Photocatalytic Oxidation Processes for Toluene Oxidation over TiO2 Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
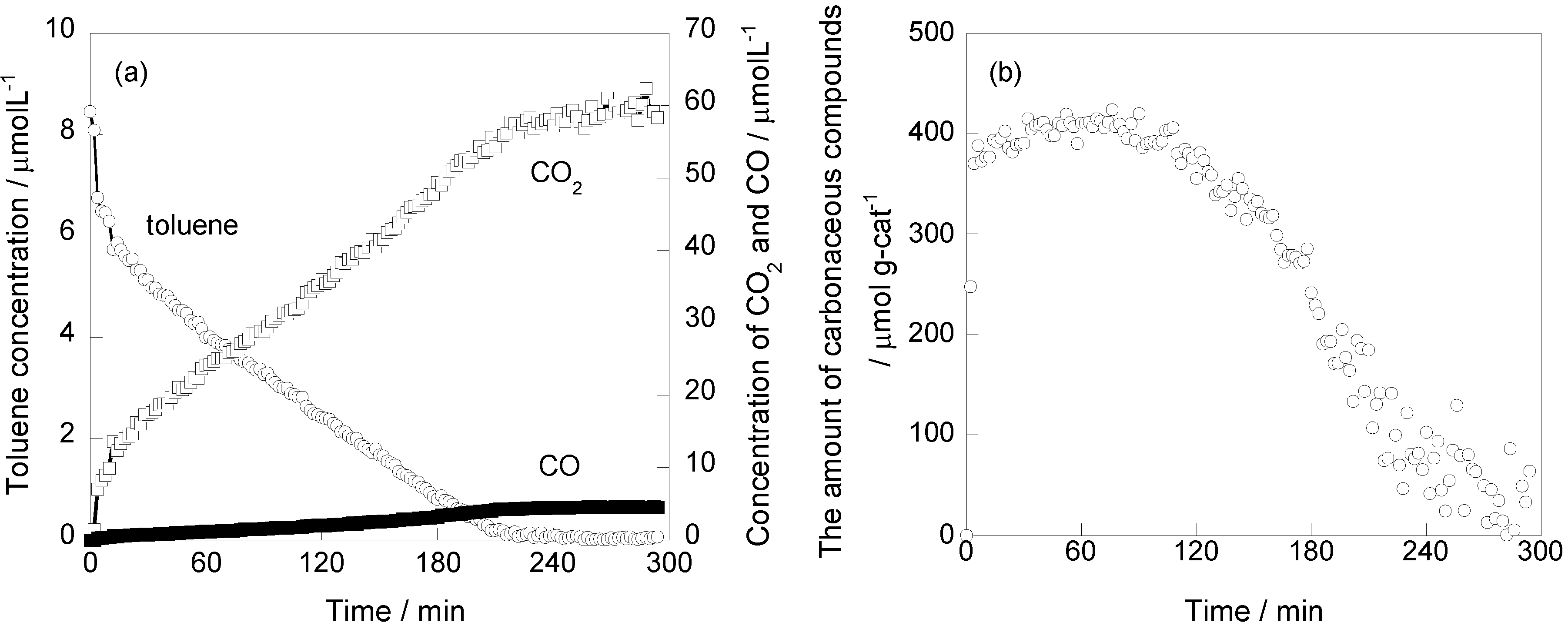
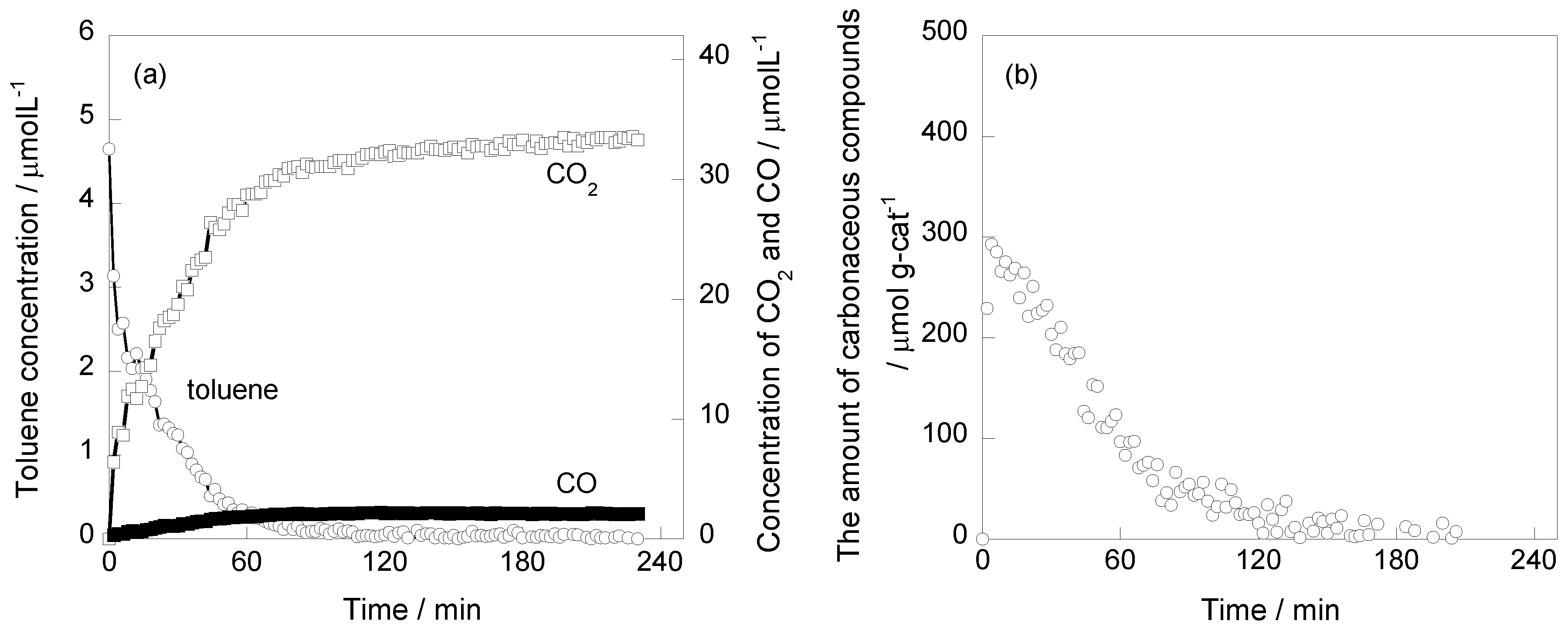
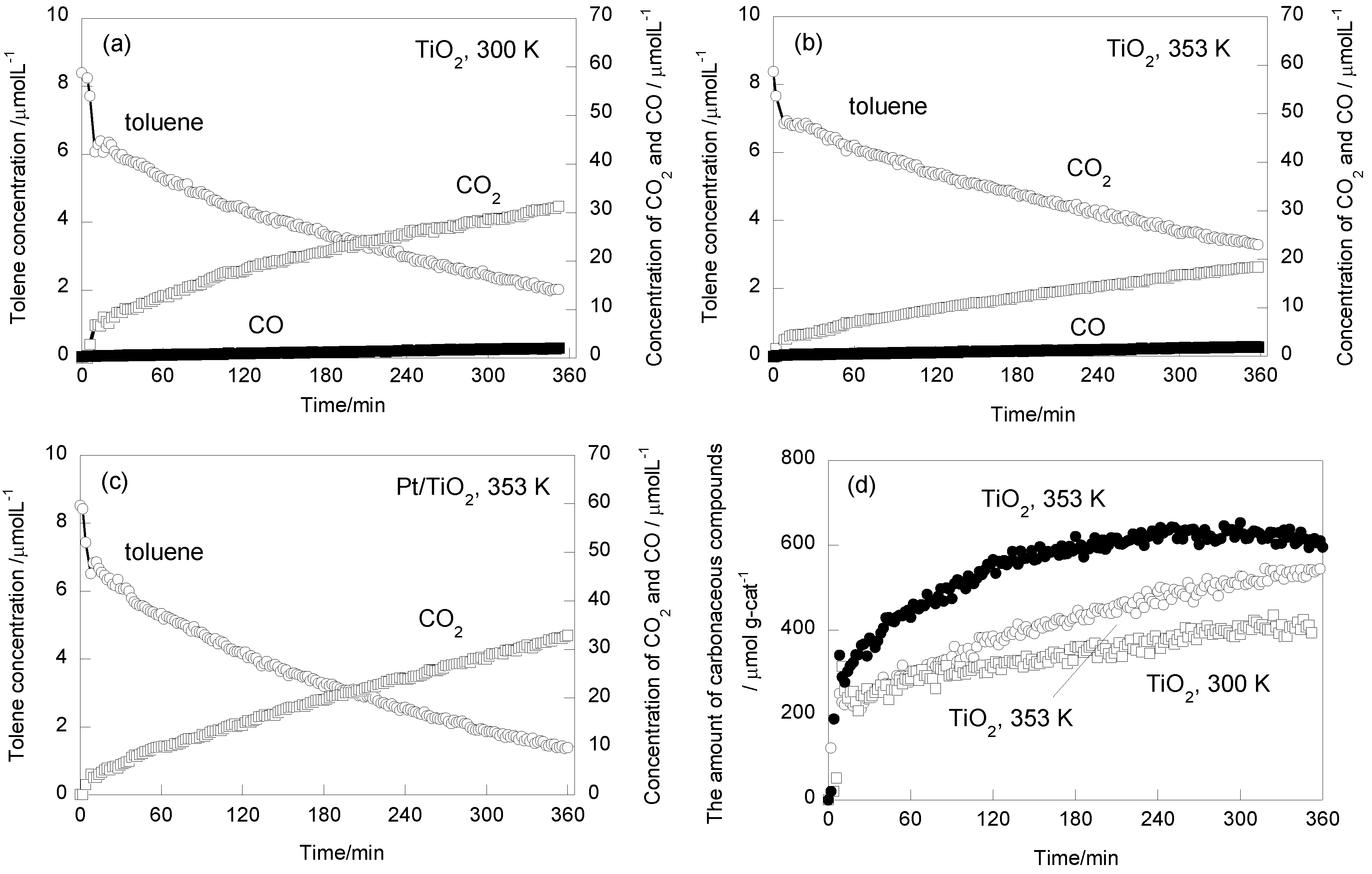
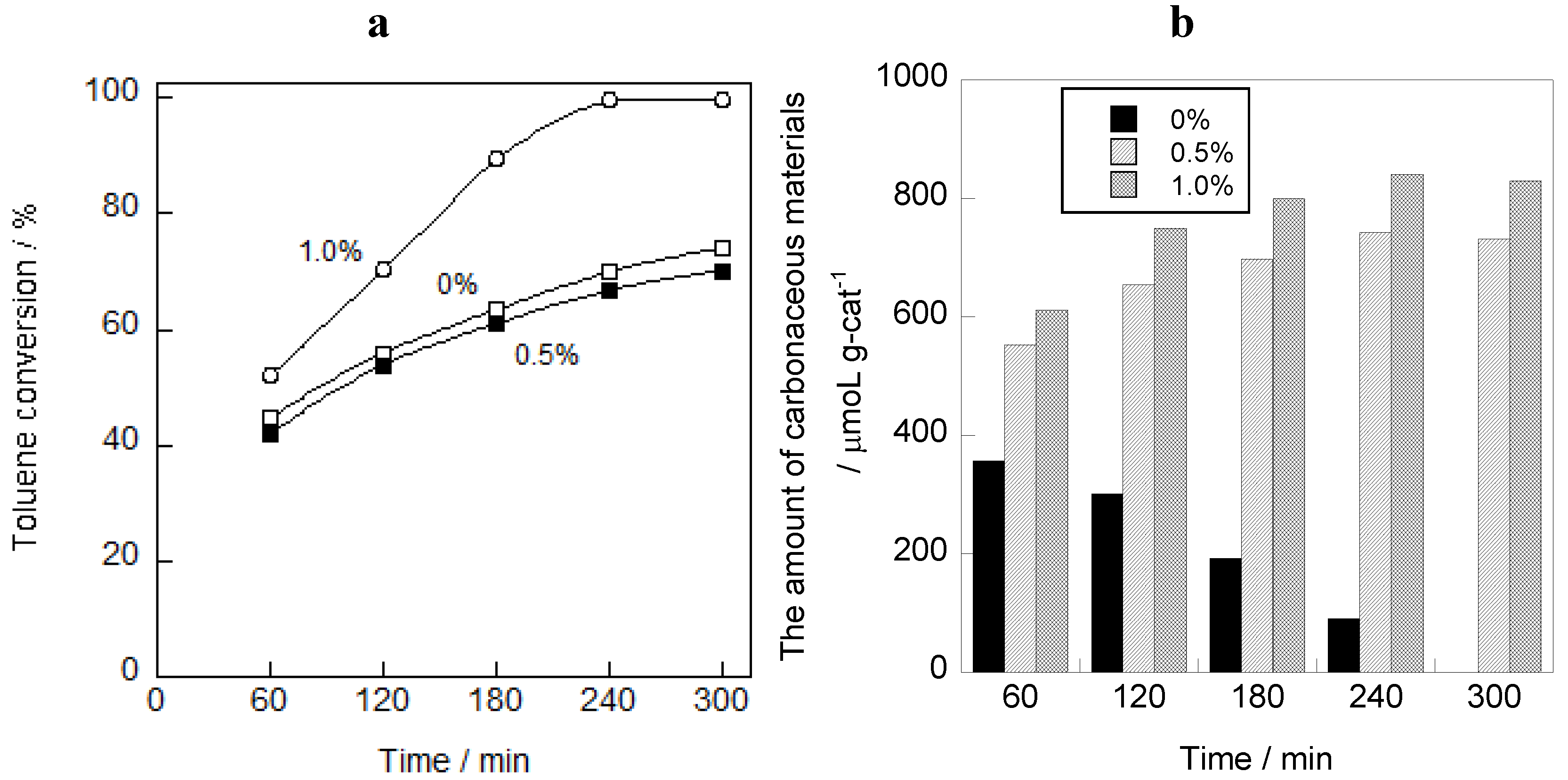
2.1. Photooxidation Behavior of Toluene on TiO2

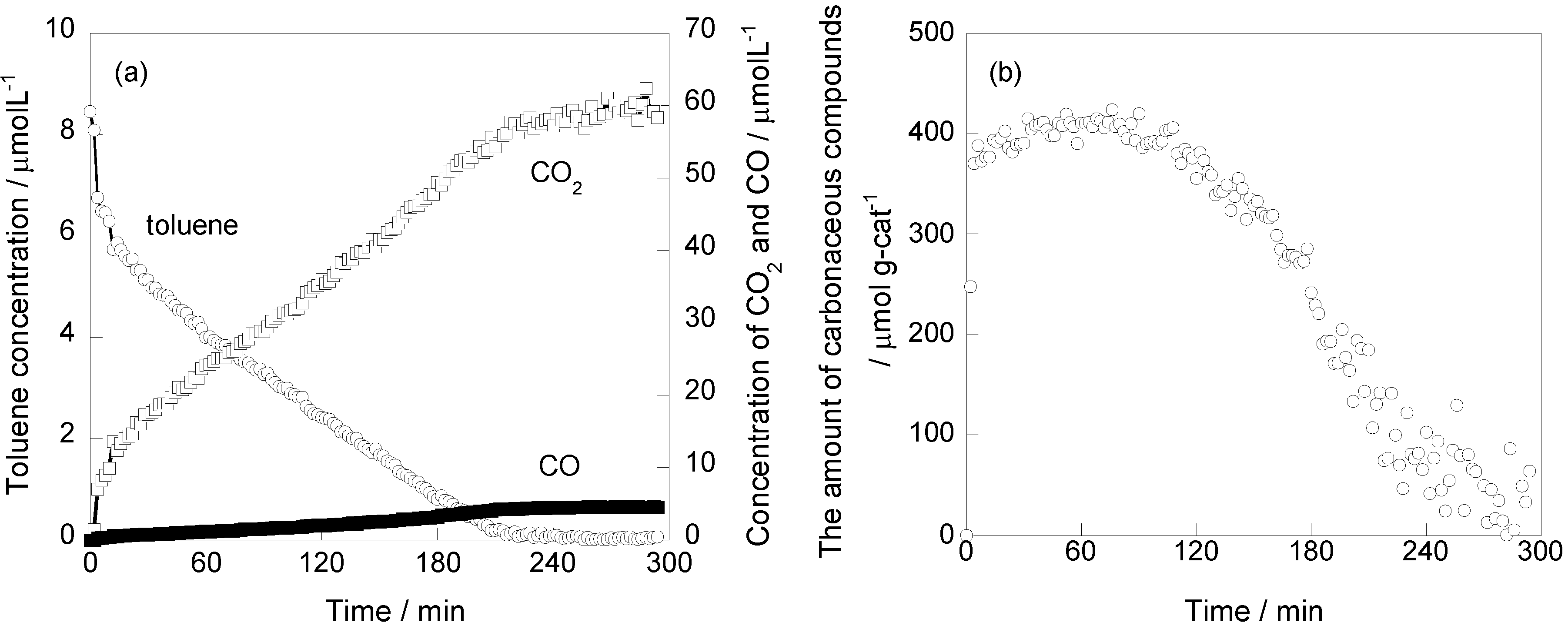
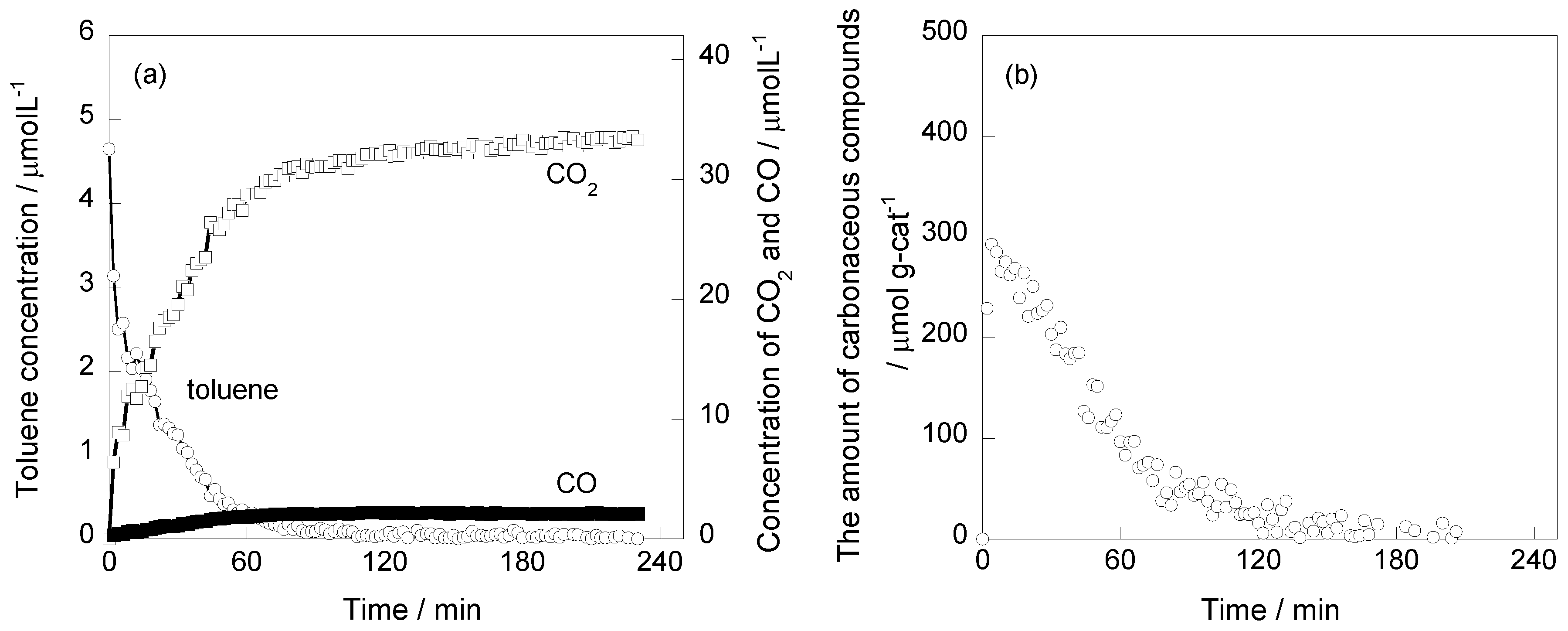


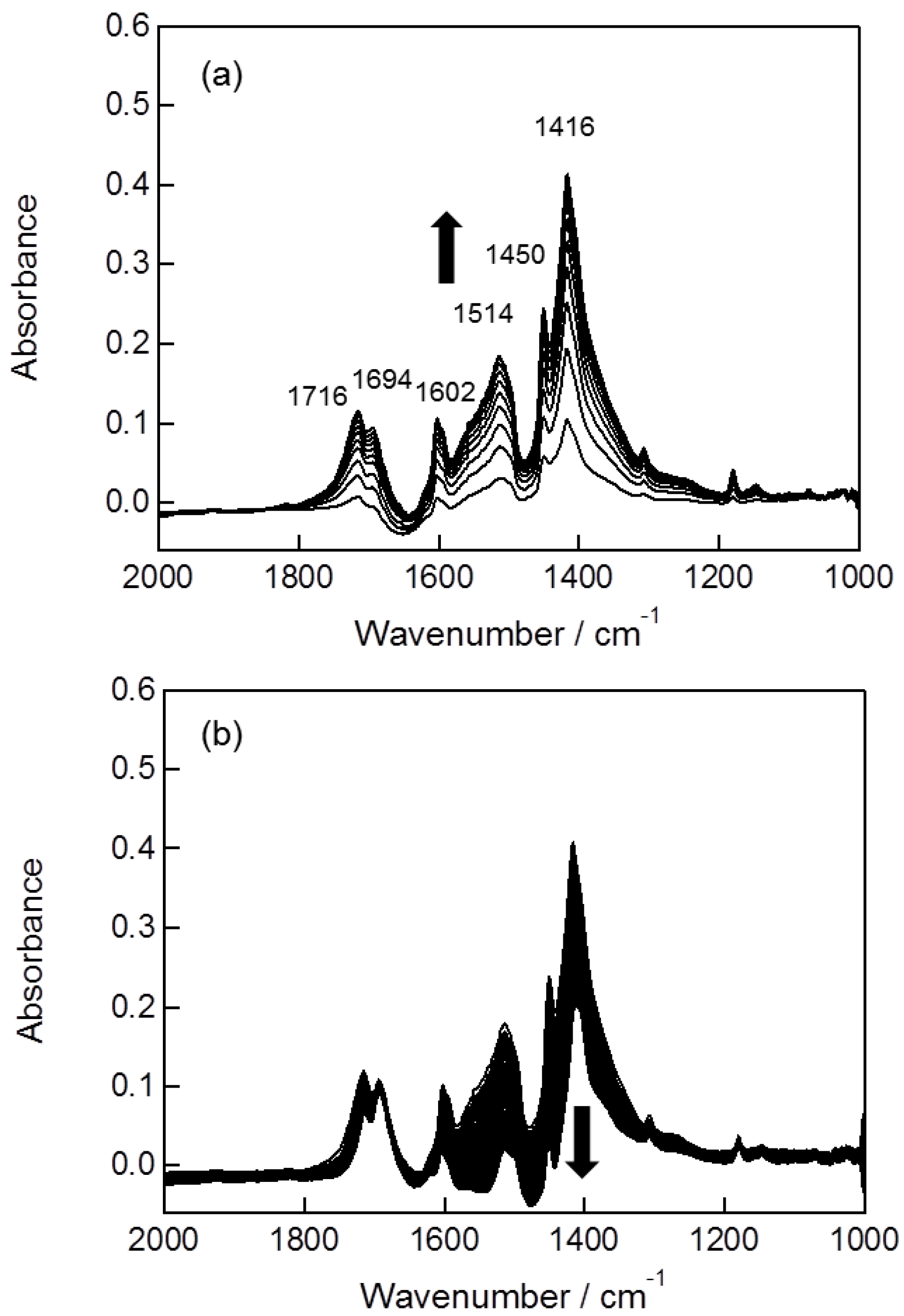
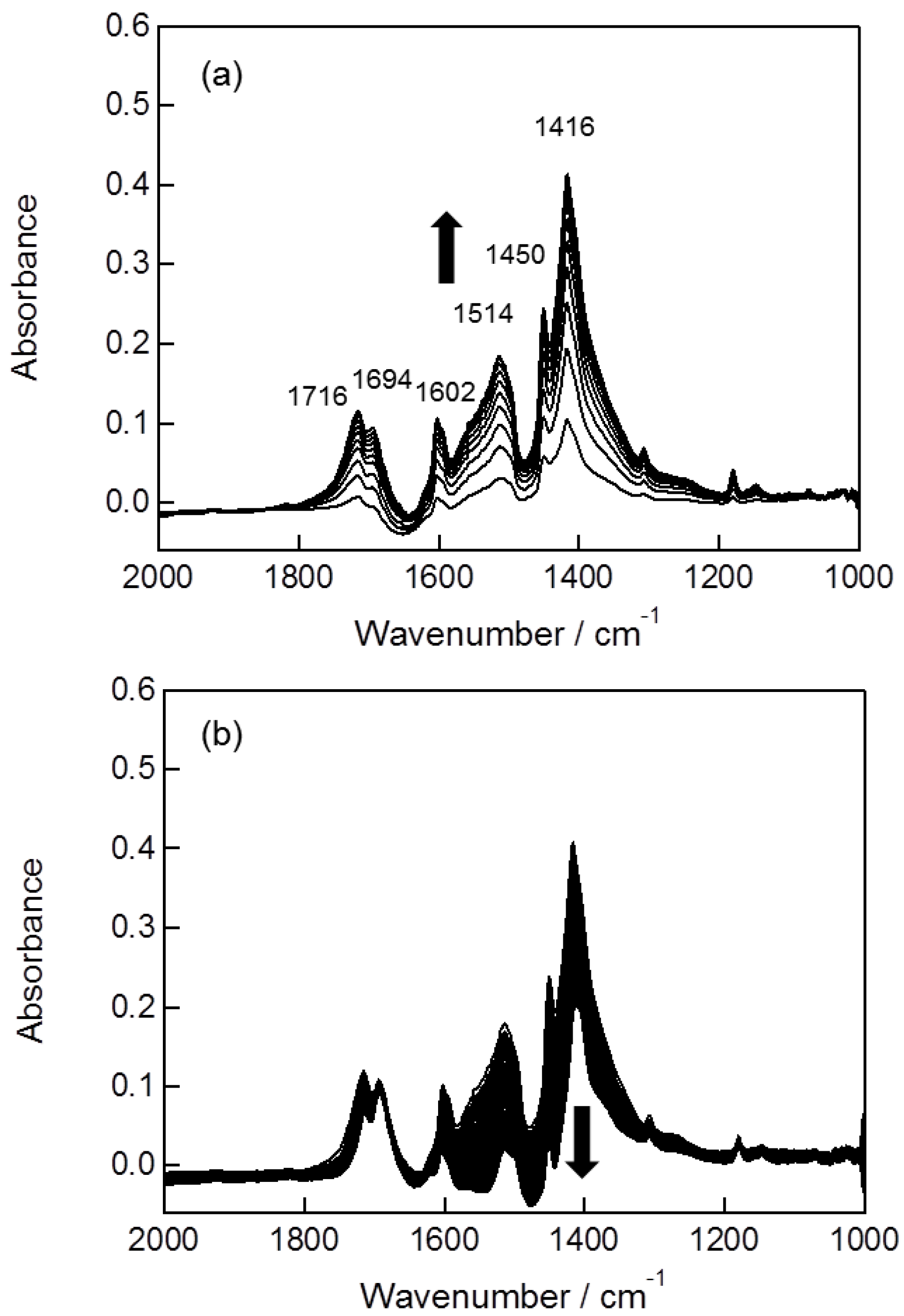
2.2. In Situ FTIR Studies for Toluene Photooxidation

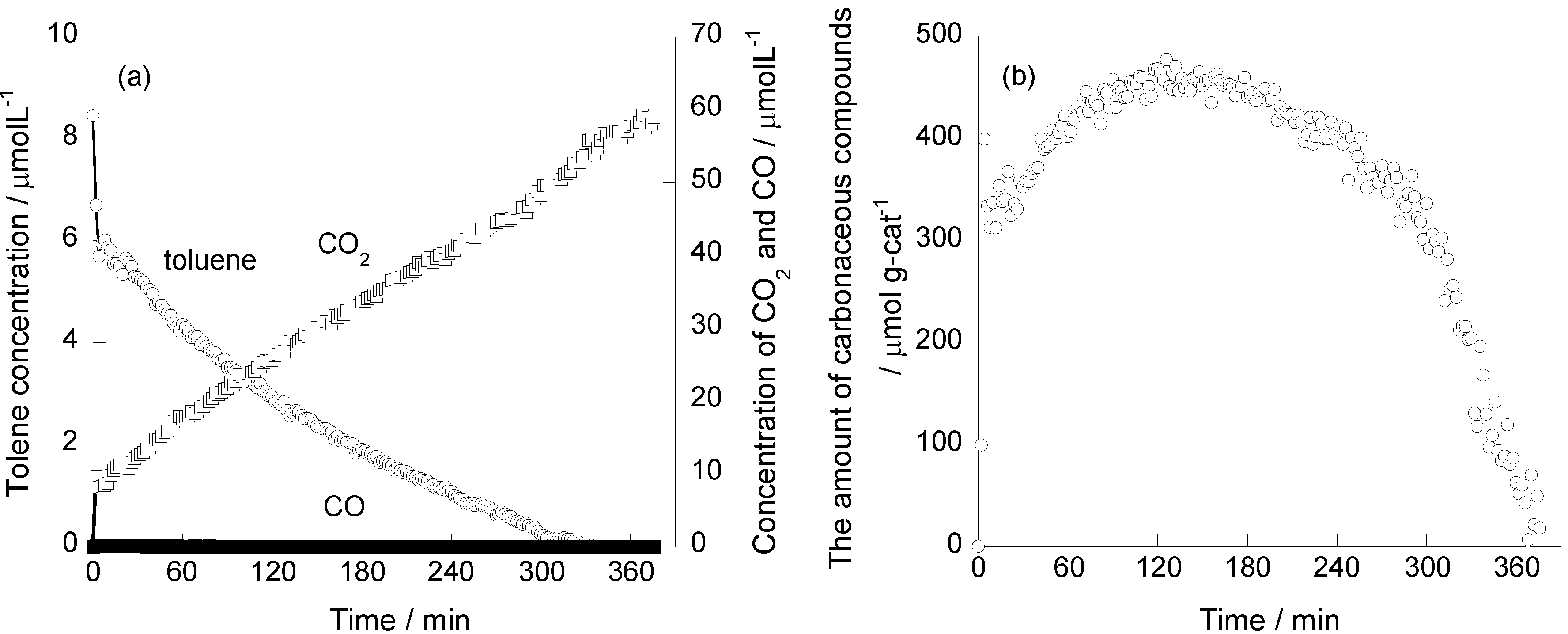
2.3. Effect of Pt Deposition and Catalyst Heating


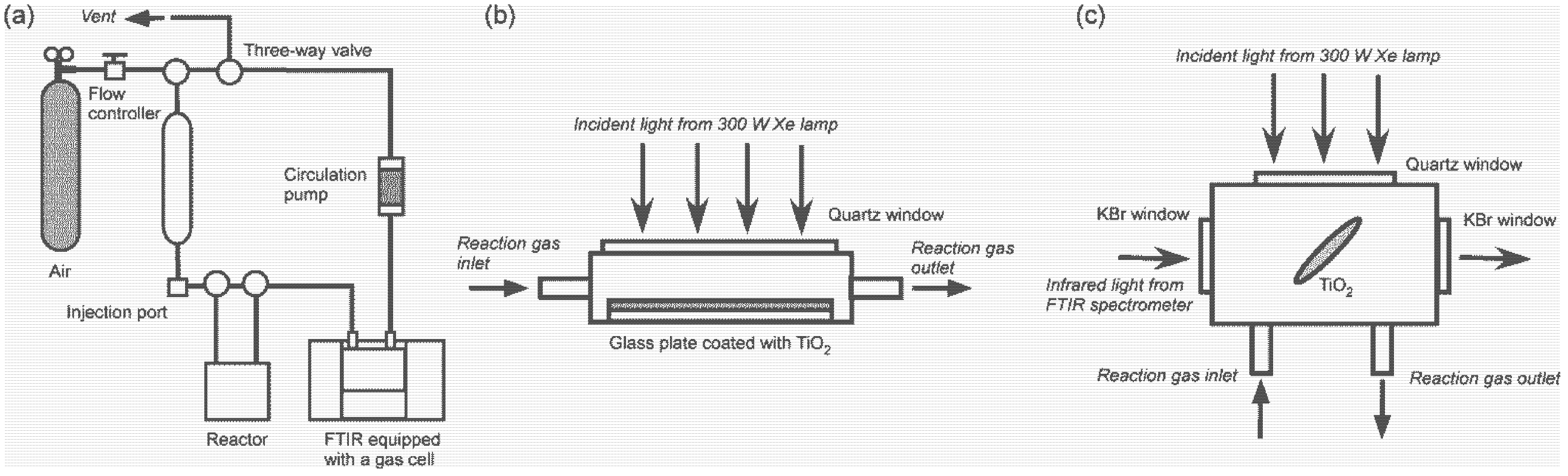
3. Experimental Section
3.1. Catalyst Materials
3.2. Photocatalytic Test Reaction

3.3. FTIR Studies
4. Conclusions
Conflict of Interest
References
- Linsebigler, A.L.; Lu, G.; Yates, J.T., Jr. Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results. Chem. Rev. 1995, 95, 735–758. [Google Scholar] [CrossRef]
- Hoffman, M.R.; Martin, S.T.; Choi, W.Y.; Bahnemann, D.W. Environmental applications of semiconductor photocatalysis. Chem. Rev. 1995, 95, 69–96. [Google Scholar]
