1. Introduction
Volatile organic compounds (VOCs) are organic chemical compounds such as aldehydes, ketones, and other light weight hydrocarbons. Since they have relatively high vapor pressures under ambient conditions, they vaporized easily and diffuse into the atmosphere. Some VOCs are harmful to human health and the environment, as recognized to cause sick building syndrome, multiple chemical sensitivity, and air pollution such as photo-chemical smog and ground-level ozone [
1,
2].
Among the VOCs, toluene is widely used as an organic solvent for paints, printing inks, adhesives, and antiseptics due to its excellent ability to dissolve organic substances. However, toluene has an unpleasant odor and causes sick building syndrome by evaporating into the atmosphere. To protect our health and the environment from such noxious influences, it is necessary to remove toluene released into the atmosphere as much as possible.
Generally, catalysts for VOCs oxidation such as toluene, acetaldehyde, and ethylene are classified into supported noble metals, metal oxides, and mixtures of noble metals and metal oxides [
3,
4,
5,
6,
7,
8]. The catalysis is affected by the influence of dispersed state of active compound and promoters, crystal structure, amount of active species, and surface area of the catalyst [
7,
9]. Temperature is also an important parameter that affects the VOCs removal by oxidation catalysis. In addition, the properties of the VOC to be purified also play a key role in the selection of the catalyst and the determination of the reaction temperature [
10].
In several VOCs oxidation catalysts, CeO
2 and CeO
2-ZrO
2 solid solutions can work effective promoters when they are in conjunction with platinum and metal oxides. In particular, some active platinum catalysts supported on them have been reported for VOCs oxidation [
11,
12,
13,
14,
15,
16,
17,
18,
19]. CeO
2 and CeO
2-ZrO
2 solid solutions can release and store oxygen corresponding to the surrounding atmosphere, and these properties make them attractive components as promoters of the automotive exhaust three-way catalysts [
11].
In our previous studies, furthermore, we found that employing such solid solutions, which have high oxygen release and storage properties, as promoters was effective to establish complete oxidation of VOCs at moderate temperatures [
16,
17,
18]. In particular, introduction of a small amount of SnO
2, which has been well-known as a typical n-type semiconductor with electronic conduction [
20], into the CeO
2-ZrO
2 lattice considerably facilitated the VOCs oxidation [
19]. However, thermal stability of the toluene oxidation catalysis on the Pt/CeO
2-ZrO
2-SnO
2 catalysts was not enough due to readily-reducibility of SnO
2.
In this study, we focused on zinc oxide (ZnO), also known as a stable n-type semiconductor [
21] and a 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst was prepared. Since ZnO has excellent thermal stability, it is expected that thermal stability of the catalyst support will be increased by introducing ZnO into the CeO
