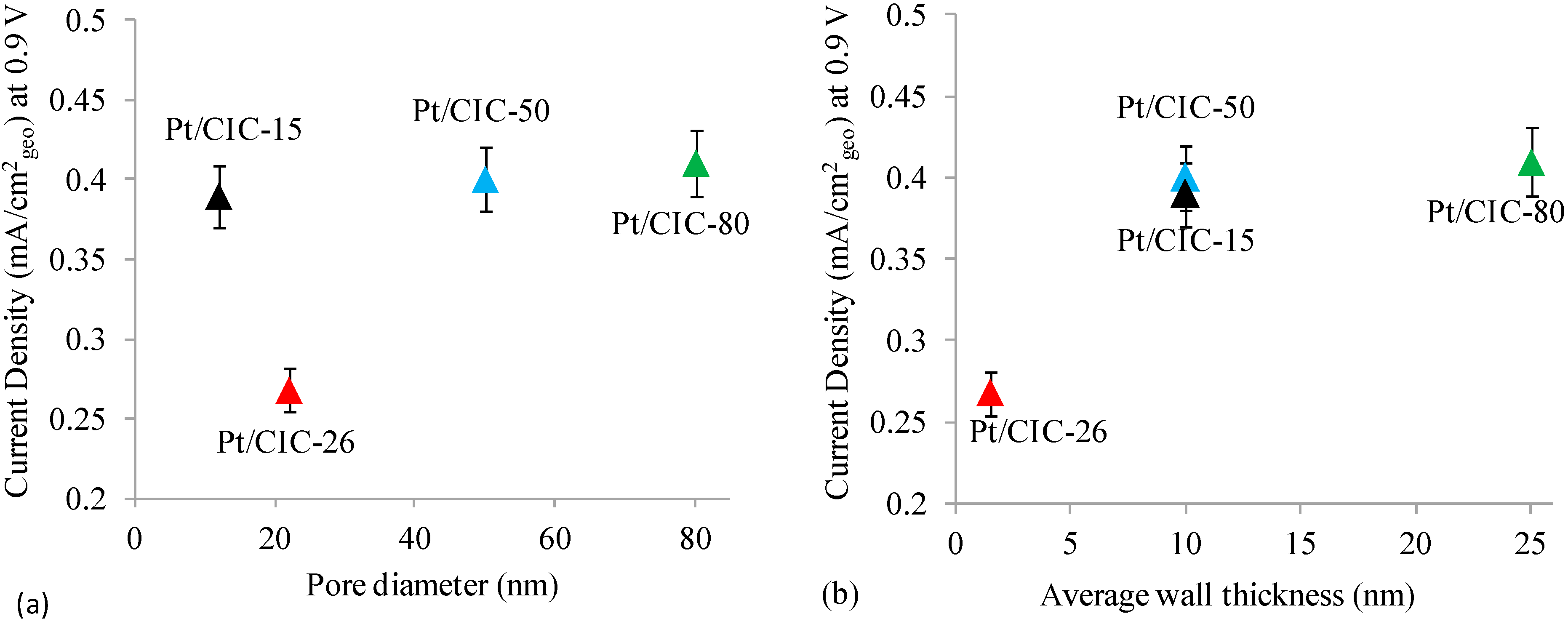
2.3. Wall Thickness of CIC Supports
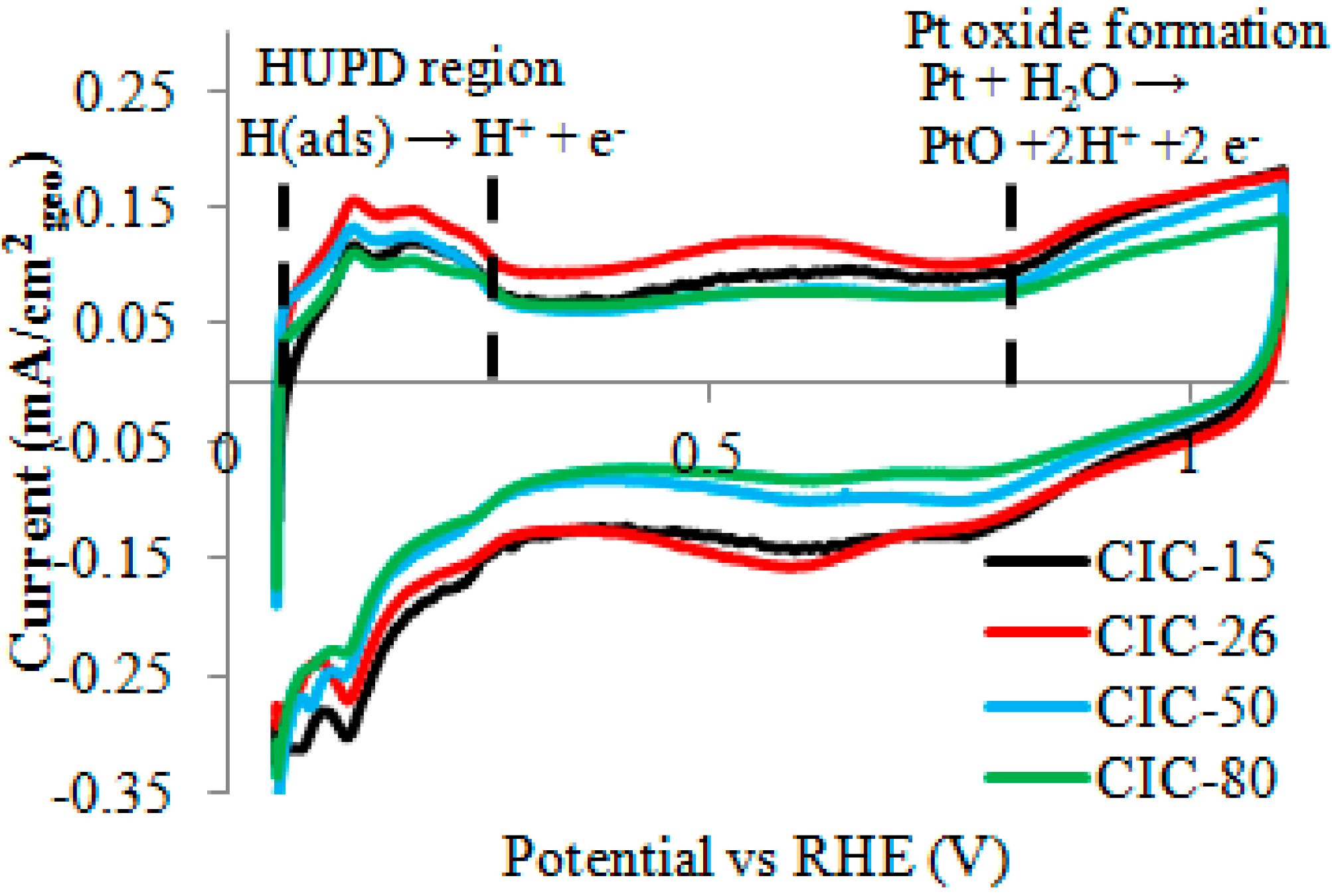
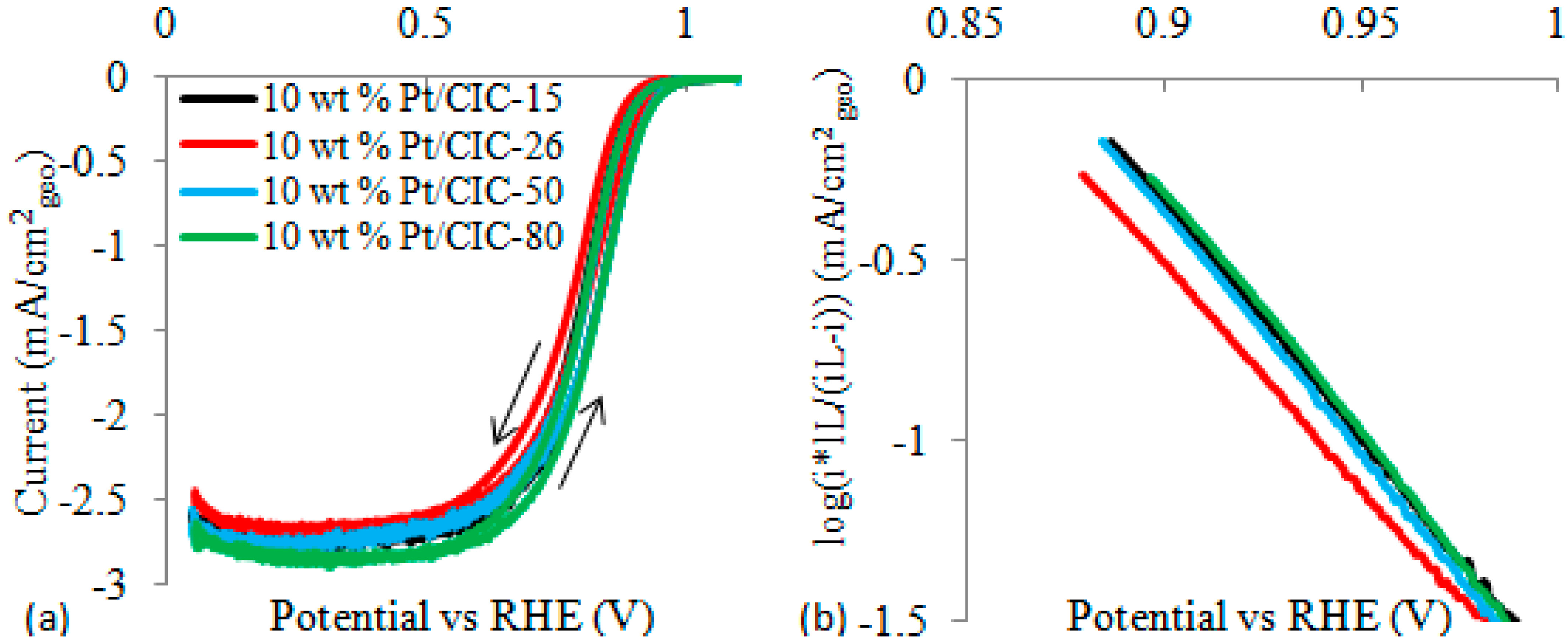
2.4. Effect of CIC Nanostructure on ORR at Pt/CIC Catalysts
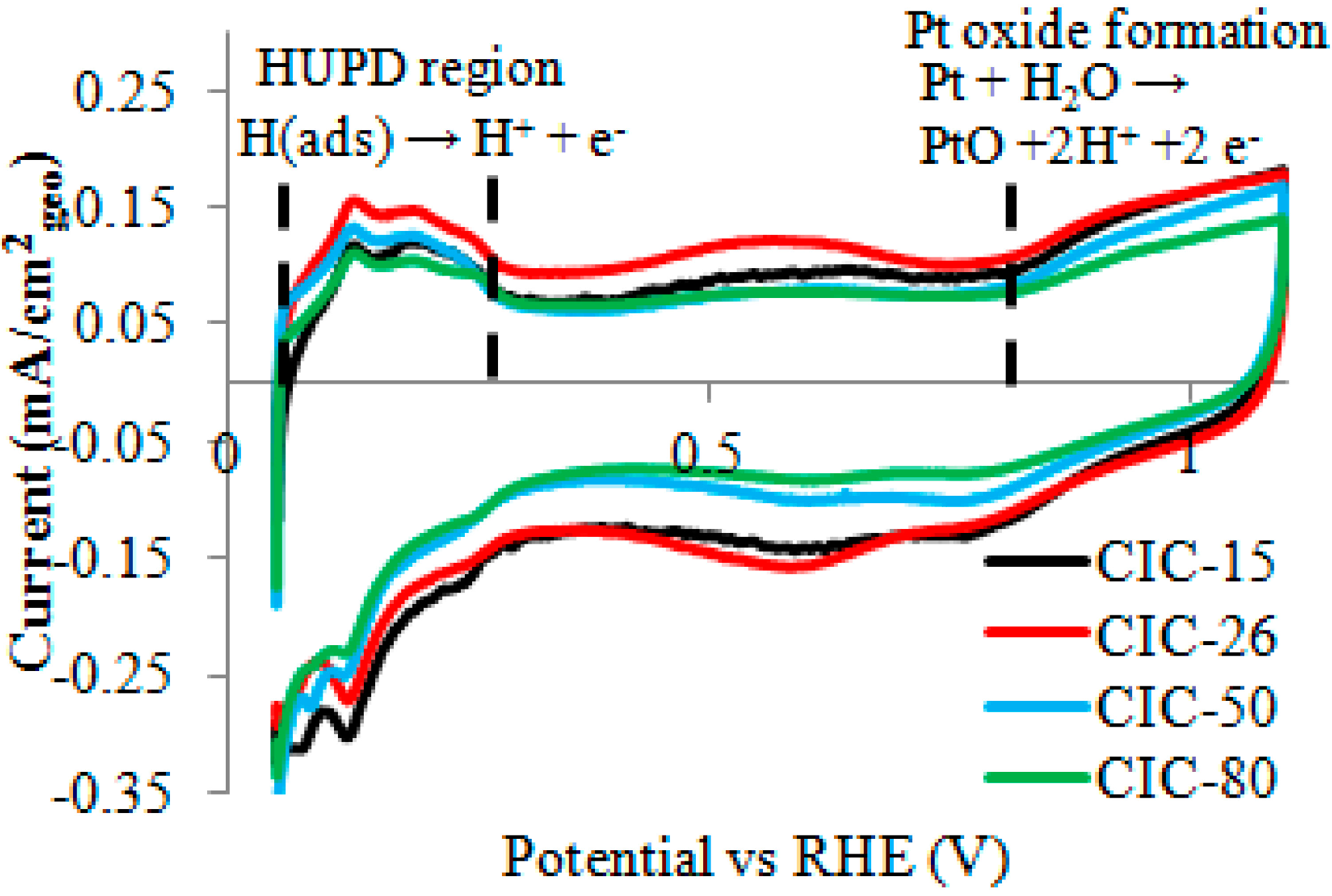
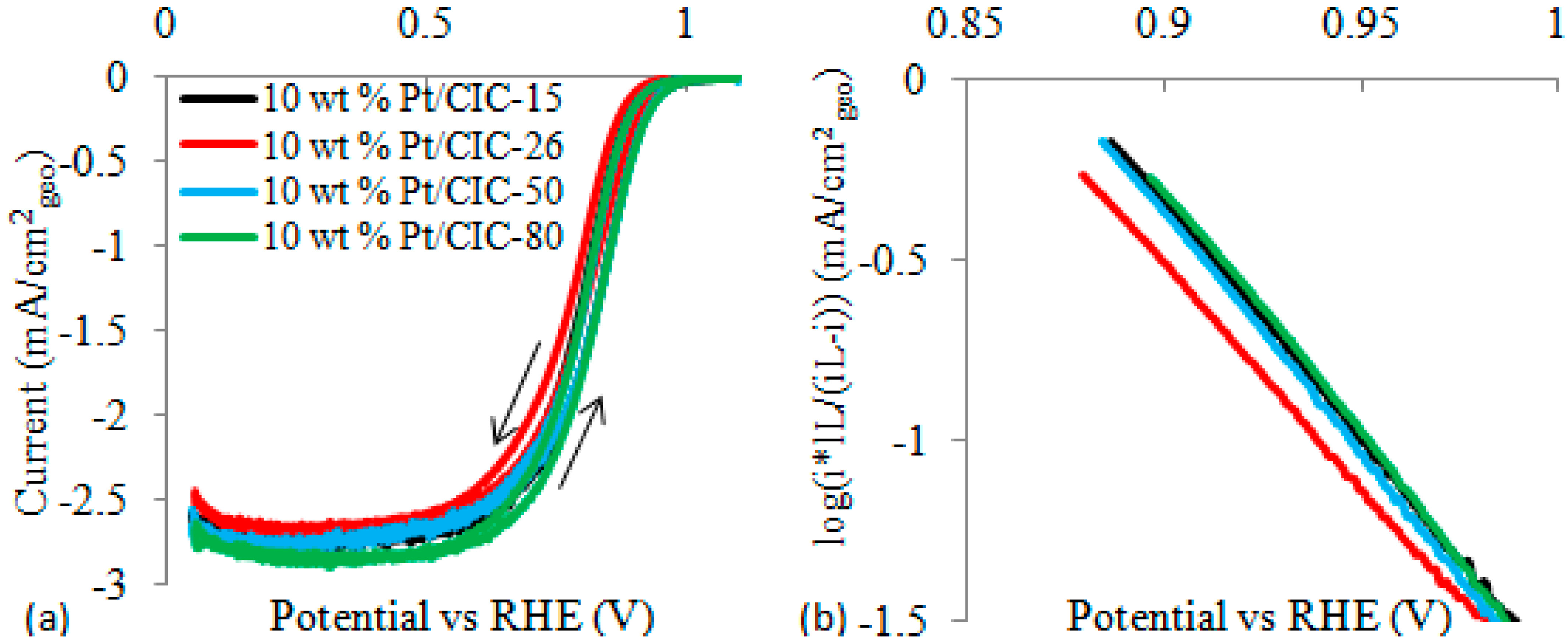
| Sample | ECSAa (m2/g) ± 10% | ORR Current at 0.9 V b (mA/cm2) | Tafel Slope (mV/dec) |
|---|---|---|---|
| 10 wt. % Pt/CIC-15 | 45 | 0.40 | 70 |
| 10 wt. % Pt/CIC-26 | 55 | 0.32 | 85 |
| 10 wt. % Pt/CIC-50 | 55 | 0.40 | 70 |
| 10 wt. % Pt/CIC-80 | 45 | 0.40 | 70 |
2.5. Verification of Effect of Carbon Wall Thickness on ORR Activity by Theoretical Modeling

| Catalysts | Experimental ORR Tafel Slope (Figure 8) a | Modelled Tafel Slope b (Figure 10) |
|---|---|---|
| 10 wt. % Pt/CIC-15 | 70 | 80 |
| 10 wt. % Pt/CIC-26 | 85 | 75 |
| 10 wt. % Pt/CIC-50 | 70 | 71 |
| 10 wt. % Pt/CIC-80 | 70 | 70 |
3. Experimental Section
3.1. Synthesis of Silica Colloid Imprinted Carbons (CICs)
3.2. Pt Loading of CIC-X (Where X Defines Pore Diameter) Supports
3.3. Electrochemical Evaluation of Oxygen Reduction Activity at Pt/CIC Catalysts
3.4. Catalyst Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barbir, F.; Yazici, S. Status and development of PEM fuel cell technology. Int. J. Energy Res. 2008, 32, 369–378. [Google Scholar] [CrossRef]
- Barbir, F. PEM Fuel Cells: Theory and Practice; Elsevier Academic Press: San Diego, CA, USA, 2005. [Google Scholar]
- Gasteiger, H.A.; Panels, J.E.; Yan, S.G. Dependence of PEM fuel cell performance on catalyst loading. J. Power Source 2004, 127, 162–171. [Google Scholar] [CrossRef]
