Enzymatic Catalysis at Interfaces—Heterophase Systems as Substrates for Enzymatic Action

{kind=link}
{kind=link}
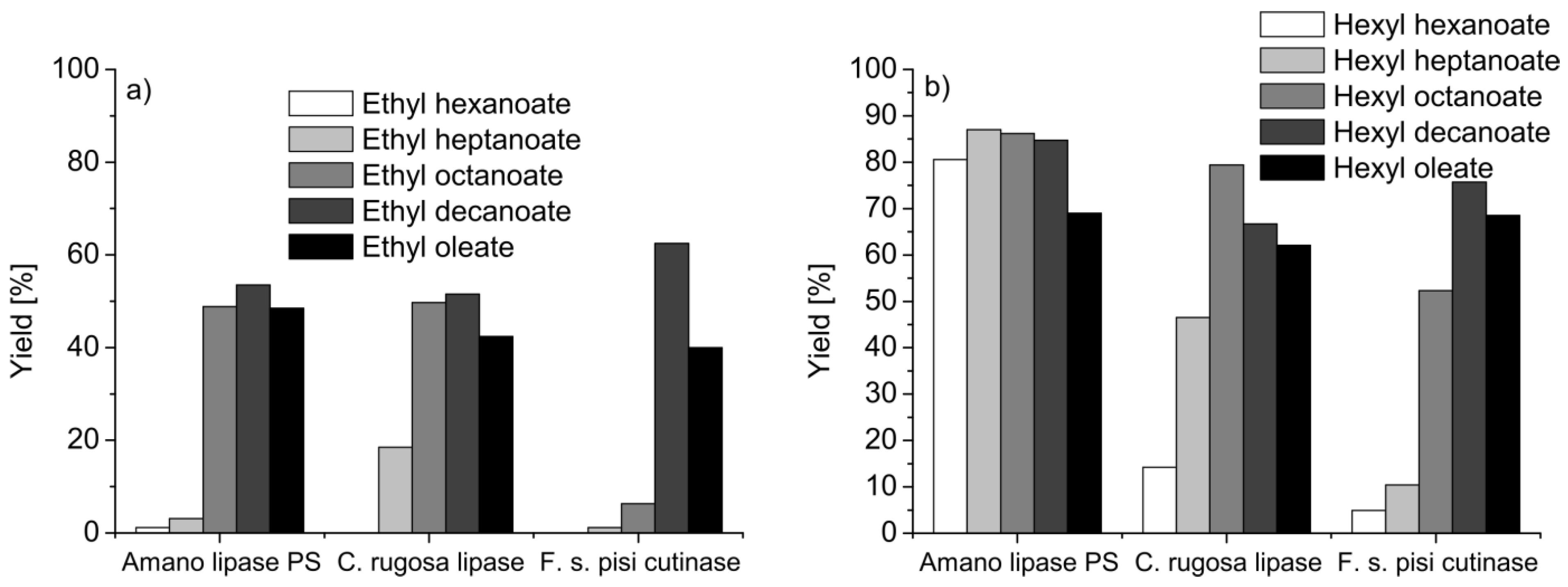{kind=link}
{kind=link}
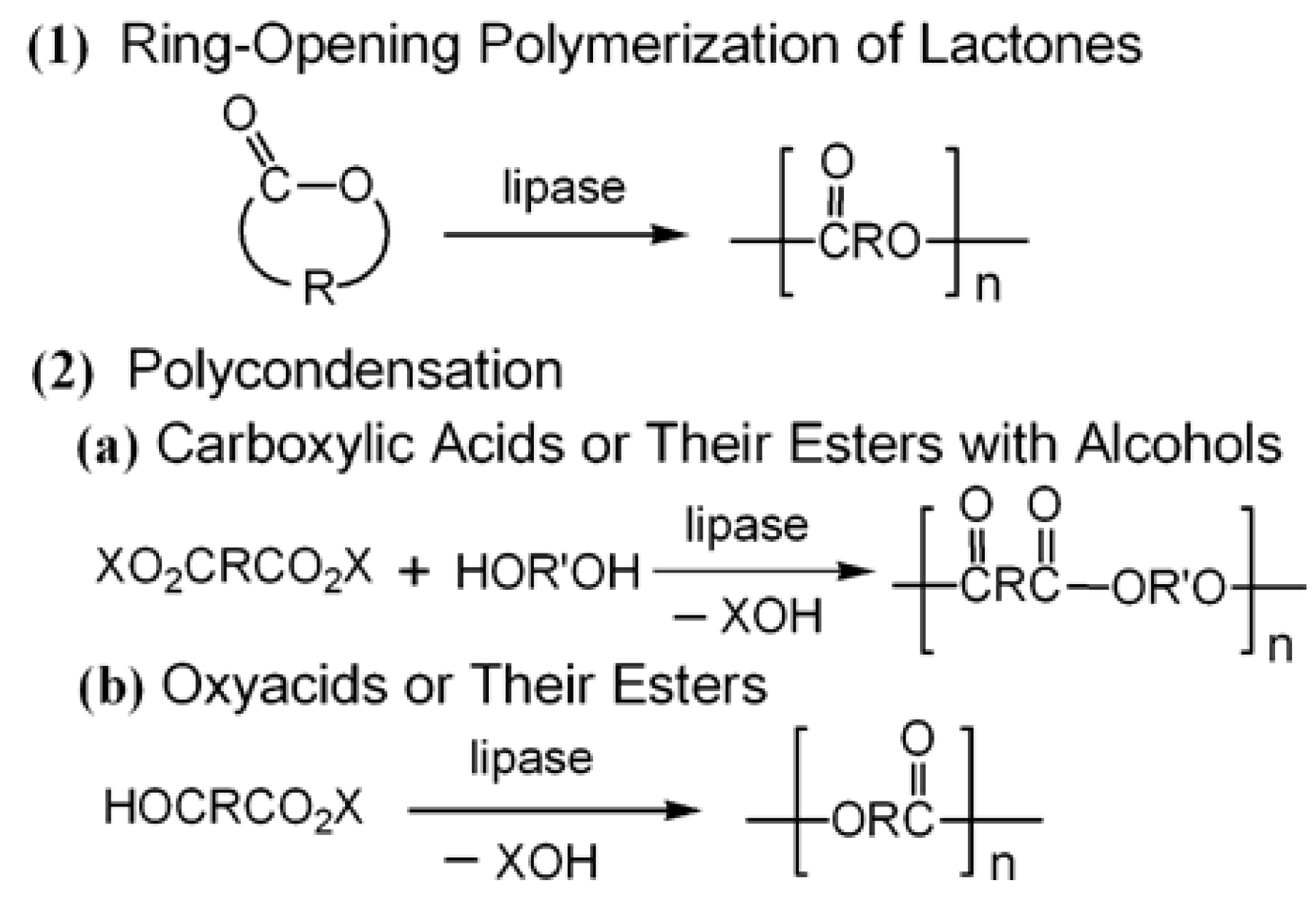{kind=link}
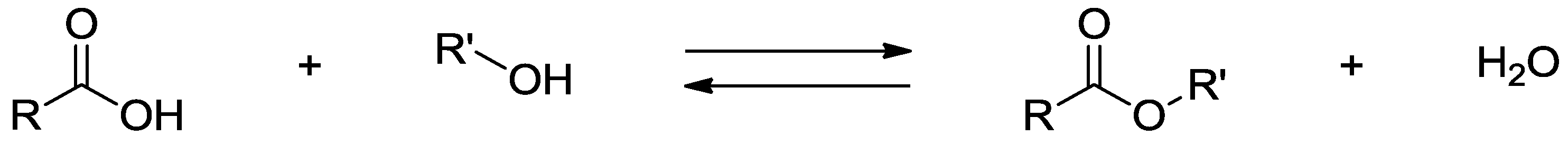{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Features of Miniemulsions
3. Enzyme Located in the Disperse Phase
4. Enzyme Located in the Continuous Phase
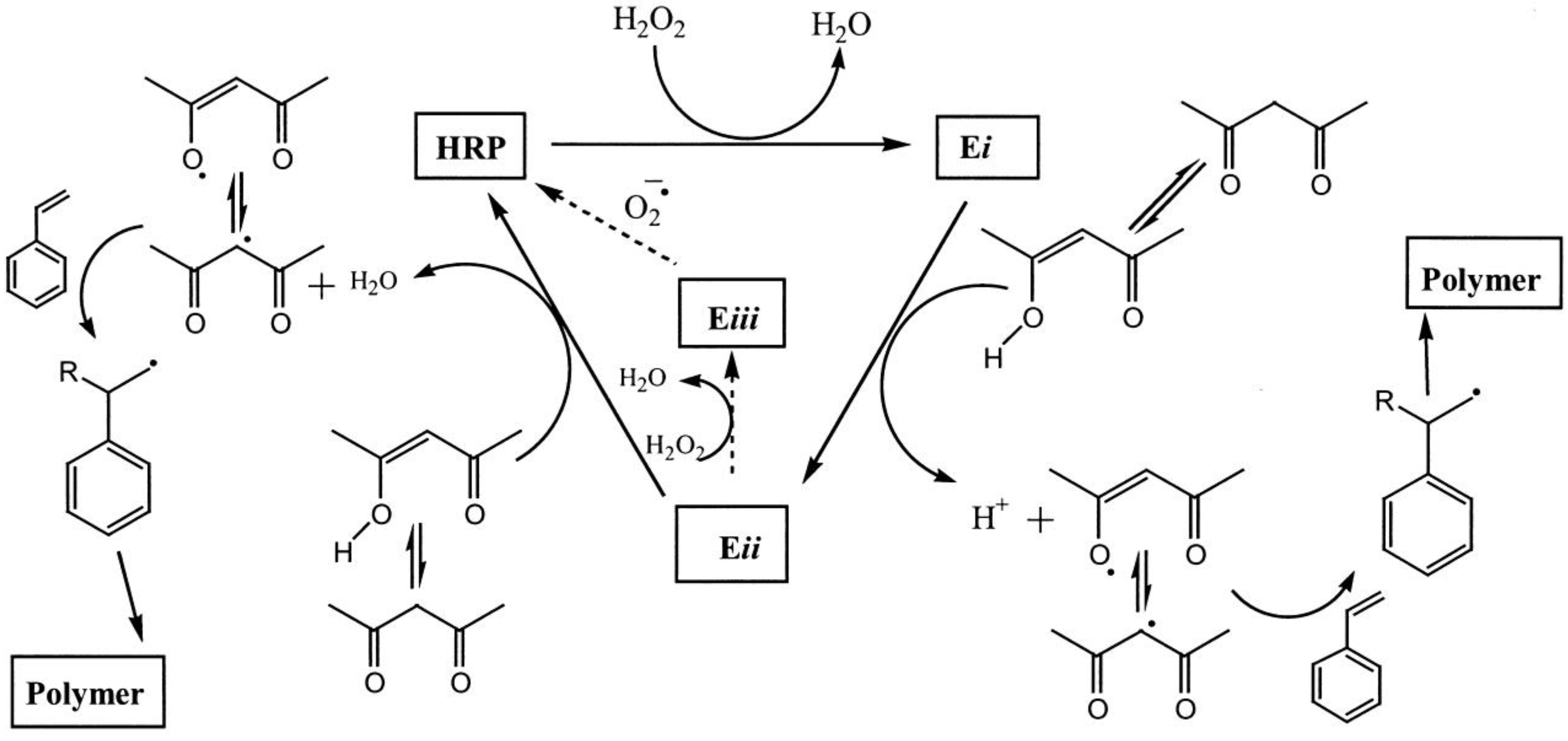
5. Enzyme Located at the Interface
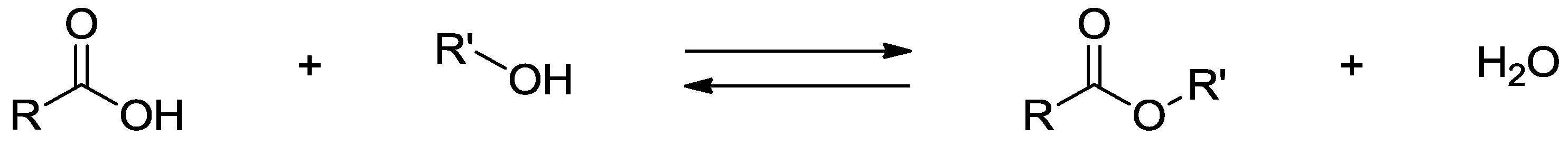
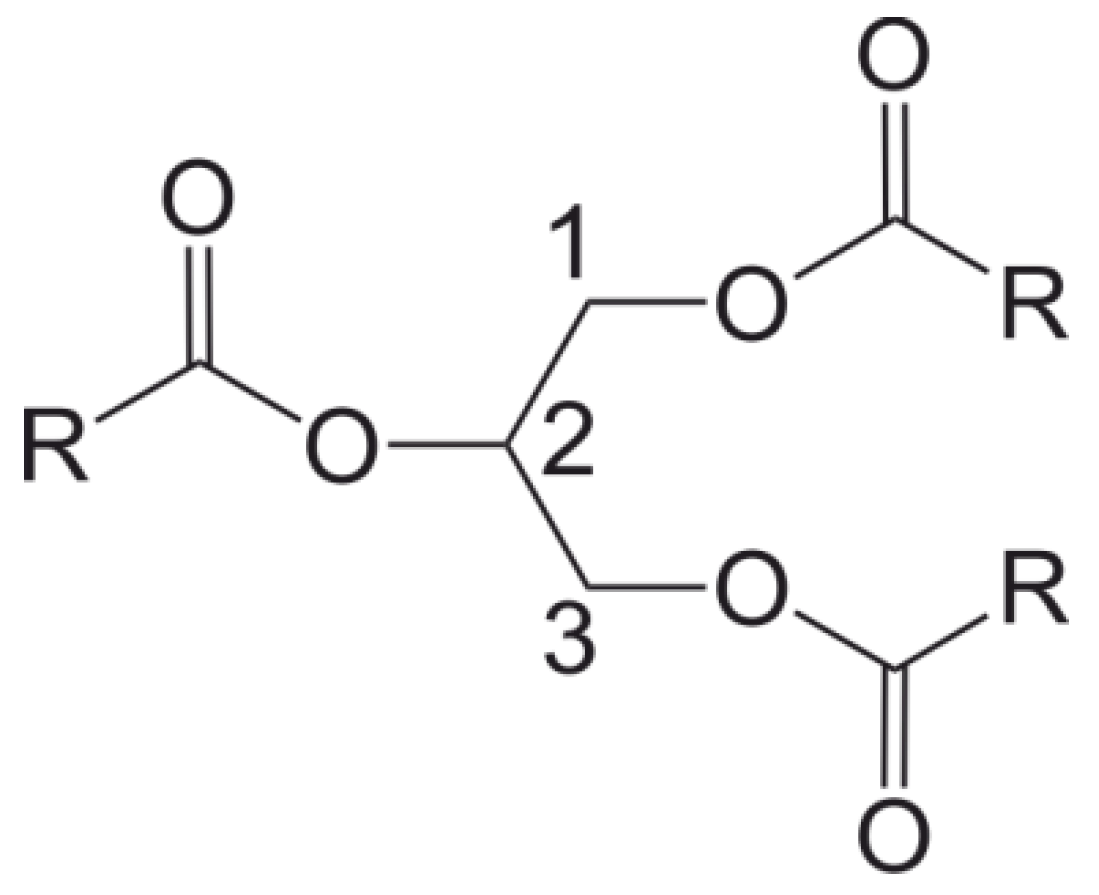
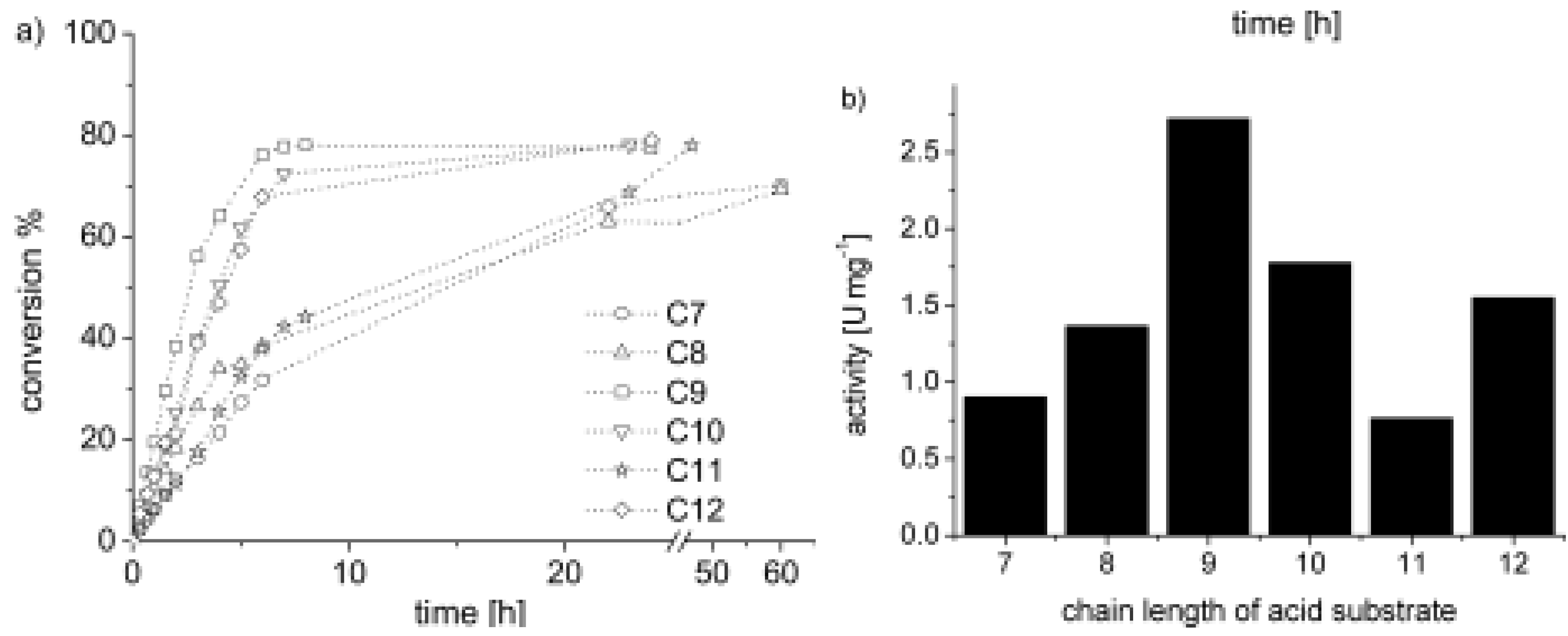
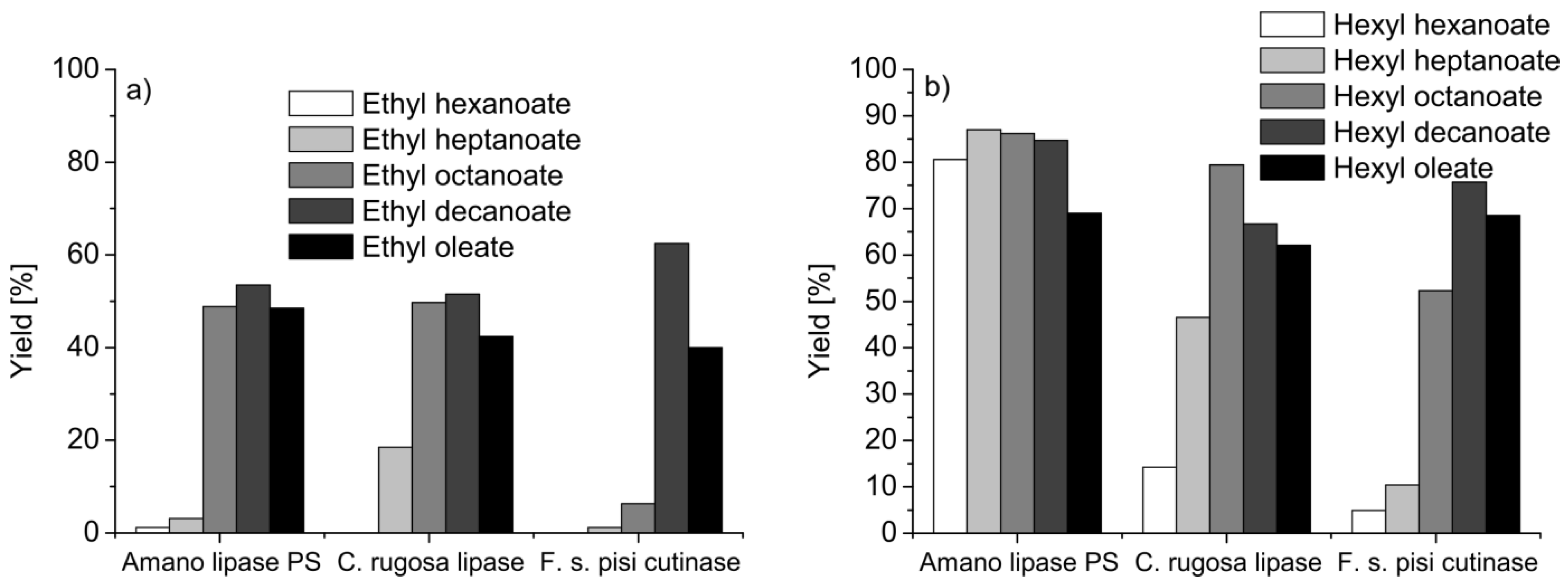
5.1. Small Molecules
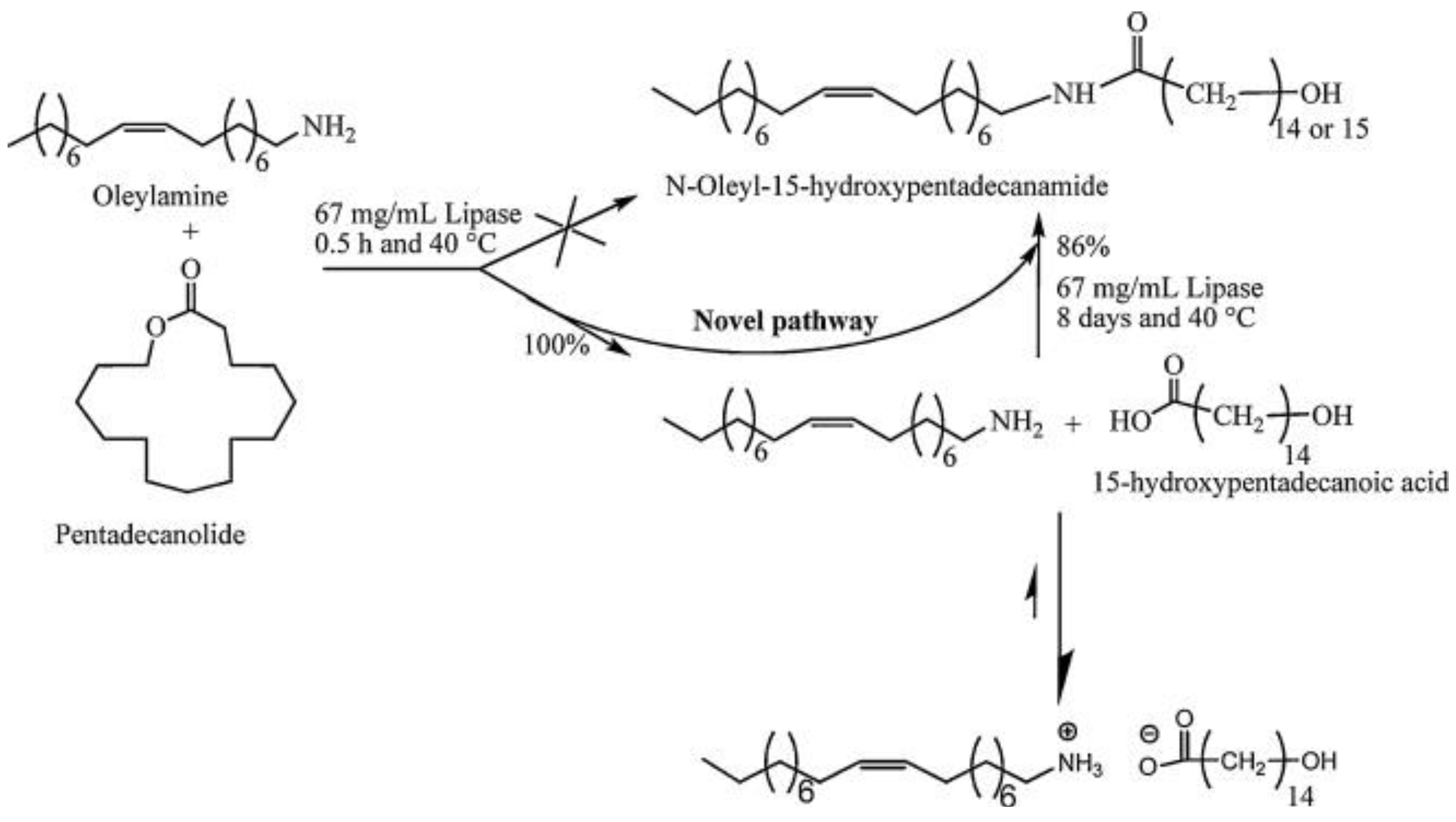
5.2. Polymers
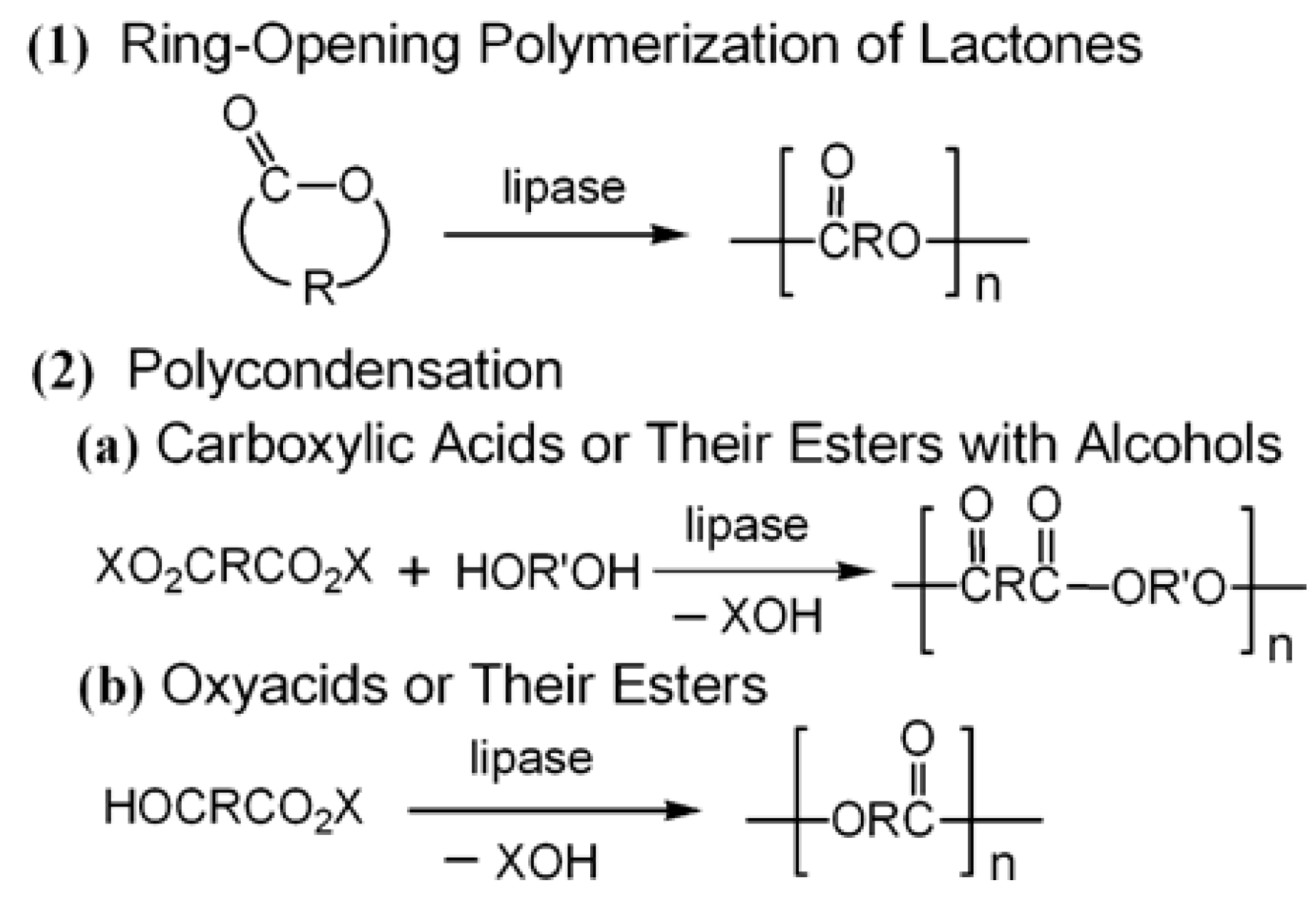
6. Enzyme Cleavable Nanostructures
7. Concluding Remarks
Conflict of Interest
References
- Beloqui, A.; de María, P.D.; Golyshin, P.N.; Ferrer, M. Recent trends in industrial microbiology. Curr. Opin. Microbiol. 2008, 11, 240–248. [Google Scholar] [CrossRef]
- Nestl, B.M.; Nebel, B.A.; Hauer, B. Recent progress in industrial biocatalysis. Curr. Opin. Chem. Biol. 2011, 15, 187–193. [Google Scholar] [CrossRef]
- Clouthier, C.M.; Pelletier, J.N. Expanding the organic toolbox: A guide to integrating biocatalysis in synthesis. Chem. Soc. Rev. 2012, 41, 1585–1605. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar]
- Illanes, A.; Cauerhff, A.; Wilson, L.; Castro, G.R. Recent trends in biocatalysis engineering. Bioresour. Technol. 2012, 115, 48–57. [Google Scholar] [CrossRef]
- Simon, M.-O.; Li, C.-J. Green chemistry oriented organic synthesis in water. Chem. Soc. Rev. 2012, 41, 1415–1427. [Google Scholar] [CrossRef]
- Landfester, K. Synthesis of colloidal particles in miniemulsions. Annu. Rev. Mater. Res. 2006, 36, 231–279. [Google Scholar] [CrossRef]