2-ZrO
2 lattice without significant decrease in the catalytic activity. Therefore, the catalytic toluene oxidation activity of the catalyst was investigated and the calcination temperature dependence on the toluene oxidation activity was characterized.
2. Results and Discussion
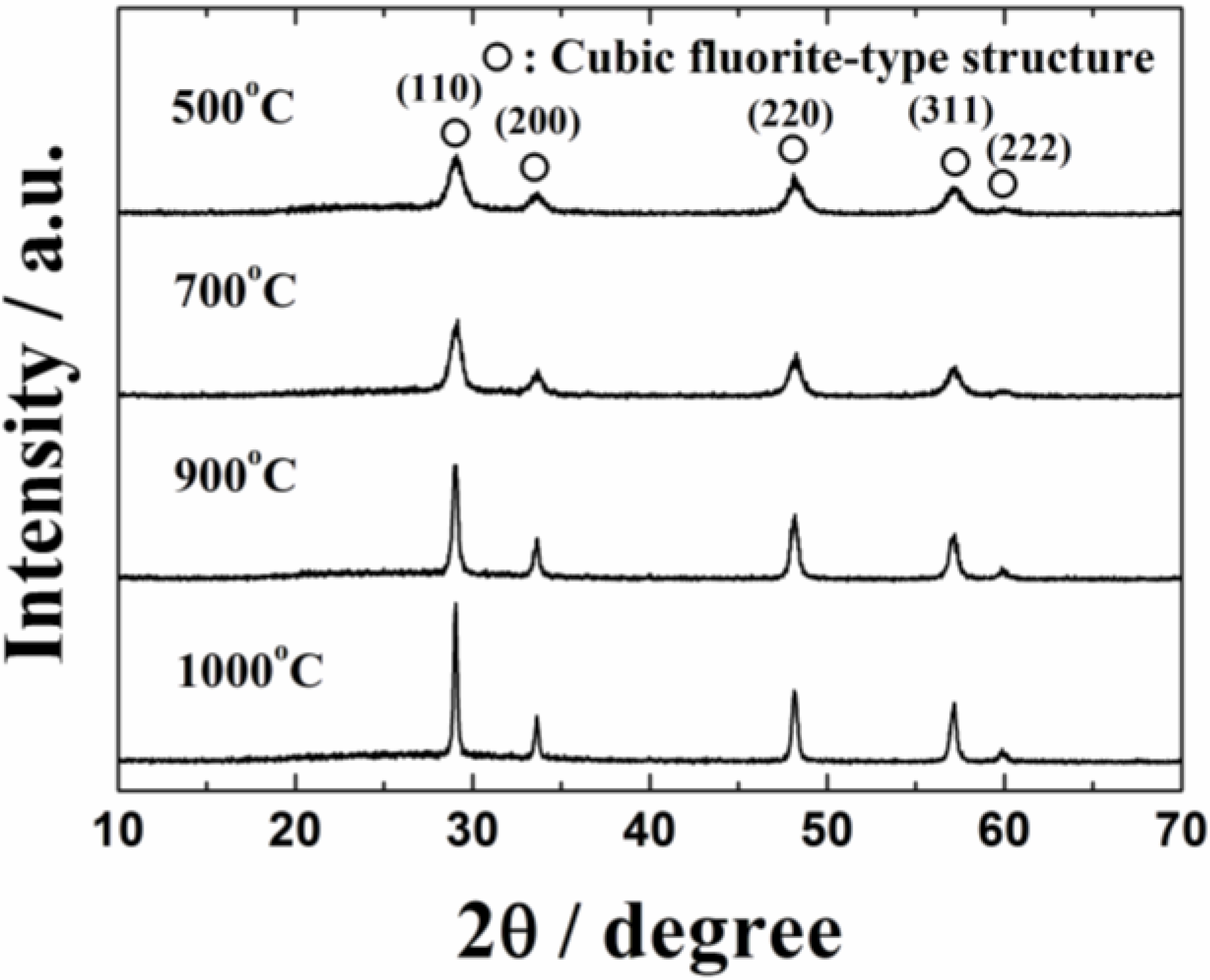
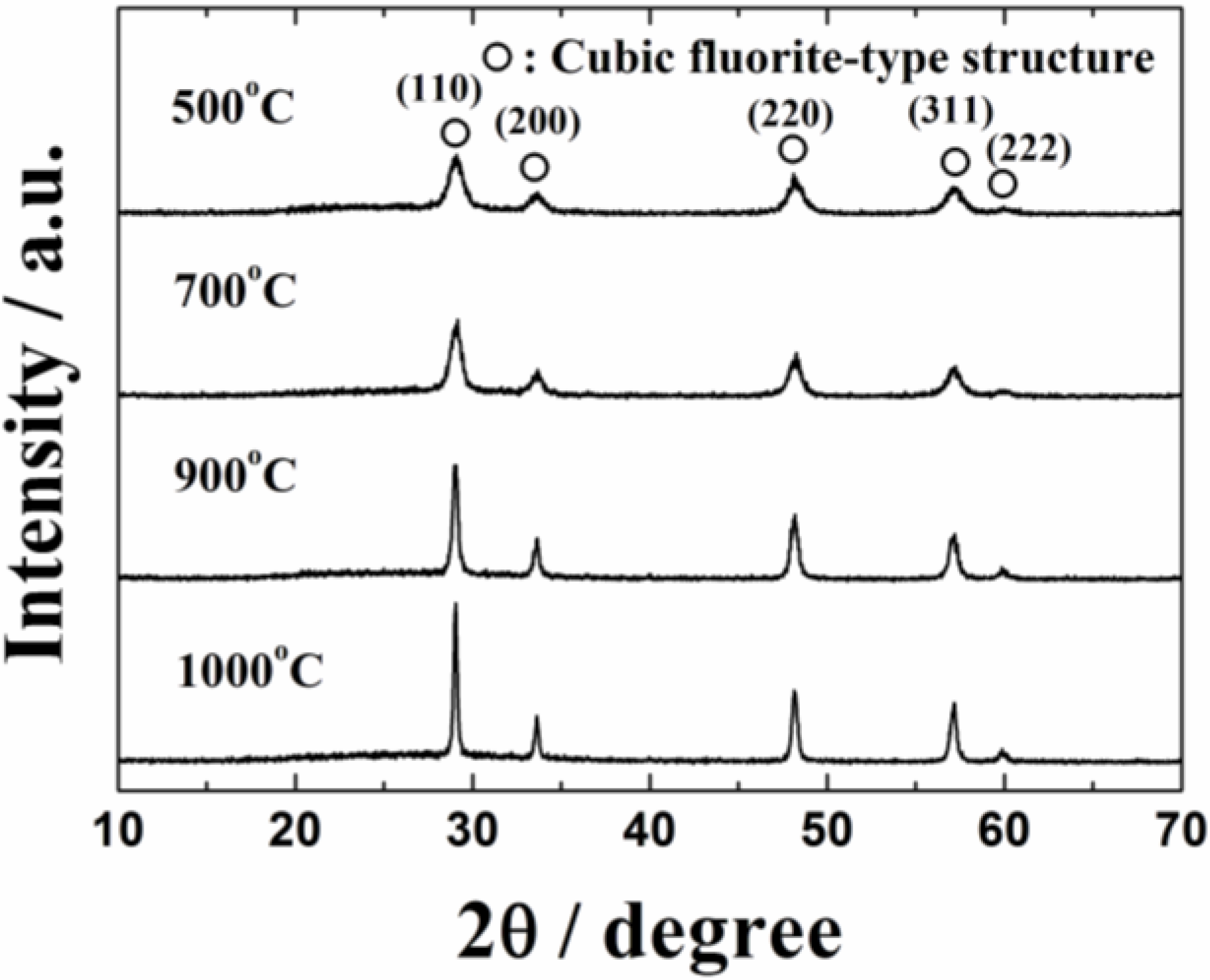
Figure 1 shows the X-ray powder diffraction (XRD) patterns of the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalysts calcined at 500, 700, 900 and 1000 °C. The XRD patterns of the catalysts contain diffraction peaks of only the cubic fluorite-type oxide, and no peaks corresponding to platinum were observed probably due to its low content. The diffraction peaks assigned to the cubic fluorite-type structure were steady and no peak shift was observed regardless of the calcination temperature, indicating that platinum was supported on the surface of the Ce
0.76Zr
0.19Zn
0.05O
1.95 support without forming solid solutions. The composition of the catalyst was confirmed by the X-ray fluorescence spectrometer (XRF) analysis to be in good agreement with their stoichiometric value. The BET specific surface area of the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalysts decreased from 33 m
2 g
−1 to 0.9 m
2 g
−1 with increasing the calcination temperature, as summarized in
Table 1. A drop in platinum dispersion was also observed with raising calcination temperature.
Figure 1.
X-ray powder diffraction (XRD) patterns of the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalysts calcined at 500, 700, 900 and 1000 °C.
Figure 1.
X-ray powder diffraction (XRD) patterns of the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalysts calcined at 500, 700, 900 and 1000 °C.
Table 1.
Brunauer-Emmett-Teller (BET) surface area of the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalysts.
Table 1.
Brunauer-Emmett-Teller (BET) surface area of the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalysts.
| Catalyst composition | Calcination temperature (°C) | BET surface area (m2 g−1) | Pt dispersion (%) |
|---|
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 500 | 33 | 63 |
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 700 | 21 | 22 |
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 900 | 4.5 | 2.3 |
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 1000 | 0.9 | 2.0 |
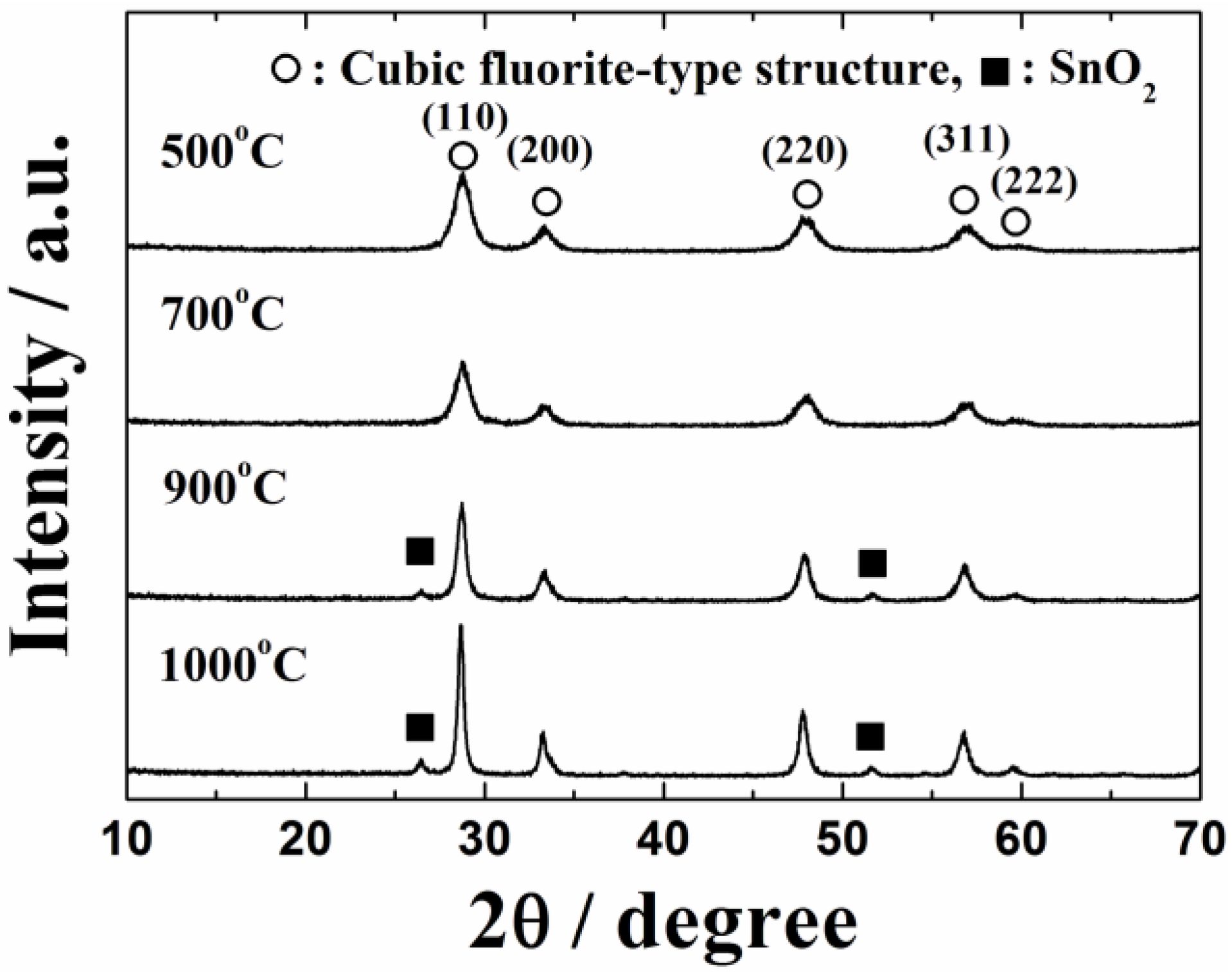
On the other hand, in the case of 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0 catalysts, in which SnO
2 was introduced into CeO
2-ZrO
2 instead of ZnO, small SnO
2 peaks were observed as the secondary impurity phase for the samples calcined at 900 and 1000 °C, as depicted in
Figure 2. From these results, it has been evidenced that the thermal stability of the zinc-doped catalyst (0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95) is higher than that of the tin-doped catalyst (0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0).