- Herranz, J.; Jaouen, F.; Lefèvre, M.; Kramm, U.I.; Proietti, E.; Dodelet, J.-P.; Bogdanoff, P.; Fiechter, S.; Abs-Wurmbach, I.; Bertrand, P.; et al. Unveiling N-protonation and anion-binding effects on Fe/N/C catalysts for O2 reduction in proton-exchange-membrane fuel cells. J. Phys. Chem. C 2011, 115, 16087–16097. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.H.; Kwon, K.; You, D.J.; Pak, C.; Chang, H.; Kim, J.M. Preparation of high loading Pt nanoparticles on ordered mesoporous carbon with a controlled Pt size and its effects on oxygen reduction and methanol oxidation reactions. Electrochim. Acta 2009, 54, 5746–5753. [Google Scholar] [CrossRef]
- Wikander, K.; Ekström, H.; Palmqvist, A.E.C.; Lindbergh, G. On the influence of Pt particle size on the PEMFC cathode performance. Electrochim. Acta 2007, 52, 6848–6855. [Google Scholar] [CrossRef]
- Banham, D.; Feng, F.; Fürstenhaupt, T.; Pei, K.; Ye, S.; Birss, V. Effect of Pt-loaded carbon support nanostructure on oxygen reduction catalysis. J. Power Source 2011, 196, 5438–5445. [Google Scholar] [CrossRef]
- Ambrosio, E.P.; Francia, C.; Manzoli, M.; Penazzi, N.; Spinelli, P. Platinum catalyst supported on mesoporous carbon for PEMFC. Int. J. Hydrogen Energy 2008, 33, 3142–3145. [Google Scholar] [CrossRef]
- Antolini, E. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Sebastián, D.; Ruíz, A.G.; Suelves, I.; Moliner, R.; Lázaro, M.J.; Baglio, V.; Stassi, A.; Aricò, A.S. Enhanced oxygen reduction activity and durability of Pt catalysts supported on carbon nanofibers. Appl. Catal. B 2012, 115–116, 269–275. [Google Scholar] [CrossRef]
