1. Introduction
Research on polymerization using metallocene catalysts began with the identification of the structure of ferrocene by Wilkins and Fischer in 1952 and was further developed by Kaminsky in the 1980s. In the case of copolymerization using metallocene catalyst, preparing polymers with narrow molecular weight distribution, high stereo-regularity and uniform comonomer incorporation is much easier than is the case when using a conventional Ziegler-Natta catalyst. That is the reason why many studies have been performed regarding coordination polymerization using metallocene catalysts [
1].
Polyolefins are difficult to adhere and compatible with other polar polymers, because they are non-polar. The needs for improving compatibility and adhesion of polyolefins have been very high in the industry.
Theoretically, there are three possible approaches to the functionalization of polyolefins. They include (a) direct polymerization of α-olefins with functional monomers; (b) chemical modification of the preformed polymer and (c) a reactive copolymer approach that entails incorporating reactive comonomers that can be selectively and effectively interconverted into functional groups.
The third approach is a relatively new one. However, studies on the polyolefins with reactive groups have been limited, even though it may be possible to use the terpolymers to develop new high-functional polyolefins [
2,
3,
4,
5]. T. C. Chung
et al. reported on the terpolymerization of ethylene, 1-octene and
p-methylstyrene (
p-MS) using [C
5Me
4(SiMe
2NtBu)]TiCl
2 metallocene catalyst [
6,
7].
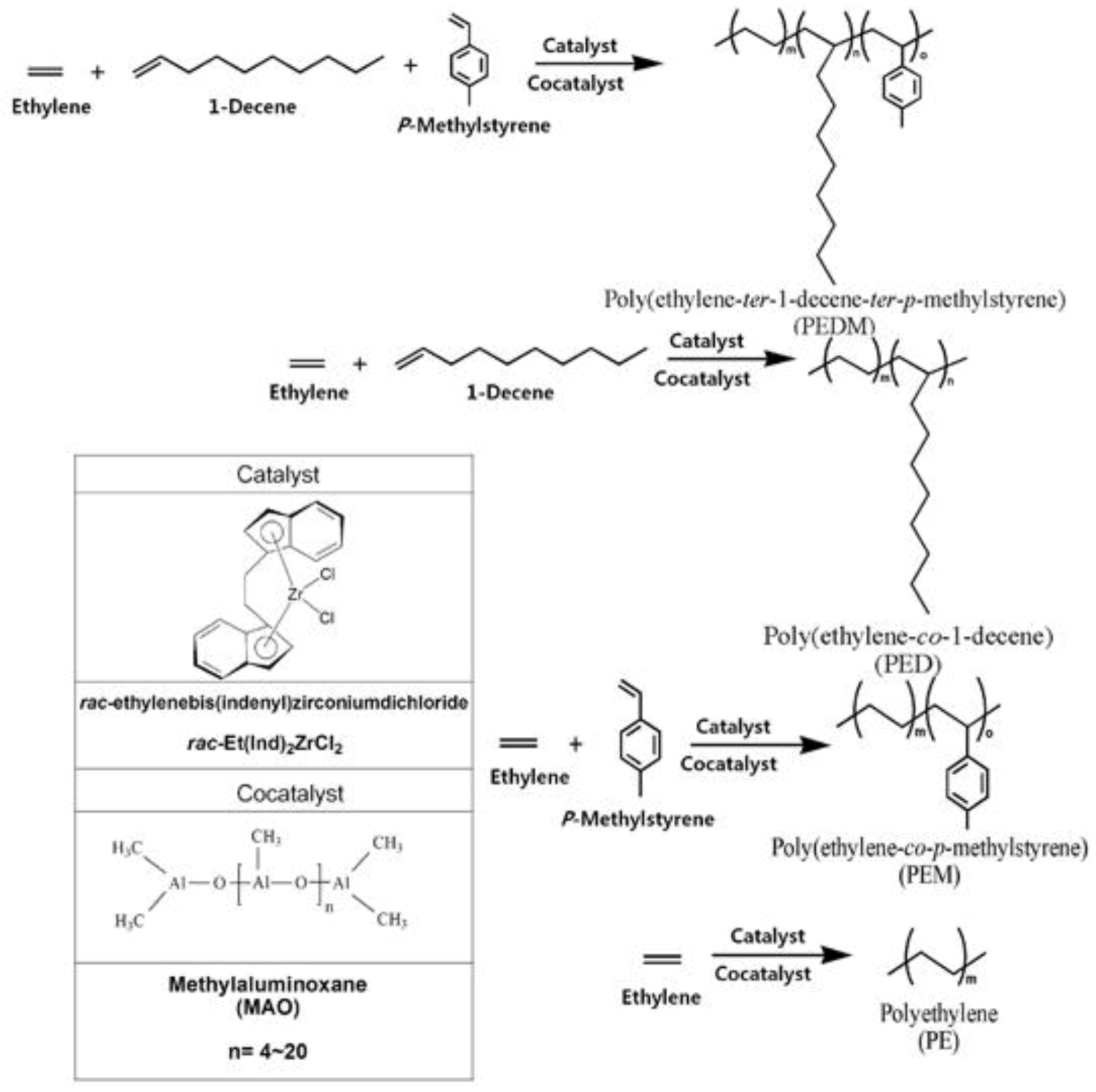
In this study, we prepared polyethylene (PE), poly(ethylene-co-1-decene) (PED), poly(ethylene-co-p-methylstyrene) (PEM) and poly(ethylene-ter-1-decene-ter-p- methylstyrene) (PEDM) using a bridged metallocene catalyst and a cocatalyst system. We then characterized and compared with various properties of the polymers.
2. Results and Discussion
2.1. Homo-, Co- and Ter-Polymerization
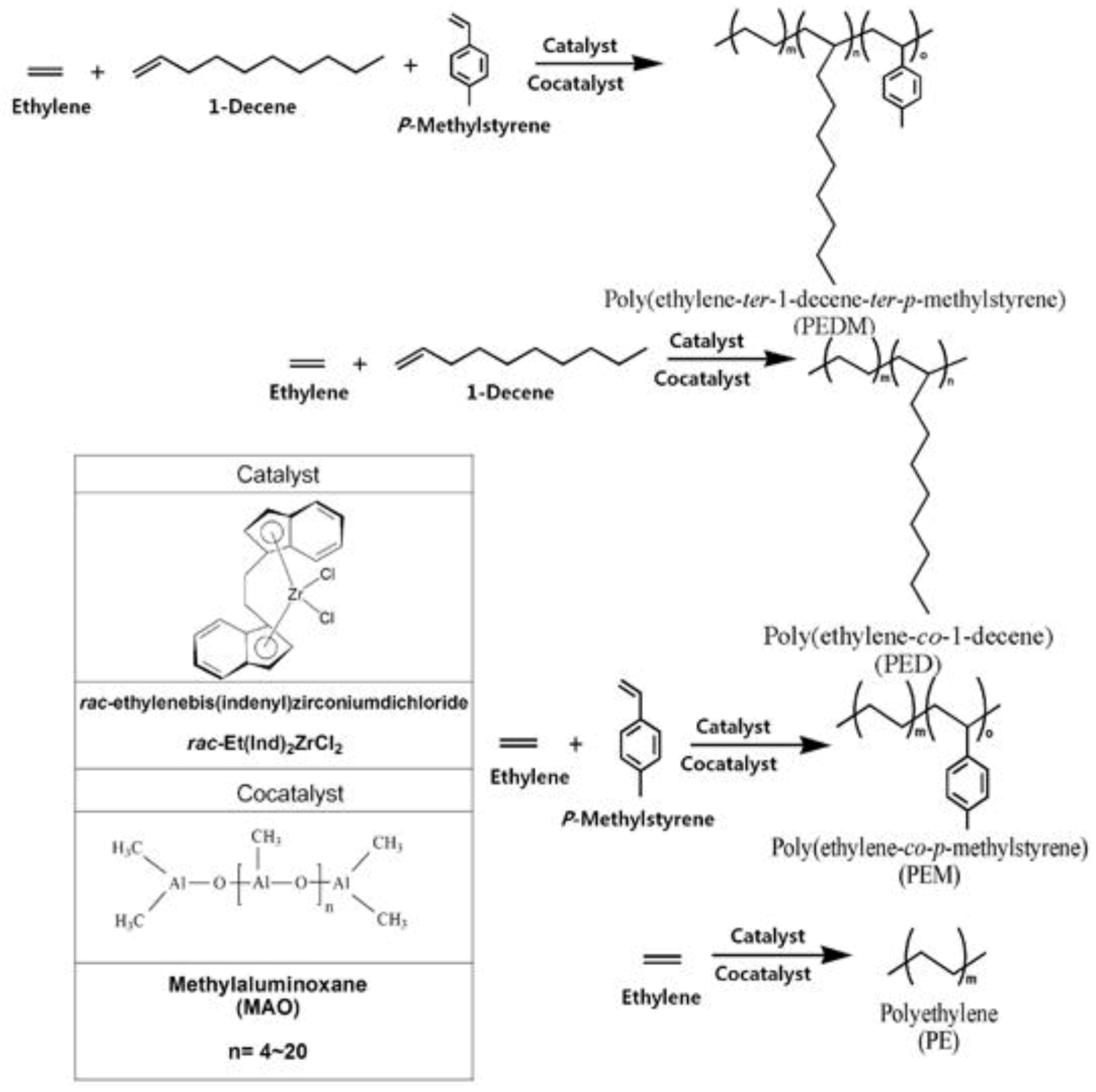
Figure 1 shows a scheme for the synthesis of PEDM, PED, PEM, PE using
rac-Et(Ind)
2ZrCl
2 and methylaluminoxane (MAO) as a catalyst and cocatalyst, respectively. Generally, the most interesting aspect of these zirconocene–MAO catalytic systems is the production of random copolymers with higher catalytic activities. The C2 symmetric metallocene catalyst,
rac-Et(Ind)
2ZrCl
2, has a relatively electron-rich indenyl ligand. This electron-rich ligand stabilize transition metals in catalyst with cationic character, which leads to a higher catalytic activity for
rac-Et(Ind)
2ZrCl
2, compared to that of other complexes.