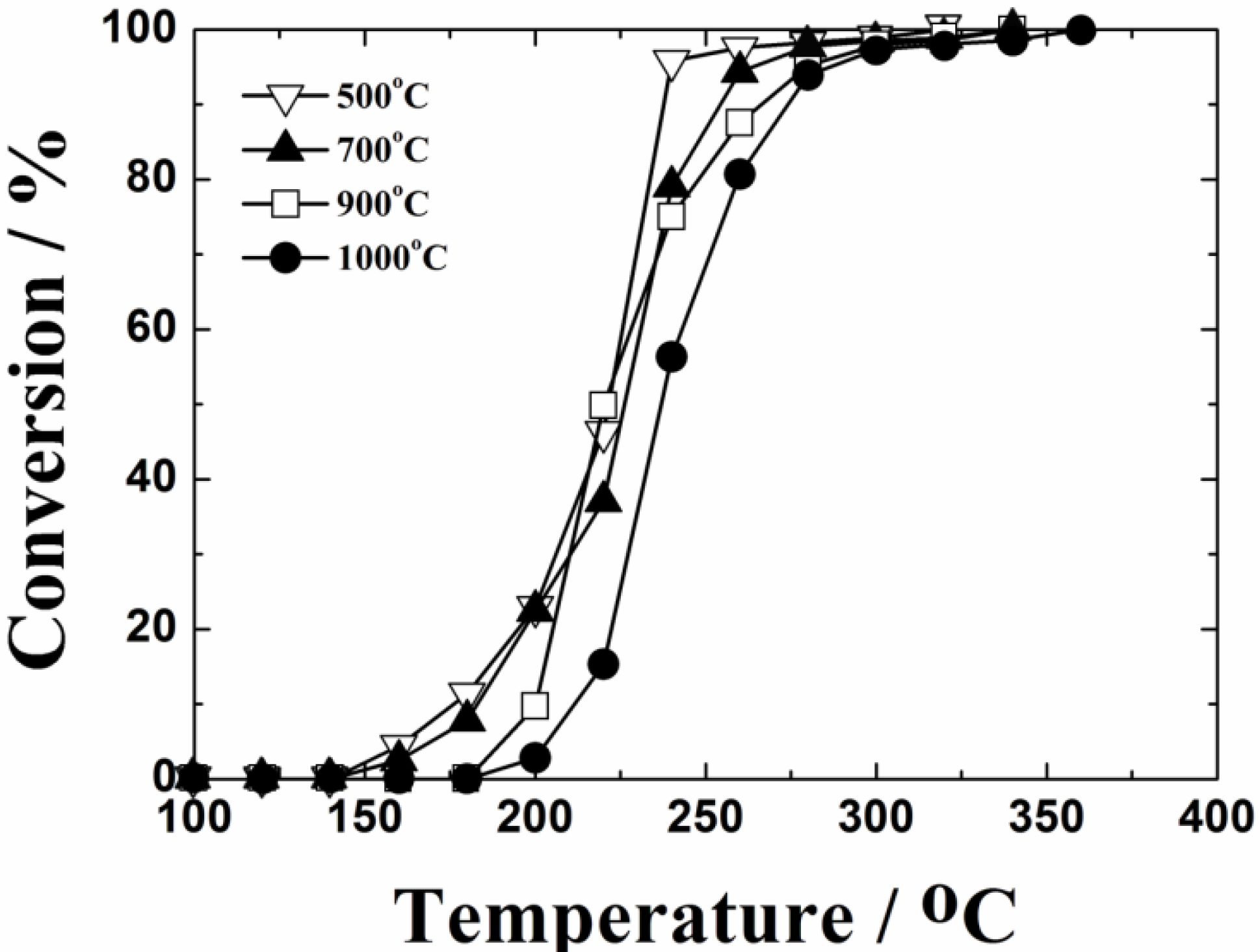
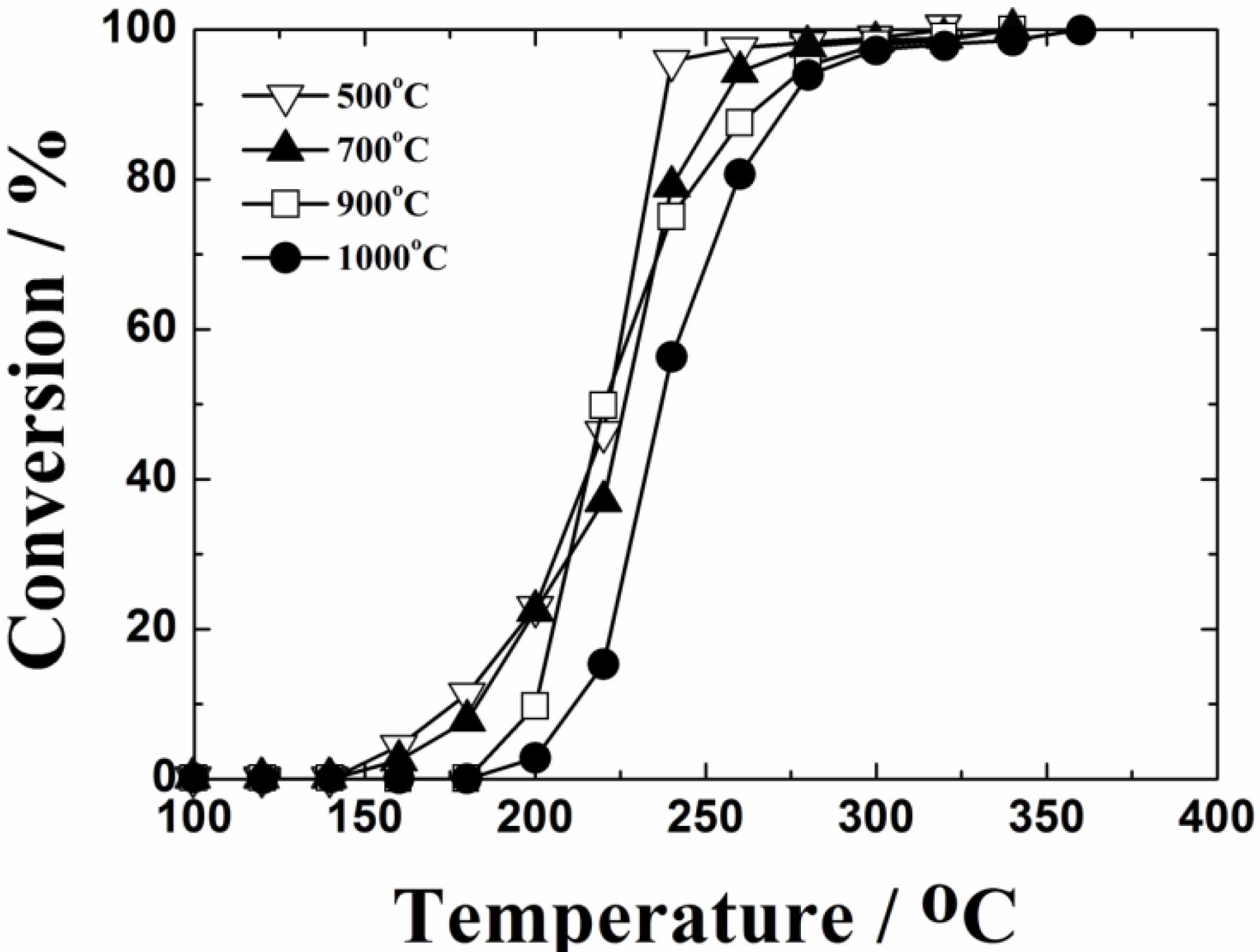
Figure 3 shows the temperature dependencies of toluene oxidation over the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalysts calcined at 500, 700, 900 and 1000 °C. It was confirmed that only CO
2 and steam were produced by the complete oxidation of toluene, and neither CO nor toluene-derived compounds were detected as by-products with a gas chromatography-mass spectrometer. Toluene was completely oxidized at 320 °C on the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst calcined at 500 °C. The toluene oxidation activity decreased with increasing the calcination temperature, because of the decrease in the BET specific surface area. However, significant deactivation was not recognized in the present 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst, and toluene was completely oxidized at 360 °C even after calcination at 1000 °C.
Figure 2.
XRD patterns of the 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 catalysts calcined at 500, 700, 900 and 1000 °C.
Figure 2.
XRD patterns of the 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 catalysts calcined at 500, 700, 900 and 1000 °C.
Figure 3.
Temperature dependencies of toluene oxidation on the 0.4 wt%Pt/Ce0.76Zr0.19 Zn0.05O1.95 catalysts calcined at 500, 700, 900 and 1000 °C.
Figure 3.
Temperature dependencies of toluene oxidation on the 0.4 wt%Pt/Ce0.76Zr0.19 Zn0.05O1.95 catalysts calcined at 500, 700, 900 and 1000 °C.
The catalytic activity and thermal stability of 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 was compared with those of 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0 and a conventional dopant-free 0.4 wt%Pt/Ce