- Sebastián, D.; Lázaro, M.J.; Suelves, I.; Moliner, R.; Baglio, V.; Stassi, A.; Aricò, A.S. The influence of carbon nanofiber support properties on the oxygen reduction behavior in proton conducting electrolyte-based direct methanol fuel cells. Int. J. Hydrogen Energy 2012, 37, 6253–6260. [Google Scholar] [CrossRef]
- Zhang, J.; Tang, S.; Liao, L.; Yu, W.; Li, J.; Seland, F.; Haarberg, G.M. Improved catalytic activity of mixed platinum catalysts supported on various carbon nanomaterials. J. Power Source 2014, 267, 706–713. [Google Scholar] [CrossRef]
- Celorrio, V.; Flórez-Montaño, J.; Moliner, R.; Pastor, E.; Lázaro, M.J. Fuel cell performance of Pt electrocatalysts supported on carbon nanocoils. Int. J. Hydrog. Energy 2014, 39, 5371–5377. [Google Scholar] [CrossRef]
- Banham, D.; Feng, F.; Pei, K.; Ye, S.; Birss, V. Effect of carbon support nanostructure on the oxygen reduction activity of Pt/C catalysts. J. Mater. Chem. A 2013, 1, 2812–2820. [Google Scholar] [CrossRef]
- Pei, K.; Banham, D.; Feng, F.; Fürstenhaupt, T.; Ye, S.; Birss, V. Oxygen reduction activity dependence on the mesoporous structure of imprinted carbon supports. Electrochem. Commun. 2010, 12, 1666–1669. [Google Scholar] [CrossRef]
- Serp, P.; Figueiredo, J.L. Carbon Materials for Catalysis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Shao, Y.; Liu, J.; Wang, Y.; Lin, Y. Novel catalyst support materials for PEM fuel cells: Current status and future prospects. J. Mater. Chem. 2009, 19, 46–59. [Google Scholar] [CrossRef]
- Shao, Y.; Yin, G.; Wang, J.; Gao, Y.; Shi, P. Multi-walled carbon nanotubes based Pt electrodes prepared with in situ ion exchange method for oxygen reduction. J. Power Source 2006, 161, 47–53. [Google Scholar] [CrossRef]
