
Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells



Abstract
:
1. Introduction
2. Strategies to Synthesize Carbon Supported Nanocatalysts
2.1. Heterogeneous Catalysis: The Major Emergency for Supported Nanomaterials Design
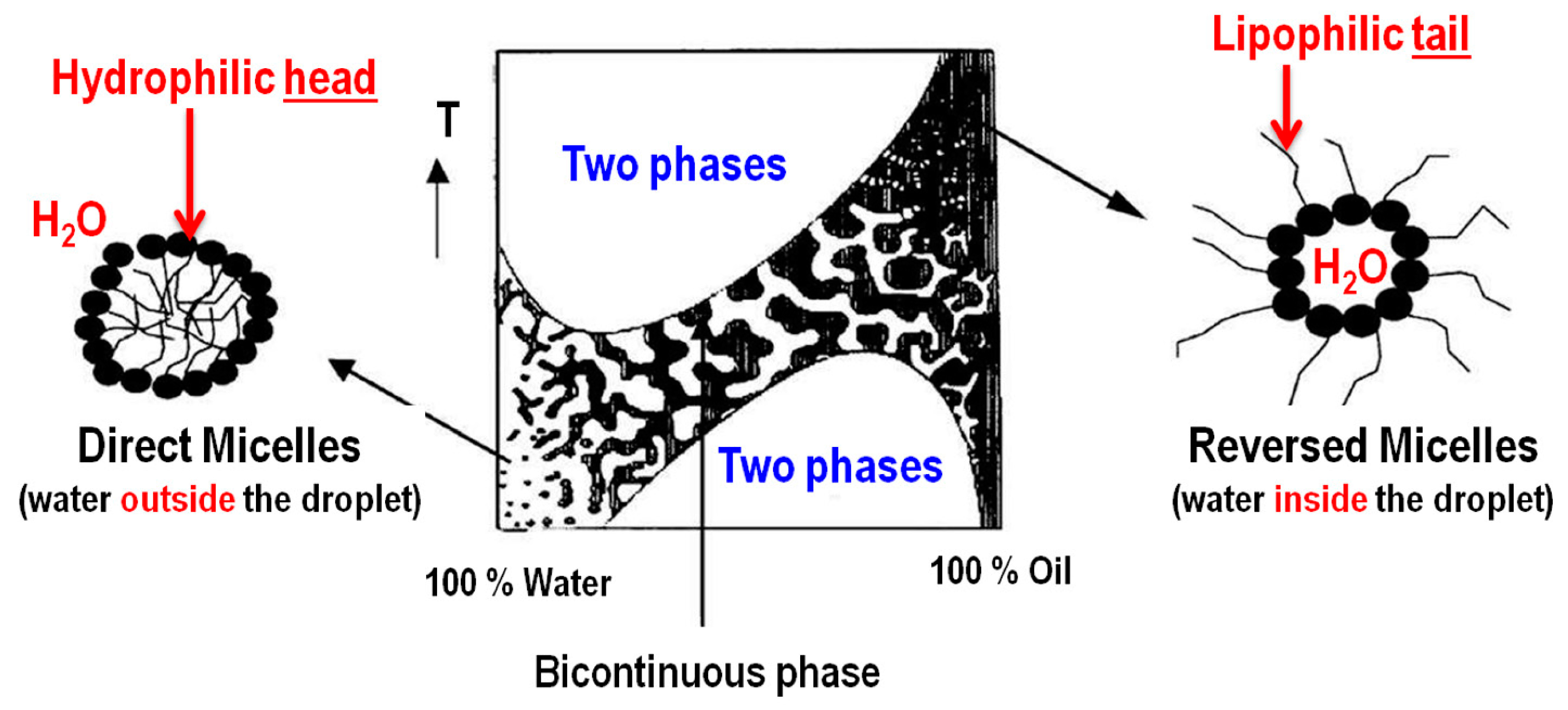
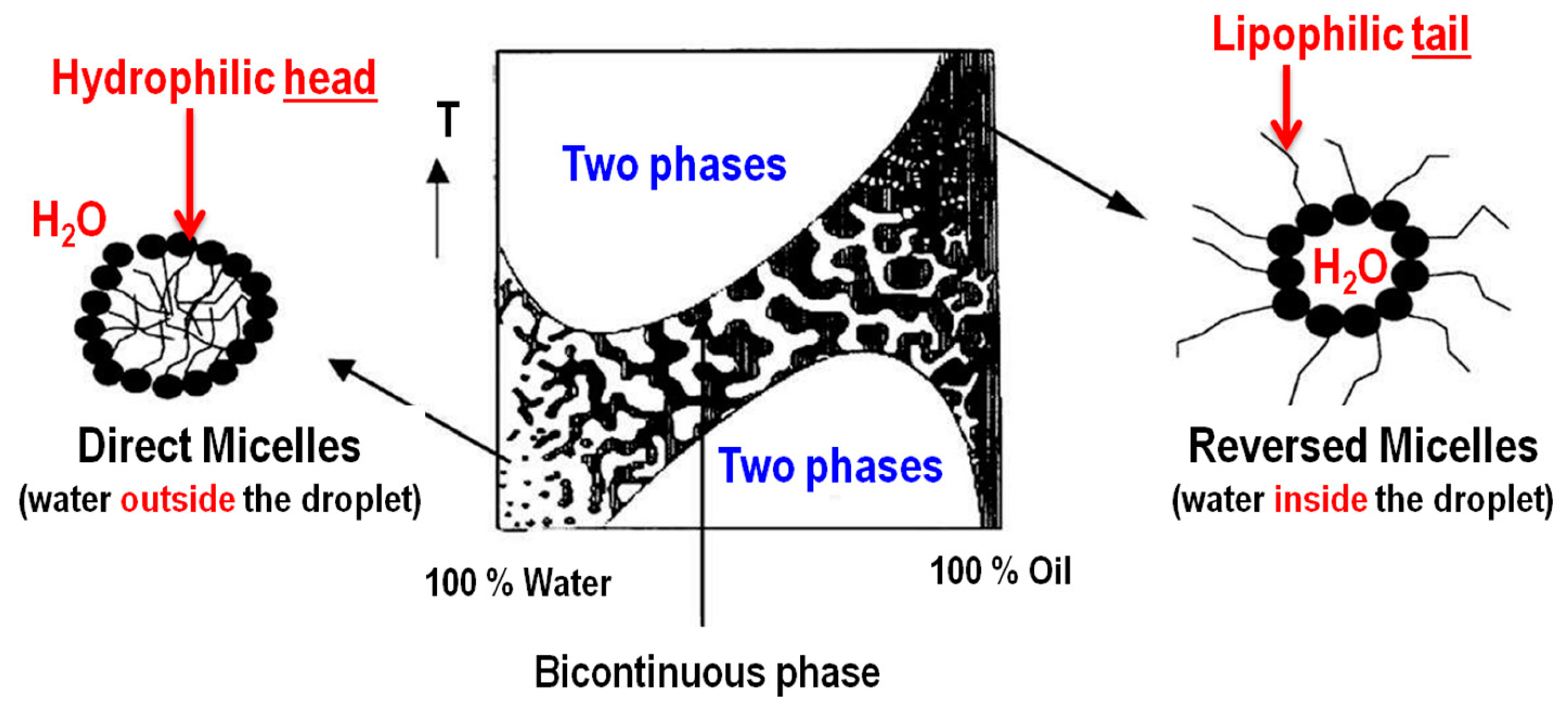
2.2. Water-in-Oil (w/o) Microemulsion Method
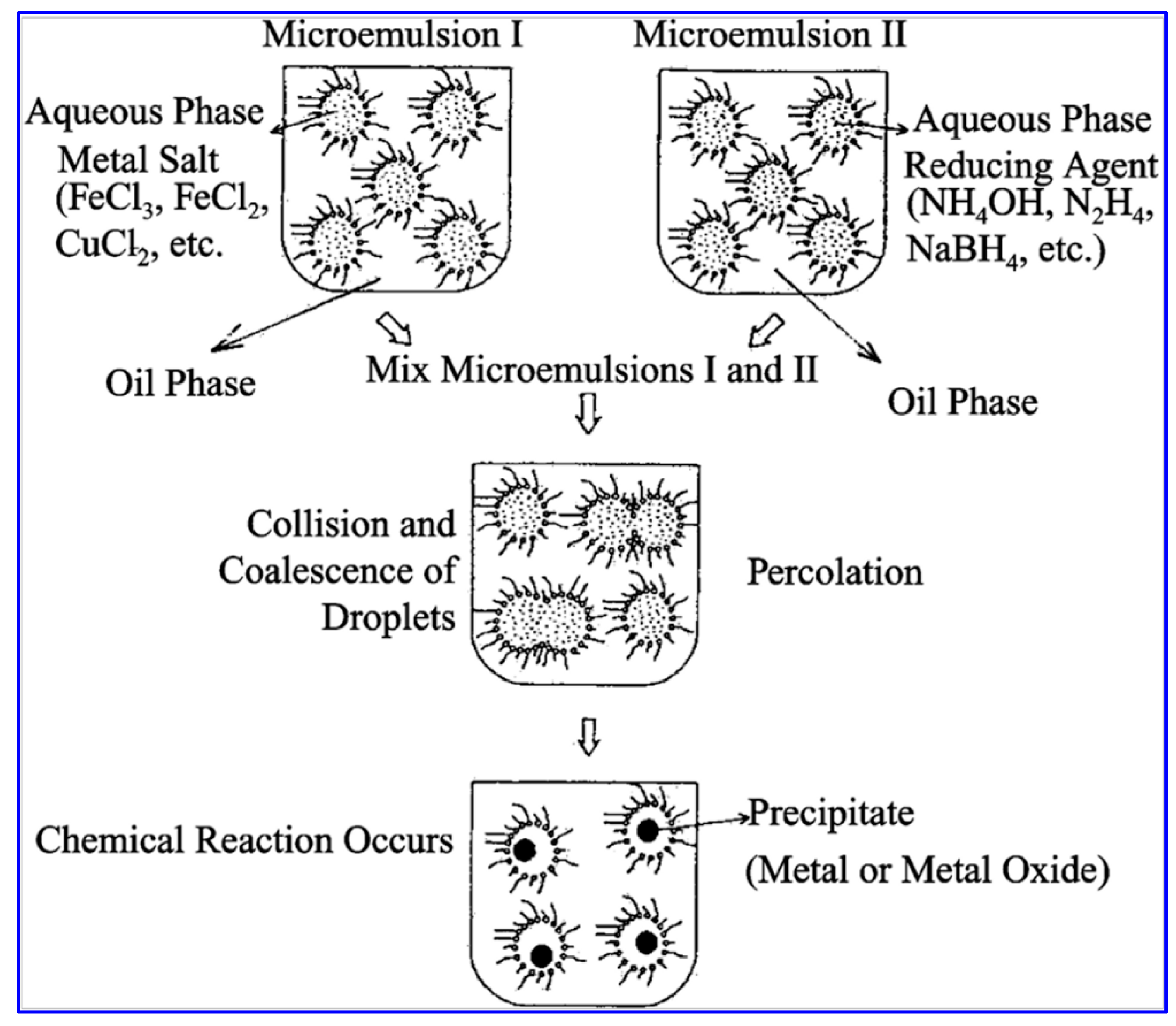
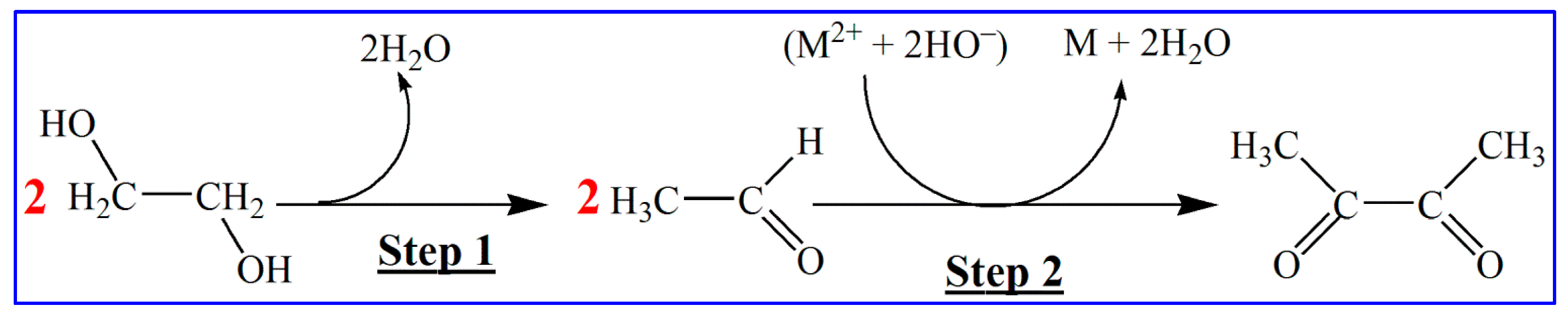
- volume of organic solvent (n-heptane) per synthesis reactor: 27.35 mL [50],

2.3. Polyol Method

2.4. Impregnation-Reduction Process
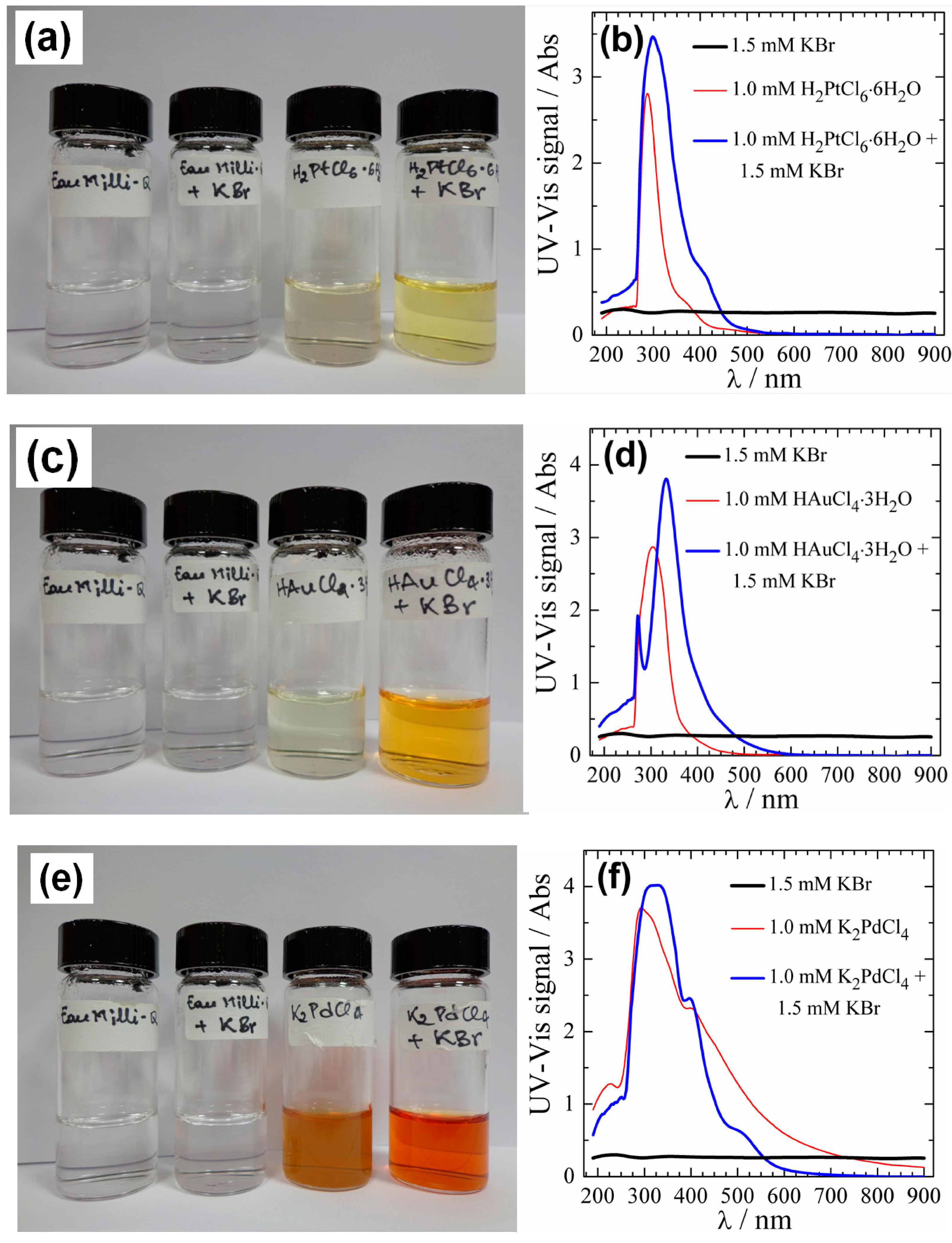
2.5. Bromide Anion Exchange (BAE) Method
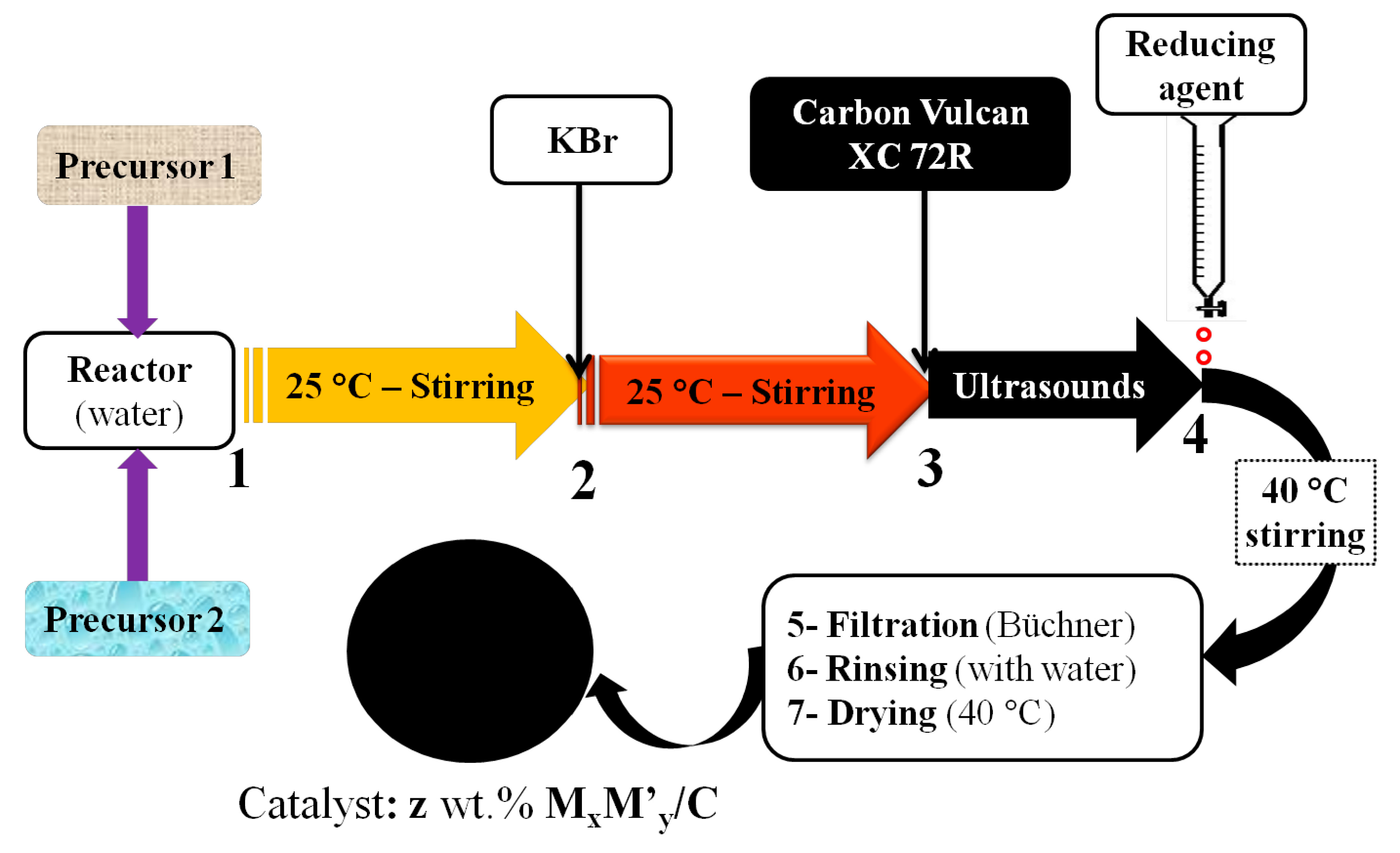

2.6. Other Synthetic Routes
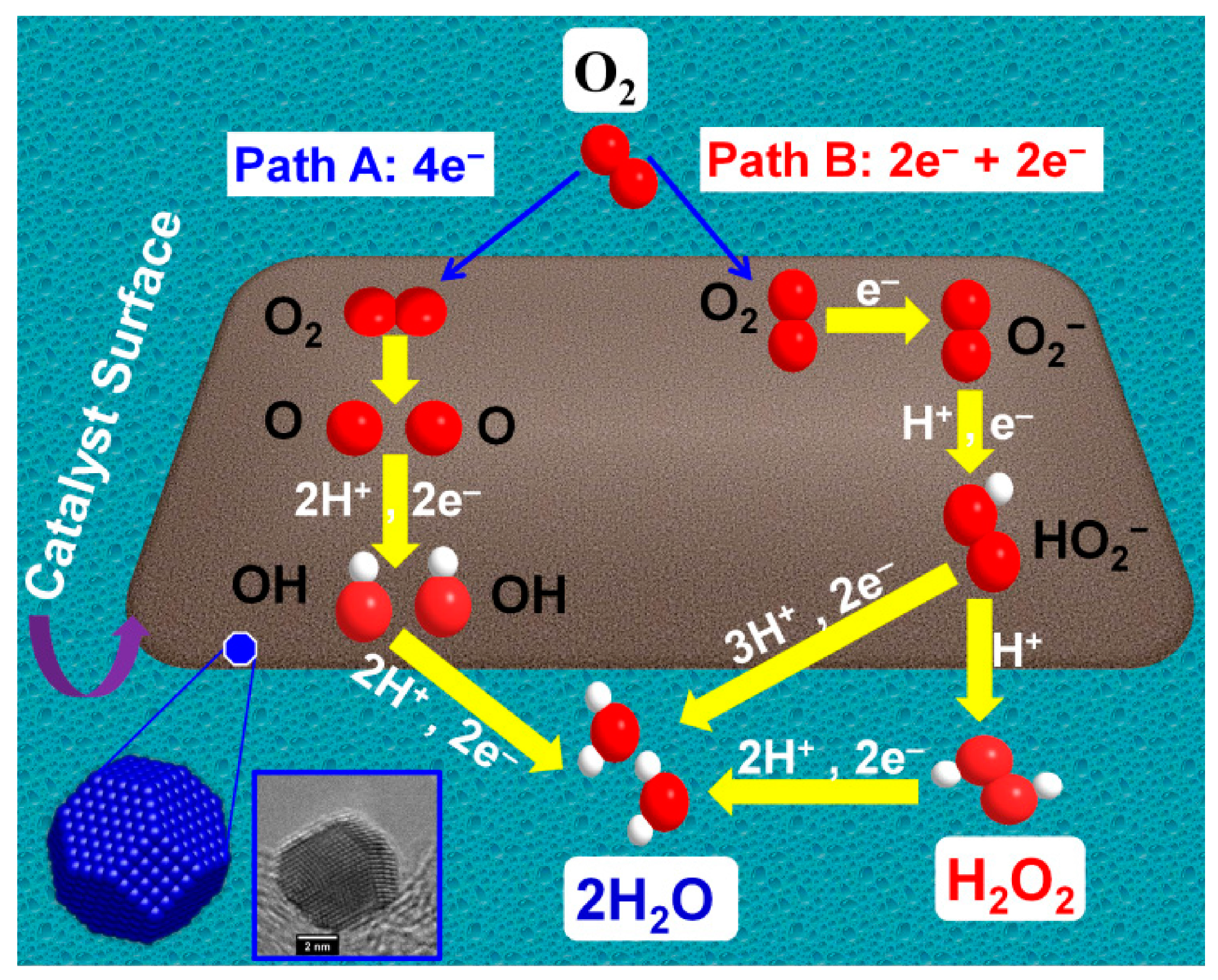
3. Application of Carbon-Supported Nanocatalysts toward the ORR
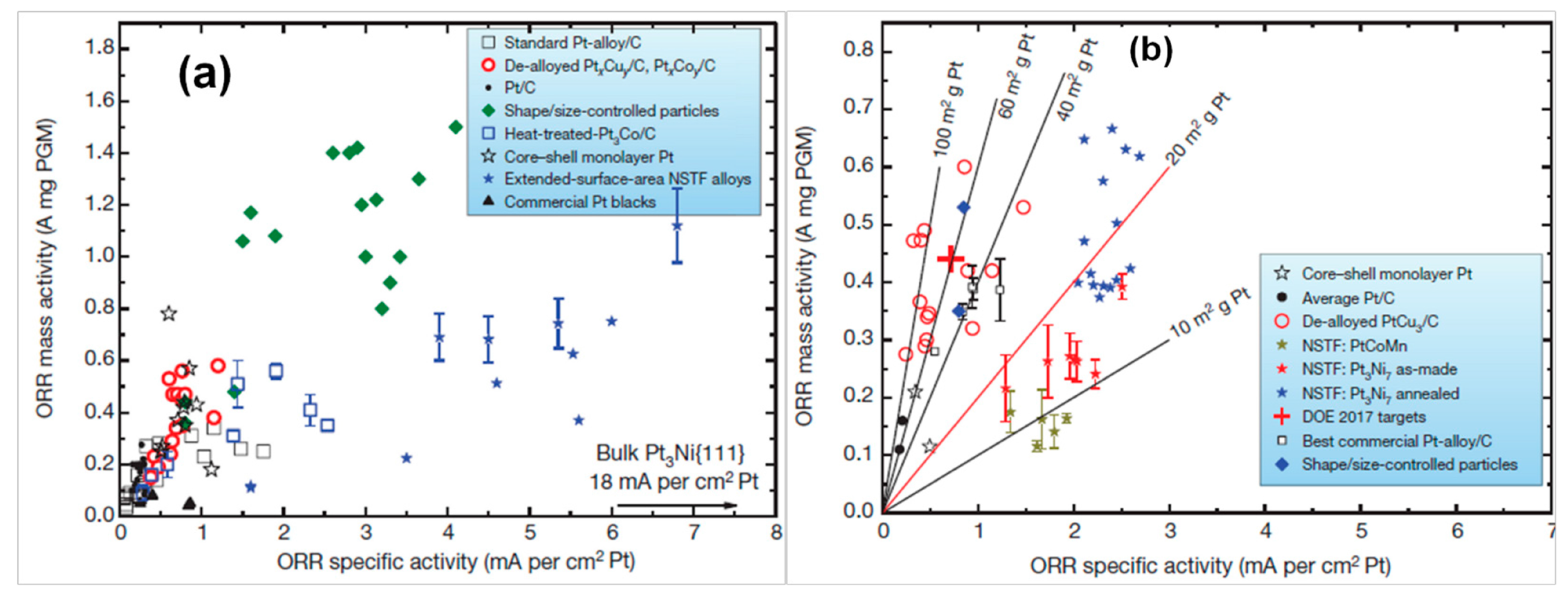
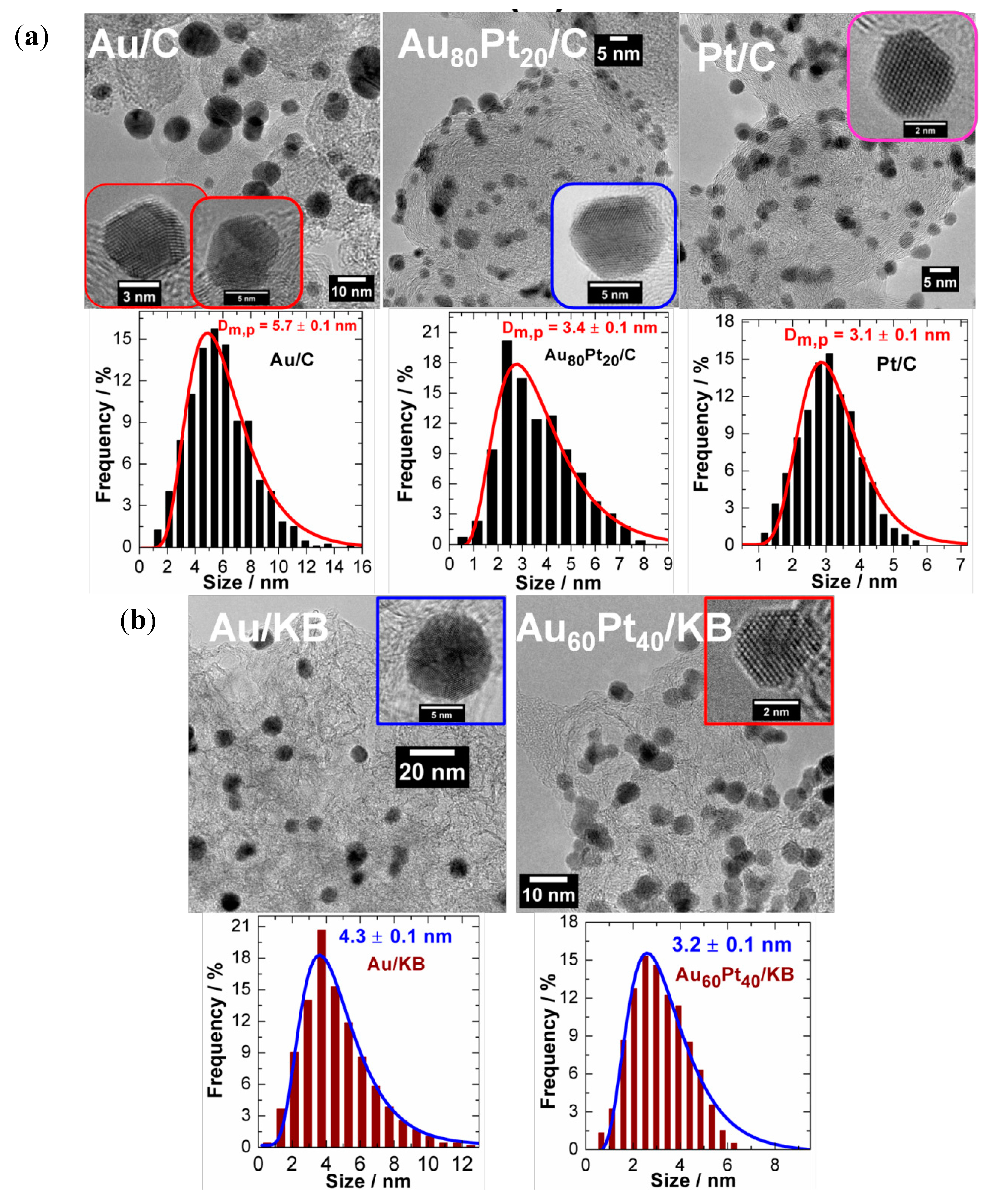
3.1. ORR Activity on Various Carbon Supported Nanoparticles Prepared from w/o Method
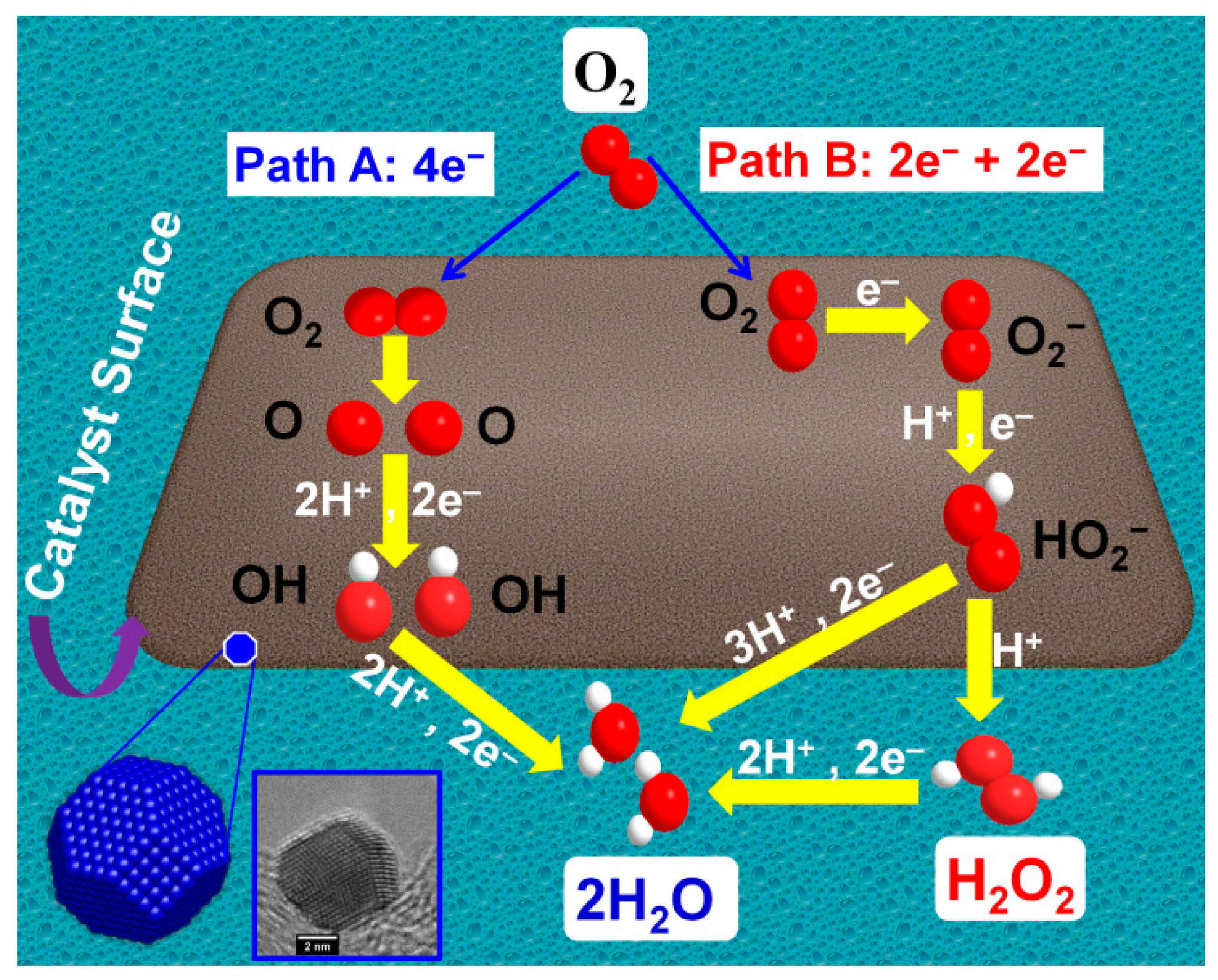
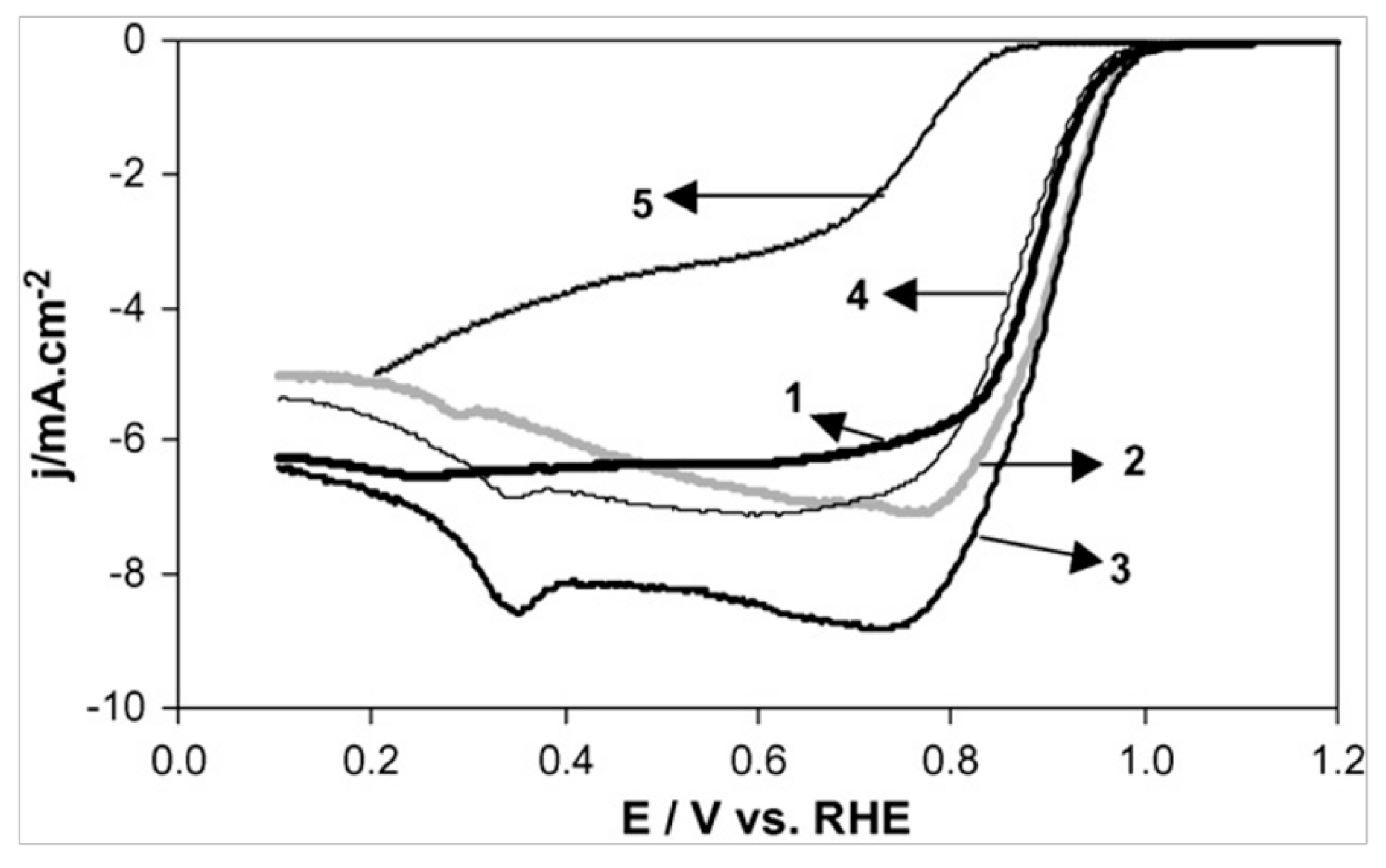
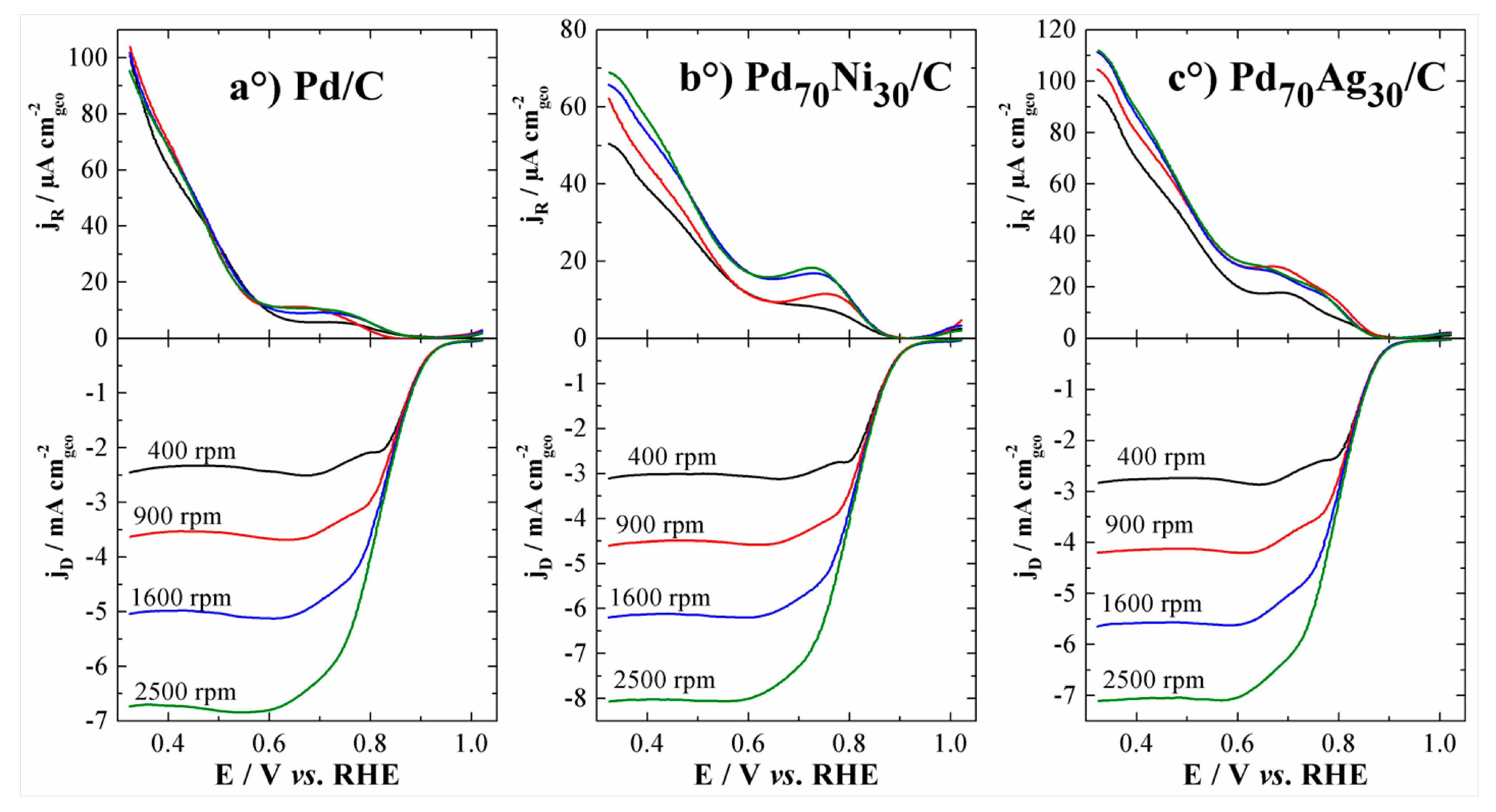
3.2. ORR Activity on Various Carbon Supported Nanoparticles Prepared from the Polyol Method
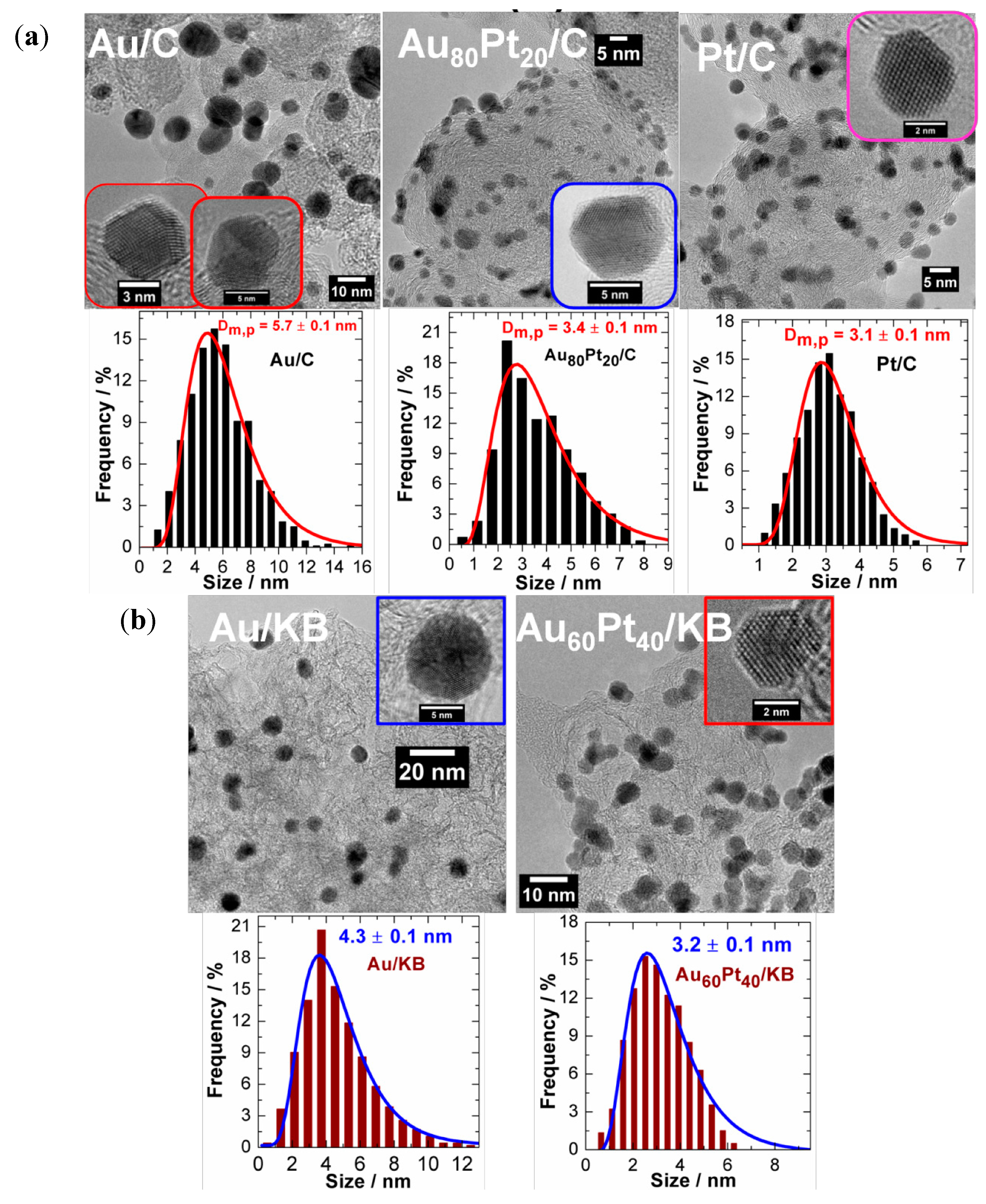
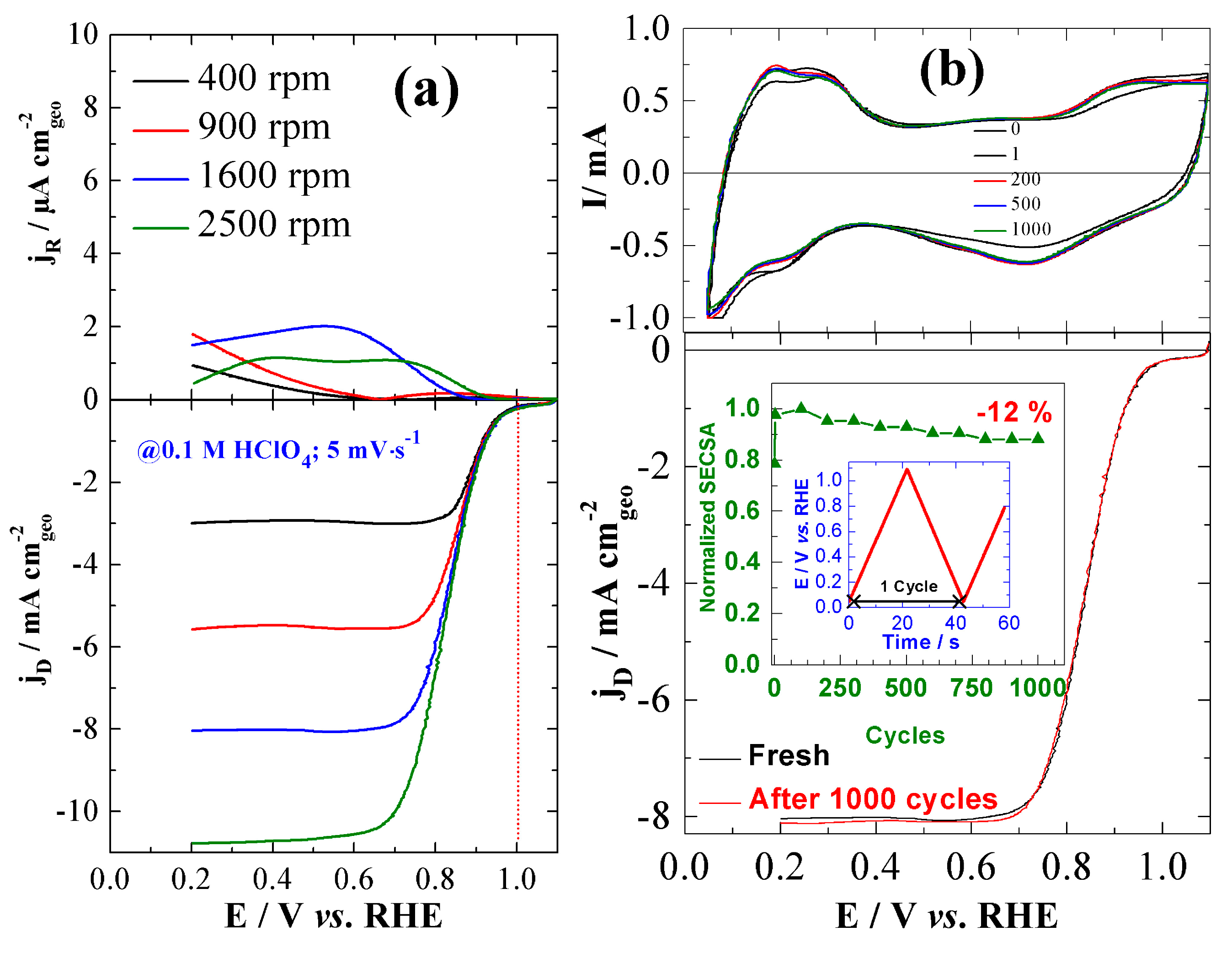
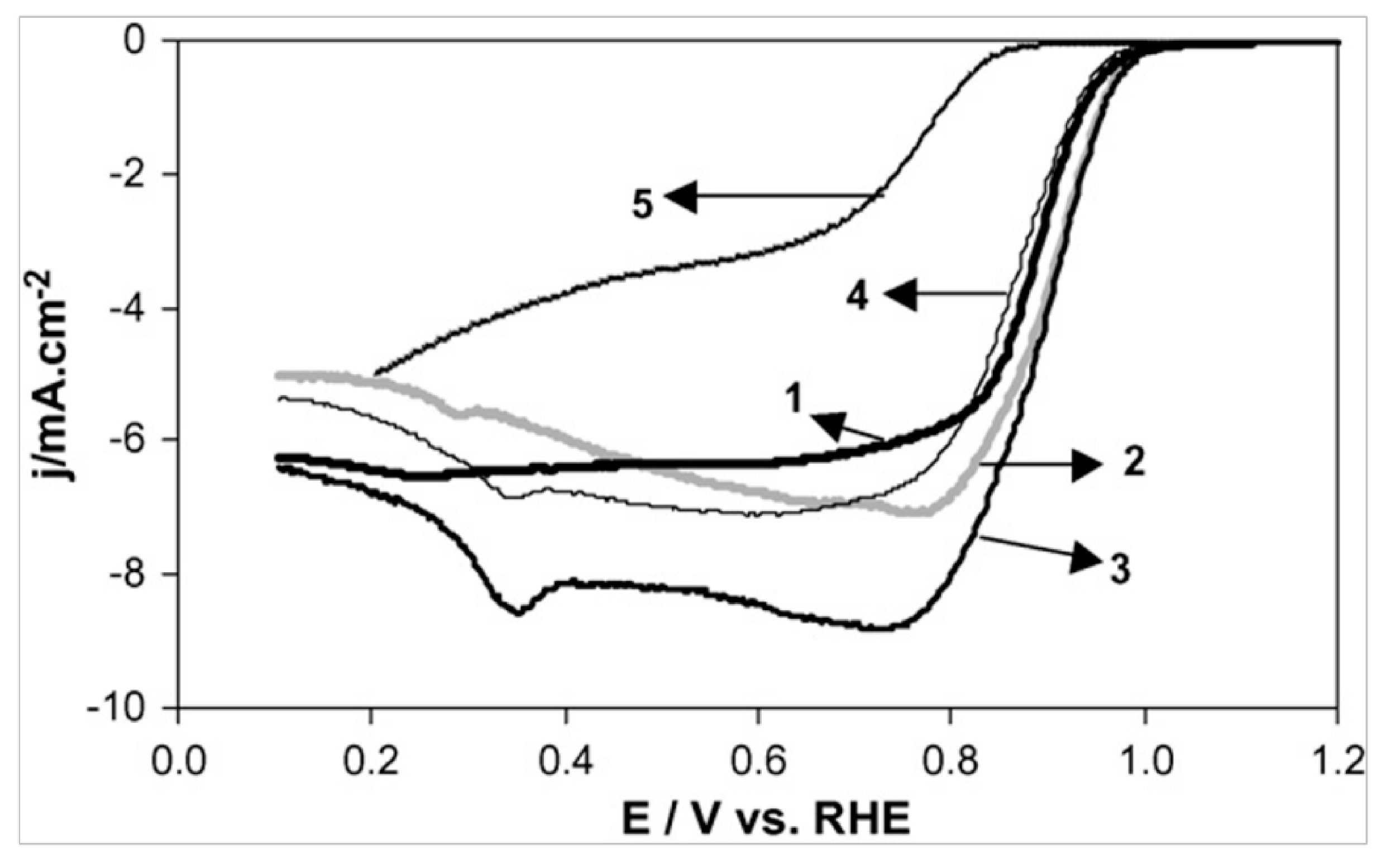
3.3. ORR Activity on Pt/C Electrocatalyst Synthesized from BAE Method


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Electrolyte | OCP | E1/2 | jk (mA cm−2Pt) | Tafel slope (mV dec−1) | j0 (× 10−3 mA cm−2) | nex | Method And Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| At (V vs. RHE) | |||||||||||
| V vs. RHE | 0.90 | 0.85 | Low | High | Low | High | |||||
| 50 wt% Pt/C | 0.2 M NaOH | 1.05 | 0.85 | 0.60 | - | 81 | 126 | 1.2 | 16.8 | 4 | w/o [33] |
| 50 wt% Pt80Bi20/C | 1.05 | 0.87 | 1.14 | - | 62 | 127 | 0.3 | 23.2 | 4 | ||
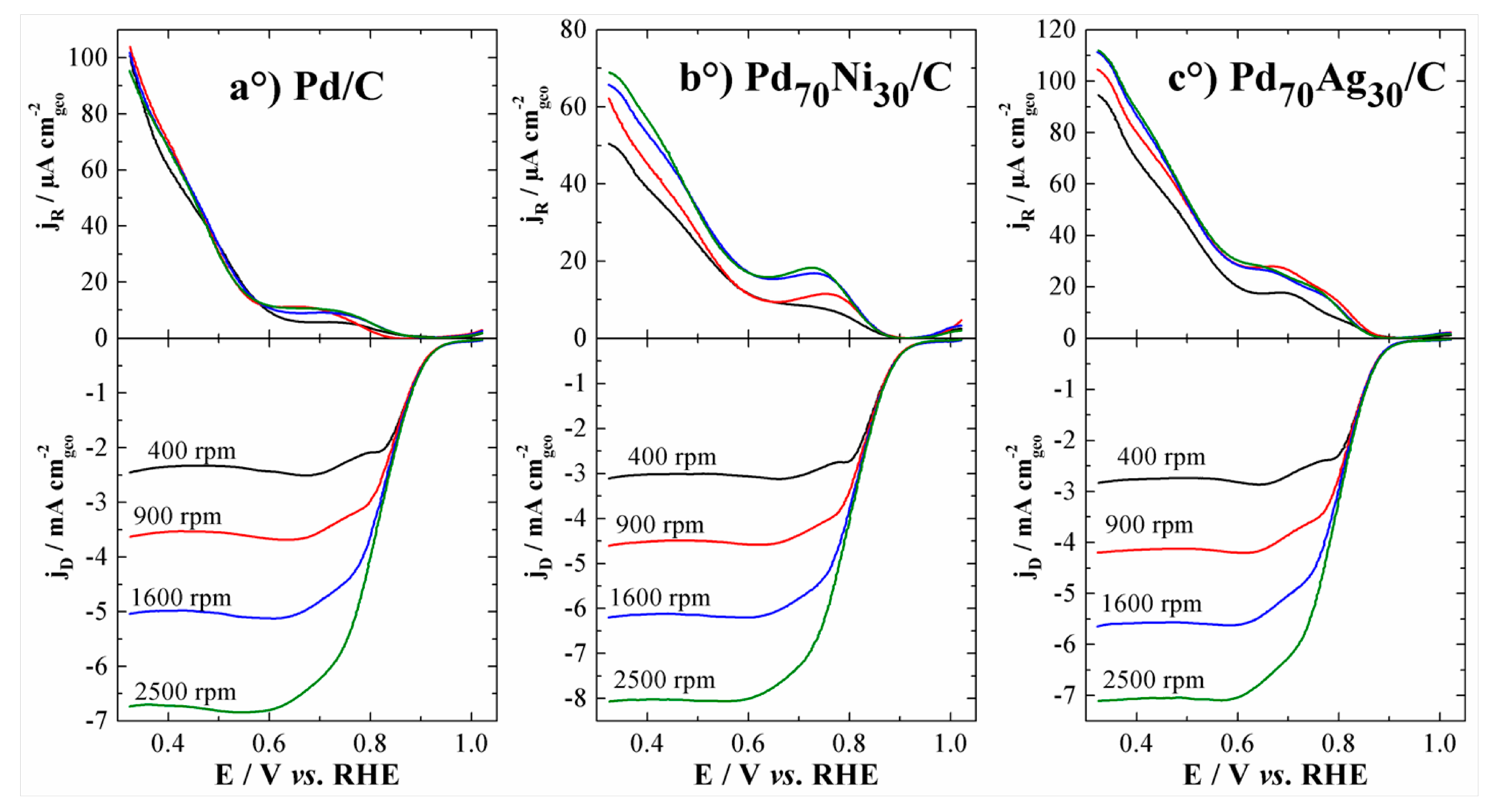
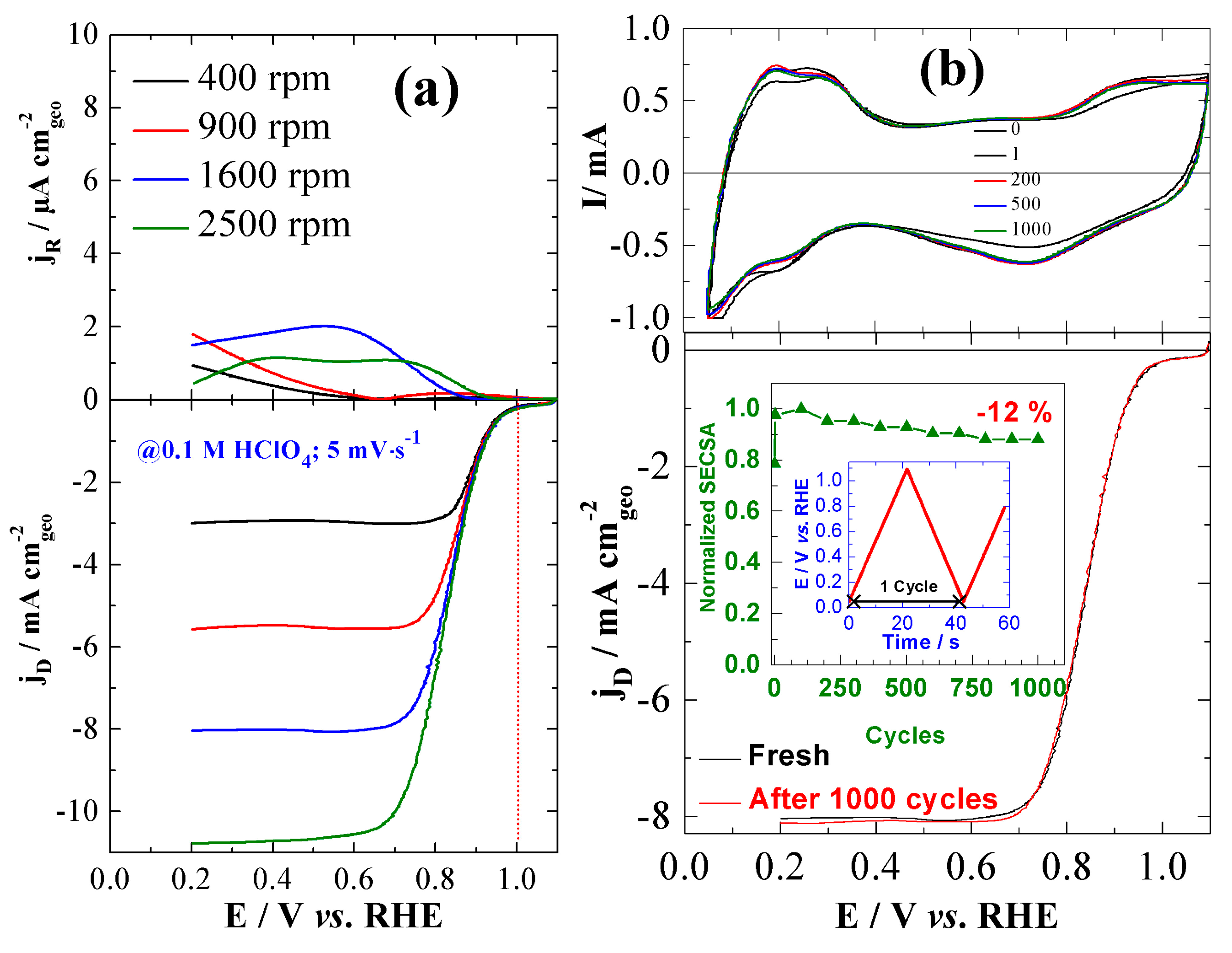
| 20 wt% Pd/C | 1 M NaOH | 0.95 | 0.83 | - | 0.07 | 89 | 162 | - | 11.1 | 3.9 | w/o [56] |
| 20 wt% Pd70Ni30/C | 0.95 | 0.85 | - | 0.06 | 76 | 133 | - | 1.63 | 3.8 | ||
| 20 wt% Pd/C | 0.5 M H2SO4 | 0.80 | 0.70 | - | - | 60 | 120 | - | - | 4 | Polyol [162] |
| 20 wt% Pt/C | 0.925 | 0.84 | - | - | - | - | - | - | 4 | Polyol [152] | |
| 20 wt% Pt-Co/C | 0.980 | 0.85 | - | - | - | - | - | - | 4 | Polyol [152] | |
| 20 wt% Pt-Cr/C | 0.985 | 0.85 | - | - | - | - | - | - | 4 | Polyol [152] | |
| 20 wt% Ag/C | 0.1 M NaOH | 0.95 | 0.80 | - | - | - | 6.3 | - | 157 | 3.9 | w/o [178] |
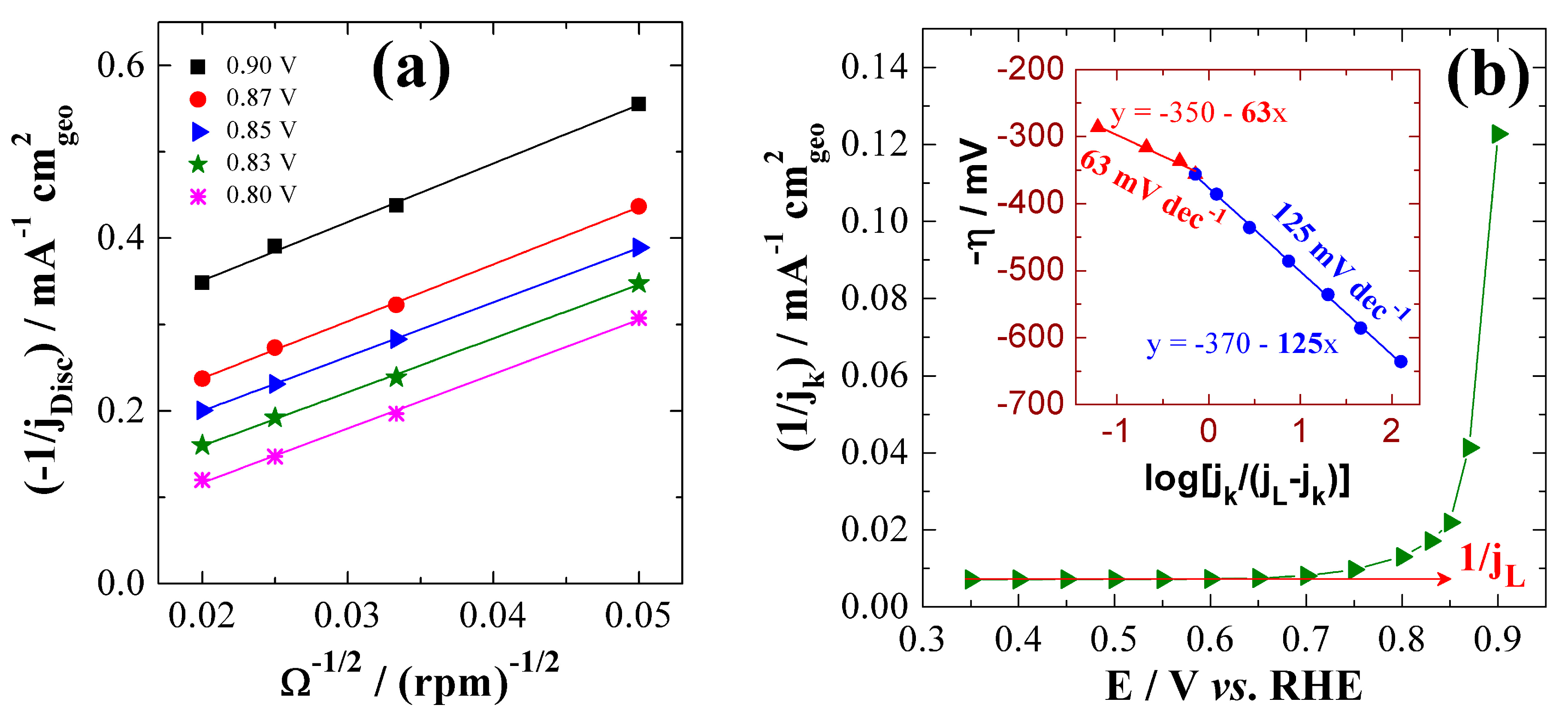
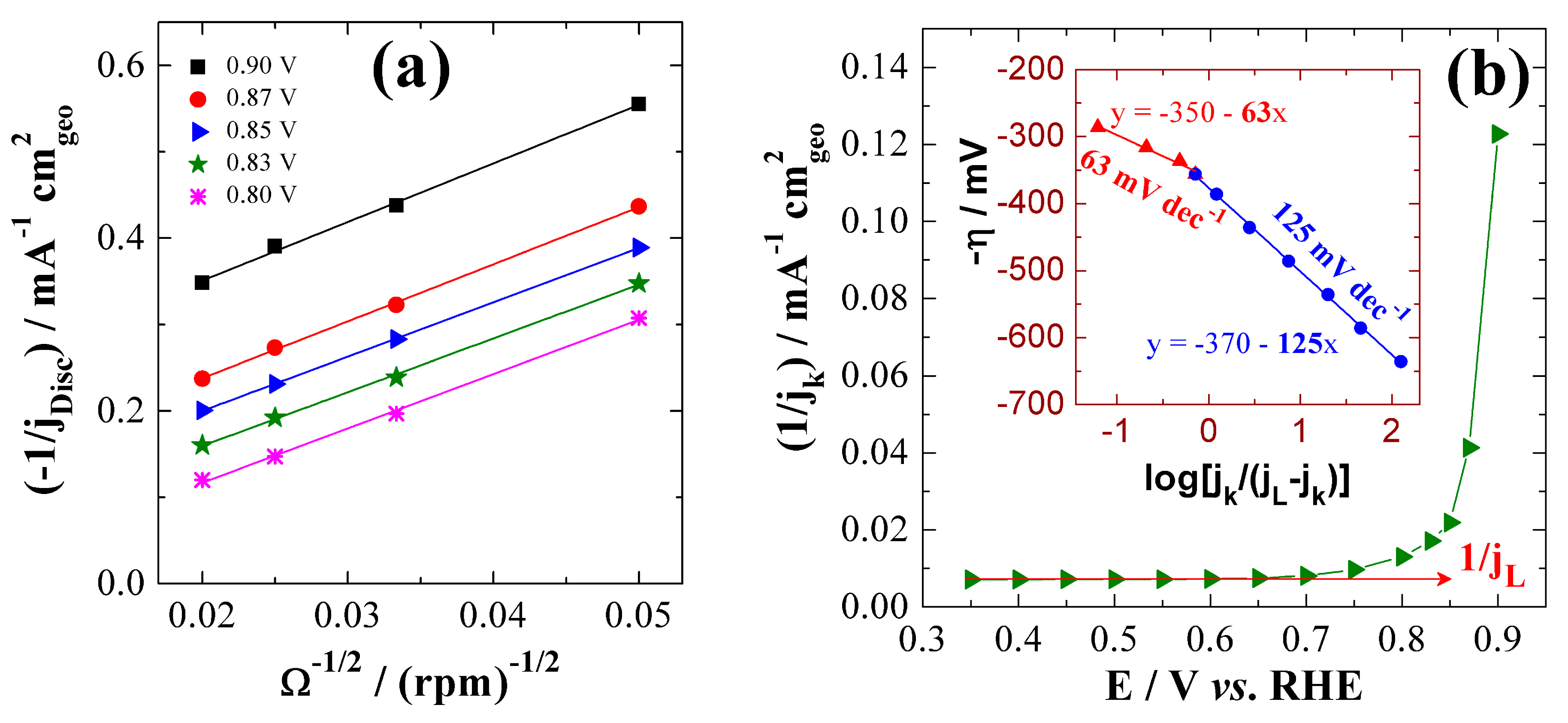
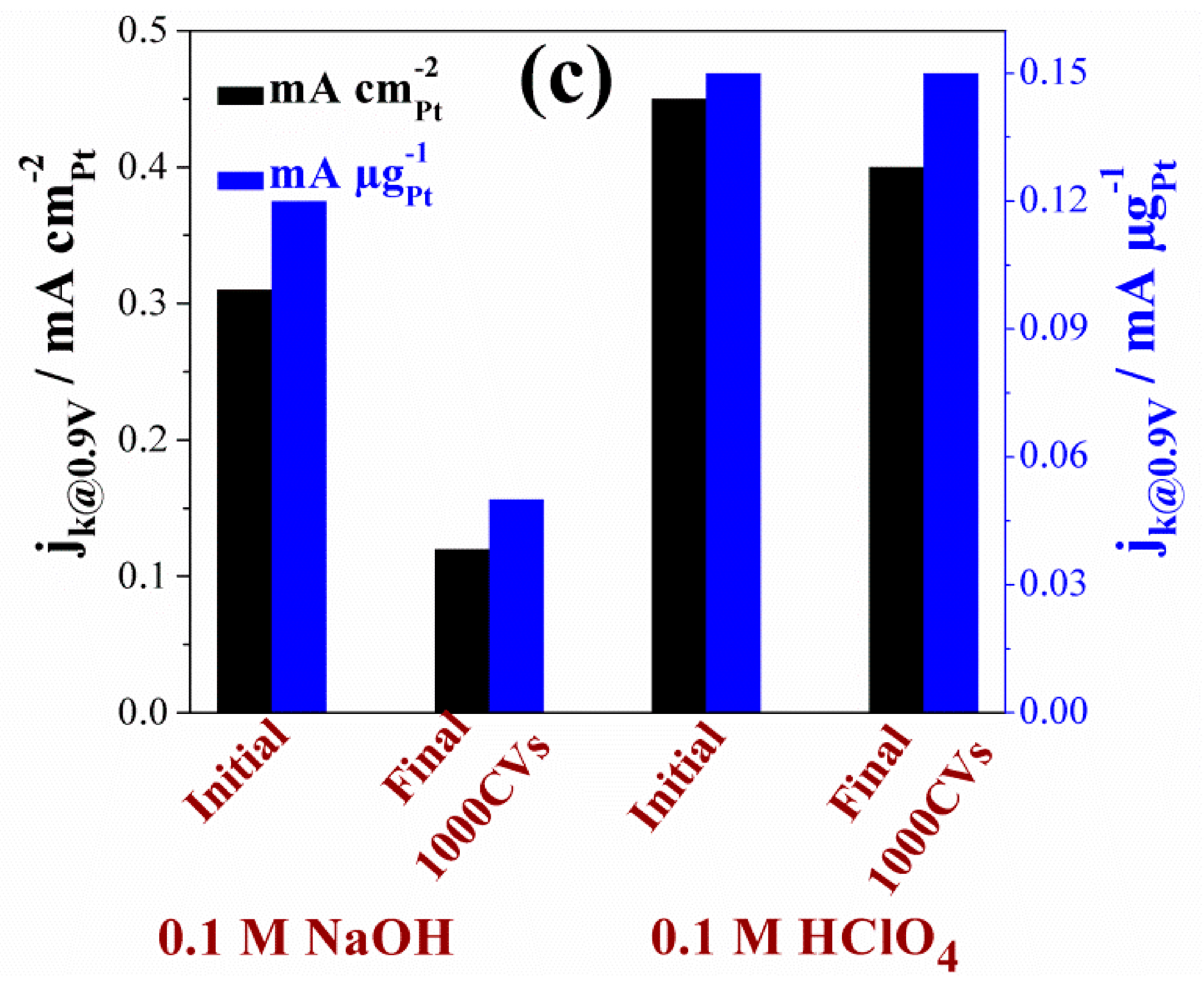
| 20 wt% Pt/C | 0.1 M HClO4 | 1.09 | 0.85 | 0.45 | 1.01 | 67 | 130 | 0.45 | 179 | 4.0 | BAE Here |
| 0.1 M NaOH | 1.08 | 0.85 | 0.31 | 0.77 | 69 | 126 | 0.64 | 122 | 4.0 | ||
4. Summary and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marković, N.M.; Ross, P.N., Jr. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. [Google Scholar] [CrossRef]
- Wieckowski, A.; Savinova, E.R.; Vayenas, C.G. Catalysis and Electrocatalysis at Nanoparticle Surfaces; Marcel Dekker, Inc.: New York, NY, USA, 2003; p. 970. [Google Scholar]
- Stamenkovic, V.R.; Mun, B.S.; Arenz, M.; Mayrhofer, K.J.J.; Lucas, C.A.; Wang, G.; Ross, P.N.; Markovic, N.M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Markovic, N.M.; Gasteiger, H.A.; Ross, P.N. Oxygen Reduction on Platinum Low-Index Single-Crystal Surfaces in Sulfuric Acid Solution: Rotating Ring-Pt(hkl) Disk Studies. J. Phys. Chem. 1995, 99, 3411–3415. [Google Scholar] [CrossRef]
- Nesselberger, M.; Ashton, S.; Meier, J.C.; Katsounaros, I.; Mayrhofer, K.J.J.; Arenz, M. The Particle Size Effect on the Oxygen Reduction Reaction Activity of Pt Catalysts: Influence of Electrolyte and Relation to Single Crystal Models. J. Am. Chem. Soc. 2011, 133, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Peles, A.; Shoemaker, K. Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity. Nano Lett. 2011, 11, 3714–3719. [Google Scholar] [CrossRef] [PubMed]
- Anastasopoulos, A.; Davies, J.C.; Hannah, L.; Hayden, B.E.; Lee, C.E.; Milhano, C.; Mormiche, C.; Offin, L. The Particle Size Dependence of the Oxygen Reduction Reaction for Carbon-Supported Platinum and Palladium. ChemSusChem 2013, 6, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Fernandez, P.; Masini, F.; McCarthy, D.N.; Strebel, C.E.; Friebel, D.; Deiana, D.; Malacrida, P.; Nierhoff, A.; Bodin, A.; Wise, A.M.; et al. Mass-selected nanoparticles of PtxY as model catalysts for oxygen electroreduction. Nat. Chem. 2014, 6, 732–738. [Google Scholar] [PubMed]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Gan, L.; Li, H.-H.; Yu, S.-H.; Heggen, M.; Strasser, P. Octahedral PtNi Nanoparticle Catalysts: Exceptional Oxygen Reduction Activity by Tuning the Alloy Particle Surface Composition. Nano Lett. 2012, 12, 5885–5889. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kang, Y.; Huo, Z.; Zhu, Z.; Huang, W.; Xin, H.L.; Snyder, J.D.; Li, D.; Herron, J.A.; Mavrikakis, M.; et al. Highly Crystalline Multimetallic Nanoframes with Three-Dimensional Electrocatalytic Surfaces. Science 2014, 343, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Corti, H.; Gonzalez, E.R. Direct Alcohol Fuel Cells: Materials, Performance, Durability and Applications; Springer: Dordrecht, The Netherlands, 2014; p. 370. [Google Scholar]