0.79Zr
0.21O
2.0 catalyst. The complete oxidation temperatures of toluene over these catalysts are summarized in
Table 2. Among the catalysts calcined at 500 °C, 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0 was the most active catalyst. However, this catalyst was significantly deactivated after the calcination at 1000 °C and the toluene oxidation temperature was increased up to 460 °C, which was higher by 100 °C than that of 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95. The 0.4 wt%Pt/Ce
0.79Zr
0.21O
2.0 catalyst had the lowest activities at both calcination temperatures. As a result, it is evident that the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst has the highest thermal stability for toluene oxidation among these catalysts.
Table 2.
Complete oxidation temperature of toluene on the catalysts calcined at 500 and 1000 °C.
Table 2.
Complete oxidation temperature of toluene on the catalysts calcined at 500 and 1000 °C.
| Catalyst composition | Calcination temperature (°C) | Complete oxidation temperature of toluene (°C) |
|---|
| 0.4 wt%Pt/Ce0.79Zr0.21O2.0 | 500 | 340 |
| 0.4 wt%Pt/Ce0.79Zr0.21O2.0 | 1000 | 480 |
| 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 | 500 | 260 |
| 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 | 1000 | 460 |
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 500 | 320 |
| 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 | 1000 | 360 |
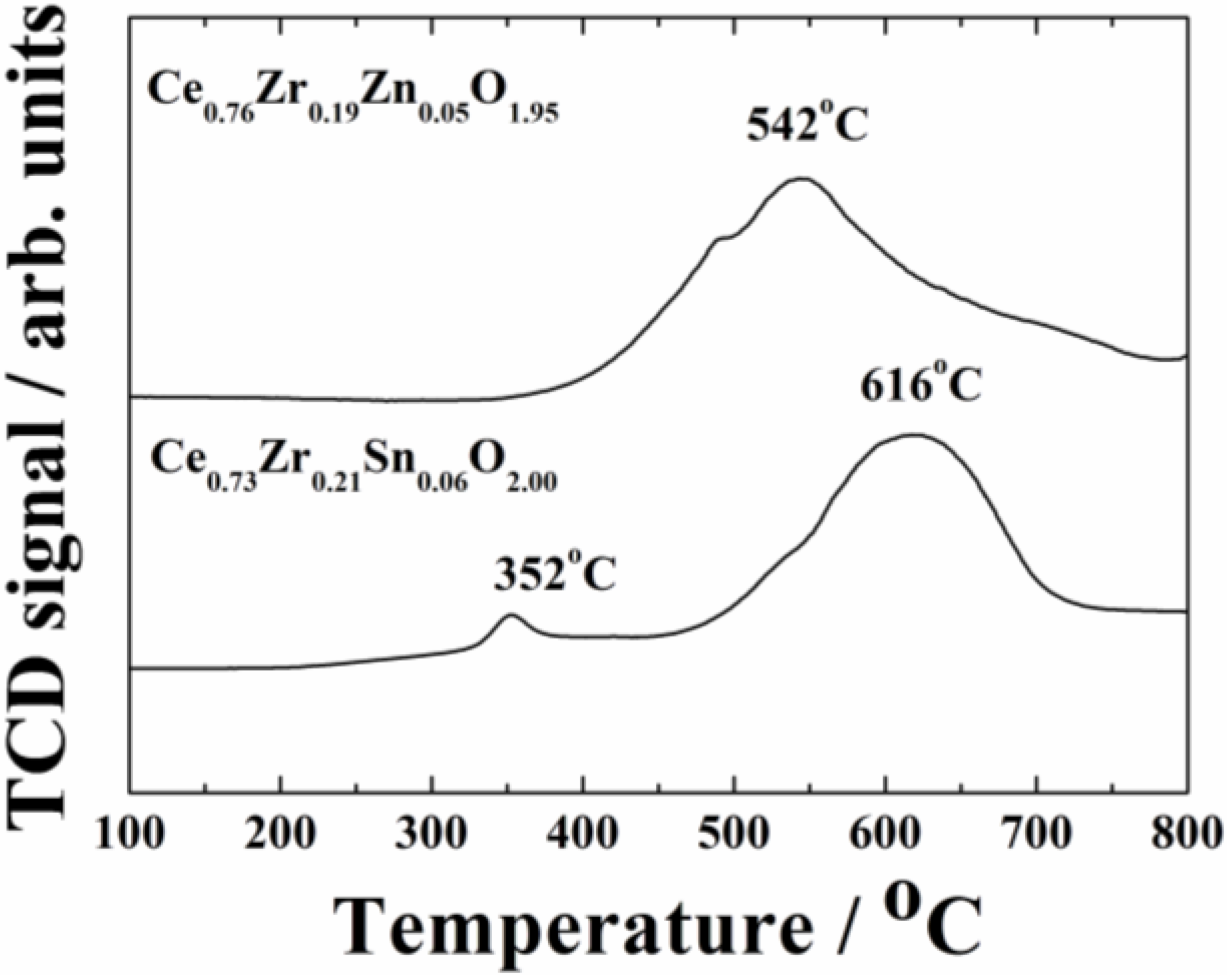
The results in