- Saha, M.S.; Li, R.; Sun, X.; Ye, S. 3-D composite electrodes for high performance PEM fuel cells composed of Pt supported on nitrogen-doped carbon nanotubes grown on carbon paper. Electrochem. Commun. 2009, 11, 438–441. [Google Scholar] [CrossRef]
- Vijayaraghavan, G.; Stevenson, K.J. Synergistic assembly of dendrimer-templated platinum catalysts on nitrogen-doped carbon nanotube electrodes for oxygen reduction. Langmuir 2007, 23, 5279–5282. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-L.; Chen, L.-C.; Su, M.-C.; Hong, L.-S.; Chyan, O.; Hsu, C.-Y.; Chen, K.-H.; Chang, T.-F.; Chang, L. Ultrafine platinum nanoparticles uniformly dispersed on arrayed CNx nanotubes with high electrochemical activity. Chem. Mater. 2005, 17, 3749–3753. [Google Scholar] [CrossRef]
- Li, Y.-H.; Hung, T.-H.; Chen, C.-W. A first-principles study of nitrogen- and boron-assisted platinum adsorption on carbon nanotubes. Carbon 2009, 47, 850–855. [Google Scholar] [CrossRef]
- An, W.; Turner, C.H. Chemisorption of transition-metal atoms on boron- and nitrogen-doped carbon nanotubes: Energetics and geometric and electronic structures. J. Phys. Chem. C 2009, 113, 7069–7078. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Liu, H.; Li, R.; Sun, X.; Ye, S.; Knights, S. Enhanced stability of Pt electrocatalysts by nitrogen doping in CNTs for PEM fuel cells. Electrochem. Commun. 2009, 11, 2071–2076. [Google Scholar] [CrossRef]