- Landfester, K. Miniemulsion polymerization and the structure of polymer and hybrid nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 4488–4507. [Google Scholar] [CrossRef]
- Landfester, K.; Weiss, C.K. Encapsulation by Miniemulsion Polymerization. Adv. Polym. Sci. 2010, 229, 1–49. [Google Scholar] [CrossRef]
- Weiss, C.K.; Landfester, K. Miniemulsion Polymerization as a Means to Encapsulate Organic and Inorganic Materials. Adv. Polym. Sci. 2010, 233, 185–236. [Google Scholar] [CrossRef]
- Munoz-Espi, R.; Weiss, C.K.; Landfester, K. Inorganic nanoparticles prepared in miniemulsion. Curr. Opin. Colloid Interface Sci. 2012, 17, 212–224. [Google Scholar] [CrossRef]
- Holmberg, K. Organic and bioorganic reactions in microemulsions. Adv. Colloid Interface Sci. 1994, 51, 137–174. [Google Scholar] [CrossRef]
- Derango, R.A.; Chiang, L.C.; Dowbenko, R.; Lasch, J.G. Enzyme-mediated polymerization of acrylic monomers. Biotechnol. Tech. 1992, 6, 523–526. [Google Scholar] [CrossRef]
- Kalra, B.; Gross, R.A. Horseradish peroxidase mediated free radical polymerization of methyl methacrylate. Biomacromolecules 2000, 1, 501–505. [Google Scholar] [CrossRef]
- Singh, A.; Ma, D.; Kaplan, D.L. Enzyme-Mediated free radical polymerization of styrene. Biomacromolecules 2000, 1, 592–596. [Google Scholar] [CrossRef]
- Qi, G.; Jones, C.W.; Schork, F.J. Enzyme-initiated miniemulsion polymerization. Biomacromolecules 2006, 7, 2927–2930. [Google Scholar] [CrossRef]
- Kohri, M.; Kobayashi, A.; Fukushima, H.; Kojima, T.; Taniguchi, T.; Saito, K.; Nakahira, T. Enzymatic miniemulsion polymerization of styrene with a polymerizable surfactant. Polym. Chem. 2012, 3, 900–906. [Google Scholar] [CrossRef]