- Agrios, A.G.; Pichat, P. State of the art and perspectives on materials and applications of photocatalysis over TiO2. J. Appl. Electrochem. 2005, 35, 655–663. [Google Scholar] [CrossRef]
- Chen, X.; Mao, S.S. Titanium dioxide nanomaterials: Synthesis, properties, modifications, and applications. Chem. Rev. 2007, 107, 2891–2959. [Google Scholar] [CrossRef]
- Ollis, D.F. Photocatalytic purification and remediation of contaminated air and water. C. R. Acad. Sci. Ser. 2000, 3, 405–411. [Google Scholar]
- Wang, S.; Ang, H.M.; Tade, M.O. Volatile organic compounds in indoor environment and photocatalytic oxidation: State of the art. Environ. Int. 2007, 33, 694–705. [Google Scholar] [CrossRef]
- Ibusuki, T.; Takeuchi, K. Toluene oxidation on U.V.-irradiated titanium dioxide with and without O2, NO2 or H2O at ambient temperature. Atmos. Environ. 1986, 20, 1711–1715. [Google Scholar] [CrossRef]
- Obee, T.N.; Brown, R.T. TiO2 photocatalysis for indoor air applications: effects of humidity and trace contaminant levels on the oxidation rates of formaldehyde, toluene, and 1,3-butadiene. Environ. Sci. Technol. 1995, 29, 1223–1231. [Google Scholar] [CrossRef]