- Joo, S.H.; Choi, S.J.; Oh, I.; Kwak, J.; Liu, Z.; Terasaki, O.; Ryoo, R. Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles. Nature 2001, 412, 169. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.; Berthon-Fabry, S.; Achard, P.; Chatenet, M.; Pradourat, A.; Chainet, E. Highly dispersed platinum on carbon aerogels as supported catalysts for PEM fuel cell-electrodes: Comparison of two different synthesis paths. J. Non-Cryst. Solids 2004, 350, 88–96. [Google Scholar] [CrossRef]
- Ambrosio, E.; Francia, C.; Gerbaldi, C.; Penazzi, N.; Spinelli, P.; Manzoli, M.; Ghiotti, G. Mesoporous carbons as low temperature fuel cell platinum catalyst supports. J. Appl. Electrochem. 2008, 38, 1019–1027. [Google Scholar] [CrossRef]
- Lebedeva, N.P.; Booij, A.S.; Janssen, G.J. Cathodes for proton-exchange-membrane fuel cells based on ordered mesoporous carbon supports. ECS Trans. 2008, 16, 2083–2092. [Google Scholar]
- Su, F.; Poh, C.K.; Tian, Z.; Xu, G.; Koh, G.; Wang, Z.; Liu, Z.; Lin, J. Electrochemical behavior of Pt nanoparticles supported on meso- and microporous carbons for fuel cells. Energy Fuels 2010, 24, 3727–3732. [Google Scholar] [CrossRef]
- Li, Z.; Jaroniec, M. Colloidal imprinting: A novel approach to the synthesis of mesoporous carbons. J. Am. Chem. Soc. 2001, 123, 9208–9209. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Fukuoka, Y.; Sugawara, Y.; Ohara, H.; Ohta, A. Improved preparation process of very-low-platinum-loading electrodes for polymer electrolyte fuel cells. J. Electrochem. Soc. 1998, 145, 3708–3713. [Google Scholar] [CrossRef]
- Uchida, M.; Fukuoka, Y.; Sugawara, Y.; Eda, N.; Ohta, A. Effects of microstructure of carbon support in the catalyst layer on the performance of polymer-electrolyte fuel cells. J. Electrochem. Soc. 1996, 143, 2245–2252. [Google Scholar] [CrossRef]
