Alcohol Dehydrogenation with a Dual Site Ruthenium, Boron Catalyst Occurs at Ruthenium

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
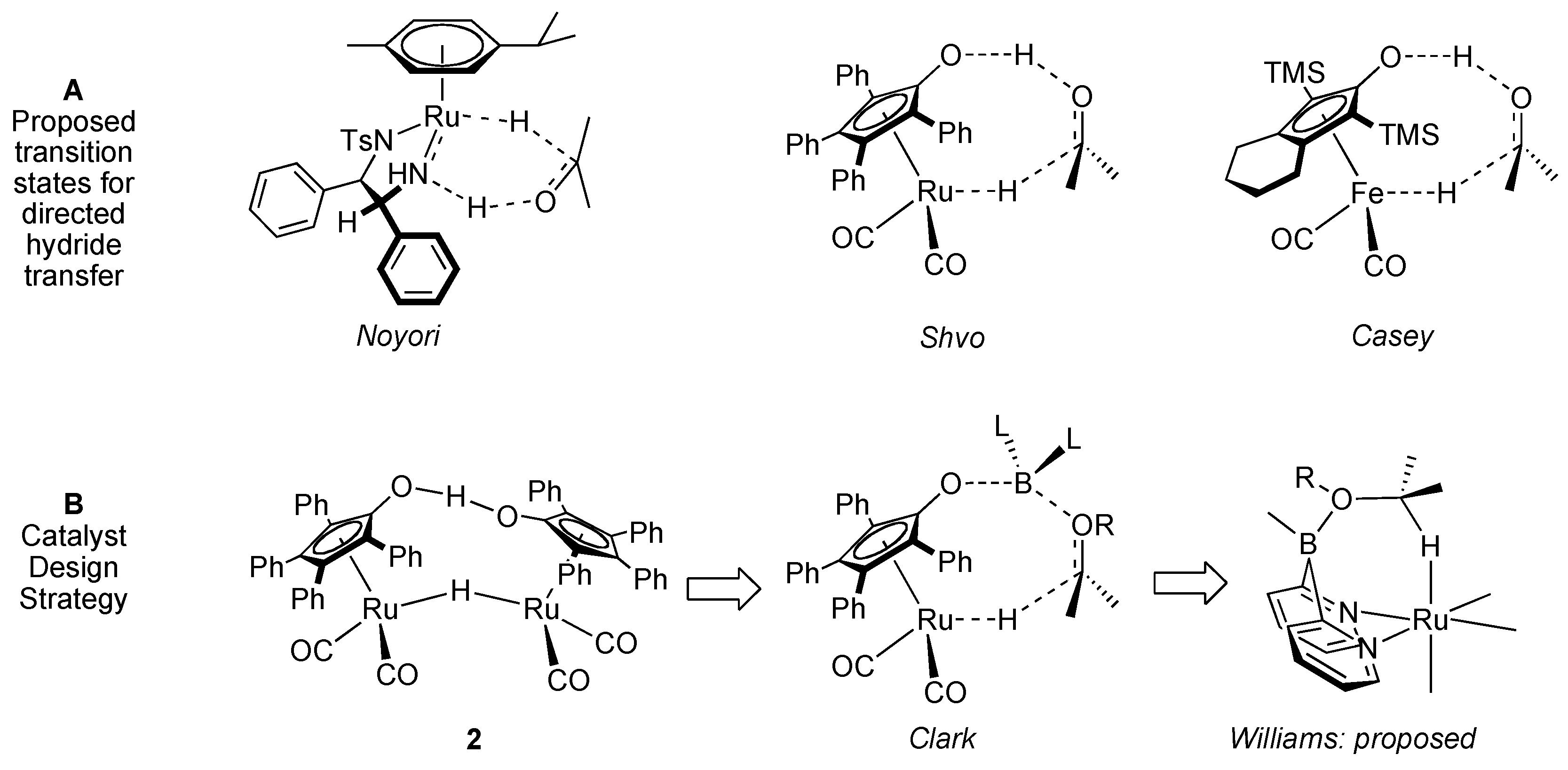