- Huang, X.; Zhao, Z.; Chen, Y.; Zhu, E.; Li, M.; Duan, X.; Huang, Y. A Rational Design of Carbon-Supported Dispersive Pt-Based Octahedra as Efficient Oxygen Reduction Reaction Catalysts. Energy Environ. Sci. 2014, 7, 2957–2962. [Google Scholar] [CrossRef]
- Wang, D.; Xin, H.L.; Hovden, R.; Wang, H.; Yu, Y.; Muller, D.A.; DiSalvo, F.J.; Abruña, H.D. Structurally ordered intermetallic platinum–cobalt core–shell nanoparticles with enhanced activity and stability as oxygen reduction electrocatalysts. Nat. Mater. 2013, 12, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.N.; Radmilovic, V.; Markovic, N.M. Physical and Electrochemical Characterization of Bimetallic Nanoparticle Electrocatalysts. In Catalysis and Electrocatalysis at Nanoparticle Surfaces; Wieckowski, A., Savinova, E.R., Vayenas, C.G., Eds.; CRC Press: New York, NY, USA, 2003; pp. 311–342. [Google Scholar]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- De Volder, M.F. L.; Tawfick, S.H.; Baughman, R.H.; Hart, A.J. Carbon Nanotubes: Present and Future Commercial Applications. Science 2013, 339, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.; Jacques, D.; Qian, D.; Rantell, T. Multiwall Carbon Nanotubes: Synthesis and Application. Acc. Chem. Res. 2002, 35, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current Progress on the Chemical Modification of Carbon Nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef] [PubMed]
- Che, A.-F.; Germain, V.; Cretin, M.; Cornu, D.; Innocent, C.; Tingry, S. Fabrication of free-standing electrospun carbon nanofibers as efficient electrode materials for bioelectrocatalysis. New J. Chem. 2011, 35, 2848–2853. [Google Scholar] [CrossRef]
- Andersen, S.M.; Borghei, M.; Lund, P.; Elina, Y.-R.; Pasanen, A.; Kauppinen, E.; Ruiz, V.; Kauranen, P.; Skou, E.M. Durability of carbon nanofiber (CNF) & carbon nanotube (CNT) as catalyst support for Proton Exchange Membrane Fuel Cells. Solid State Ionics 2013, 231, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Shulaker, M.M.; Hills, G.; Patil, N.; Wei, H.; Chen, H.-Y.; Wong, H.S.P.; Mitra, S. Carbon nanotube computer. Nature 2013, 501, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Viry, L.; Maugey, M.; Poulin, P.; Mano, N. Engineering hybrid nanotube wires for high-power biofuel cells. Nat. Commun. 2010, 1, 3. [Google Scholar] [PubMed]
- Zebda, A.; Gondran, C.; le Goff, A.; Holzinger, M.; Cinquin, P.; Cosnier, S. Mediatorless high-power glucose biofuel cells based on compressed carbon nanotube-enzyme electrodes. Nat. Commun. 2011, 2, 370. [Google Scholar] [CrossRef] [PubMed]
- Sinfelt, J.H.; Lam, Y.L.; Cusumano, J.A.; Barnett, A.E. Nature of ruthenium-copper catalysts. J. Catal. 1976, 42, 227–237. [Google Scholar] [CrossRef]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Scherer, G.G.; Kötz, R.; Wokaun, A. Nanoparticles in Energy Technology: Examples from Electrochemistry and Catalysis. Angew. Chem. Int. Ed. 2005, 44, 2190–2209. [Google Scholar] [CrossRef]
- Katsounaros, I.; Cherevko, S.; Zeradjanin, A.R.; Mayrhofer, K.J.J. Oxygen Electrochemistry as a Cornerstone for Sustainable Energy Conversion. Angew. Chem. Int. Ed. 2014, 53, 102–121. [Google Scholar] [CrossRef]
- Hoar, T.; Schulman, J. Transparent water-in-oil dispersions: the oleopathic hydro-micelle. Nature 1943, 152, 102–103. [Google Scholar] [CrossRef]
- Boutonnet, M.; Kizling, J.; Stenius, P.; Maire, G. The preparation of monodisperse colloidal metal particles from microemulsions. Colloids Surf. 1982, 5, 209–225. [Google Scholar] [CrossRef]
- Schwuger, M.-J.; Stickdorn, K.; Schomaecker, R. Microemulsions in Technical Processes. Chem. Rev. 1995, 95, 849–864. [Google Scholar] [CrossRef]
- Bock, C.; Halvorsen, H.; MacDougall, B. Catalyst Synthesis Techniques. In PEM Fuel Cell Electrocatalysts and Catalyst Layers; Zhang, J., Ed.; Springer-Verlag: London, UK, 2008; pp. 447–485. [Google Scholar]
- Demarconnay, L.; Coutanceau, C.; Léger, J.M. Study of the oxygen electroreduction at nanostructured PtBi catalysts in alkaline medium. Electrochim. Acta 2008, 53, 3232–3241. [Google Scholar] [CrossRef]
- Simões, M.; Baranton, S.; Coutanceau, C. Electrooxidation of Sodium Borohydride at Pd, Au, and PdxAu1−x Carbon-Supported Nanocatalysts. J. Phys. Chem. C 2009, 113, 13369–13376. [Google Scholar] [CrossRef]
- Roychowdhury, C.; Matsumoto, F.; Mutolo, P.F.; Abruña, H.D.; DiSalvo, F.J. Synthesis, Characterization, and Electrocatalytic Activity of PtBi Nanoparticles Prepared by the Polyol Process. Chem. Mater. 2005, 17, 5871–5876. [Google Scholar] [CrossRef]
- Holade, Y.; Morais, C.; Arrii-Clacens, S.; Servat, K.; Napporn, T.W.; Kokoh, K.B. New Preparation of PdNi/C and PdAg/C Nanocatalysts for Glycerol Electrooxidation in Alkaline Medium. Electrocatalysis 2013, 4, 167–178. [Google Scholar] [CrossRef]
- Holade, Y.; Morais, C.; Napporn, T.W.; Servat, K.; Kokoh, K.B. Electrochemical Behavior of Organics Oxidation on Palladium-Based Nanocatalysts Synthesized from Bromide Anion Exchange. ECS Trans. 2014, 58, 25–35. [Google Scholar] [CrossRef]