- Hurt, R.; Krammer, G.; Crawford, G.; Jian, K.; Rulison, C. Polyaromatic assembly mechanisms and structure selection in carbon materials. Chem. Mater. 2002, 14, 4558–4565. [Google Scholar] [CrossRef]
- Joo, J.B.; Kim, P.; Kim, W.; Yi, J. Preparation of Pt supported on mesoporous carbons for the reduction of oxygen in polymer electrolyte membrane fuel cell (PEMFC). J. Electroceram. 2006, 17, 713–718. [Google Scholar] [CrossRef]
- Fang, B.; Kim, J.H.; Yu, J.-S. Colloid-imprinted carbon with superb nanostructure as an efficient cathode electrocatalyst support in proton exchange membrane fuel cell. Electrochem. Commun. 2008, 10, 659–662. [Google Scholar] [CrossRef]
- Banham, D.; Feng, F.; Furstenhaupt, T.; Ye, S.; Birss, V. First time investigation of Pt nanocatalysts deposited inside carbon mesopores of controlled length and diameter. J. Mater. Chem. 2012, 22, 7164–7171. [Google Scholar] [CrossRef]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, 1st ed.; Springer: Dordrecht, The Netherlands, 2006; p. 347. [Google Scholar]
- Haynes, J.M. Pore size analysis according to the kelvin equation. Mater. Construct. 1973, 6, 209–213. [Google Scholar] [CrossRef]
- Mooney, M. The viscosity of a concentrated suspension of spherical particles. J. Colloid Sci. 1951, 6, 162–170. [Google Scholar] [CrossRef]
- Li, Z.; Jaroniec, M. Synthesis and adsorption properties of colloid-imprinted carbons with surface and volume mesoporosity. Chem. Mater. 2003, 15, 1327–1333. [Google Scholar] [CrossRef]