Design of a Dual Site Ruthenium, Boron Catalyst
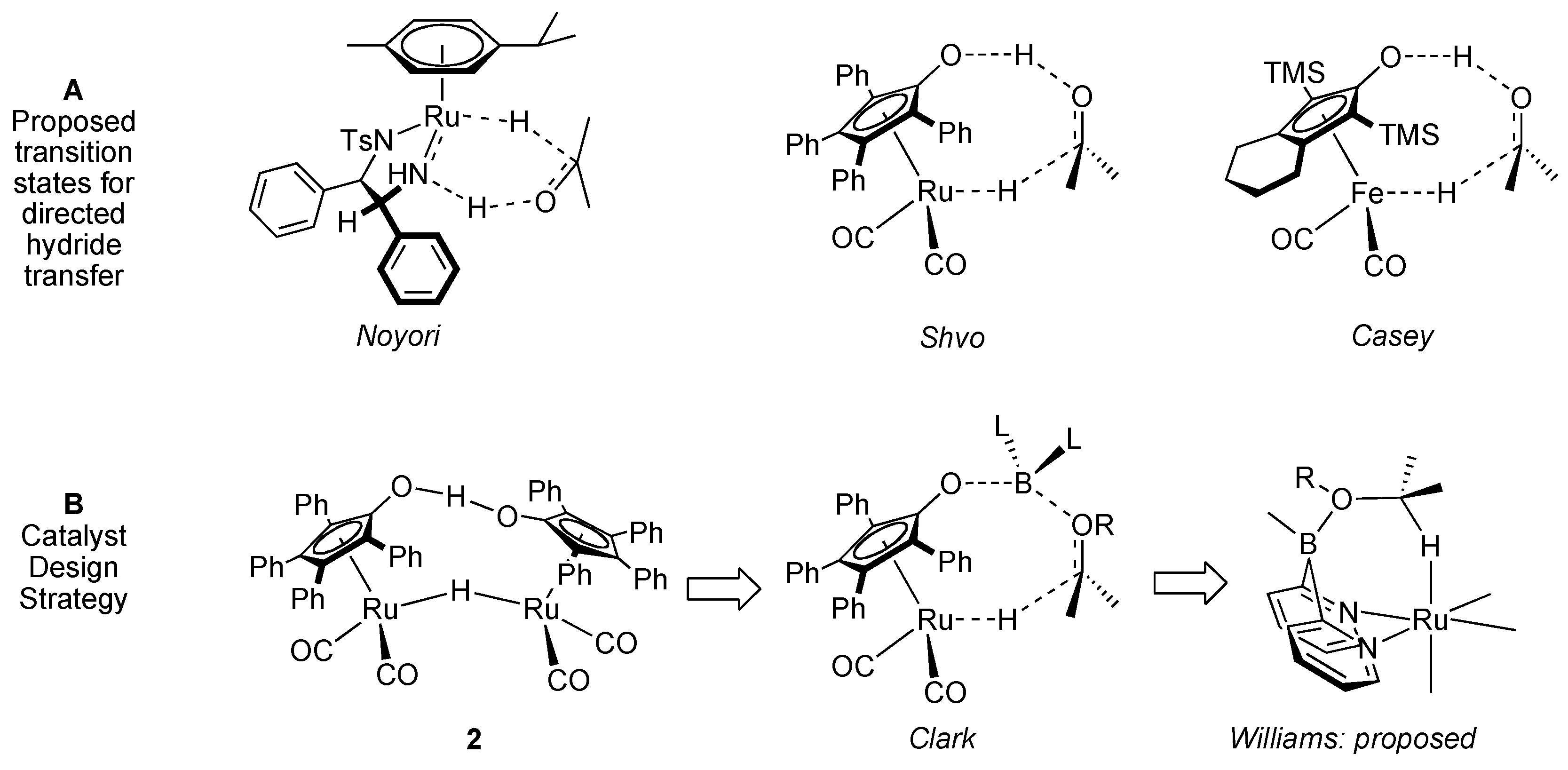