- Ameen, M.M.; Raupp, G.B. Reversible catalyst deactivation in the photocatalytic oxidation of dilute O-xylene in air. J. Catal. 1999, 184, 112–122. [Google Scholar] [CrossRef]
- Cao, L.X.; Gao, Z.; Suib, S.L.; Obee, T.N.; Hay, S.O.; Freihaut, J.D. Photocatalytic oxidation of toluene on nanoscale TiO2 catalysts: Studies of deactivation and regeneration. J. Catal. 1999, 184, 112–122. [Google Scholar] [CrossRef]
- D'Hennezel, O.; Pichat, P.; Ollis, D.F. Benzene and toluene gas-phase photocatalytic degradation over H2O and HCL pretreated TiO2 by-products and mechanisms. J. Photochem. Photobiol. A 1998, 118, 197–204. [Google Scholar] [CrossRef]
- Mendez-Roman, R.; Cardona-Martinez, N. Relationship between the formation of surface species and catalyst deactivation during the gas-phase photocatalytic oxidation of toluene. Catal. Today 1998, 40, 353–365. [Google Scholar] [CrossRef]
- Maira, A.J.; Coronado, J.M.; Augugliaro, V.; Yeung, K.L.; Conesa, J.C.; Soria, J. Fourier transform infrared study of the performance of nanostructured TiO2 particles for the photocatalytic oxidation of gaseous toluene. J. Catal. 2001, 202, 413–420. [Google Scholar]