- Tonda-Mikiela, P.; Napporn, T.W.; Morais, C.; Servat, K.; Chen, A.; Kokoh, K.B. Synthesis of Gold-Platinum Nanomaterials Using Bromide Anion Exchange-Synergistic Electroactivity toward CO and Glucose Oxidation. J. Electrochem. Soc. 2012, 159, H828–H833. [Google Scholar] [CrossRef]
- Lahmani, M.; Bréchignac, C.; Houdy, P. Les Nanosciences: 2. Nanomatériaux et Nanochimie, 2nd ed.; Belin: Paris, France, 2012; p. 732. (In French) [Google Scholar]
- Schulman, J.H.; Stoeckenius, W.; Prince, L.M. Mechanism of Formation and Structure of Micro Emulsions by Electron Microscopy. J. Phys. Chem. 1959, 63, 1677–1680. [Google Scholar] [CrossRef]
- Schulman, J.H.; Friend, J.A. Light scattering investigation of the structure of transparent oil-water disperse systems. II. J. Colloid Sci. 1949, 4, 497–509. [Google Scholar] [CrossRef]
- Clausse, M.; Peyrelasse, J.; Heil, J.; Boned, C.; Lagourette, B. Bicontinuous structure zones in microemulsions. Nature 1981, 293, 636–638. [Google Scholar] [CrossRef]
- Danielsson, I.; Lindman, B. The definition of microemulsion. Colloids Surf. 1981, 3, 391–392. [Google Scholar] [CrossRef]
- Eriksson, S.; Nylén, U.; Rojas, S.; Boutonnet, M. Preparation of catalysts from microemulsions and their applications in heterogeneous catalysis. Appl. Catal. A 2004, 265, 207–219. [Google Scholar] [CrossRef]
- Capek, I. Preparation of metal nanoparticles in water-in-oil (w/o) microemulsions. Adv. Colloid Int. Sci. 2004, 110, 49–74. [Google Scholar] [CrossRef]
- Ingelsten, H.H.; Bagwe, R.; Palmqvist, A.; Skoglundh, M.; Svanberg, C.; Holmberg, K.; Shah, D.O. Kinetics of the Formation of Nano-Sized Platinum Particles in Water-in-Oil Microemulsions. J. Colloid Int. Sci. 2001, 241, 104–111. [Google Scholar] [CrossRef]
- Solla-Gullón, J.; Rodes, A.; Montiel, V.; Aldaz, A.; Clavilier, J. Electrochemical characterisation of platinum–palladium nanoparticles prepared in a water-in-oil microemulsion. J. Electroanal. Chem. 2003, 554–555, 273–284. [Google Scholar]
- Solla-Gullón, J.; Vidal-Iglesias, F.J.; Montiel, V.; Aldaz, A. Electrochemical characterization of platinum–ruthenium nanoparticles prepared by water-in-oil microemulsion. Electrochim. Acta 2004, 49, 5079–5088. [Google Scholar] [CrossRef]
- Habrioux, A. Préparation et caractérisation de nanoparticules à base d’or et de platine pour l’anode d’une biopile glucose/dioxygène. Ph.D. Thesis, University of Poitiers, France, October 2009. [Google Scholar]
- Simões, M. Développement d’électrocatalyseurs anodiques plurimétalliques nanostructurés pour une application en pile à combustible à membrane alcaline solide (SAMFC). Ph.D. Thesis, University of Poitiers, France, March 2011. [Google Scholar]
- Liu, H.; Song, C.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D.P. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Habrioux, A.; Diabaté, D.; Rousseau, J.; Napporn, T.; Servat, K.; Guétaz, L.; Trokourey, A.; Kokoh, K.B. Electrocatalytic Activity of Supported Au–Pt Nanoparticles for CO Oxidation and O2 Reduction in Alkaline Medium. Electrocatalysis 2010, 1, 51–59. [Google Scholar] [CrossRef]
- Habrioux, A.; Sibert, E.; Servat, K.; Vogel, W.; Kokoh, K.B.; Alonso-Vante, N. Activity of Platinum−Gold Alloys for Glucose Electrooxidation in Biofuel Cells. J. Phys. Chem. B 2007, 111, 10329–10333. [Google Scholar] [CrossRef] [PubMed]
- Habrioux, A.; Vogel, W.; Guinel, M.; Guetaz, L.; Servat, K.; Kokoh, B.; Alonso-Vante, N. Structural and electrochemical studies of Au-Pt nanoalloys. Phys. Chem. Chem. Phys. 2009, 11, 3573–3579. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-L.; Chen, D.-H.; Huang, T.-C. Preparation of Au/Pt Bimetallic Nanoparticles in Water-in-Oil Microemulsions. Chem. Mater. 2001, 13, 599–606. [Google Scholar] [CrossRef]
- Diabaté, D.; Napporn, T.W.; Servat, K.; Habrioux, A.; Arrii-Clacens, S.; Trokourey, A.; Kokoh, K.B. Kinetic Study of Oxygen Reduction Reaction on Carbon Supported Pd-Based Nanomaterials in Alkaline Medium. J. Electrochem. Soc. 2013, 160, H302–H308. [Google Scholar] [CrossRef]
- Holade, Y.; Morais, C.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Enhancing the available specific surface area of carbon supports to boost the electroactivity of nanostructured Pt catalysts. Phys. Chem. Chem. Phys. 2014, 16, 25609–25620. [Google Scholar] [CrossRef] [PubMed]
- Grolleau, C.; Coutanceau, C.; Pierre, F.; Léger, J.M. Effect of potential cycling on structure and activity of Pt nanoparticles dispersed on different carbon supports. Electrochim. Acta 2008, 53, 7157–7165. [Google Scholar] [CrossRef]
- Solla-Gullón, J.; Montiel, V.; Aldaz, A.; Clavilier, J. Electrochemical characterisation of platinum nanoparticles prepared by microemulsion: How to clean them without loss of crystalline surface structure. J. Electroanal. Chem. 2000, 491, 69–77. [Google Scholar] [CrossRef]
- Napporn, T.; Habrioux, A.; Rousseau, J.; Servat, K.; Léger, J.-M.; Kokoh, B. Effect of the Cleaning Step on the Morphology of Gold Nanoparticles. Electrocatalysis 2011, 2, 24–27. [Google Scholar] [CrossRef]
- Fievet, F.; Lagier, J.P.; Blin, B.; Beaudoin, B.; Figlarz, M. Homogeneous and heterogeneous nucleations in the polyol process for the preparation of micron and submicron size metal particles. Solid State Ionics 1989, 32–33(Part 1), 198–205. [Google Scholar]
- Bonet, F.; Guéry, C.; Guyomard, D.; Herrera Urbina, R.; Tekaia-Elhsissen, K.; Tarascon, J.M. Electrochemical reduction of noble metal compounds in ethylene glycol. Int. J. Inorg. Mater. 1999, 1, 47–51. [Google Scholar] [CrossRef]