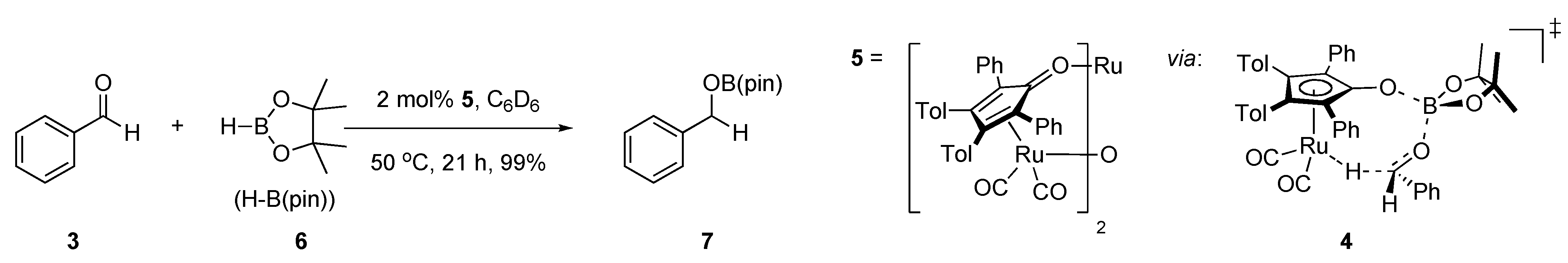
2. Results and Discussion
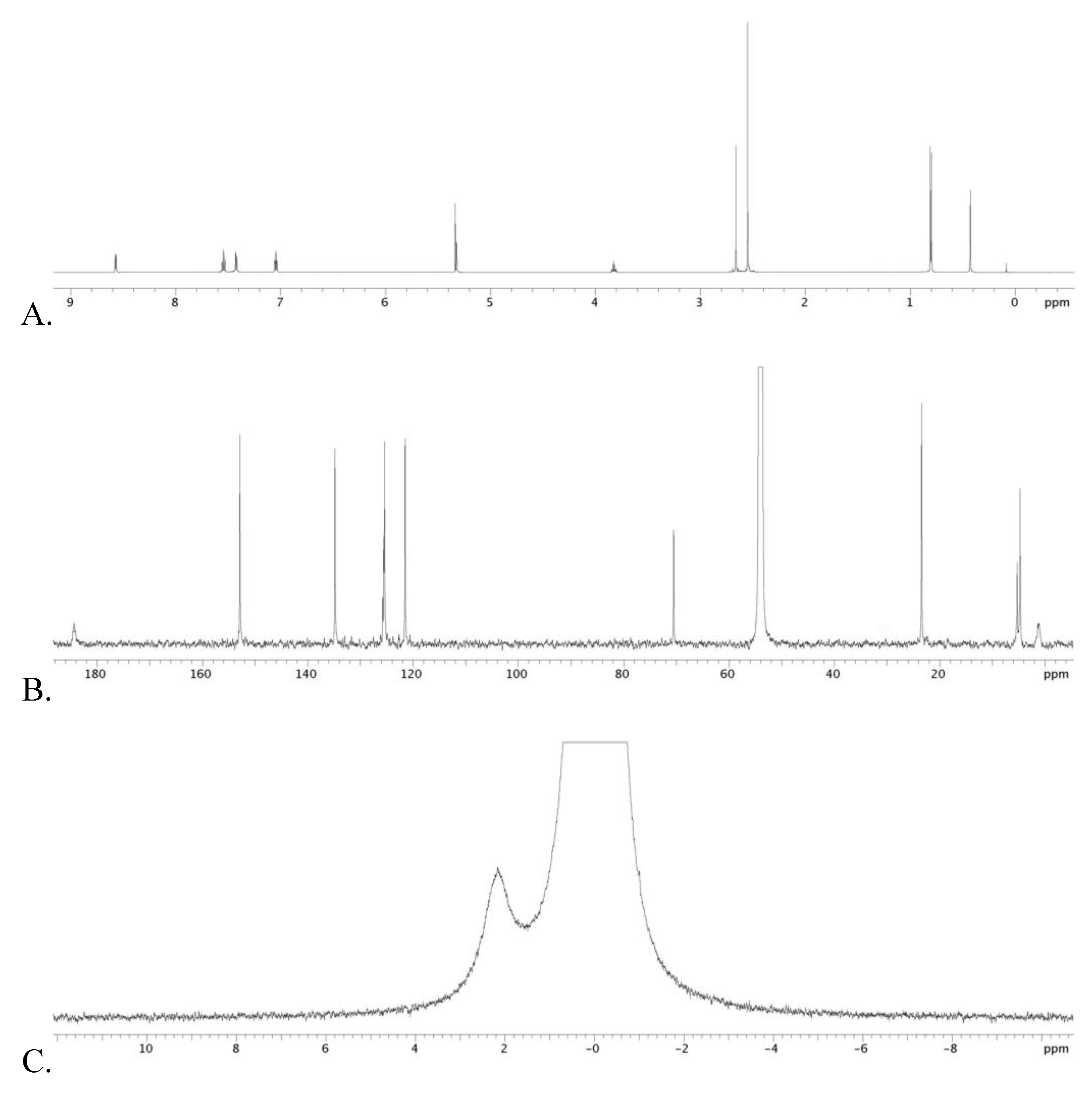