- Kohri, M.; Kobayashi, A.; Fukushima, H.; Taniguchi, T.; Nakahira, T. Effect of surfactant type on enzymatic miniemulsion polymerization using horseradish peroxidase as a catalyst. Chem. Lett. 2012, 41, 1131–1133. [Google Scholar]
- Bechthold, N.; Landfester, K. Kinetics of miniemulsion polymerization as revealed by calorimetry. Macromolecules 2000, 33, 4682–4689. [Google Scholar] [CrossRef]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147–148, 237–250. [Google Scholar]
- Rogalska, E.; Cudrey, C.; Ferrato, F.; Verger, R. Stereoselective hydrolysis of triglycerides by animal and microbial lipases. Chirality 1993, 5, 24–30. [Google Scholar] [CrossRef]
- Schmid, R.D.; Verger, R. Lipases: Interfacial enzymes with attractive applications. Angew. Chem. Int. Ed. 1998, 37, 1609–1633. [Google Scholar]
- Wilde, P.J.; Chu, B.S. Interfacial colloidal aspects of lipid digestion. Adv. Colloid Interface Sci. 2011, 165, 14–22. [Google Scholar] [CrossRef]
- Okumura, S.; Iwai, M.; Tsujisaka, Y. Synthesis of various kinds of esters by 4 microbial lipases. Biochim. Biophys. Acta 1979, 575, 156–165. [Google Scholar] [CrossRef]
- Eggers, D.K.; Blanch, H.W.; Prausnitz, J.M. Extractive catalysis: Solvent effects on equilibria of enzymatic reactions in two-phase systems. Enzyme Microb. Technol. 1989, 11, 84–89. [Google Scholar] [CrossRef]
- Bornscheuer, U.T. Lipase-catalyzed syntheses of monoacylglycerols. Enzyme Microb. Technol. 1995, 17, 578–586. [Google Scholar] [CrossRef]
- Castro, G.R.; Knubovets, T. Homogeneous biocatalysis in organic solvents and water-organic mixtures. Crit. Rev. Biotechnol. 2003, 23, 195–231. [Google Scholar]
- Biasutti, M.A.; Abuin, E.B.; Silber, J.J.; Correa, N.M.; Lissi, E.A. Kinetics of reactions catalyzed by enzymes in solutions of surfactants. Adv. Colloid Interface Sci. 2008, 136, 1–24. [Google Scholar] [CrossRef]