- Marci, G.; Addamo, M.; Augugliaro, V.; Coluccia, S.; García-López, E.; Loddo, V.; Martra, G.; Palmisano, L.; Schiavello, M. Photocatalytic oxidation of toluene on irradiated TiO2: Comparison of degradation performance in humidified air, in water and in water containing a zwitterionic surfactant. J. Photochem. Photobiol. A 2003, 160, 105–114. [Google Scholar] [CrossRef]
- Blount, M.C.; Falconer, J.L. Steady-state surface species during toluene photocatalysis. Appl. Catal. B 2002, 139, 39–50. [Google Scholar]
- Debono, O.; Thevenet, F.; Gravejat, P.; Hequet, V.; Raillard, C.; Lecoq, L.; Locoge, N. Toluene photocatalytic oxidation at ppbv levels: Kinetic investigation and carbon balance determination. Appl. Catal. B 2011, 106, 600–608. [Google Scholar] [CrossRef]
- Vildozo, D.; Portel, R.; Ferronato, C.; Chovelon, J.M. Photocatalytic oxidation of 2-propanol/toluene binary mixtures at indoor air concentration levels. Appl. Catal. B 2011, 107, 347–354. [Google Scholar] [CrossRef]
- Sleiman, M.; Conchon, P.; Ferronato, C.; Chovelon, J.M. Photocatalytic oxidation of toluene at indoor air levels (ppbv): Towards a better assessment of conversion, reaction intermediates and mineralization. Appl. Catal. B 2009, 86, 159–165. [Google Scholar] [CrossRef]
- Thompson, T.L.; Yates, J.T., Jr. TiO2-based photocatalysis: Surface defects, oxygen and charge transfer. Top. Catal. 2005, 35, 197–210. [Google Scholar] [CrossRef]
- Thompson, T.L.; Yates, J.T., Jr. Surface science studies of the photoactivation of TiO2-new photochemical processes. Chem. Rev. 2006, 106, 4428–4453. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Fu, X.Z.; Zeltner, W.A.; Anderson, M.A. The gas-phase photocatalytic mineralization of benzene on porous titania-based catalysts. Appl. Catal. B 1995, 6, 209–224. [Google Scholar] [CrossRef]
- Li, Y.; Huang, J.C.; Peng, T.; Xu, J.; Zhao, X.J. Photothermocatalytic synergetic effect leads to high efficient detoxification of benzene on TiO2 and Pt/TiO2 nanocomposite. ChemCatChem 2010, 2, 1082–1087. [Google Scholar] [CrossRef]