- Bock, C.; Paquet, C.; Couillard, M.; Botton, G.A.; MacDougall, B.R. Size-Selected Synthesis of PtRu Nano-Catalysts: Reaction and Size Control Mechanism. J. Am. Chem. Soc. 2004, 126, 8028–8037. [Google Scholar] [CrossRef] [PubMed]
- Skrabalak, S.E.; Wiley, B.J.; Kim, M.; Formo, E.V.; Xia, Y. On the Polyol Synthesis of Silver Nanostructures: Glycolaldehyde as a Reducing Agent. Nano Lett. 2008, 8, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lim, B.; Lee, E.P.; Xia, Y. Shape-controlled synthesis of platinum nanocrystals for catalytic and electrocatalytic applications. Nano Today 2009, 4, 81–95. [Google Scholar] [CrossRef]
- Xiong, Y.; Xia, Y. Shape-Controlled Synthesis of Metal Nanostructures: The Case of Palladium. Adv. Mater. 2007, 19, 3385–3391. [Google Scholar] [CrossRef]
- Grzelczak, M.; Perez-Juste, J.; Mulvaney, P.; Liz-Marzan, L.M. Shape control in gold nanoparticle synthesis. Chem. Soc. Rev. 2008, 37, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Daimon, H.; Onodera, T.; Koda, T.; Sun, S. A General Approach to the Size- and Shape-Controlled Synthesis of Platinum Nanoparticles and Their Catalytic Reduction of Oxygen. Angew. Chem. Int. Ed. 2008, 47, 3588–3591. [Google Scholar] [CrossRef]
- Alia, S.M.; Zhang, G.; Kisailus, D.; Li, D.; Gu, S.; Jensen, K.; Yan, Y. Porous Platinum Nanotubes for Oxygen Reduction and Methanol Oxidation Reactions. Adv. Funct. Mater. 2010, 20, 3742–3746. [Google Scholar] [CrossRef]
- Lee, I.; Delbecq, F.; Morales, R.; Albiter, M.A.; Zaera, F. Tuning selectivity in catalysis by controlling particle shape. Nat. Mater. 2009, 8, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, G.; Beaumont, S.K.; Humphrey, S.M.; Antonetti, C.; Lambert, R.M. Sonogashira Coupling Catalyzed by Gold Nanoparticles: Does Homogeneous or Heterogeneous Catalysis Dominate? ChemCatChem 2010, 2, 1444–1449. [Google Scholar] [CrossRef]
- Campbell, C.T.; Parker, S.C.; Starr, D.E. The Effect of Size-Dependent Nanoparticle Energetics on Catalyst Sintering. Science 2002, 298, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Biacchi, A.J.; Schaak, R.E. The Solvent Matters: Kinetic versus Thermodynamic Shape Control in the Polyol Synthesis of Rhodium Nanoparticles. ACS Nano 2011, 5, 8089–8099. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, F.; Connor, S.; Somorjai, G.A.; Yang, P. Pt Nanocrystals: Shape Control and Langmuir−Blodgett Monolayer Formation. J. Phys. Chem. B 2004, 109, 188–193. [Google Scholar] [CrossRef]
- Susut, C.; Nguyen, T.D.; Chapman, G.B.; Tong, Y. Particle Size Limit for Concomitant Tuning of Size and Shape of Platinum Nanoparticles. J. Cluster Sci. 2007, 18, 773–780. [Google Scholar] [CrossRef]
- Lee, E.; Murthy, A.; Manthiram, A. Comparison of the stabilities and activities of Pt–Ru/C and Pt3–Sn/C electrocatalysts synthesized by the polyol method for methanol electro-oxidation reaction. J. Electroanal. Chem. 2011, 659, 168–175. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, S.M.; Green, S.; Tompsett, G.A.; Lee, S.H.; Huber, G.W.; Kim, W.B. Highly active and stable PtRuSn/C catalyst for electrooxidations of ethylene glycol and glycerol. Appl. Catal. B: Env. 2011, 101, 366–375. [Google Scholar] [CrossRef]
- Wang, L.; Zhai, J.-J.; Jiang, K.; Wang, J.-Q.; Cai, W.-B. Pd–Cu/C electrocatalysts synthesized by one-pot polyol reduction toward formic acid oxidation: Structural characterization and electrocatalytic performance. Int. J. Hydrogen Energ. 2015, 40, 1726–1734. [Google Scholar] [CrossRef]
- Wang, G.; Takeguchi, T.; Muhamad, E.N.; Yamanaka, T.; Sadakane, M.; Ueda, W. Preparation of Well-Alloyed PtRu/C Catalyst by Sequential Mixing of the Precursors in a Polyol Method. J. Electrochem. Soc. 2009, 156, B1348. [Google Scholar]
- Joseyphus, R.J.; Matsumoto, T.; Takahashi, H.; Kodama, D.; Tohji, K.; Jeyadevan, B. Designed synthesis of cobalt and its alloys by polyol process. J. Solid State Chem. 2007, 180, 3008–3018. [Google Scholar] [CrossRef]
- Susut, C.; Tong, Y.J. Size-Dependent Methanol Electro-oxidation Activity of Pt Nanoparticles with Different Shapes. Electrocatalysis 2011, 2, 75–81. [Google Scholar] [CrossRef]
- González-Quijano, D.; Pech-Rodríguez, W.J.; Escalante-García, J.I.; Vargas-Gutiérrez, G.; Rodríguez-Varela, F.J. Electrocatalysts for ethanol and ethylene glycol oxidation reactions. Part I: Effects of the polyol synthesis conditions on the characteristics and catalytic activity of Pt–Sn/C anodes. Int. J. Hydrogen Energ. 2014, 39, 16676–16685. [Google Scholar] [CrossRef]
- Jiang, L.; Hsu, A.; Chu, D.; Chen, R. Ethanol electro-oxidation on Pt/C and PtSn/C catalysts in alkaline and acid solutions. Int. J. Hydrogen Energy 2010, 35, 365–372. [Google Scholar] [CrossRef]
- Lee, S.; Kim, H.J.; Choi, S.M.; Seo, M.H.; Kim, W.B. The promotional effect of Ni on bimetallic PtNi/C catalysts for glycerol electrooxidation. Appl. Catal. A 2012, 429–430, 39–47. [Google Scholar]
- Nghia, N.V.; Truong, N.N.K.; Thong, N.M.; Hung, N.P. Synthesis of Nanowire-Shaped Silver by Polyol Process of Sodium Chloride. Int. J. Mater. Chem. 2012, 2, 75–78. [Google Scholar] [CrossRef]