Table 2 suggest that the thermal stability of the catalytic activity was improved by the presence of zinc. To identify the reason for the positive effect of the zinc doping, temperature programmed reduction (TPR) spectra were measured for Ce
0.76Zr
0.19Zn
0.05O
1.95 and Ce
0.73Zr
0.21Sn
0.06O
2.0 after the calcination at 1000 °C, and their oxygen release behaviors were compared as shown in
Figure 4. For the Ce
0.76Zr
0.19Zn
0.05O
1.95 support, the strong reduction peaks were observed at 542 °C and 841 °C, while for the Ce
0.73Zr
0.21Sn
0.06O
2.0 one, a small and a main reduction peaks were observed at 352 °C and 616 °C, respectively. It is noteworthy that the main reduction peak for the former at 542 °C was lower by approximately 80 °C than that of the latter (616 °C), and introduction of ZnO into the CeO
2-ZrO
2 lattice was obviously more effective than that of SnO
2 to inhibit the thermal degradation of the catalyst support. Therefore, the complete toluene oxidation temperature for 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 (360 °C) became lower than that of 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0 (460 °C) after heating at 1000 °C. From a comprehensive consideration of the XRD, catalysis, and TPR results, it was found that thermal stability of the support is particularly important to preserve the activity even after high-temperature firing.
Figure 4.
Temperature programmed reduction (TPR) profiles of the Ce0.76Zr0.19Zn0.05O1.95 and Ce0.73Zr0.21Sn0.06O2.0 supports calcined at 1000 °C.
Figure 4.
Temperature programmed reduction (TPR) profiles of the Ce0.76Zr0.19Zn0.05O1.95 and Ce0.73Zr0.21Sn0.06O2.0 supports calcined at 1000 °C.