- Einaga, H.; Futamura, S.; Ibusuki, T. Photocatalytic decomposition of benzene over TiO2 in a humidified airstream. Phys. Chem. Chem. Phys. 1999, 1, 4903–4908. [Google Scholar] [CrossRef]
- Einaga, H.; Futamura, S.; Ibusuki, T. Complete oxidation of benzene in gas phase by platinized titania photocatalysts. Environ. Sci. Technol. 2001, 35, 1880–1884. [Google Scholar]
- Einaga, H.; Futamura, S.; Ibusuki, T. Heterogeneous photocatalytic oxidation of benzene, toluene, cyclohexene and cyclohexane in humidified air, comparison of decomposition behavior on photoirradiated TiO2 catalyst. Appl. Catal. B 2002, 38, 215–225. [Google Scholar] [CrossRef]
- Einaga, H.; Ibusuki, T.; Futamura, S. Improvement of catalyst durability by deposition of Rh on TiO2 in photooxidation of aromatic compounds. Environ. Sci. Technol. 2004, 38, 285–289. [Google Scholar] [CrossRef]
- Einaga, H.; Ibusuki, T.; Futamura, S. Photocatalytic oxidation of benzene in air. J. Solar Energy Eng. 2004, 126, 789–793. [Google Scholar] [CrossRef]
- Suh, M.; Bagus, P.S.; Pak, S.; Rosynek, M.P.; Lunsford, J.H. Reactions of hydroxyl radicals on titania, silica, alumina, and gold surfaces. J. Phys. Chem. B 2000, 104, 2736–2742. [Google Scholar] [CrossRef]