- Chee, S.-S.; Lee, J.-H. Synthesis of tin nanoparticles through modified polyol process and effects of centrifuging and drying on nanoparticles. Trans. Nonferrous Met. Soc. China 2012, 22, s707–s711. [Google Scholar] [CrossRef]
- Salgado, J.R.C.; Antolini, E.; Gonzalez, E.R. Structure and Activity of Carbon-Supported Pt−Co Electrocatalysts for Oxygen Reduction. J. Phys. Chem. B 2004, 108, 17767–17774. [Google Scholar] [CrossRef]
- Alvarez, G.F.; Mamlouk, M.; Senthil Kumar, S.M.; Scott, K. Preparation and characterisation of carbon-supported palladium nanoparticles for oxygen reduction in low temperature PEM fuel cells. J. Appl. Electrochem. 2011, 41, 925–937. [Google Scholar] [CrossRef]
- Pradeep, T.; Anshup. Noble metal nanoparticles for water purification: A critical review. Thin Solid Films 2009, 517, 6441–6478. [Google Scholar] [CrossRef]
- Nashner, M.S.; Frenkel, A.I.; Somerville, D.; Hills, C.W.; Shapley, J.R.; Nuzzo, R.G. Core Shell Inversion during Nucleation and Growth of Bimetallic Pt/Ru Nanoparticles. J. Am. Chem. Soc. 1998, 120, 8093–8101. [Google Scholar] [CrossRef]
- Takasu, Y.; Fujiwara, T.; Murakami, Y.; Sasaki, K.; Oguri, M.; Asaki, T.; Sugimoto, W. Effect of Structure of Carbon‐Supported PtRu Electrocatalysts on the Electrochemical Oxidation of Methanol. J. Electrochem. Soc. 2000, 147, 4421–4427. [Google Scholar] [CrossRef]
- Rahsepar, M.; Pakshir, M.; Piao, Y.; Kim, H. Preparation of Highly Active 40 wt.% Pt on Multiwalled Carbon Nanotube by Improved Impregnation Method for Fuel Cell Applications. Fuel Cells 2012, 12, 827–834. [Google Scholar] [CrossRef]
- Knani, S.; Chirchi, L.; Baranton, S.; Napporn, T.W.; Léger, J.-M.; Ghorbel, A. A methanol–Tolerant carbon supported Pt–Sn cathode catalysts. Int. J. Hydrogen Energy 2014, 39, 9070–9079. [Google Scholar] [CrossRef]
- Coutanceau, C.; Brimaud, S.; Lamy, C.; Léger, J.M.; Dubau, L.; Rousseau, S.; Vigier, F. Review of different methods for developing nanoelectrocatalysts for the oxidation of organic compounds. Electrochim. Acta 2008, 53, 6865–6880. [Google Scholar] [CrossRef]
- Knani, S.; Chirchi, L.; Napporn, W.T.; Baranton, S.; Léger, J.M.; Ghorbel, A. Promising ternary Pt–Co–Sn catalyst for the oxygen reduction reaction. J. Electroanal. Chem. 2015, 738, 145–153. [Google Scholar] [CrossRef]
- Miller, H.A.; Bevilacqua, M.; Filippi, J.; Lavacchi, A.; Marchionni, A.; Marelli, M.; Moneti, S.; Oberhauser, W.; Vesselli, E.; Innocenti, M.; et al. Nanostructured Fe-Ag electrocatalysts for the oxygen reduction reaction in alkaline media. J. Mater. Chem. A 2013, 1, 13337–13347. [Google Scholar] [CrossRef]
- Choi, S.M.; Yoon, J.S.; Kim, H.J.; Nam, S.H.; Seo, M.H.; Kim, W.B. Electrochemical benzene hydrogenation using PtRhM/C (M = W, Pd, or Mo) electrocatalysts over a polymer electrolyte fuel cell system. Appl. Catal. A 2009, 359, 136–143. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, S.M.; Nam, S.H.; Seo, M.H.; Kim, W.B. Effect of Rh content on carbon-supported PtRh catalysts for dehydrogenative electrooxidation of cyclohexane to benzene over polymer electrolyte membrane fuel cell. Appl. Catal. A 2009, 352, 145–151. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, S.M.; Nam, S.H.; Seo, M.H.; Kim, W.B. Carbon-supported PtNi catalysts for electrooxidation of cyclohexane to benzene over polymer electrolyte fuel cells. Catal. Today 2009, 146, 9–14. [Google Scholar] [CrossRef]
- Longoni, G.; Chini, P. Synthesis and chemical characterization of platinum carbonyl dianions [Pt3(CO)6]n2− (n = apprx.10, 6, 5, 4, 3, 2, 1). A new series of inorganic oligomers. J. Am. Chem. Soc. 1976, 98, 7225–7231. [Google Scholar] [CrossRef]
- Holade, Y.; Morais, C.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Toward the Electrochemical Valorization of Glycerol: Fourier Transform Infrared Spectroscopic and Chromatographic Studies. ACS Catal. 2013, 3, 2403–2411. [Google Scholar] [CrossRef]
- Holade, Y.; MacVittie, K.; Conlon, T.; Guz, N.; Servat, K.; Napporn, T.W.; Kokoh, K.B.; Katz, E. Pacemaker Activated by an Abiotic Biofuel Cell Operated in Human Serum Solution. Electroanalysis 2014, 26, 2445–2457. [Google Scholar] [CrossRef]
- Holade, Y.; Engel, A.B.; Tingry, S.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Insights on Hybrid Glucose Biofuel Cell Based on Bilirubin Oxidase Cathode and Gold-Based Nanomaterials Anode. ChemElectroChem 2014, 1, 1976–1987. [Google Scholar] [CrossRef]
- Lim, B.; Kobayashi, H.; Camargo, P.C.; Allard, L.; Liu, J.; Xia, Y. New insights into the growth mechanism and surface structure of palladium nanocrystals. Nano Res. 2010, 3, 180–188. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, M.; Xiong, Y.; Lim, B.; Xia, Y. Shape-Controlled Synthesis of Pd Nanocrystals and Their Catalytic Applications. Acc. Chem. Res. 2013, 46, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.R.; Personick, M.L.; Zhang, J.; Mirkin, C.A. Defining Rules for the Shape Evolution of Gold Nanoparticles. J. Am. Chem. Soc. 2012, 134, 14542–14554. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, Y.; Li, Y.; Zhou, H.; Duan, X.; Huang, Y. Synthesis of PtPd Bimetal Nanocrystals with Controllable Shape, Composition, and Their Tunable Catalytic Properties. Nano Lett. 2012, 12, 4265–4270. [Google Scholar] [CrossRef] [PubMed]