Table 1 reports the results of homo-, co- and ter-polymerization using a metallocene catalyst/cocatalyst system under the same conditions of catalyst content, Al/Zr molar ratio, polymerization time and polymerization temperature. The catalytic activities of PED and PEDM were higher than that of PE. The catalytic activity of PED went up 2.7 fold values more than that of PE. This was attributed to the "positive” comonomer (1-decene) effect: (1) enhanced solubility of the copolymer (PED) in comparison with that of the homopolymer (PE), which favored monomer diffusion to the active center; (2) comonomer activation of new catalyst sites by increased affinity for the metallocene catalyst; and (3) increased solubility of the monomer in the liquid phase, due to the presence of the comonomer, which increased its insertion rate [
8].
In the case of the terpolymer, the catalytic activity is lower than that of the copolymer (PED). This may have been due to the “negative comonomer (
p-MS) effect.” We can infer that, after the incorporation of
p-MS into the terpolymer, its bulkiness made further insertion of ethylene and 1-decene difficult. This could also be confirmed by the very low catalytic activity of PEM, which was a copolymer of ethylene and
p-MS, as shown in
Table 1.
Figure 1.
Scheme for the synthesis of homo-, co- and ter-polymer.
Figure 1.
Scheme for the synthesis of homo-, co- and ter-polymer.
Comparing with the experimental results of T. C. Chung
et al. [
6], where they synthesized poly(ethylene-
ter-1-octene-
ter-p-MS), the composition of 1-octene in the terpolymer was higher than that of PEDM. It can be explained by the higher chain length of 1-decene than that of 1-octene. Also, composition of p-MS in the terpolymer was more than five-times that of PEDM. This means the bulky aromatic monomer is difficult to insert into
rac-Et(Ind)
2ZrCl
2, which has a narrower bond angle than [C
5Me
4(SiMe
2NtBu)]TiCl
2.
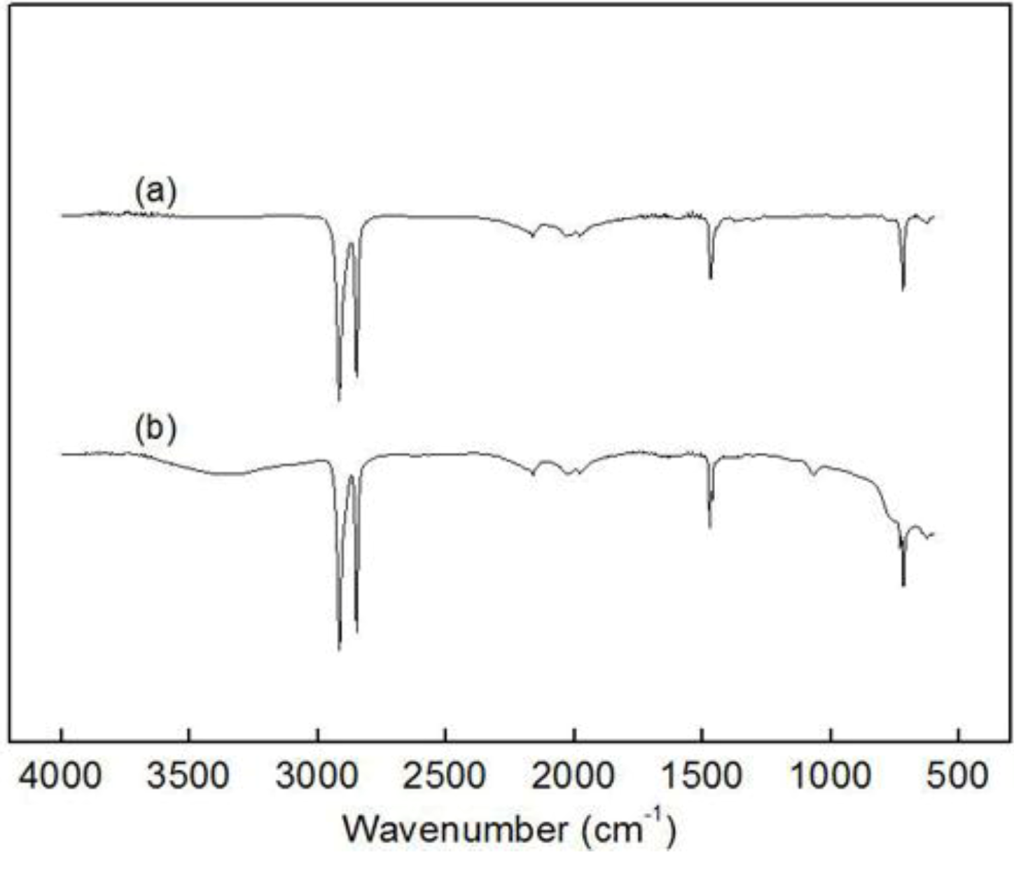
Figure 2.
Fourier transform infrared (FT-IR) spectra of (a) insoluble parts after Soxhlet processing with hexane for poly(ethylene-ter-1-decene-ter-p- methylstyrene) (PEDM) and (b) polyethylene (PE).
Figure 2.
Fourier transform infrared (FT-IR) spectra of (a) insoluble parts after Soxhlet processing with hexane for poly(ethylene-ter-1-decene-ter-p- methylstyrene) (PEDM) and (b) polyethylene (PE).
Table 1.
Results for homo-, co- and ter-polymerization.
Table 1.
Results for homo-, co- and ter-polymerization.