- Hernández-Alonso, M.D.; Tejedor-Tejedor, I.; Coronado, J.M.; Anderson, M.A. Operando FTIR study of the photocatalytic oxidation of methylcyclohexane and toluene in air over TiO2-ZrO2 thin films: Influence of the aromaticity of the target molecule on deactivation. Appl. Catal. B 2011, 101, 283–293. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Z.; Zhao, Q.; Liu, S. FT-IR study of the photocatalytic degradation of gaseous toluene over UV-irradiated TiO2 microballs: Enhanced performance by hydrothermal treatment in alkaline solution. Appl. Surf. Sci. 2011, 257, 4709–4714. [Google Scholar]
- Augugliaro, V.; Coluccia, S.; Loddo, V.; Marchese, L.; Martra, G.; Palmisano, L.; Schiavello, M. Photocatalytic Oxidation of Gaseous Toluene on Anatase TiO2 Catalyst: Mechanistic Aspects and FT-IR Investigation. Appl. Catal. B 1999, 20, 15–27. [Google Scholar] [CrossRef]
- Martra, G.; Coluccia, S.; Marchese, L.; Augugliaro, V.; Loddo, V.; Palmisano, L.; Schiavello, M. The role of H2O in the photocatalytic oxidation of toluene in vapour phase on anatase TiO2 catalyst a FTIR study. Catal. Today 1999, 53, 695–702. [Google Scholar] [CrossRef]