- Monot, F.; Borzeix, F.; Bardin, M.; Vandecasteele, J.-P. Enzymatic esterification in organic media: Role of water and organic solvent in kinetics and yield of butyl butyrate synthesis. Appl Microb. Biotechnol. 1991, 35, 759–765. [Google Scholar]
- Carvalho, C.M.L.; Aires-Barros, M.R.; Cabral, J.M.S. Cutinase: From molecular level to bioprocess development. Biotechnol. Bioeng. 1999, 66, 17–34. [Google Scholar]
- Rona, P.; Ammon, R. Tests on enzymatic ester-hydrolysis and ester-synthesis. Biochem. Z. 1932, 249, 446–454. [Google Scholar]
- Sym, E.A. On the esterase-effect. III. Biochem. Z. 1933, 258, 304–324. [Google Scholar]
- de Lima, A.P.D.; Aschenbrenner, E.M.; Oliveira, S.; Doucet, J.-B.; Weiss, C.K.; Ziener, U.; Fonseca, L.P.; Ricardo, N.M.P.S.; de Freitas, L.L.; Petzhold, C.L.; et al. Towards regioselective enzymatic hydrolysis and glycerolysis of tricaprylin in miniemulsion and the direct preparation of polyurethane from the hydrolysis products. J. Mol. Catal. to be submitted for publication. 2013. [Google Scholar]
- Aschenbrenner, E.M.; Weiss, C.K.; Landfester, K. Enzymatic esterification in aqueous miniemulsions. Chem. Eur. J. 2009, 15, 2434–2444. [Google Scholar] [CrossRef]
- de Barros, D.P.C.; Fernandes, P.; Cabral, J.M.S.; Fonseca, L.P. Synthetic application and activity of cutinase in an aqueous, miniemulsion model system: Hexyl octanoate synthesis. Catal. Today 2011, 173, 95–102. [Google Scholar]
- de Barros, D.P.C.; Fonseca, L.P.; Cabral, J.M.S.; Aschenbrenner, E.M.; Weiss, C.K.; Landfester, K. Miniemulsion as efficient system for enzymatic synthesis of acid alkyl esters. Biotechnol. Bioeng. 2010, 106, 507–515. [Google Scholar] [CrossRef]
- de Barros, D.P.C.; Fonseca, L.P.; Cabral, J.M.S.; Weiss, C.K.; Landfester, K. Synthesis of alkyl esters by cutinase in miniemulsion and organic solvent media. Biotechnol. J. 2009, 4, 674–683. [Google Scholar] [CrossRef]