| NAME a | Feeding | E:D:M Feed Molar ratio | Composition | Catalytic Activity (kg of polymer/(mol·h)) | Mw d (kg/mol) | Mn d (kg/mol) | MWD d |
|---|
| E b (mol/L) | D b (mol/L) | M b (mol/L) | E b,c (mol %) | D b,c (mol %) | M b,c (mol %) |
|---|
| PE | 0.4 | 0 | 0 | 1:0:0 | 100 | 0 | 0 | 1420 | - | - | - |
| PED | 0.4 | 0.4 | 0 | 1:1:0 | 79.8 | 20.2 | 0 | 3840 | 100 | 29 | 3.4 |
| PEM | 0.4 | 0 | 0.4 | 1: 0:1 | 98.9 | 0 | 1.1 | 350 | 85 | 26 | 3.3 |
| PEDM | 0.4 | 0.4 | 0.4 | 1: 1:1 | 65.4 | 33.4 | 1.2 | 2648 | 100 | 37 | 2.7 |
2.2. Characterization
Figure 2 shows two kinds of Fourier transform infrared (FT-IR) spectra: (a) the spectrum of insoluble parts produced through the Soxhlet process using n-hexane for PEDM and (b) the spectrum of PE. The spectra of (a) and (b) have very similar peaks. This result indicated that both of the polymers were homo-polyethylene and the polyethylene, which occurred during the terpolymerization and was efficiently eliminated through the Soxhlet process. The C-H stretching peaks mutually appear near 2850 cm
−1 and 2950 cm
−1. The CH
2 bending peak and out of plane C-H bending are near 1400 cm
−1 and 720 cm
−1, respectively. We concluded with certainty that, through the Soxhlet process, we separated the by-product (homo-polyethylene) from the terpolymer.
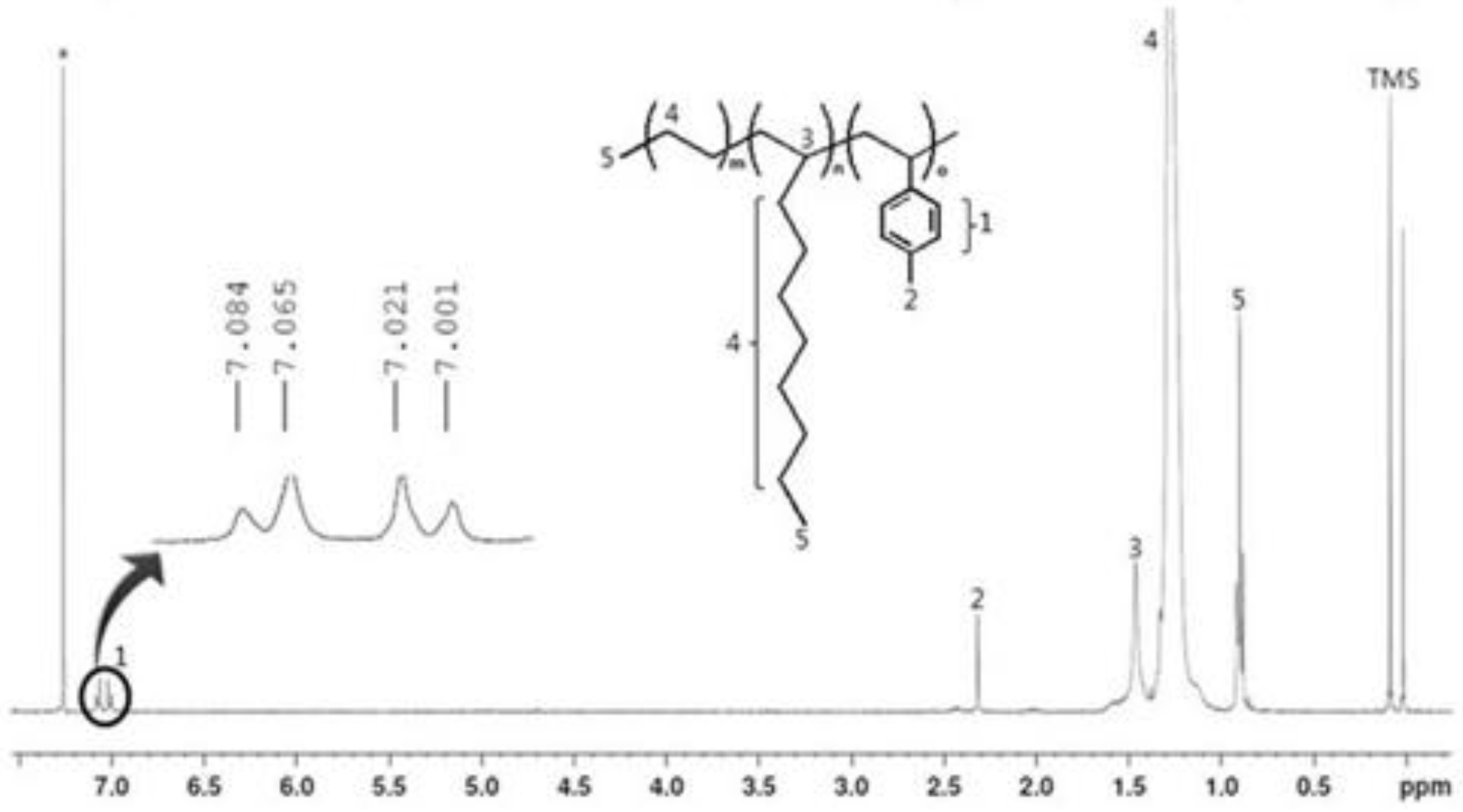
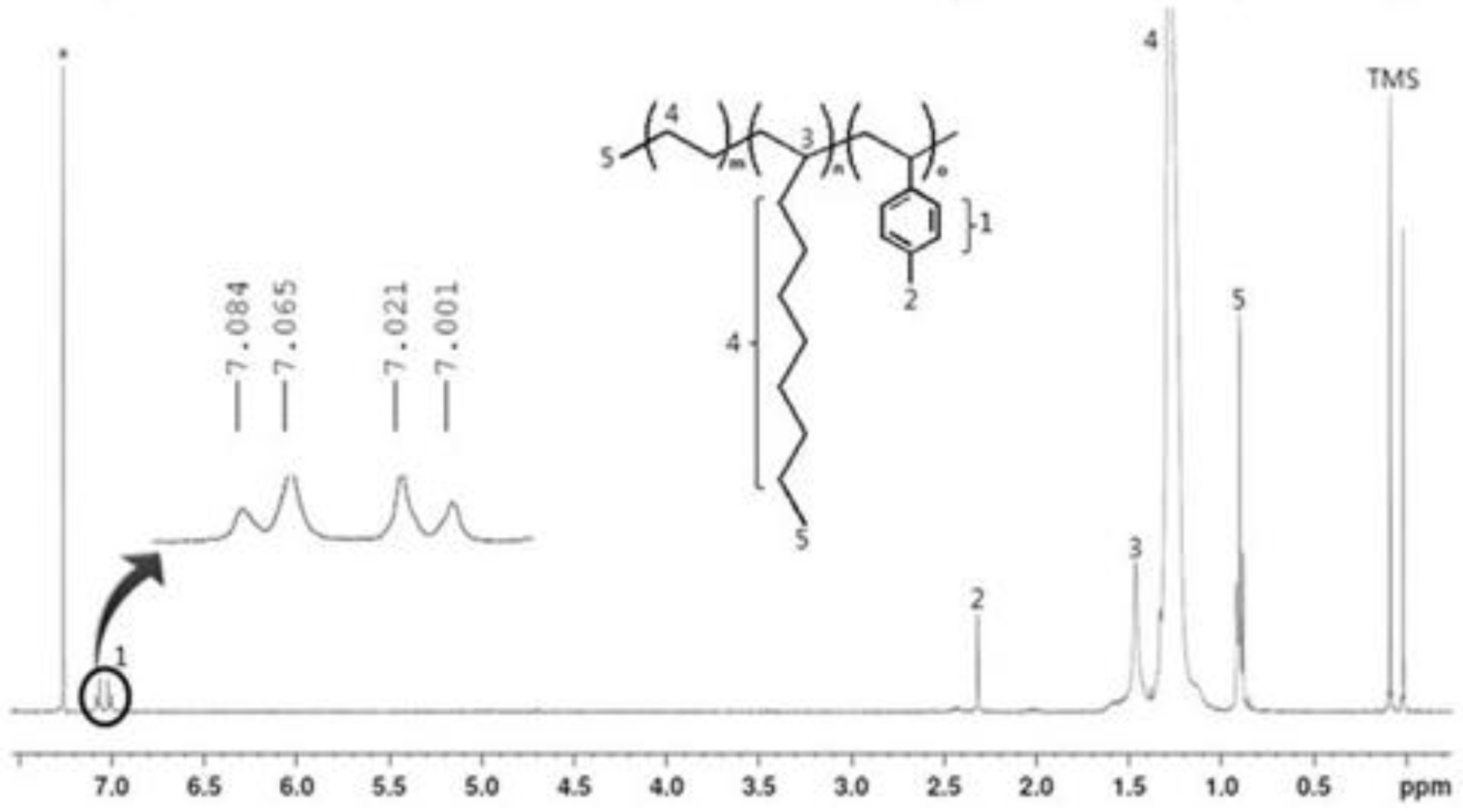
Figure 3.
1H NMR spectrum for the terpolymer (PEDM).
Figure 3.
1H NMR spectrum for the terpolymer (PEDM).
Figure 3 is the
1H NMR spectra of PEDM. The signatures near 7.0 ppm correspond to the aromatic peaks of
p-MS. The peaks for the CH, CH
2 and CH
3 of ethylene or 1-decene appear in the range 0–3 ppm. The peaks for the CH
3 of the 1-decene/ethylene and
p-MS were near 0.9 ppm and at 2.3 ppm, respectively. The peak for the CH of the 1-decene was near 1.5 ppm. Compared with the peaks for the CH
3 of
p-MS, the CH
3 of 1-decene or ethylene occupied a relatively broad area. We can confirm the insertion of the 1-decene and p-MS into the terpolymer by the characteristic peaks for the CH
3 of the
p-MS and the CH of the 1-decene.
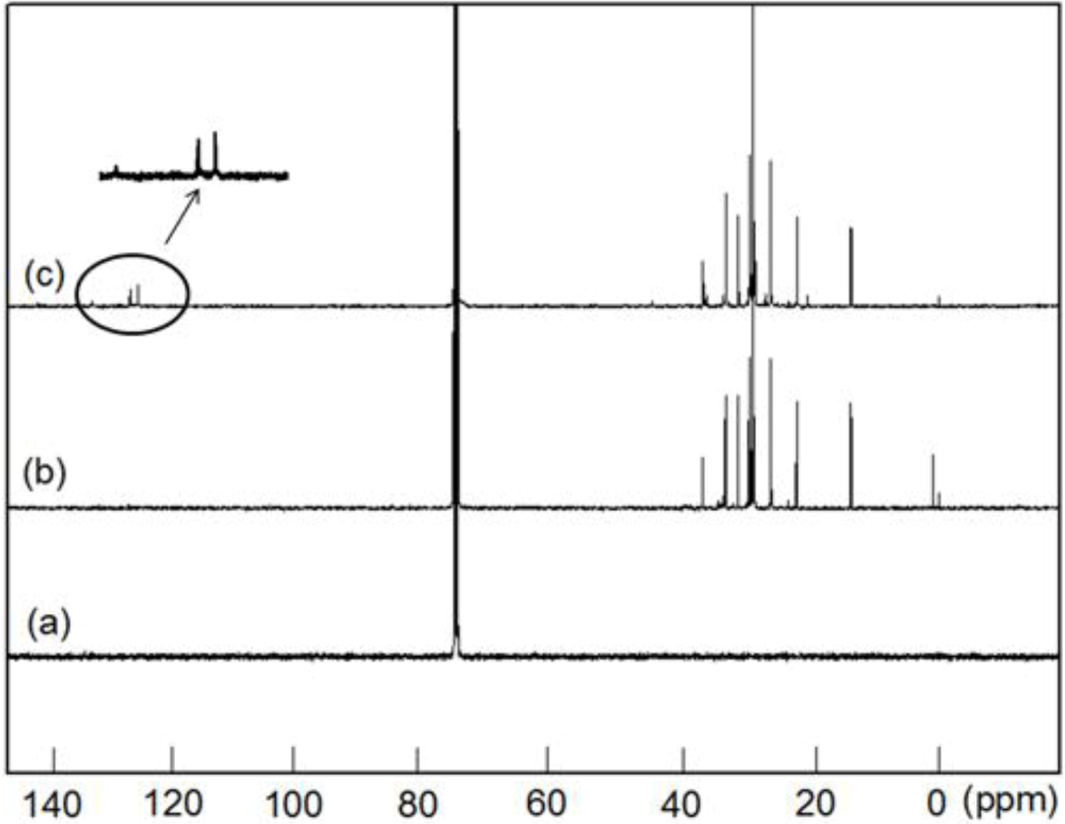
Figure 4 shows the
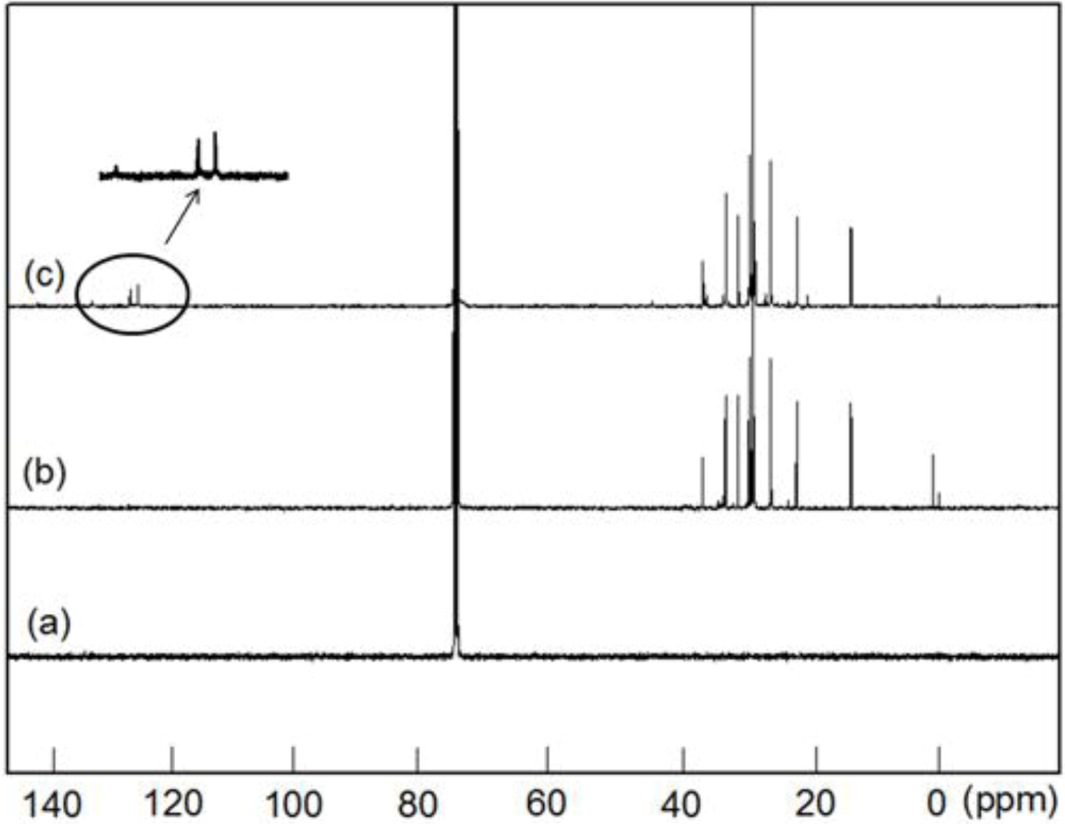
13C NMR spectra of the PE, PED and PEDM. In the case of PEDM in particular, or
tho and
meta carbon atoms are present in the benzene ring. The carbon atom at the
ortho position is
ortho plus
meta to the substituents, and the carbon atom at the
para position is
meta plus
para, being 129.6 ppm and 133.2 ppm, respectively. The peak for the CH
3 of
p-MS was detected at 21 ppm.