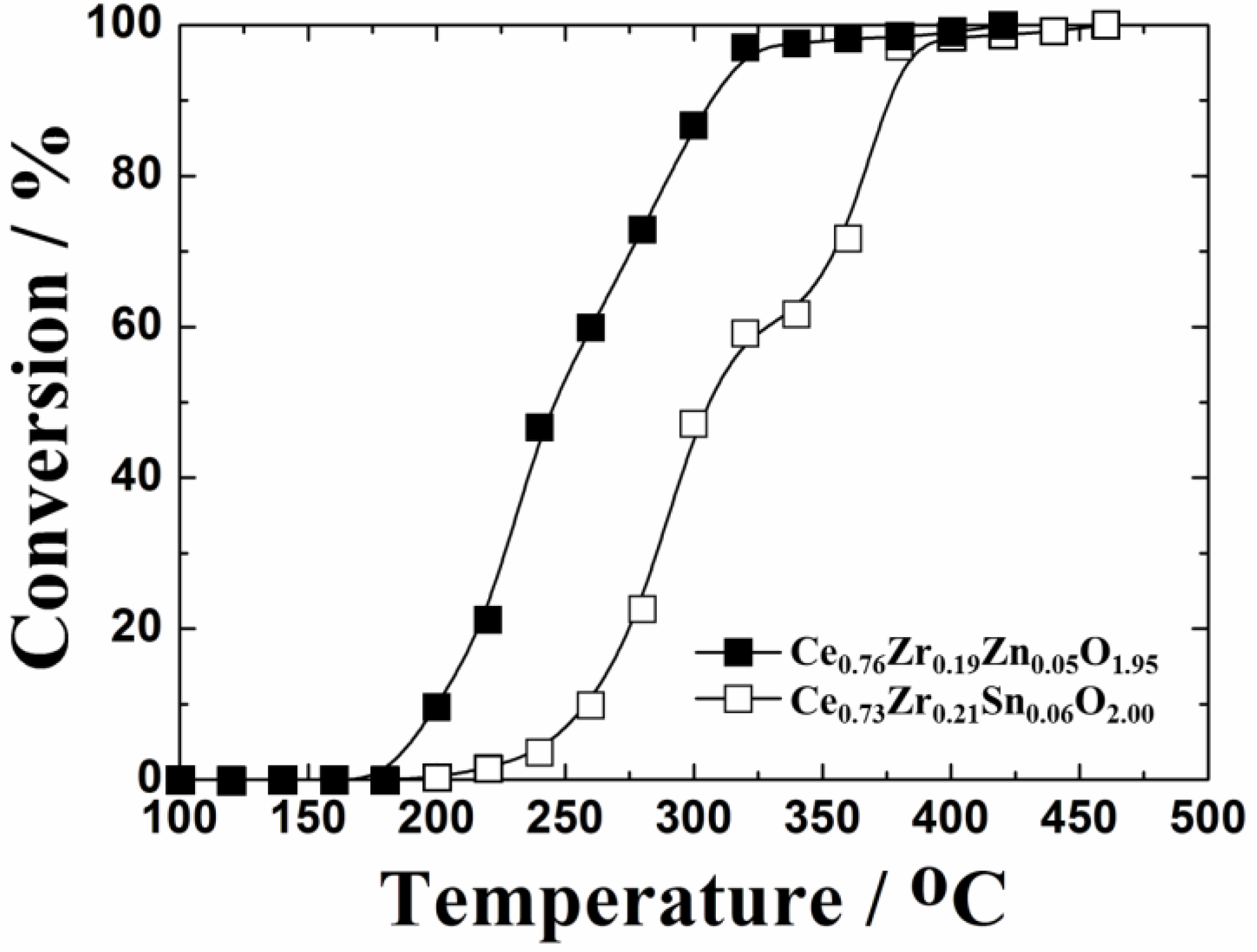
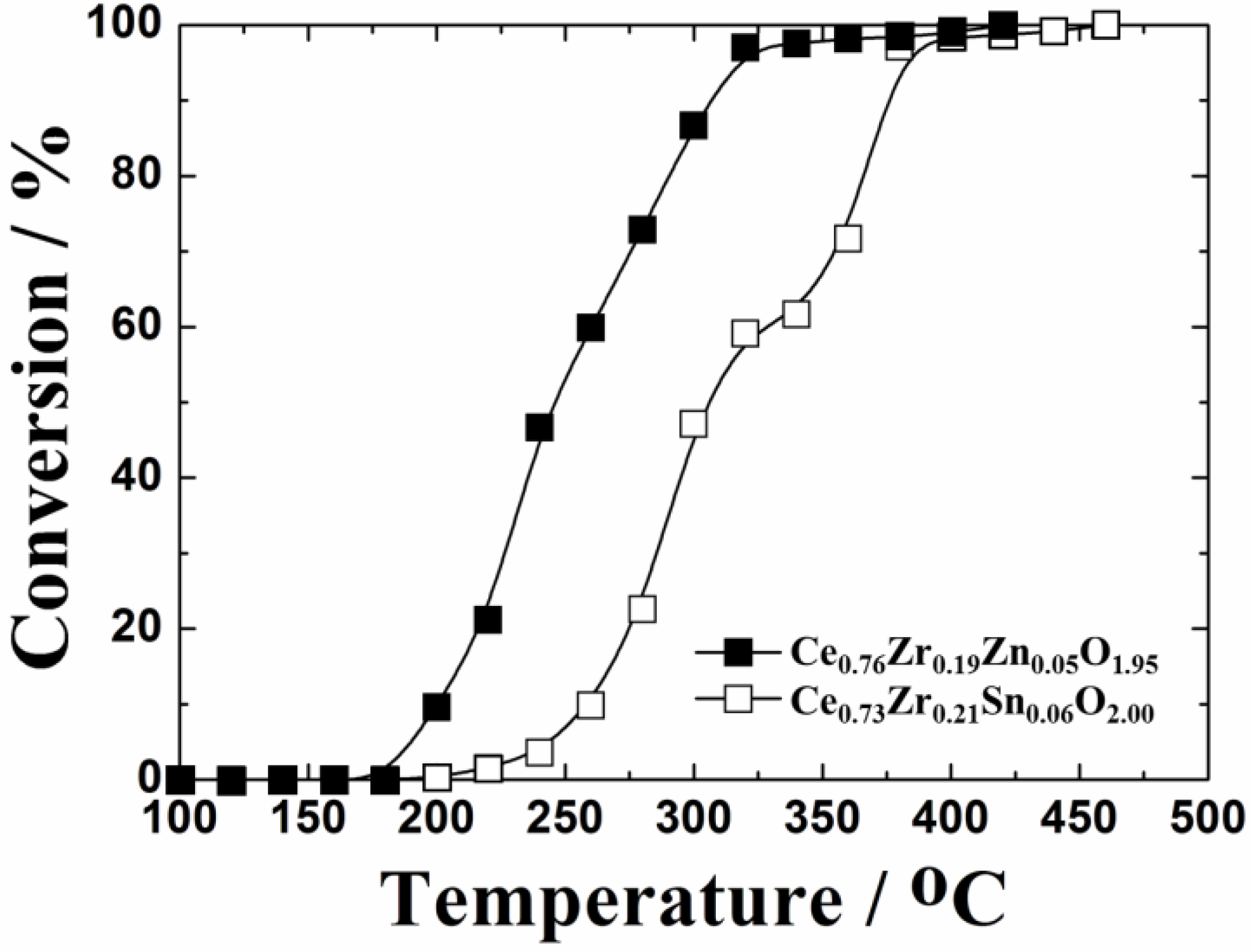
Figure 5 depicts the temperature dependencies of toluene oxidation over the Ce
0.76Zr
0.19Zn
0.05O
1.95 and Ce
0.73Zr
0.21Sn
0.06O
2.0 supports calcined at 1000 °C. It was confirmed in these cases that only CO
2 and steam were produced by the complete oxidation of toluene, and neither CO nor toluene-derived compounds were detected as by-products. The catalytic activity was remarkably promoted by the ZnO doping into the CeO
2-ZrO
2 lattice. These results correspond to the TPR results discussed above and apparently show the advantage of the ZnO doping to improve the thermal stability of the support.
Figure 5.
Temperature dependencies of toluene oxidation on Ce0.76Zr0.19Zn0.05O1.95 and Ce0.73Zr0.21Sn0.06O2.0 supports calcined at 1000 °C.
Figure 5.
Temperature dependencies of toluene oxidation on Ce0.76Zr0.19Zn0.05O1.95 and Ce0.73Zr0.21Sn0.06O2.0 supports calcined at 1000 °C.
Figure 6 shows TPR spectra of 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 and 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06 O
2.0 after the calcination at 1000 °C. The reduction onset temperature of 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 (367 °C) was higher than that of 0.4 wt%Pt/Ce
0.73Zr
0.21Sn
0.06O
2.0 (214 °C). However, reduction of the former was almost completed at 500 °C, while a main peak was observed at 572 °C for the latter: relatively larger amount of lattice oxygen was released for 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 at 500 °C or below. The difference of the reduction behavior will be responsible for higher activity of 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 after heating at 1000 °C.