- Målberg, S.; Finne-Wistrand, A.; Albertsson, A.-C. The environmental influence in enzymatic polymerization of aliphatic polyesters in bulk and aqueous mini-emulsion. Polymer 2010, 51, 5318–5322. [Google Scholar] [CrossRef]
- Taden, A.; Antonietti, M.; Landfester, K. Enzymatic polymerization towards biodegradable polyester nanoparticles. Macromol. Rapid Commun. 2003, 24, 512–516. [Google Scholar] [CrossRef]
- Groger, H.; May, O.; Husken, H.; Georgeon, S.; Drauz, K.; Landfester, K. Enantioselective enzymatic reactions in miniemulsions as efficient “nanoreactors”. Angew. Chem. Int. Ed. 2006, 45, 1645–1648. [Google Scholar] [CrossRef]
- Baile, M.; Chou, Y.J.; Saam, J.C. Direct polyesterification in aqueous emulsion. Polym. Bull. 1990, 23, 251–257. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Suda, S.; Namekawa, S. Dehydration polymerization in aqueous medium catalyzed by lipase. Chem. Lett. 1997, 26. [Google Scholar] [CrossRef]
- Suda, S.; Uyama, H.; Kobayashi, S. Dehydration polycondensation in water for synthesis of polyesters by lipase catalyst. Proc. Jpn. Acad. Ser. B 1999, 75, 201–206. [Google Scholar] [CrossRef]
- Manabe, K.; Iimura, S.; Sun, X.M.; Kobayashi, S. Dehydration reactions in water. Bronsted acid-surfactant-combined catalyst for ester, ether, thioether, and dithioacetal formation in water. J. Am. Chem. Soc. 2002, 124, 11971–11978. [Google Scholar]
- Manabe, K.; Sun, X.M.; Kobayashi, S. Dehydration reactions in water. Surfactant-type bronsted acid-catalyzed direct esterification of carboxylic acids with alcohols in an emulsion system. J. Am. Chem. Soc. 2001, 123, 10101–10102. [Google Scholar] [CrossRef]
- De Barros, D.P.C.; Fonseca, L.P.; Cabral, J.M.S.; Weiss, C.K.; Landfester, K. Biosynthesis of fatty acids alkyl esters in miniemulsion as a reaction media. New Biotechnol. 2009, 25, S116. [Google Scholar]