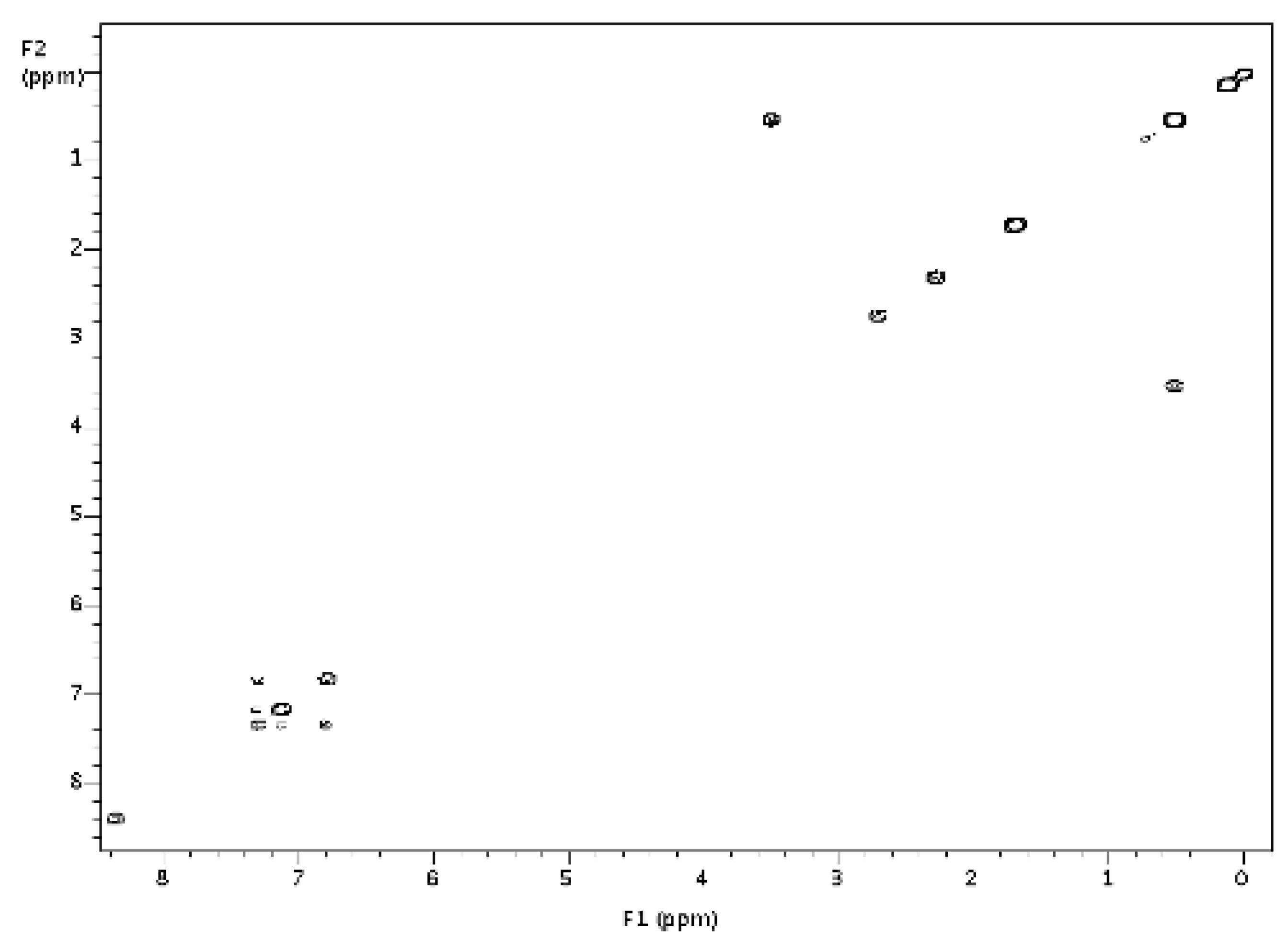
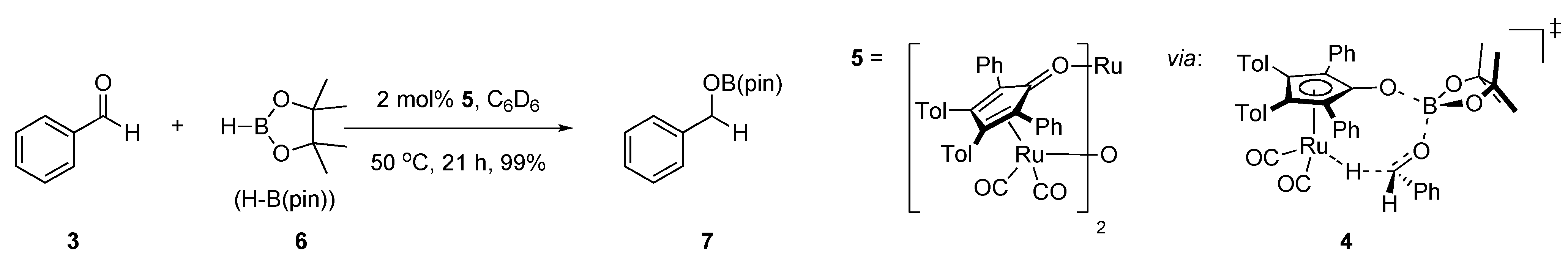