- Gierszal, K.P.; Yoon, S.B.; Yu, J.-S.; Jaroniec, M. Adsorption and structural properties of mesoporous carbons obtained from mesophase pitch and phenol-formaldehyde carbon precursors using porous templates prepared from colloidal silica. J. Mater. Chem. 2006, 16, 2819–2823. [Google Scholar] [CrossRef]
- Chai, G.S.; Yoon, S.B.; Yu, J.-S.; Choi, J.-H.; Sung, Y.-E. Ordered porous carbons with tunable pore sizes as catalyst supports in direct methanol fuel cell. J. Phys. Chem. B 2004, 108, 7074–7079. [Google Scholar] [CrossRef]
- Chen, M.-H.; Jiang, Y.-X.; Chen, S.-R.; Huang, R.; Lin, J.-L.; Chen, S.-P.; Sun, S.-G. Synthesis and durability of highly dispersed platinum nanoparticles supported on ordered mesoporous carbon and their electrocatalytic properties for ethanol oxidation. J. Phys. Chem. C 2010, 114, 19055–19061. [Google Scholar] [CrossRef]
- Kimijima, K.; Hayashi, A.; Umemura, S.; Miyamoto, J.; Sekizawa, K.; Yoshida, T.; Yagi, I. Oxygen reduction reactivity of precisely controlled nanostructured model catalysts. J. Phys. Chem. C 2010, 114, 14675–14683. [Google Scholar] [CrossRef]
- Shan, J.; Pickup, P.G. Characterization of polymer supported catalysts by cyclic voltammetry and rotating disk voltammetry. Electrochim. Acta 2000, 46, 119–125. [Google Scholar] [CrossRef]
- De Levie, R. Electrochemical Response of Porous and Rough Electrodes; John Wiley & Sons: New York, NY, USA, 1967; Volume 6. [Google Scholar]
- Perry, M.L.; Newman, J.; Cairns, E.J. Mass transport in gas-diffusion electrodes: A diagnostic tool for fuel-cell cathodes. J. Electrochem. Soc. 1998, 145, 5–15. [Google Scholar] [CrossRef]