- Holade, Y.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Electrocatalytic properties of nanomaterials synthesized from “Bromide Anion Exchange” method—Investigations of glucose and glycerol oxidation. Electrochim. Acta 2015. [Google Scholar] [CrossRef]
- Habrioux, A.; Servat, K.; Tingry, S.; Kokoh, K.B. Enhancement of the performances of a single concentric glucose/O2 biofuel cell by combination of bilirubin oxidase/Nafion cathode and Au–Pt anode. Electrochem. Commun. 2009, 11, 111–113. [Google Scholar] [CrossRef]
- Lee, W.-D.; Lim, D.-H.; Chun, H.-J.; Lee, H.-I. Preparation of Pt nanoparticles on carbon support using modified polyol reduction for low-temperature fuel cells. Int. J. Hydrogen Energy 2012, 37, 12629–12638. [Google Scholar] [CrossRef]
- Sakai, G.; Arai, T.; Matsumoto, T.; Ogawa, T.; Yamada, M.; Sekizawa, K.; Taniguchi, T. Electrochemical and ESR Study on Pt-TiOx/C Electrocatalysts with Enhanced Activity for ORR. ChemElectroChem 2014, 1, 366–370. [Google Scholar] [CrossRef]
- Teranishi, T.; Kurita, R.; Miyake, M. Shape Control of Pt Nanoparticles. J. Inorg. Organomet. Polym. 2000, 10, 145–156. [Google Scholar] [CrossRef]
- Mirdamadi-Esfahani, M.; Mostafavi, M.; Keita, B.; Nadjo, L.; Kooyman, P.; Remita, H. Bimetallic Au-Pt nanoparticles synthesized by radiolysis: Application in electro-catalysis. Gold Bull. 2010, 43, 49–56. [Google Scholar] [CrossRef]
- Rivadulla, J.F.; Vergara, M.C.; Blanco, M.C.; López-Quintela, M.A.; Rivas, J. Optical Properties of Platinum Particles Synthesized in Microemulsions. J. Phys. Chem. B 1997, 101, 8997–9004. [Google Scholar] [CrossRef]
- Lim, B.; Jiang, M.; Tao, J.; Camargo, P.H.C.; Zhu, Y.; Xia, Y. Shape-Controlled Synthesis of Pd Nanocrystals in Aqueous Solutions. Adv. Funct. Mater. 2009, 19, 189–200. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Yan, J.-M.; Wang, H.-L.; Ping, Y.; Jiang, Q. Pd/C Synthesized with Citric Acid: An Efficient Catalyst for Hydrogen Generation from Formic Acid/Sodium Formate. Sci. Rep. 2012, 2, 598. [Google Scholar] [PubMed]
- Klotz, P.; Feldberg, S.; Newman, L. Mixed-ligand complexes of palladium(II) with bromide and chloride in acetonitrile. Inorg. Chem. 1973, 12, 164–168. [Google Scholar] [CrossRef]
- Ksar, F.; Ramos, L.; Keita, B.; Nadjo, L.; Beaunier, P.; Remita, H. Bimetallic Palladium−Gold Nanostructures: Application in Ethanol Oxidation. Chem. Mater. 2009, 21, 3677–3683. [Google Scholar] [CrossRef]
- Imbeault, R.; Reyter, D.; Garbarino, S.; Roué, L.; Guay, D. Metastable AuxRh100–x Thin Films Prepared by Pulsed Laser Deposition for the Electrooxidation of Methanol. J. Phys. Chem. C 2012, 116, 5262–5269. [Google Scholar] [CrossRef]
- Bonneman, H.; Brijoux, W.; Brinkmann, R.; Fretzen, R.; Joussen, T.; Koppler, R.; Korall, B.; Neiteler, P.; Richter, J. Preparation, characterization, and application of fine metal particles and metal colloids using hydrotriorganoborates. J. Mol. Catal. 1994, 86, 129–177. [Google Scholar] [CrossRef]
- Bönnemann, H.; Braun, G.A. Enantioselective Hydrogenations on Platinum Colloids. Angew. Chem. Int. Ed. 1996, 35, 1992–1995. [Google Scholar] [CrossRef]
- Choudhury, N.A.; Raman, R.K.; Sampath, S.; Shukla, A.K. An alkaline direct borohydride fuel cell with hydrogen peroxide as oxidant. J. Power Sources 2005, 143, 1–8. [Google Scholar] [CrossRef]
- Ma, J.; Choudhury, N.A.; Sahai, Y. A comprehensive review of direct borohydride fuel cells. Renew. Sustain. Energy Rev. 2010, 14, 183–199. [Google Scholar] [CrossRef]
- Wroblowa, H.S.; Yen Chi, P.; Razumney, G. Electroreduction of oxygen: A new mechanistic criterion. J. Electroanal. Chem. Interf. Electrochem. 1976, 69, 195–201. [Google Scholar] [CrossRef]
- Acres, G.J.K.; Frost, J.C.; Hards, G.A.; Potter, R.J.; Ralph, T.R.; Thompsett, D.; Burstein, G.T.; Hutchings, G.J. Electrocatalysts for fuel cells. Catal. Today 1997, 38, 393–400. [Google Scholar] [CrossRef]
- Xing, W.; Yin, G.; Zhang, J. Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; p. 322. [Google Scholar]
- Srinivasan, S.; Ticianelli, E.A.; Derouin, C.R.; Redondo, A. Advances in solid polymer electrolyte fuel cell technology with low platinum loading electrodes. J. Power Sources 1988, 22, 359–375. [Google Scholar] [CrossRef]
- Wilson, M.S.; Valerio, J.A.; Gottesfeld, S. Low platinum loading electrodes for polymer electrolyte fuel cells fabricated using thermoplastic ionomers. Electrochim. Acta 1995, 40, 355–363. [Google Scholar] [CrossRef]
- Gloaguen, F.; Andolfatto, F.; Durand, R.; Ozil, P. Kinetic study of electrochemical reactions at catalyst-recast ionomer interfaces from thin active layer modelling. J. Appl. Electrochem. 1994, 24, 863–869. [Google Scholar] [CrossRef]
- Coutanceau, C.; Croissant, M.J.; Napporn, T.; Lamy, C. Electrocatalytic reduction of dioxygen at platinum particles dispersed in a polyaniline film. Electrochim. Acta 2000, 46, 579–588. [Google Scholar] [CrossRef]
- Li, B.; Amiruddin, S.; Prakash, J. A Kinetic Study of Oxygen Reduction Reaction on Palladium-Nickel Alloy Surfaces. ECS Trans. 2008, 6, 139–144. [Google Scholar]
- Oh, H.-S.; Oh, J.-G.; Kim, H. Modification of polyol process for synthesis of highly platinum loaded platinum–carbon catalysts for fuel cells. J. Power Sources 2008, 183, 600–603. [Google Scholar] [CrossRef]
- Lebègue, E.; Baranton, S.; Coutanceau, C. Polyol synthesis of nanosized Pt/C electrocatalysts assisted by pulse microwave activation. J. Power Sources 2011, 196, 920–927. [Google Scholar] [CrossRef]