It is logical to expect that the methyl group substitution at the
para-position will have very little effect on the chemical shifts of methylene and methine carbons in the polymer backbone. In addition to the two chemical shifts (21.01 and 29.8 ppm), corresponding to the methyl group from
p-MS and methylene group from ethylene, respectively, there are three peaks (27.74, 37.01 and 45.77 ppm) corresponding to methylene and methane group from
p-MS units, which are separated by multiple ethylene units along the polymer chain [
9].
Figure 4.
13C-NMR spectra of (a) PE; (b) poly(ethylene-co-1-decene) (PED) and (c) PEDM.
Figure 4.
13C-NMR spectra of (a) PE; (b) poly(ethylene-co-1-decene) (PED) and (c) PEDM.
2.3. DMA
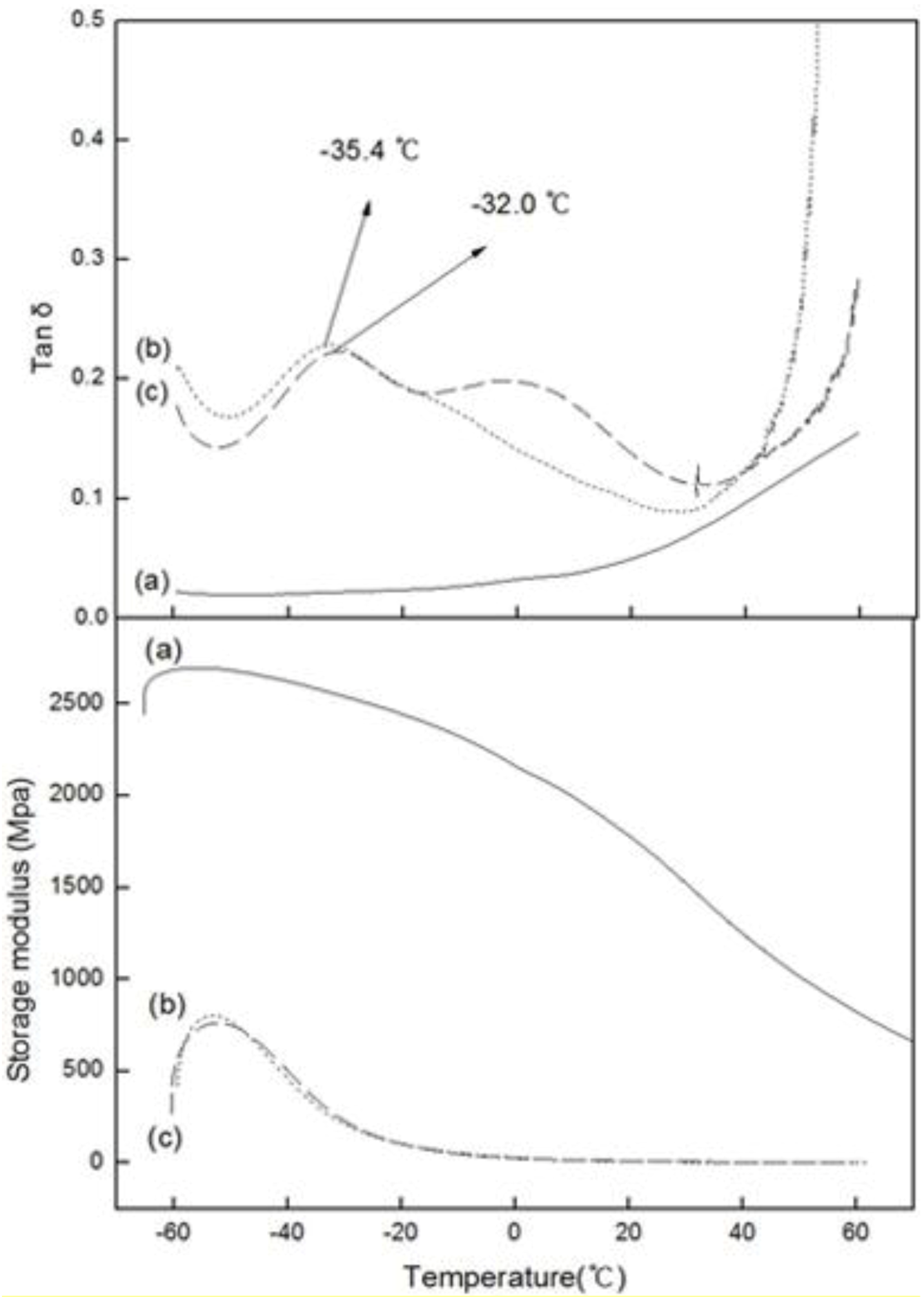
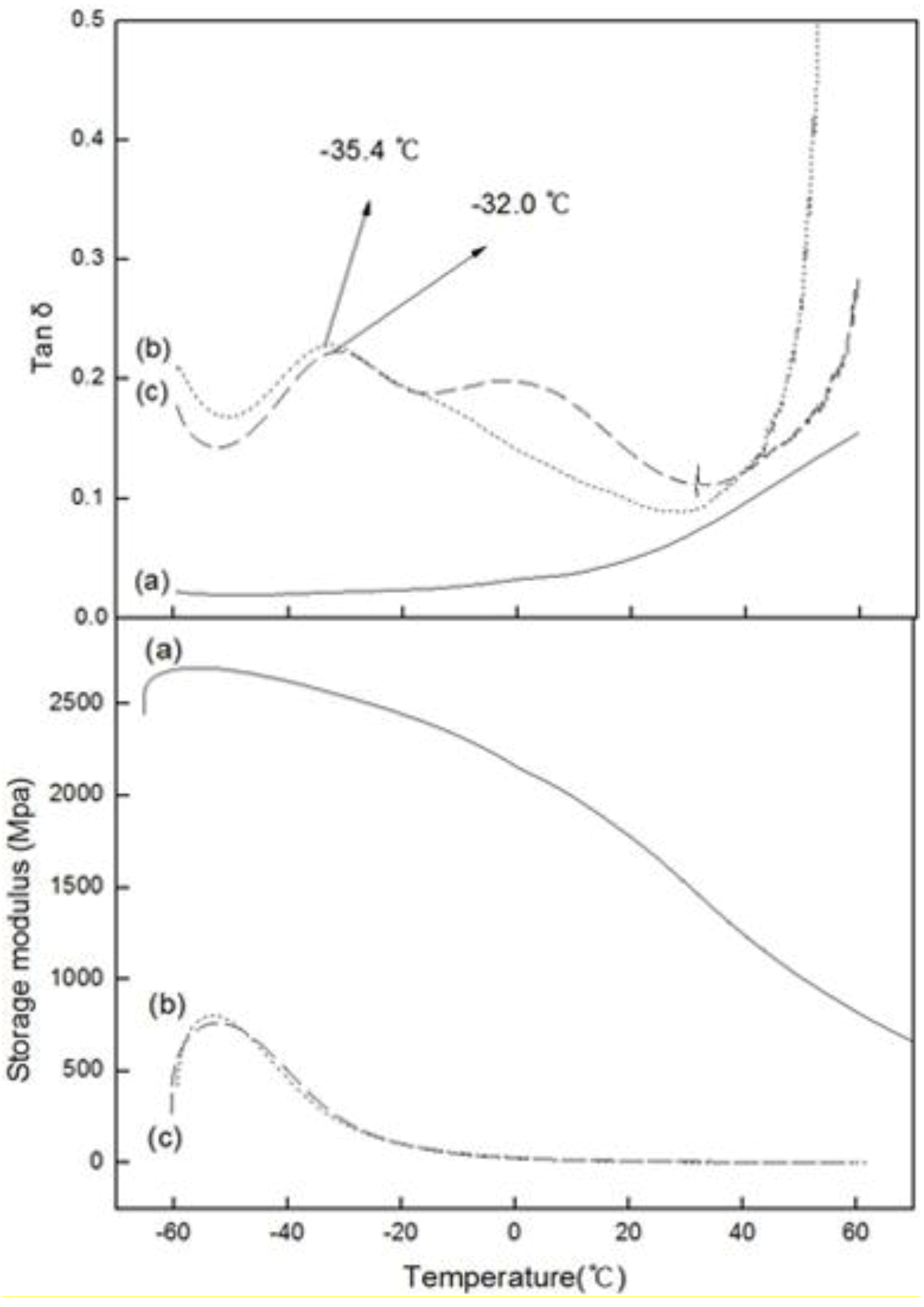
Figure 5 shows the variation in the loss tangent (tan δ) and dynamic storage moduli (E’) of PE, PED and PEDM, according to the temperature.
Figure 5.
Tan δ and storage moduli versus temperature fort (a) PE; (b) PED and (c) PEDM.
Figure 5.
Tan δ and storage moduli versus temperature fort (a) PE; (b) PED and (c) PEDM.
DMA is a very sensitive method for measuring the
Tg value of polymers. Side-chain or main-chain motion in specific regions of a polymer and local mode relaxations, which cannot be monitored by DSC, can be observed using DMA [
10].
Tg is the dominant transition in amorphous polymers or semi-crystalline polymers that are composed of amorphous regions. Because
Tg occurs at the highest temperature among other transitions, it is also called the α-transition.
Figure 5 show that the
Tg value of PEDM and PED were –32.0 °C and –35.4 °C, respectively. The
Tg value of PEDM was higher than that of PED, which could be ascribed to the rigid molecular
p-MS feature in the terpolymer.
The dynamic storage modulus (E’) is approximately similar to the Young’s modulus, elastic modulus or stiffness [
11]. The Young’s or elastic modulus is related to the crystallinity of the polymer. The storage moduli of PEDM and PED were much lower than that of PE, which may have been due to the very low crystallinity of PED and PEDM compared with PE.