- Banham, D.W.; Soderberg, J.N.; Birss, V.I. Pt/carbon catalyst layer microstructural effects on measured and predicted tafel slopes for the oxygen reduction reaction. J. Phys. Chem. C 2009, 113, 10103–10111. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Srinivasan, S. Fuel Cells: Their Electrochemistry; McGraw-Hill: New York, NY, USA, 1969. [Google Scholar]
- Soderberg, J.N.; Co, A.C.; Sirk, A.H.C.; Birss, V.I. Impact of porous electrode properties on the electrochemical transfer coefficient. J. Phys. Chem. B 2006, 110, 10401–10410. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Hurwitz, H.D.; Bockris, J.O.M. Fundamental equations of electrochemical kinetics at porous gas-diffusion electrodes. J. Chem. Phys. 1967, 46, 3108–3122. [Google Scholar] [CrossRef]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using imod. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, D.N. Dual-axis tomography: An approach with alignment methods that preserve resolution. J. Struct. Biol. 1997, 120, 343–352. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banham, D.; Feng, F.; Fürstenhaupt, T.; Pei, K.; Ye, S.; Birss, V. Novel Mesoporous Carbon Supports for PEMFC Catalysts. Catalysts 2015, 5, 1046-1067. https://doi.org/10.3390/catal5031046
Banham D, Feng F, Fürstenhaupt T, Pei K, Ye S, Birss V. Novel Mesoporous Carbon Supports for PEMFC Catalysts. Catalysts. 2015; 5(3):1046-1067. https://doi.org/10.3390/catal5031046
Chicago/Turabian StyleBanham, Dustin, Fangxia Feng, Tobias Fürstenhaupt, Katie Pei, Siyu Ye, and Viola Birss. 2015. "Novel Mesoporous Carbon Supports for PEMFC Catalysts" Catalysts 5, no. 3: 1046-1067. https://doi.org/10.3390/catal5031046