- Brunel, L.; Denele, J.; Servat, K.; Kokoh, K.B.; Jolivalt, C.; Innocent, C.; Cretin, M.; Rolland, M.; Tingry, S. Oxygen transport through laccase biocathodes for a membrane-less glucose/O2 biofuel cell. Electrochem. Commun. 2007, 9, 331–336. [Google Scholar] [CrossRef]
- Silvert, P.Y.; Tekaia-Elhsissen, K. Synthesis of monodisperse submicronic gold particles by the polyol process. Solid State Ionics 1995, 82, 53–60. [Google Scholar] [CrossRef]
- Joseyphus, R.J.; Kodama, D.; Matsumoto, T.; Sato, Y.; Jeyadevan, B.; Tohji, K. Role of polyol in the synthesis of Fe particles. J. Magn. Magn. Mater. 2007, 310, 2393–2395. [Google Scholar] [CrossRef]
- Couto, G.G.; Klein, J.J.; Schreiner, W.H.; Mosca, D.H.; de Oliveira, A.J.A.; Zarbin, A.J.G. Nickel nanoparticles obtained by a modified polyol process: Synthesis, characterization, and magnetic properties. J. Colloid Int. Sci. 2007, 311, 461–468. [Google Scholar] [CrossRef]
- Tzitzios, V.; Basina, G.; Gjoka, M.; Alexandrakis, V.; Georgakilas, V.; Niarchos, D.; Boukos, N.; Petridis, D. Chemical synthesis and characterization of hcp Ni nanoparticles. Nanotechnology 2006, 17, 3750–3755. [Google Scholar] [CrossRef]
- Kalyan Kamal, S.S.; Sahoo, P.K.; Premkumar, M.; Rama Rao, N.V.; Jagadeesh Kumar, T.; Sreedhar, B.; Singh, A.K.; Ram, S.; Chandra Sekhar, K. Synthesis of cobalt nanoparticles by a modified polyol process using cobalt hydrazine complex. J. Alloys Compd. 2009, 474, 214–218. [Google Scholar] [CrossRef]
- Ducamp-Sanguesa, C.; Herrera-Urbina, R.; Figlarz, M. Synthesis and characterization of fine and monodisperse silver particles of uniform shape. J. Solid State Chem. 1992, 100, 272–280. [Google Scholar] [CrossRef]
- Donati, I.; Travan, A.; Pelillo, C.; Scarpa, T.; Coslovi, A.; Bonifacio, A.; Sergo, V.; Paoletti, S. Polyol Synthesis of Silver Nanoparticles: Mechanism of Reduction by Alditol Bearing Polysaccharides. Biomacromolecules 2009, 10, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Blosi, M.; Albonetti, S.; Dondi, M.; Martelli, C.; Baldi, G. Microwave-assisted polyol synthesis of Cu nanoparticles. J. Nanopart. Res. 2011, 13, 127–138. [Google Scholar] [CrossRef]
- Cuya Huaman, J.L.; Sato, K.; Kurita, S.; Matsumoto, T.; Jeyadevan, B. Copper nanoparticles synthesized by hydroxyl ion assisted alcohol reduction for conducting ink. J. Mater. Chem. 2011, 21, 7062–7069. [Google Scholar] [CrossRef]
- Jo, Y.H.; Jung, I.; Choi, C.S.; Kim, I.; Lee, H.M. Synthesis and characterization of low temperature Sn nanoparticles for the fabrication of highly conductive ink. Nanotechnology 2011, 22, 225701. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Toda, T.; Igarashi, H.; Uchida, H.; Watanabe, M. Enhancement of the Electroreduction of Oxygen on Pt Alloys with Fe, Ni, and Co. J. Electrochem. Soc. 1999, 146, 3750–3756. [Google Scholar] [CrossRef]
- Xiong, L.; Manthiram, A. Effect of Atomic Ordering on the Catalytic Activity of Carbon Supported PtM (M = Fe, Co, Ni, and Cu) Alloys for Oxygen Reduction in PEMFCs. J. Electrochem. Soc. 2005, 152, A697–A703. [Google Scholar] [CrossRef]
- Yang, H.; Vogel, W.; Lamy, C.; Alonso-Vante, N. Structure and Electrocatalytic Activity of Carbon-Supported Pt−Ni Alloy Nanoparticles Toward the Oxygen Reduction Reaction. J. Phys. Chem. B 2004, 108, 11024–11034. [Google Scholar] [CrossRef]
- Santiago, E.I.; Varanda, L.C.; Villullas, H.M. Carbon-Supported Pt−Co Catalysts Prepared by a Modified Polyol Process as Cathodes for PEM Fuel Cells. J. Phys. Chem. C 2007, 111, 3146–3151. [Google Scholar] [CrossRef]
- Rao, C.V.; Reddy, A.L.M.; Ishikawa, Y.; Ajayan, P.M. Synthesis and electrocatalytic oxygen reduction activity of graphene-supported Pt3Co and Pt3Cr alloy nanoparticles. Carbon 2011, 49, 931–936. [Google Scholar] [CrossRef]
- Liu, C.; Wu, X.; Klemmer, T.; Shukla, N.; Yang, X.; Weller, D.; Roy, A.G.; Tanase, M.; Laughlin, D. Polyol Process Synthesis of Monodispersed FePt Nanoparticles. J. Phys. Chem. B 2004, 108, 6121–6123. [Google Scholar] [CrossRef] [PubMed]
- Venkateswara Rao, C.; Viswanathan, B. ORR Activity and Direct Ethanol Fuel Cell Performance of Carbon-Supported Pt−M (M = Fe, Co, and Cr) Alloys Prepared by Polyol Reduction Method. J. Phys. Chem. C 2009, 113, 18907–18913. [Google Scholar] [CrossRef]
- Min, M.-k.; Cho, J.; Cho, K.; Kim, H. Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications. Electrochim. Acta 2000, 45, 4211–4217. [Google Scholar] [CrossRef]
- Léger, J.M. Preparation and activity of mono- or bi-metallic nanoparticles for electrocatalytic reactions. Electrochim. Acta 2005, 50, 3123–3129. [Google Scholar] [CrossRef]
- Tseng, C.-J.; Lo, S.-T.; Lo, S.-C.; Chu, P.P. Characterization of Pt-Cu binary catalysts for oxygen reduction for fuel cell applications. Mater. Chem. Phys. 2006, 100, 385–390. [Google Scholar] [CrossRef]
- Xiong, L.; Kannan, A.M.; Manthiram, A. Pt–M (M = Fe, Co, Ni and Cu) electrocatalysts synthesized by an aqueous route for proton exchange membrane fuel cells. Electrochem. Commun. 2002, 4, 898–903. [Google Scholar] [CrossRef]
- Cambanis, G.; Chadwick, D. Platinum-vanadium carbon supported catalysts for fuel cell applications. Appl. Catal. 1986, 25, 191–198. [Google Scholar] [CrossRef]