- de Barros, D.P.C.; Fernandes, P.; Cabral, J.M.S.; Fonseca, L.P. Operational stability of cutinase in organic solvent system: Model esterification of alkyl esters. J. Chem. Technol. Biotechnol. 2010, 85, 1553–1560. [Google Scholar] [CrossRef]
- De Barros, D.P.C.; Azevedo, A.M.; Cabral, J.M.S.; Fonseca, L.P. Optimization of flavor esters synthesis by fusarium solani pisi cutinase. J. Food Biochem. 2012, 36, 275–284. [Google Scholar]
- de Barros, D.P.C.; Fonseca, L.P.; Fernandes, P.; Cabral, J.M.S.; Mojovic, L. Biosynthesis of ethyl caproate and other short ethyl esters catalyzed by cutinase in organic solvent. J. Mol. Catal. B 2009, 60, 178–185. [Google Scholar] [CrossRef]
- Cunnah, P.J.; Aires-Barros, M.R.; Cabral, J.M.S. Esterification and transesterification catalysed by cutinase in reverse micelles of ctab for the synthesis of short chain esters. Biocatal. Biotransform. 1996, 14, 125–146. [Google Scholar] [CrossRef]
- Pinto-Sousa, A.M.C.; Cabral, J.M.S.; Aires-Barros, M.R. Ester Synthesis by a recombinant cutinase in reversed micelles of a natural phospholipid. Biocatal. Biotransform. 1994, 9, 169–179. [Google Scholar] [CrossRef]
- Gotor, V. Non-conventional hydrolase chemistry: Amide and carbamate bond formation catalyzed by lipases. Biorg. Med. Chem. 1999, 7, 2189–2197. [Google Scholar] [CrossRef]
- Gotor-Fernández, V.; Busto, E.; Gotor, V. Candida antarctica lipase b: An ideal biocatalyst for the preparation of nitrogenated organic compounds. Adv. Synth. Catal. 2006, 348, 797–812. [Google Scholar] [CrossRef]
- Ragupathy, L.; Pluhar, B.; Ziener, U.; Keller, H.; Dyllick-Brenzinger, R.; Landfester, K. Enzymatic aminolysis of lactones in aqueous miniemulsion: Catalysis through a novel pathway. J. Mol. Catal. B 2010, 62, 270–276. [Google Scholar] [CrossRef]