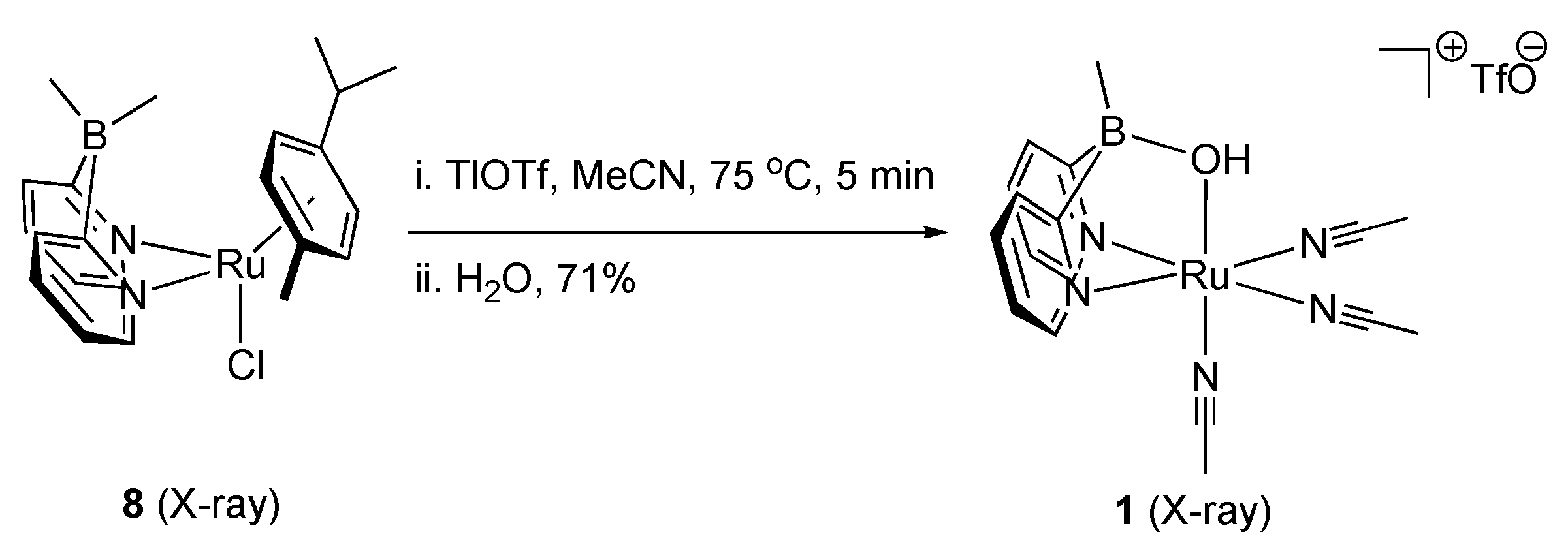
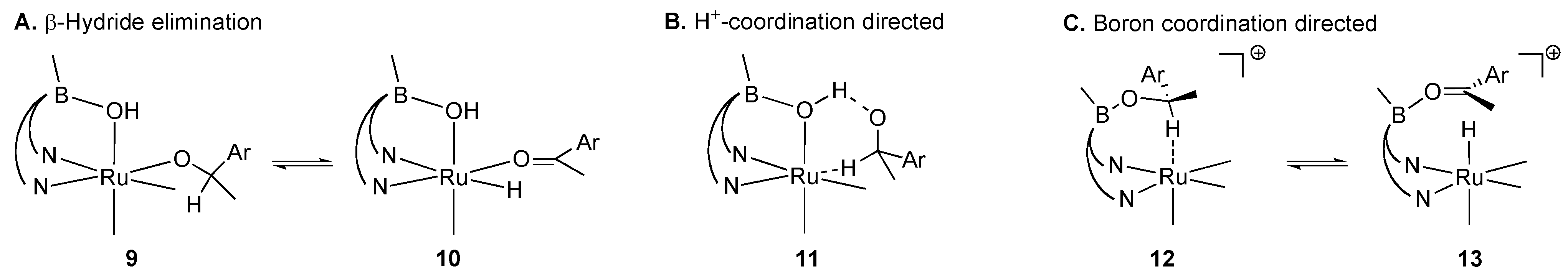