2.4. DSC
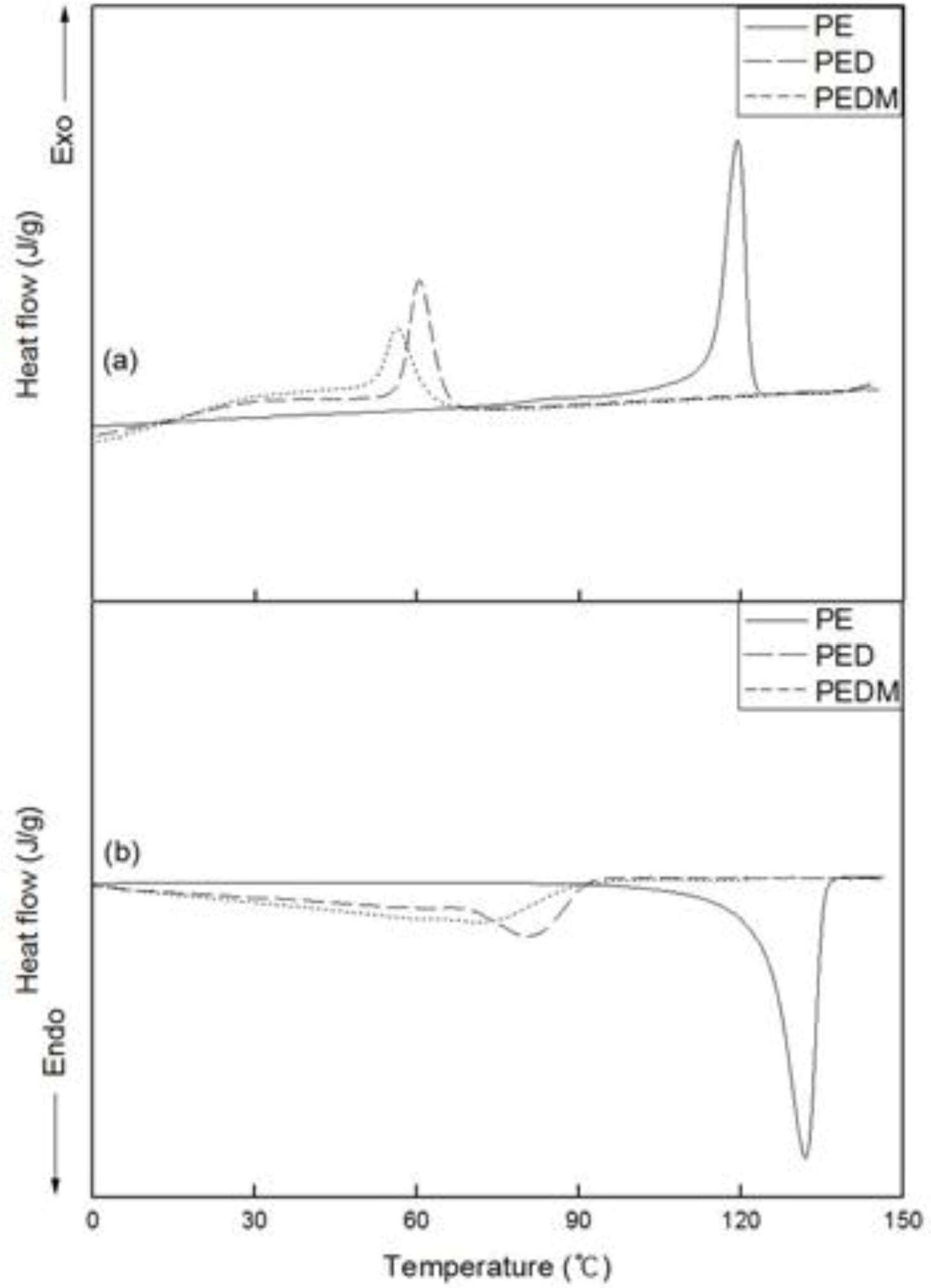
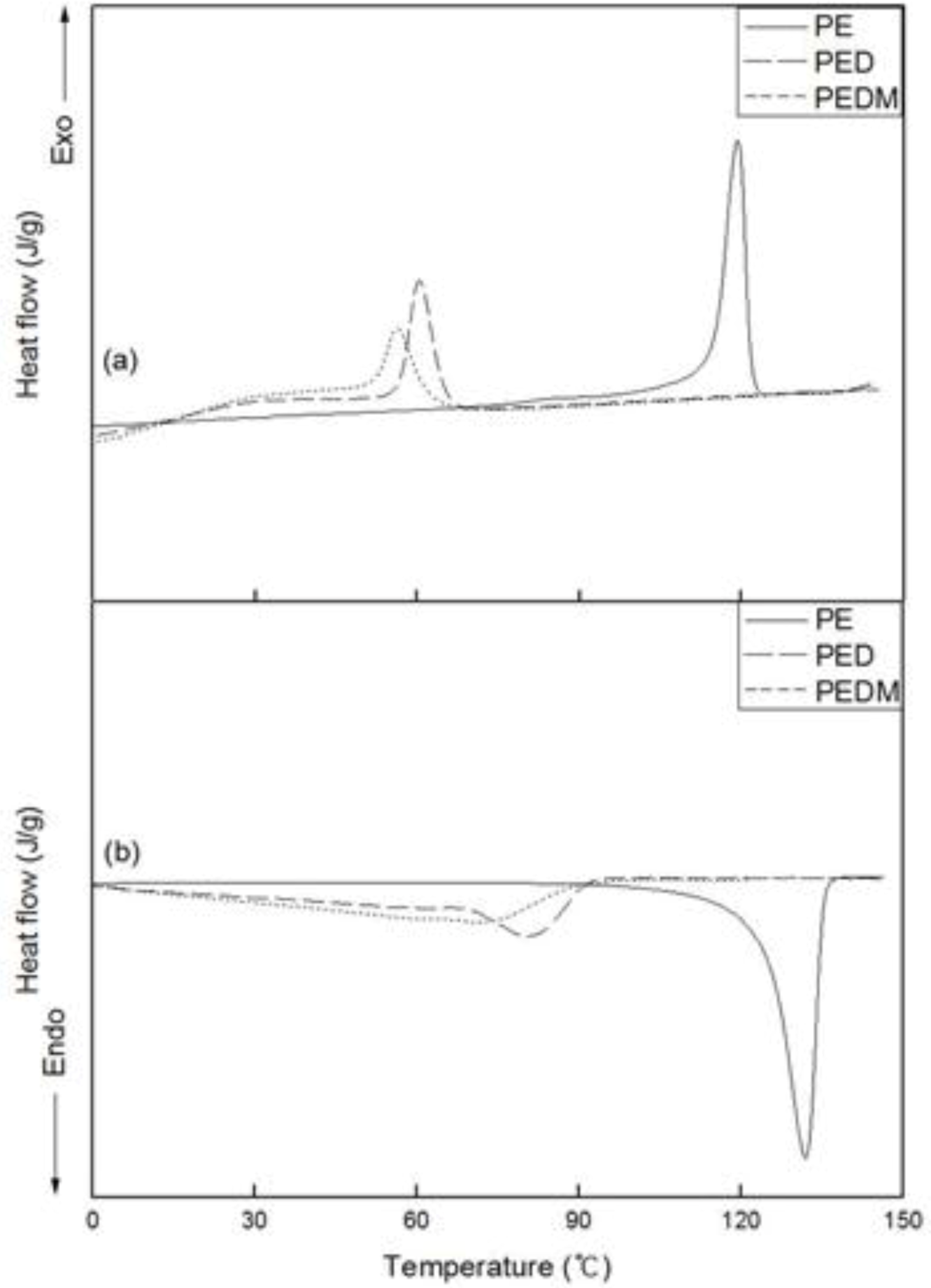
Figure 6 shows the DSC thermograms for PE, PED and PEDM. The variation in the second cooling and third heating curves of the polymers is shown in the upper and lower areas, respectively.
Figure 6.
DSC (a) cooling and (b) melting thermograms of PE, PED and PEDM.
Figure 6.
DSC (a) cooling and (b) melting thermograms of PE, PED and PEDM.
We confirmed the crystallization temperatures (Tc) and melting temperatures (Tm) of the polymers. The Tm value of PE was around 135 °C, which is similar to that of commercial high-density polyethylene.
As expected, both the Tm and Tc values of PED and PEDM showed important decreases, compared with those of PE. PEDM has higher value of Tm than PED, which was possibly due to the high crystallinity of PEDM and the contribution of the p-MS in the PEDM. We can explain this contribution of the p-MS using the experimental results of T. C. Chung et al., who explored the thermal properties of the ethylene-p-MS copolymer. They reported a melting point of 115 °C for p-MS content of 1.1 mol % in the copolymer, which was very similar to p-MS content for our PEDM sample. This means that through the incorporation of p-MS in the terpolymer, we were able to raise the Tm value of the ethylene-1-decene copolymer. Tc of the polymers showed trends very similar to their Tm.
Table 2 summarizes the
Tm,
Tc, Δ
Hm and crystallinity values measured by DSC. The crystallinities were calculated from their enthalpies of fusion. Since we were able to confirm the enthalpy of melting (Δ
Hm) of PED than that of PEDM, the crystallinity of PEDM was higher than that of PED.
Table 2.
Tm, Tc, ΔHm and crystallinities of PE, PED and PEDM.
Table 2.