- Gross, R.A.; Ganesh, M.; Lu, W. Enzyme-catalysis breathes new life into polyester condensation polymerizations. Trends Biotechnol. 2010, 28, 435–443. [Google Scholar] [CrossRef]
- Kobayashi, S. Recent developments in lipase-catalyzed synthesis of polyesters. Macromol. Rapid Commun. 2009, 30, 237–266. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: an opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic polymerization. Chem. Rev. 2001, 101, 3793–3818. [Google Scholar] [CrossRef]
- Namekawa, S.; Uyama, H.; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of lactones in water. Polym. J. 1998, 30, 269–271. [Google Scholar] [CrossRef]
- Nallani, M.; de Hoog, H.-P.M.; Cornelissen, J.J.L.M.; Palmans, A.R.A.; van Hest, J.C.M.; Nolte, R.J.M. Polymersome nanoreactors for enzymatic ring-opening polymerization. Biomacromolecules 2007, 8, 3723–3728. [Google Scholar] [CrossRef]
- Panlawan, P.; Luangthongkam, P.; Wiemann, L.O.; Sieber, V.; Marie, E.; Durand, A.; Inprakhon, P. Lipase-catalyzed interfacial polymerization of ω-pentadecalactone in aqueous biphasic medium: A mechanistic study. J. Mol. Catal. B 2013, 88, 69–76. [Google Scholar] [CrossRef]
- Gardella, J.A.; Novak, F.P.; Hercules, D.M. Static secondary ion mass-spectrometry for study of surface hydrolysis of poly(tert-butyl methacrylate). Anal. Chem. 1984, 56, 1371–1375. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Birkinshaw, C. Hydrolysis of poly (n-butylcyanoacrylate) nanoparticles using esterase. Polym. Degrad. Stable 2002, 78, 7–15. [Google Scholar] [CrossRef]
- Sullivan, C.O.; Birkinshaw, C. In vitro degradation of insulin-loaded poly (n-butylcyanoacrylate) nanoparticles. Biomaterials 2004, 25, 4375–4382. [Google Scholar] [CrossRef]