Figure 6.
TPR profiles of 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 and 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 calcined at 1000 °C.
Figure 6.
TPR profiles of 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 and 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 calcined at 1000 °C.
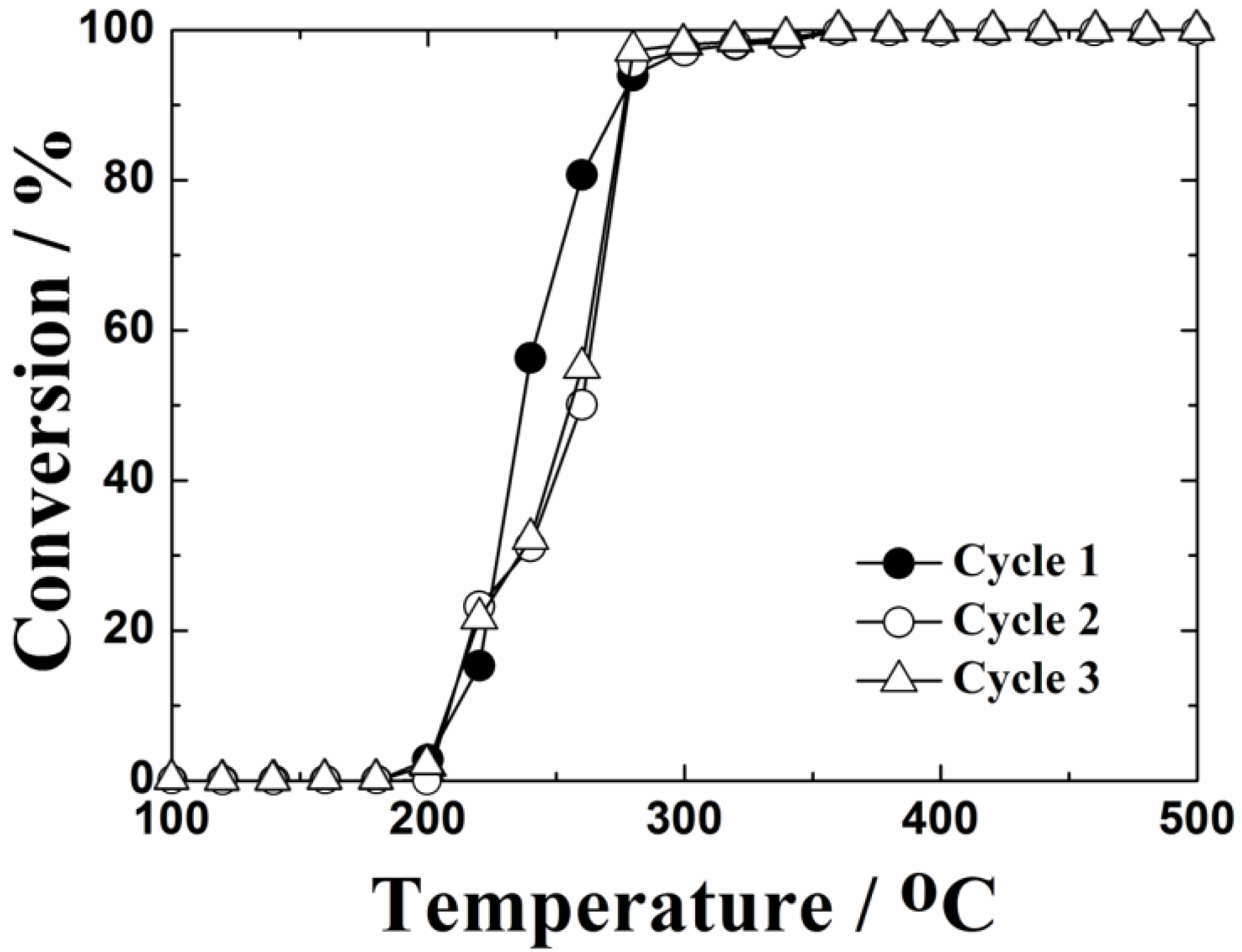
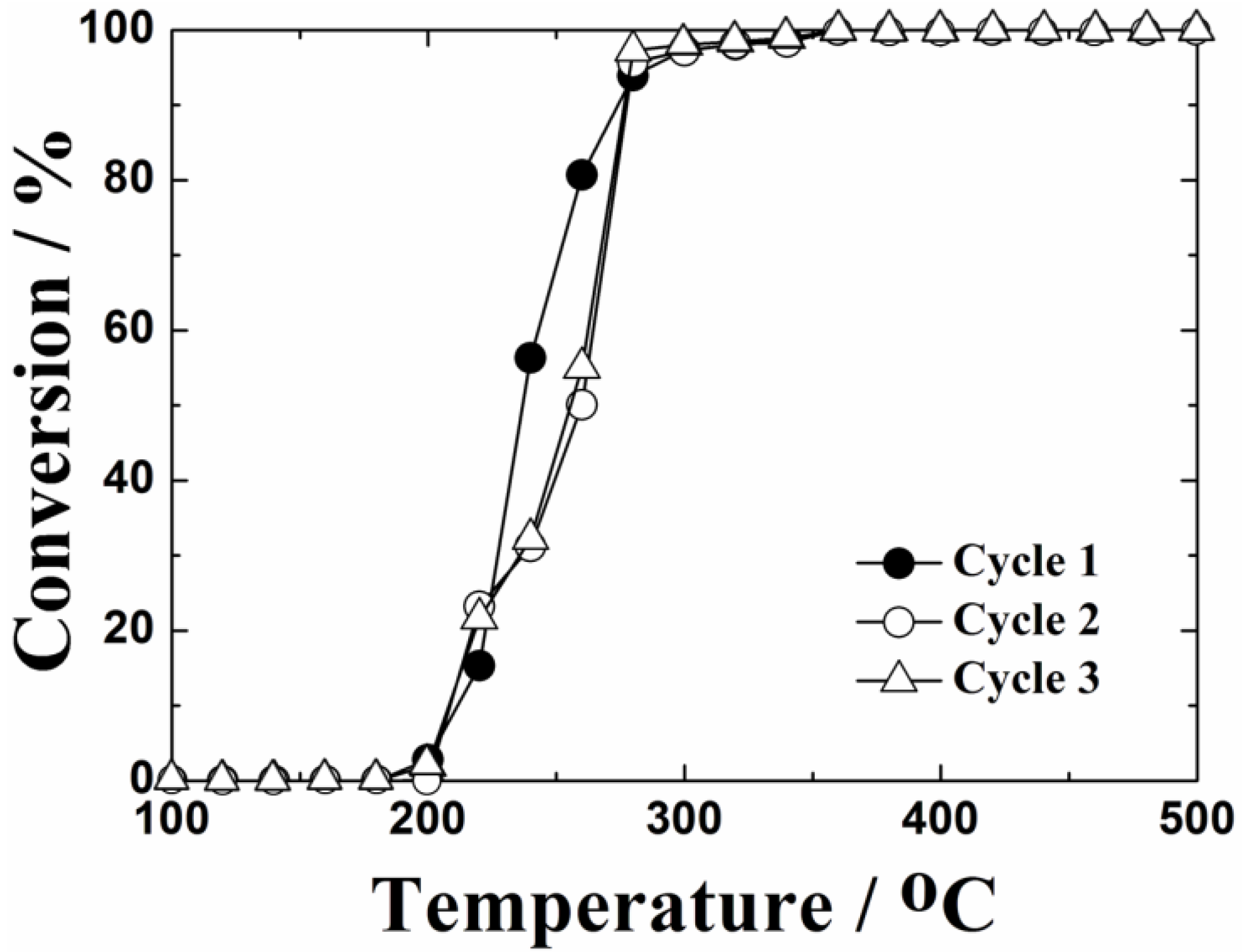
The catalytic test up to 1000 °C was repeated three times to evaluate thermal stability of the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst. As demonstrated in
Figure 7, it was confirmed that the catalyst was stable and was not deactivated throughout the repeated use of the catalyst: 100% conversion of toluene to CO
2 and steam was sustained and carbon deposition was not observed. Furthermore, neither CO nor H
2 were detected as by-products by gas chromatography mass spectrometry and there were no side reactions such as imperfect combustion and partial oxidation. As a result, it became obvious that the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst has high thermal stability for toluene oxidation.
Figure 7.
Measurement of the thermal stability by the temperature dependencies of toluene oxidation over the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalyst calcined at 1000 °C.
Figure 7.
Measurement of the thermal stability by the temperature dependencies of toluene oxidation over the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalyst calcined at 1000 °C.