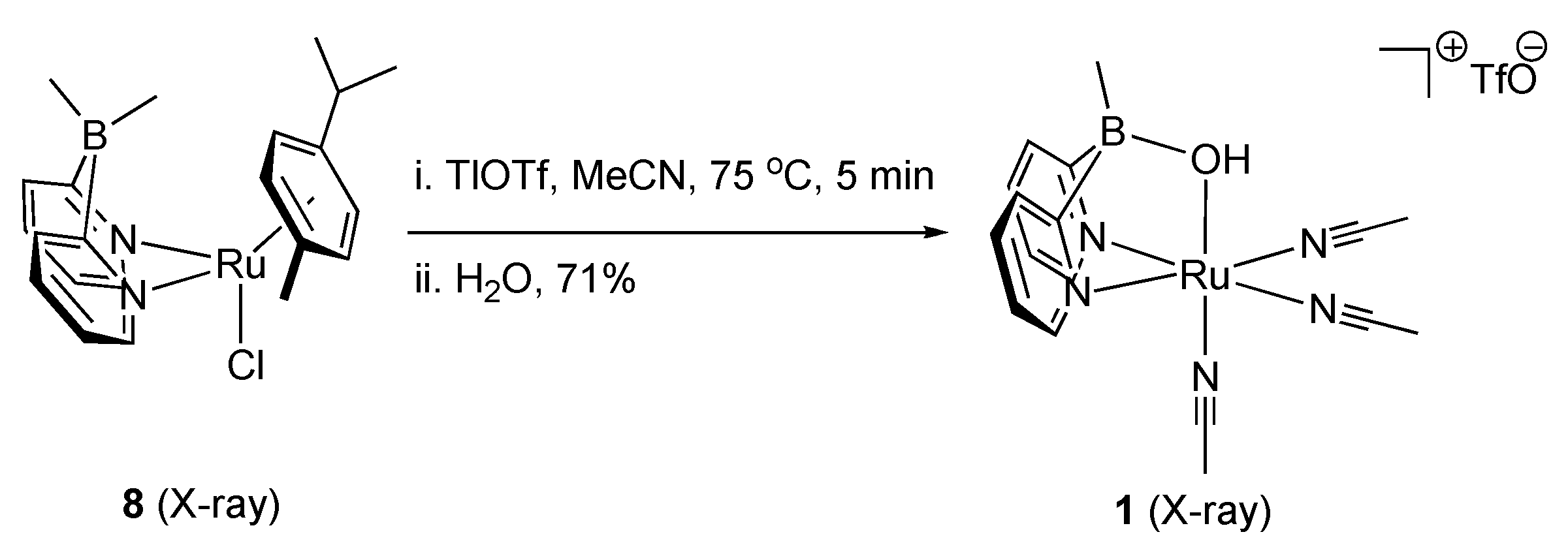
2.1. Stoichiometric Studies
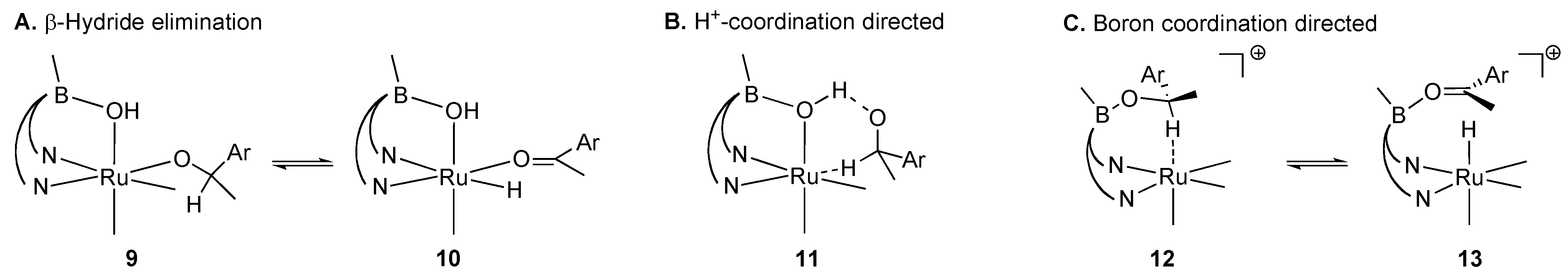