- Deveau, P.A.; Arsac, F.; Thivel, P.X.; Ferronato, C.; Delpech, F.; Chovelon, J.M.; Kaluzny, P.; Monnetd, C. Different methods in TiO2 photodegradation mechanism studies: Gaseous and TiO2-adsorbed phases. J. Hazard. Mater. 2007, 144, 692–697. [Google Scholar] [CrossRef]
- Einaga, H.; Harada, M.; Futamura, S.; Ibusuki, T. Generation of active sites for CO photooxidation on TiO2 by platinum deposition. J. Phys. Chem. B 2003, 107, 9290–9297. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Einaga, H.; Mochiduki, K.; Teraoka, Y. Photocatalytic Oxidation Processes for Toluene Oxidation over TiO2 Catalysts. Catalysts 2013, 3, 219-231. https://doi.org/10.3390/catal3010219
Einaga H, Mochiduki K, Teraoka Y. Photocatalytic Oxidation Processes for Toluene Oxidation over TiO2 Catalysts. Catalysts. 2013; 3(1):219-231. https://doi.org/10.3390/catal3010219
Chicago/Turabian StyleEinaga, Hisahiro, Keisuke Mochiduki, and Yasutake Teraoka. 2013. "Photocatalytic Oxidation Processes for Toluene Oxidation over TiO2 Catalysts" Catalysts 3, no. 1: 219-231. https://doi.org/10.3390/catal3010219