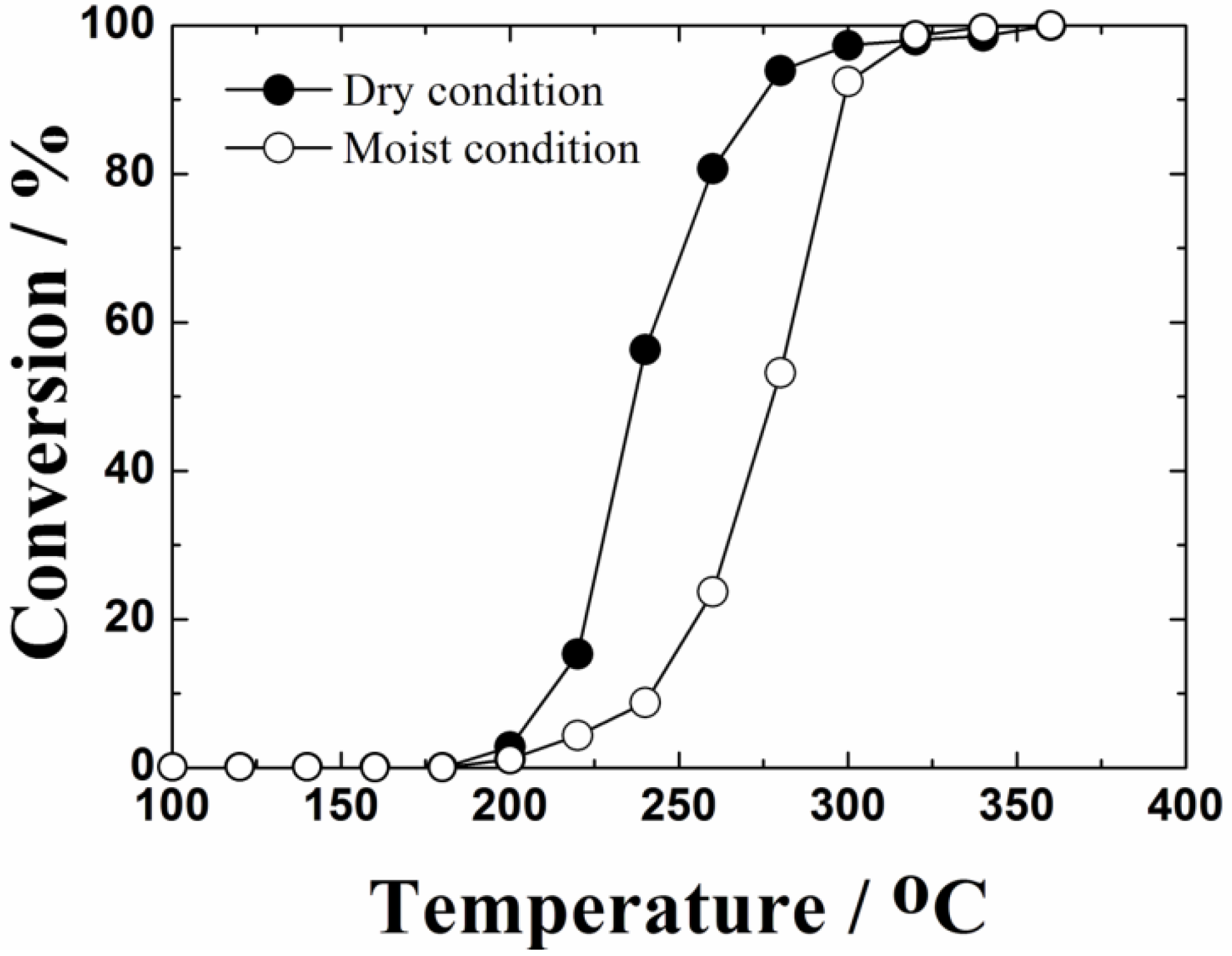
Figure 8.
Temperature dependencies of toluene combustion to CO2 and steam on the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalyst calcined at 1000 °C. Open (○) and closed (●) symbols correspond to data obtained in moist (saturated water vapor at 0 °C; 0.6 vol%) and dry atmospheres, respectively.
Figure 8.
Temperature dependencies of toluene combustion to CO2 and steam on the 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalyst calcined at 1000 °C. Open (○) and closed (●) symbols correspond to data obtained in moist (saturated water vapor at 0 °C; 0.6 vol%) and dry atmospheres, respectively.
For practical applications of the toluene oxidation catalysts, it is important to investigate the impact of water vapor (steam) on the catalytic activities. Steam is a product of toluene oxidation and usually acts as a catalyst poison, because water molecules are adsorbed on the surface of the catalyst. Therefore, the catalytic performance of toluene oxidation in the presence of moisture must be evaluated. The temperature dependencies of toluene oxidation on the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst calcined at 1000 °C under dry and moist conditions are shown in
Figure 8. Also in the moist condition, the reaction products confirmed by gas chromatography-mass spectrometry were only carbon dioxide and steam. Although the light off behavior of the toluene oxidation activity of the 0.4 wt%Pt/Ce
0.76Zr
0.19Zn
0.05O
1.95 catalyst was slightly decreased in the presence of saturated water vapor at 0 °C (0.60 vol%), complete oxidation temperature of toluene was maintained at 360 °C. It is extremely important for the catalysts to maintain the complete reaction temperature in the presence of moisture to put them to practical use.
3. Experimental Section
A Ce0.76Zr0.19Zn0.05O1.95 solid solution was synthesized by a co-precipitation method. Aqueous solutions of 1.0 mol dm−3 Ce(NO3)3 (9.5 cm3), 0.1 mol dm−3 ZrO(NO3)2 (23.75 cm3), and 0.1 mol dm−3 Zn(NO3)2 (6.25 cm3) were mixed with 3.0 mol dm−3 HNO3 (100 cm3) in a stoichiometric ratio, and polyvinylpyrrolidone K25 (PVP; Wako Pure Chemical Industries, Ltd., Osaka, Japan, mean molecular weight is 35,000 and mean degree of polymerization is 315) was also dissolved into the mixture. The molar ratio of total metal cations to PVP was 31.5. Then, the solution was stirred at 80 °C for 6 h. After the solvent was evaporated at 180 °C, the resulting powder was heated at 350 °C for 4 h to remove PVP and then calcined at 500 °C for 1 h. A supported 0.4 wt%Pt/Ce0.76Zr0.19Zn0.05O1.95 catalyst was prepared by impregnating the Ce0.76Zr0.19Zn0.05O1.95 support with a platinum colloid stabilized with PVP (Tanaka Kikinzoku Kogyo Co., Ltd, Tokyo, Japan). After impregnation, the catalyst was dried at 80 °C for 12 h and then finally calcined at 500, 700, 900 and 1000 °C for 4 h. For references, 0.4 wt%Pt/Ce0.79Zr0.21O2.0 and 0.4 wt%Pt/Ce0.73Zr0.21Sn0.06O2.0 catalysts were also prepared using the same procedure.
The sample compositions were analyzed using an X-ray fluorescence spectrometer (XRF; Rigaku, ZSX-100e). The crystal structures of the catalysts were identified by X-ray powder diffraction (XRD; Rigaku, SmartLab, Tokyo, Japan) using Cu-Kα radiation (40 kV, 30mA). The Brunauer-Emmett-Teller (BET) specific surface area was measured by nitrogen adsorption at −196 °C (MicromeriticsTristar 3000, Norcross GA, USA). Temperature programmed reduction (TPR) measurements were carried out under a flow of 5 vol% H2-Ar (50 cm3 min−1) at a heating rate of 5 °C min−1 and the platinum dispersion analysis was carried out by a pulse method at −50 °C using 10% CO-He (0.03 cm3) with a BELCAT-B apparatus (BEL, Osaka, Japan).
The oxidation activity for toluene was tested in a conventional fixed-bed flow reactor consisting of a 10-mm-diameter quartz glass tube. The feed gas was composed of 0.09 vol% toluene in an air balance and the rate was 20 cm3 min−1 over 0.1 g of the catalyst, where the space velocity is 12,000 dm−3 kg−1 h−1. Prior to the measurements, the catalyst was heated at 200 °C for 2 h in a flow of Ar (20 cm3 min−1) to remove water molecules adsorbed on the surface of the catalyst. The catalytic activity was evaluated in terms of toluene conversion. The gas composition after the reaction was analyzed using a gas chromatograph with a flame ionization detector (FID; Shimadzu GC-8AIF, Kyoto, Japan) in which the SunPak-A column was used for gas separation and a gas chromatograph-mass spectrometer (GC-Mass; Shimadzu GCMS-QP2010 Plus, Kyoto, Japan) in which the RTX624 column was installed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}