Tm, Tc, ΔHm and crystallinities of PE, PED and PEDM.
| NAME a | Tc b (°C) | Tm b (°C) | ΔHm b (J/g) | Crystallinity b (%) |
|---|
| PE | 118.7 | 135.0 | 171.0 | 58.2 |
| PED | 58.4 | 79.4 | 3.7 | 1.3 |
| PEDM | 58.4 | 82.7 | 4.5 | 1.5 |
2.5. WAXS
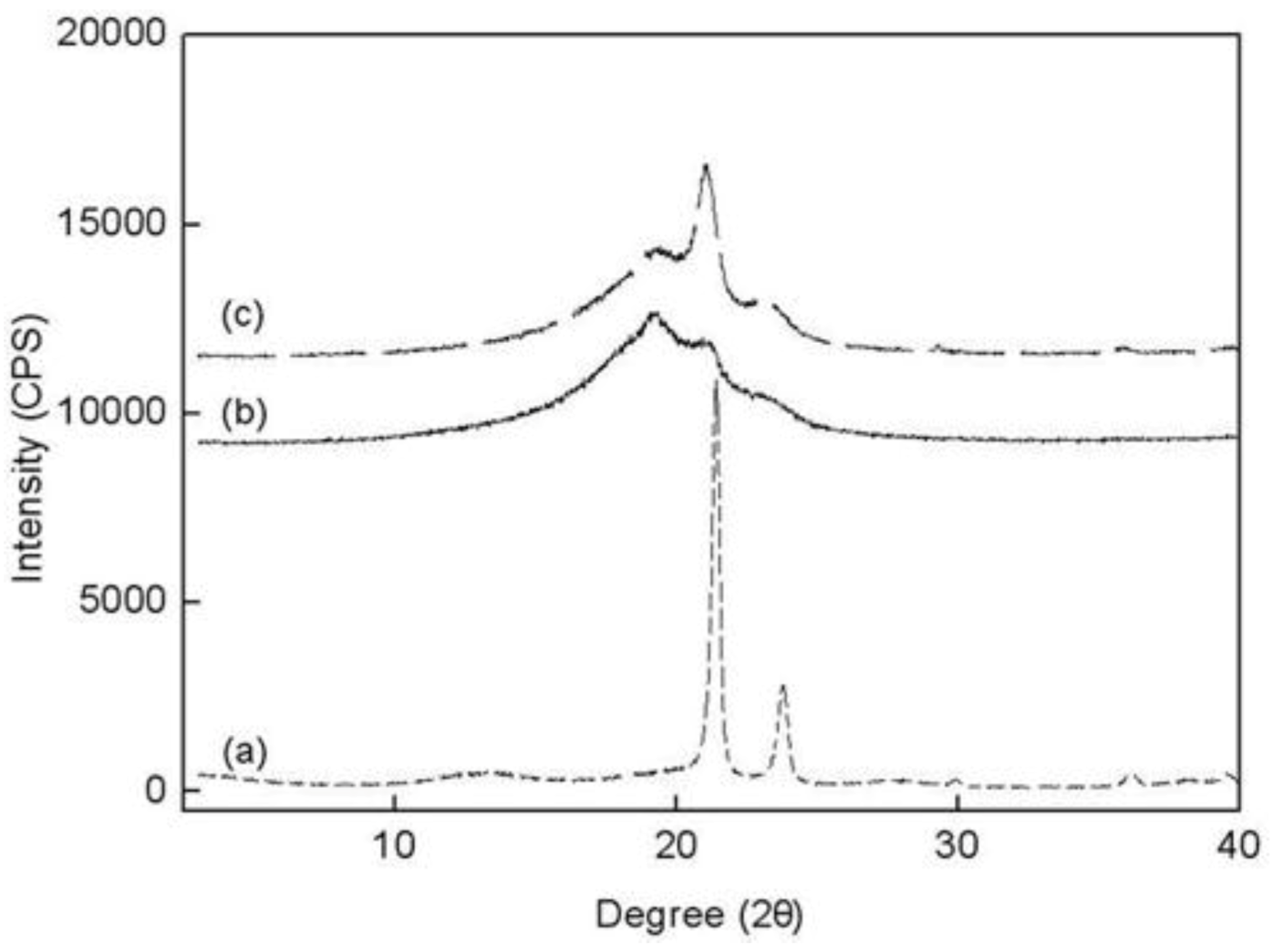
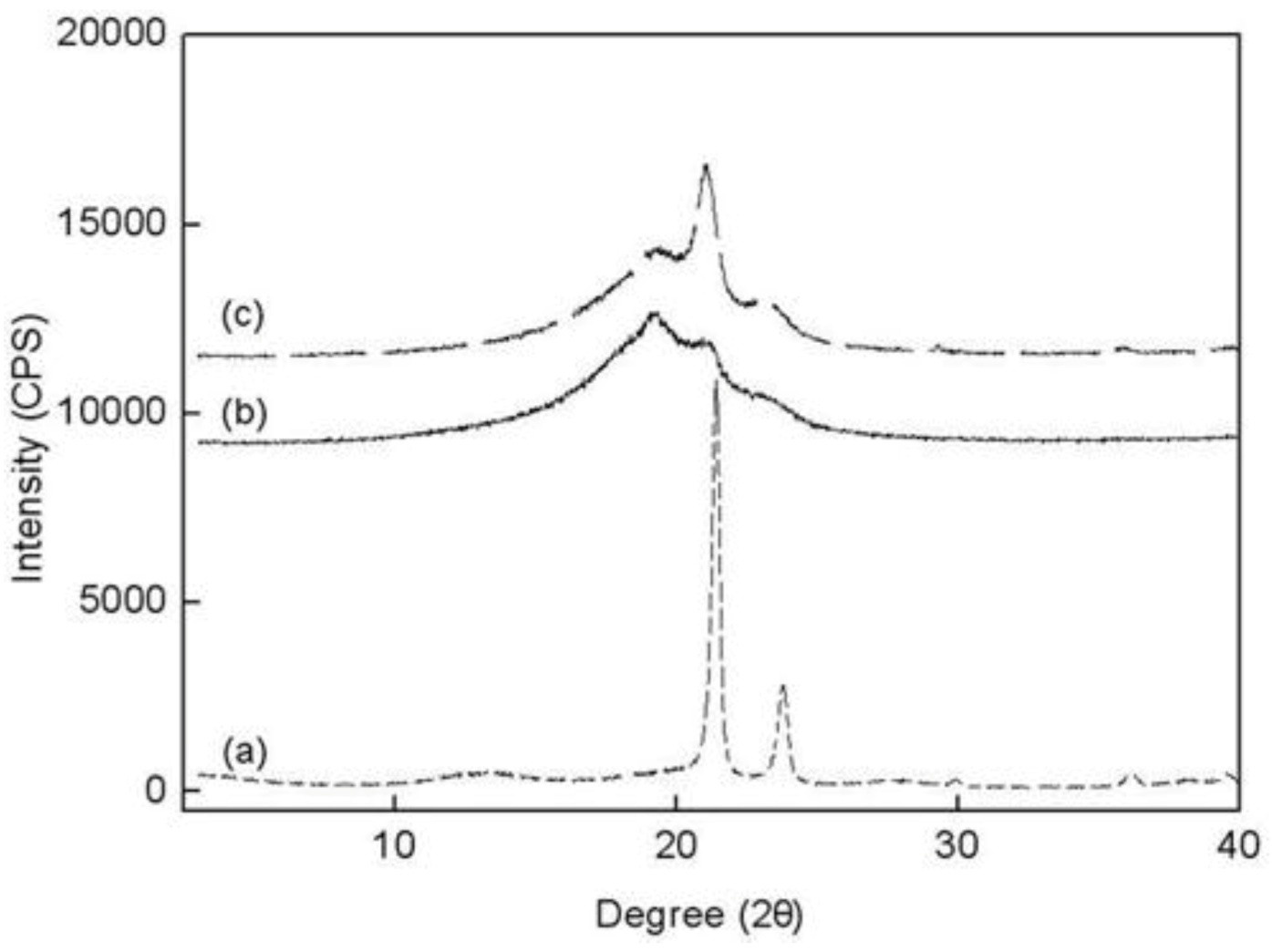
The wide-angle X-ray scattering (WAXS) data for PE, PED and PEDM are shown in
Figure 7. There are three peaks in the copolymer (PED) and terpolymer (PEDM). The broad amorphous peaks near 19.5–20° appeared were due to the contribution of 1-decene. However, the broad amorphous peaks decreased as
p-MS was inserted into the terpolymer. Two other small peaks for the copolymer and the terpolymer that were present in the 21.8° and 24.3° were identical for the crystalline homopolymer (PE). This fact indicates that the copolymer and terpolymer were partially crystalline, although the crystallinity decreased substantially with the insertion of the comonomer and termonomer.