- Vauthier, C.; Dubernet, C.; Fattal, E.; Pinto-Alphandary, H.; Couvreur, P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv. Drug Deliv. Rev. 2003, 55, 519–548. [Google Scholar] [CrossRef]
- Klinger, D.; Aschenbrenner, E.M.; Weiss, C.K.; Landfester, K. Enzymatically degradable nanogels by inverse miniemulsion copolymerization of acrylamide with dextran methacrylates as crosslinkers. Polym. Chem. 2012, 3, 204–216. [Google Scholar] [CrossRef]
- Klinger, D.; Landfester, K. Enzymatic- and light-degradable hybrid nanogels: Crosslinking of polyacrylamide with acrylate-functionalized dextrans containing photocleavable linkers. J. Polym. Sci. Part A 2012, 50, 1062–1075. [Google Scholar] [CrossRef]
- Andrieu, J.; Kotman, N.; Maier, M.; Mailander, V.; Strauss, W.S.L.; Weiss, C.K.; Landfester, K. Live monitoring of cargo release from peptide-based hybrid nanocapsules induced by enzyme cleavage. Macromol. Rapid Commun. 2012, 33, 248–253. [Google Scholar] [CrossRef]
- Maier, M.; Kotman, N.; Friedrichs, C.; Andrieu, J.; Wagner, M.; Graf, R.; Strauss, W.S.L.; Mailander, V.; Weiss, C.K.; Landfester, K. Highly site specific, protease cleavable, hydrophobic peptide-polymer nanoparticles. Macromolecules 2011, 44, 6258–6267. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Weiss, C.K.; Landfester, K. Enzymatic Catalysis at Interfaces—Heterophase Systems as Substrates for Enzymatic Action. Catalysts 2013, 3, 401-417. https://doi.org/10.3390/catal3020401
Weiss CK, Landfester K. Enzymatic Catalysis at Interfaces—Heterophase Systems as Substrates for Enzymatic Action. Catalysts. 2013; 3(2):401-417. https://doi.org/10.3390/catal3020401
Chicago/Turabian StyleWeiss, Clemens K., and Katharina Landfester. 2013. "Enzymatic Catalysis at Interfaces—Heterophase Systems as Substrates for Enzymatic Action" Catalysts 3, no. 2: 401-417. https://doi.org/10.3390/catal3020401