- Antolini, E.; Passos, R.R.; Ticianelli, E.A. Electrocatalysis of oxygen reduction on a carbon supported platinum–vanadium alloy in polymer electrolyte fuel cells. Electrochim. Acta 2002, 48, 263–270. [Google Scholar] [CrossRef]
- Mukerjee, S.; Srinivasan, S.; Soriaga, M.P.; McBreen, J. Effect of Preparation Conditions of Pt Alloys on Their Electronic, Structural, and Electrocatalytic Activities for Oxygen Reduction—XRD, XAS, and Electrochemical Studies. J. Phys. Chem. 1995, 99, 4577–4589. [Google Scholar] [CrossRef]
- Huang, M.; Li, L.; Guo, Y. Microwave heated polyol synthesis of Pt3Te/C catalysts. Electrochim. Acta 2009, 54, 3303–3308. [Google Scholar] [CrossRef]
- Chen, C.; Kang, Y.; Huo, Z.; Zhu, Z.; Huang, W.; Xin, H.L.; Snyder, J.D.; Li, D.; Herron, J.A.; Mavrikakis, M.; et al. Highly crystalline multimetallic nanoframes with three-dimensional electrocatalytic surfaces. Science 2014, 343, 1339–1343. [Google Scholar] [CrossRef] [PubMed]
- Senthil Kumar, S.M.; Soler Herrero, J.; Irusta, S.; Scott, K. The effect of pretreatment of Vulcan XC-72R carbon on morphology and electrochemical oxygen reduction kinetics of supported Pd nano-particle in acidic electrolyte. J. Electroanal. Chem. 2010, 647, 211–221. [Google Scholar] [CrossRef]
- Murthi, V.S.; Urian, R.C.; Mukerjee, S. Oxygen Reduction Kinetics in Low and Medium Temperature Acid Environment: Correlation of Water Activation and Surface Properties in Supported Pt and Pt Alloy Electrocatalysts. J. Phys. Chem. B 2004, 108, 11011–11023. [Google Scholar] [CrossRef]
- Jiang, L.; Hsu, A.; Chu, D.; Chen, R. Oxygen Reduction Reaction on Carbon Supported Pt and Pd in Alkaline Solutions. J. Electrochem. Soc. 2009, 156, B370–B376. [Google Scholar] [CrossRef]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Yam, V.W.W. Behind platinum’s sparkle. Nat. Chem. 2010, 2, 790. [Google Scholar] [CrossRef] [PubMed]
- Borup, R.; Meyers, J.; Pivovar, B.; Kim, Y.S.; Mukundan, R.; Garland, N.; Myers, D.; Wilson, M.; Garzon, F.; Wood, D.; et al. Scientific Aspects of Polymer Electrolyte Fuel Cell Durability and Degradation. Chem. Rev. 2007, 107, 3904–3951. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Fernández, P.; Troiani, H.; Martins, M.; Arenillas, A.; Camara, G. Agglomeration and Cleaning of Carbon Supported Palladium Nanoparticles in Electrochemical Environment. Electrocatalysis 2014, 5, 204–212. [Google Scholar] [CrossRef]
- Hiraoka, F.; Matsuzawa, K.; Mitsushima, S. Degradation of Pt/C Under Various Potential Cycling Patterns. Electrocatalysis 2013, 4, 10–16. [Google Scholar] [CrossRef]
- Cui, C.-H.; Yu, S.-H. Engineering Interface and Surface of Noble Metal Nanoparticle Nanotubes toward Enhanced Catalytic Activity for Fuel Cell Applications. Acc. Chem. Res. 2013, 46, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Habrioux, A.; Alonso-Vante, N. The Effect of Substrates at Cathodes in Low-temperature Fuel Cells. ChemElectroChem 2014, 1, 37–46. [Google Scholar] [CrossRef]
- Jiang, Z.-Z.; Wang, Z.-B.; Chu, Y.-Y.; Gu, D.-M.; Yin, G.-P. Ultrahigh stable carbon riveted Pt/TiO2-C catalyst prepared by in situ carbonized glucose for proton exchange membrane fuel cell. Energy Environ. Sci. 2011, 4, 728–735. [Google Scholar] [CrossRef]
- Strmcnik, D.; Kodama, K.; van der Vliet, D.; Greeley, J.; Stamenkovic, V.R.; Marković, N.M. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat. Chem. 2009, 1, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Strmcnik, D.; Escudero-Escribano, M.; Kodama, K.; Stamenkovic, V.R.; Cuesta, A.; Marković, N.M. Enhanced electrocatalysis of the oxygen reduction reaction based on patterning of platinum surfaces with cyanide. Nat. Chem. 2010, 2, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sasaki, K.; Sutter, E.; Adzic, R.R. Stabilization of Platinum Oxygen-Reduction Electrocatalysts Using Gold Clusters. Science 2007, 315, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Sepa, D.B.; Vojnovic, M.V.; Vracar, L.M.; Damjanovic, A. Apparent enthalpies of activation of electrodic oxygen reduction at platinum in different current density regions—I. Acid solution. Electrochim. Acta 1986, 31, 91–96. [Google Scholar] [CrossRef]
- Damjanovic, A. Electron transfer through thin anodic films in oxygen evolution at Pt electrodes in alkaline solutions. Electrochim. Acta 1992, 37, 2533–2539. [Google Scholar] [CrossRef]
- Demarconnay, L.; Coutanceau, C.; Léger, J.M. Electroreduction of dioxygen (ORR) in alkaline medium on Ag/C and Pt/C nanostructured catalysts—effect of the presence of methanol. Electrochim. Acta 2004, 49, 4513–4521. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holade, Y.; Sahin, N.E.; Servat, K.; Napporn, T.W.; Kokoh, K.B. Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells. Catalysts 2015, 5, 310-348. https://doi.org/10.3390/catal5010310
Holade Y, Sahin NE, Servat K, Napporn TW, Kokoh KB. Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells. Catalysts. 2015; 5(1):310-348. https://doi.org/10.3390/catal5010310
Chicago/Turabian StyleHolade, Yaovi, Nihat Ege Sahin, Karine Servat, Teko W. Napporn, and Kouakou B. Kokoh. 2015. "Recent Advances in Carbon Supported Metal Nanoparticles Preparation for Oxygen Reduction Reaction in Low Temperature Fuel Cells" Catalysts 5, no. 1: 310-348. https://doi.org/10.3390/catal5010310