2.6. Kinetics
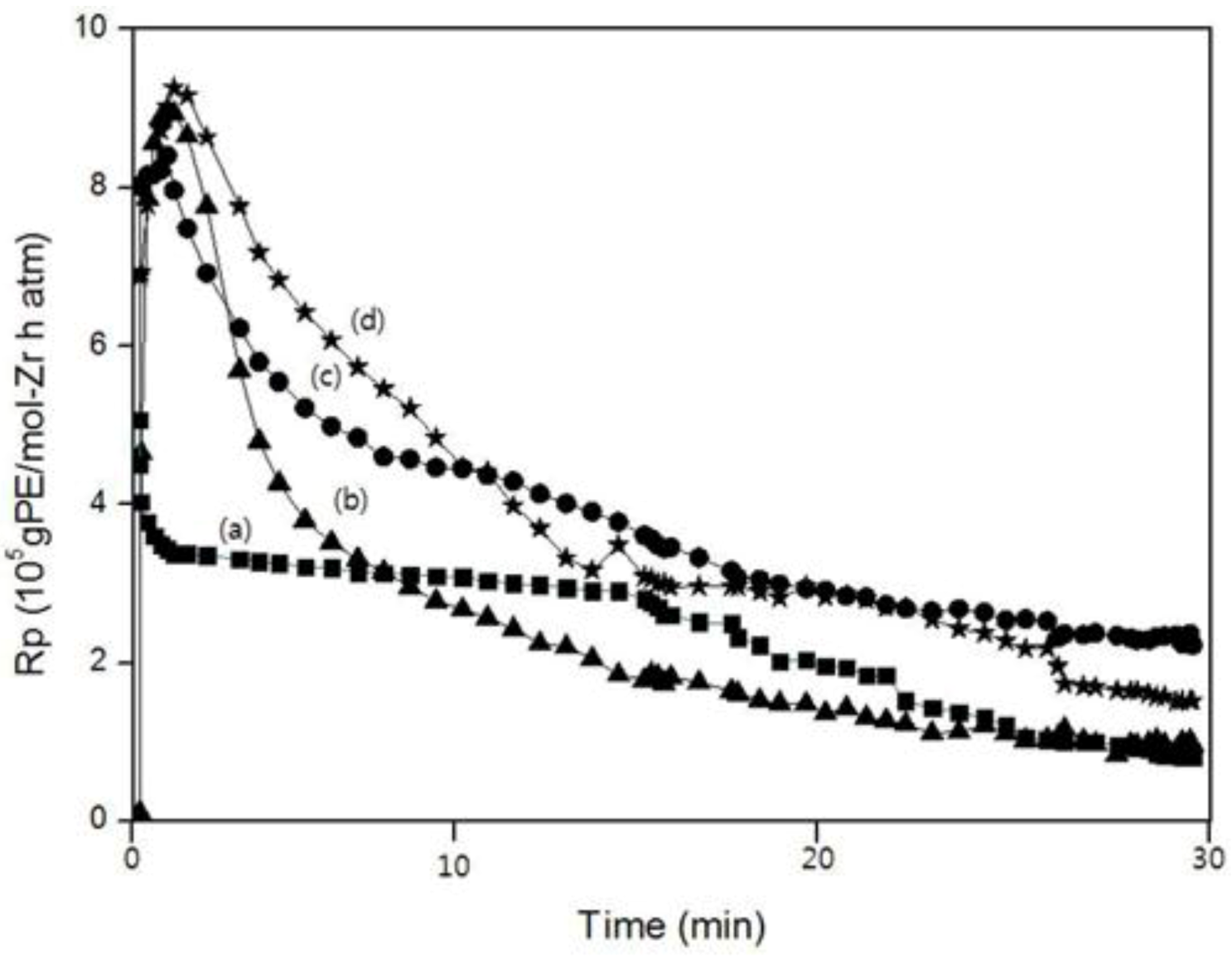
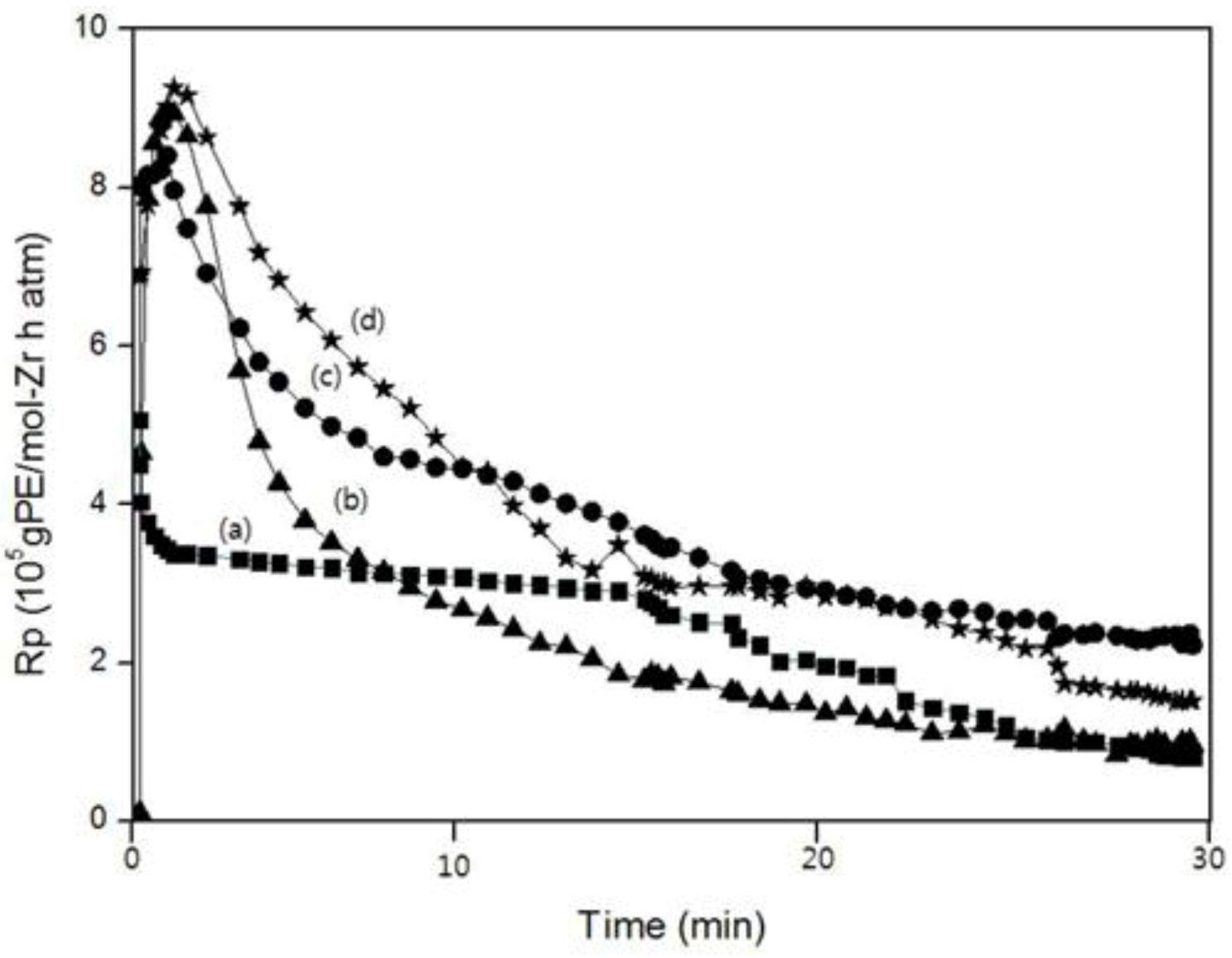
Figure 8 show the polymerization rate curves for the homo-, co- and ter-polymer. All of the polymerizations were marked by very high initial rates for which the maximum rate (R
p,
max) was reached within 1–2 min. Within this period, a pronounced change in the viscosity of the solvent was observed. Except for the polymerization curve of PEM, the reaction rates of the copolymerization and the terpolymerization were higher than that for homopolymerization at all times. This fact was ascribed to the “positive comonomer (1-decene) effect.”
In the case of PEM, although the reaction rate was higher than that of PE in the beginning, the reaction rate became lower than that of PE. The reaction rate of PEDM was higher than that of PED until 12 min. after which, the reaction rate of PEDM became lower than that of PED. These observations were ascribed to the “negative comonomer (
p-MS) effect.” This negative comonomer effect could be explained by the fact that
p-MS that was inserted into the copolymer or terpolymer disturbed additional incorporation of other monomers [
12,
13].
Figure 7.
Wide-angle X-ray scattering data for (a) PE; (b) PED and (c) PEDM at room temperature.
Figure 7.
Wide-angle X-ray scattering data for (a) PE; (b) PED and (c) PEDM at room temperature.
Figure 8.
A comparison of the polymerization rate (Rp) curves according to the polymerization time: (a) PE; (b) PEM; (c) PED and (d) PEDM.
Figure 8.
A comparison of the polymerization rate (Rp) curves according to the polymerization time: (a) PE; (b) PEM; (c) PED and (d) PEDM.
2.7. Mechanical Properties
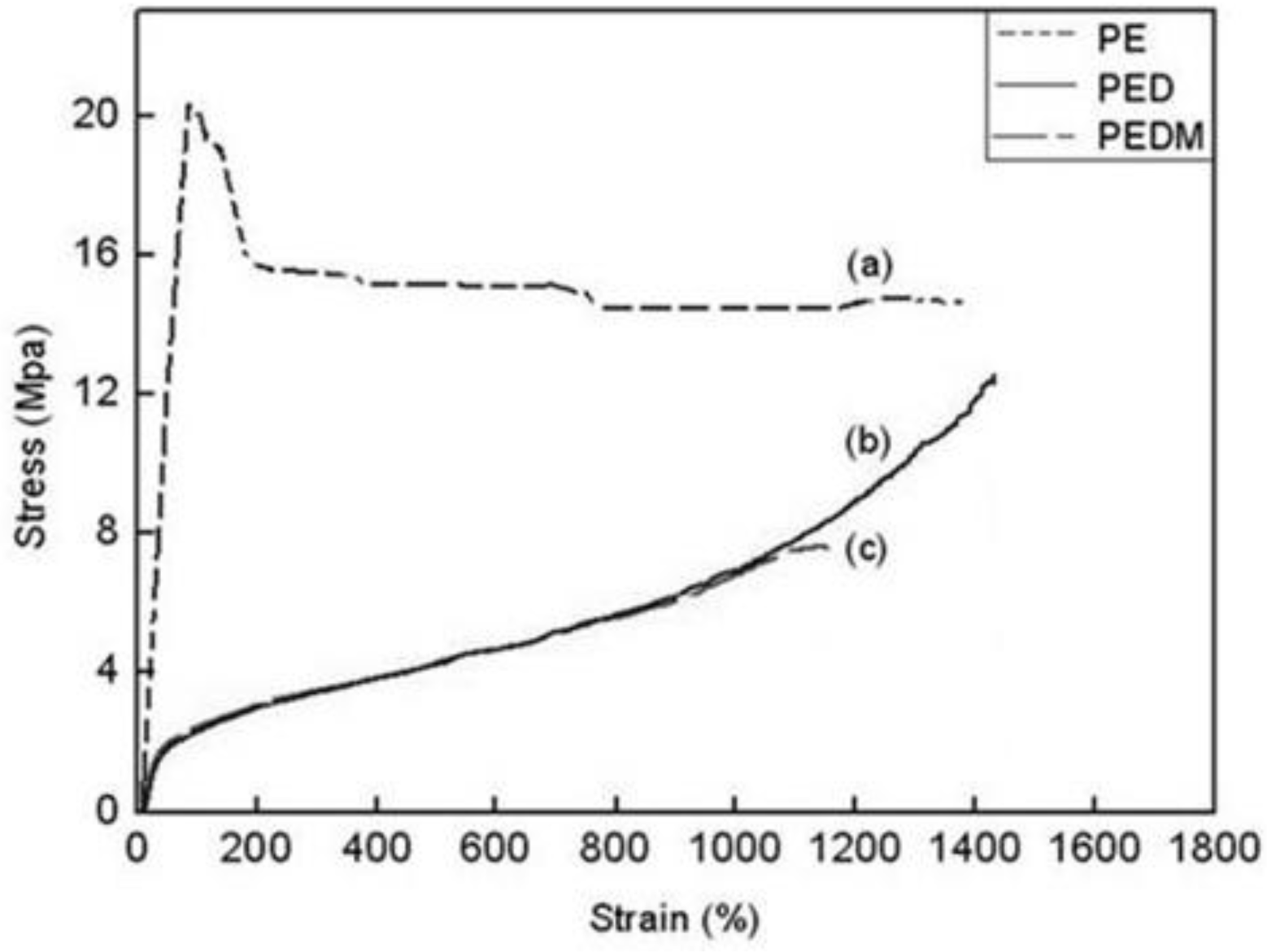
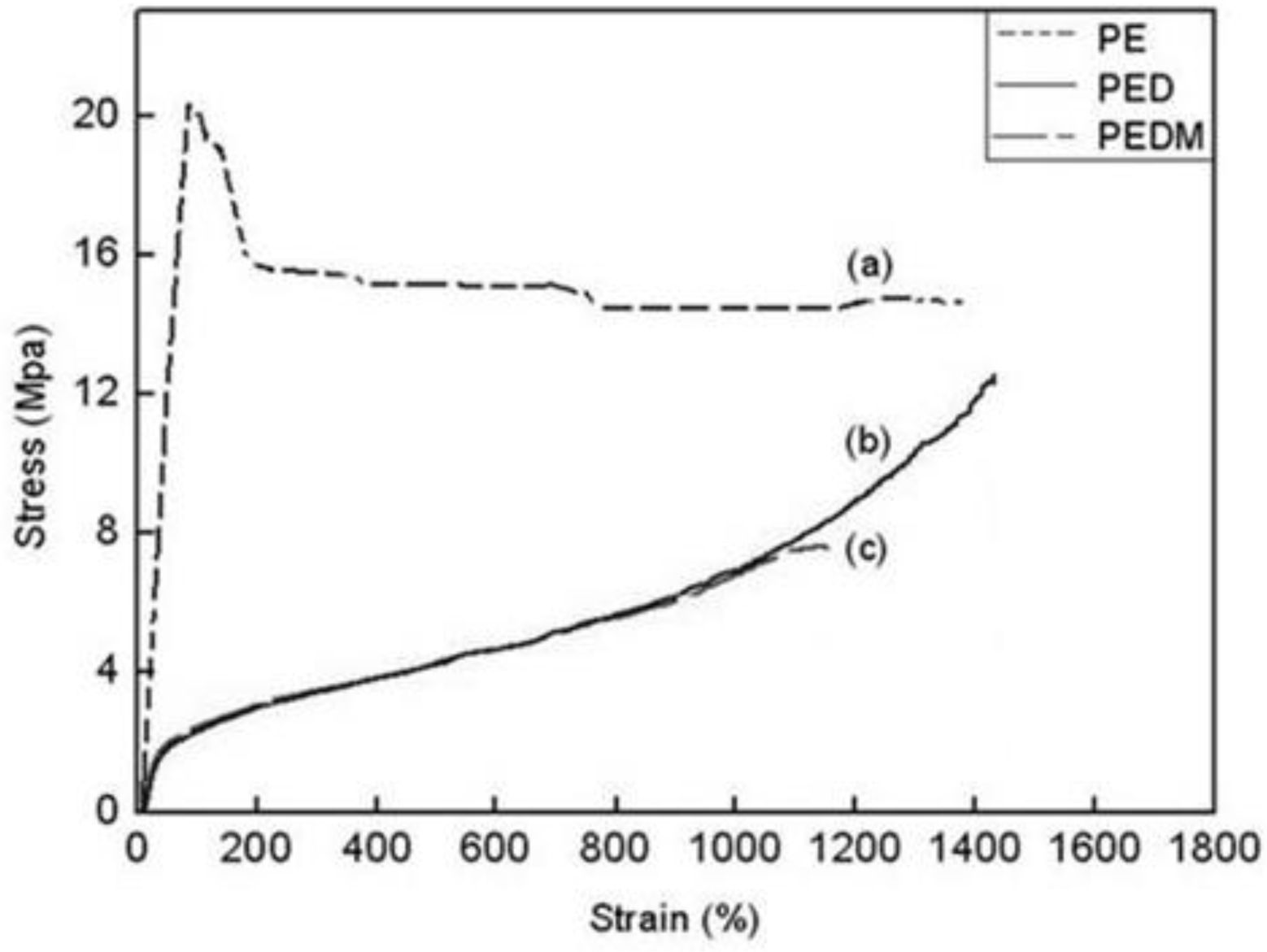
The stress-strain behaviors of PE, PED and PEDM are shown in
Figure 9. The polymers showed very different stress-strain curves. PE had a very high elastic modulus and exhibited typical ductile plastic fracturing with a marked yield point, which was related to its low degree of branching and high crystallinity. On the other hand, PED and PEDM underwent dramatically extensive plastic deformation (exceeding 1000% stain) before breaking and show elastomeric behavior with a lower modulus than that for PE. They became so rubbery that there was no longer a yield point. PEDM had higher tensile strength than PED, which was ascribed to the higher crystallinity of PEDM.
Figure 9.
Stress and strain curves of (a) PE, (b) PED and (c) PEDM.
Figure 9.
Stress and strain curves of (a) PE, (b) PED and (c) PEDM.
3. Experimental Section
3.1. Materials and Specimens
All manipulations were carried out in an inert nitrogen atmosphere. We used high-purity grade ethylene gas (Dae-myung Gas Co., cheonan, Korea) after passing it through an alumina/zeolite column. 1-decene (Aldrich, 94%), n-hexane (SAMCHUN, 99.5%,) and toluene (SAMCHUN, 99.5%) were purified by refluxing them over sodium, using benzophenone as an indicator and stored in a dry box. A solution of rac-Et(Ind)2ZrCl2 (Sigma Aldrich, MO, USA) in toluene was also prepared. MAO (Tosho, methylaluminoxane) was used as received. p-methylstyrene was distilled under reduced pressure in the presence of CaH2 after performing standard purification procedure. Specimens for the thermal, mechanical and wide-angle X-ray scattering (WAXS) analysis were prepared by compression molding in a hot press.
3.2. Polymerizations
Typically, all of the polymerization reactions were carried out in a 300 mL stainless steel autoclave with a mechanical stirrer. After the autoclave was saturated with ethylene gas, the polymerization reaction was started by the injection of the required amount of catalyst solution and MAO. After several minutes, the polymer solution was poured into a diluted HCl/EtOH solution. The resultant polymer was washed with EtOH (600 mL) and dried in vacuo.
Using a Soxhlet apparatus and n-hexane as a solvent, we separated each soluble copolymer or terpolymer from the insoluble polyethylene (some types of by-products). After drying the soluble fractions, we could finally obtain pure copolymer and terpolymer, which were completely soluble in common organic solvents, such as the hexane, toluene and tetrahydrofuran. Polymer yield and catalytic activity were determined by gravimetry.
3.3. Characterization
Fourier transformed infrared (FT-IR) analysis was performed by NICOLET 6700 (Thermo Scientific Co., Waltham, MA, USA).
13C NMR spectra were obtained at 60 °C. The equipment that was used was AVANCE 500 MHz ( Bruker, Karlsruhe, Germany). Sample solutions of the polymer were prepared in CDCl3. The deuterated solvent was used to provide an internal lock signal.
1H NMR spectra were recorded on AVANCE 500 MHz (Bruker, Karlsruhe, Germany). The NMR samples were prepared in CDCl3 solvent at 60 °C.
The molecular weight and the molecular weight distribution of the polymers were measured using PL-GPC210 (Polymer Laboratories Co., Church Stretton, UK) gel permeation chromatography fitted with Styragel (olesis guard column) HT-type columns. This test method uses commercially available polystyrene standards. The analyses were performed at 140 °C and 1.0 mL/min with 1,2,4-trichlorobenzene as a solvent.
3.4. Dynamic Mechanical Analysis
The dynamic mechanical spectra of the samples were obtained by using a dynamic mechanical analysis (DMA) machine [Seiko Co., Exstar 6000] over the temperature range –60 to 60 °C. The specimens were analyzed in tension mode at a constant frequency of 1 Hz using 0.01% strain at a heating rate of 2 °C/min.
3.5. Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) data for the terpolymer were recorded by means of a DSC7 (PERKIN ELMER Co.). Samples were heated from 0 °C to 150 °C and then cooled down at 10 °C/min to 0 °C. Following this, they were reheated at 10 °C/min to 150 °C. The crystallization temperature (
Tc), melting temperature (
Tm) and enthalpy of fusion (Δ
Hm) were derived from the second and third run curves, respectively. The measured Δ
Hm value was converted into the degree of crystallinity (1 − λ)
ΔH using 290 J/g as the enthalpy of fusion of a perfect polyethylene crystal [
14].
3.6. Wide-Angle X-ray Scattering
Wide-angle X-ray Scattering (WAXS) patterns were recorded in reflection mode at room temperature using D/MAX-2200V X-ray Diffractometer (Rigaku, Tokyo, Japan) connected to a computer. The instrument used the Cu source, and it performed measurements at 40 kV and 40 mA. The diffraction scans were collected over a period of 20 min between 2 θ values from 3.0 to 40.0° at a scan rate of 2°/min.
3.7. Mechanical Property
Elastic modulus, tensile strength and elongation at break were determined with a universal testing machine (Tinius Olsen Co. Ltd., Salford, UK, H5K-T), according to ASTM method D412 at a crosshead speed of 10 mm/min. For each film sample, measurements were made on five specimens taken from the same film, and the average of their results was reported.
4. Conclusions
Homo-, co- and ter-polymer were prepared and compared with various properties. We confirmed the synthesis of PED and PEDM using 1H NMR, 13C NMR and FT-IR analysis. Tg of PEDM was higher than that of PED, which is possibly due to the rigid molecular feature of the p-MS in the terpolymer. In the case of the storage modulus, those for PED and PEDM were much lower than that of PE, which was possibly due to the very low crystallinity of PED and PEDM. Both Tm and Tc of the PED and PEDM showed significant decreases, compared with those of PE. All of the copolymer and terpolymer were partially crystalline, although the crystallinity decreased greatly with the insertion of the comonomer and termonomer. Through a kinetic study, we ascertained that 1-decene and p-MS exerted a positive and negative comonomer effects on terpoloymerization, respectively. PEDM had higher tensile strength than PED, which was attributed to the higher crystallinity of the terpolymer.
In this study, we synthesized the terpolymer composed of ethylene, 1-decene and p-MS. 1-decene and p-MS are responsible for reducing the gas transition temperature of the terpolymer and reactive moiety, respectively, which could be easily converted into functional groups like -OH, -NH, -COOH, anhydrides and halogens under mild conditions. We have therefore opened up the possibility of developing new high-functional elastomers in the future.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}