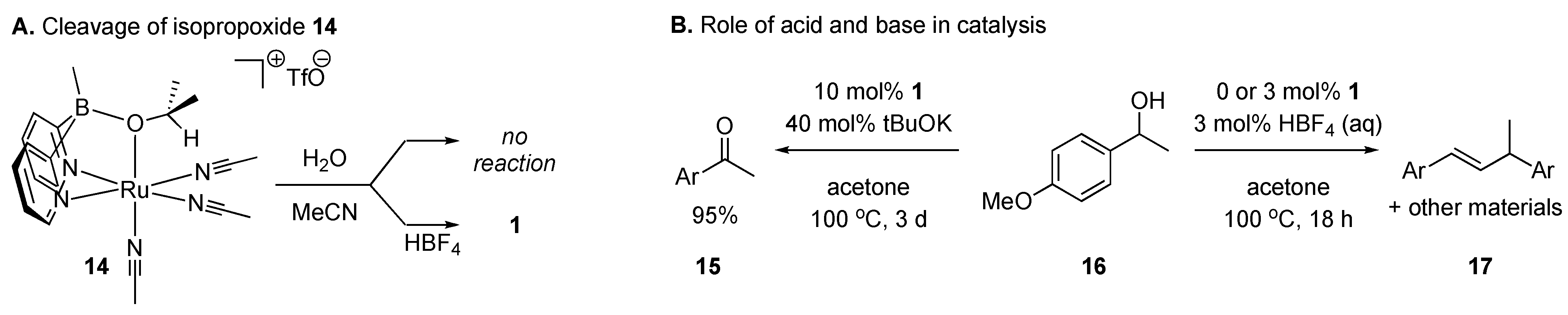
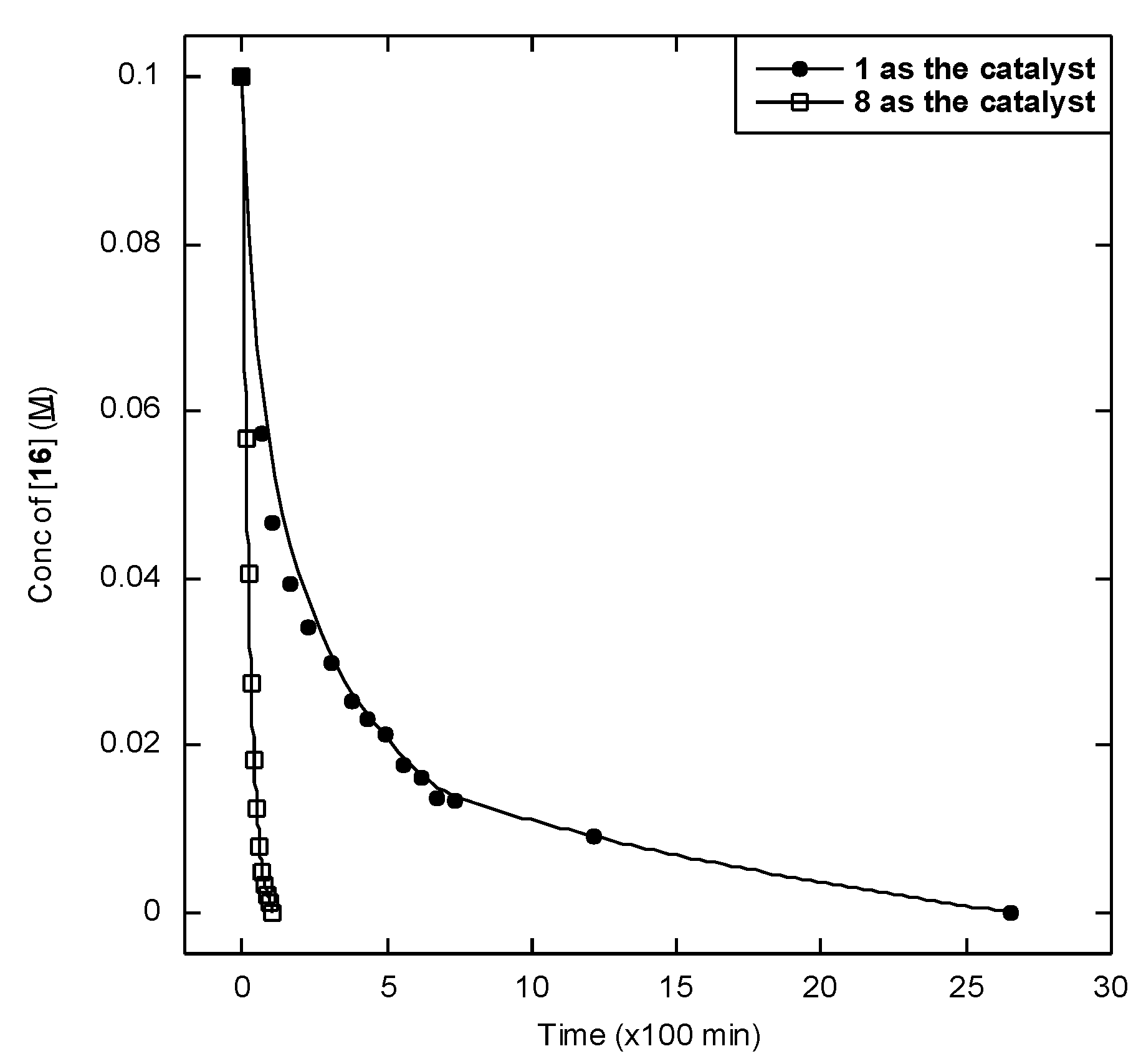
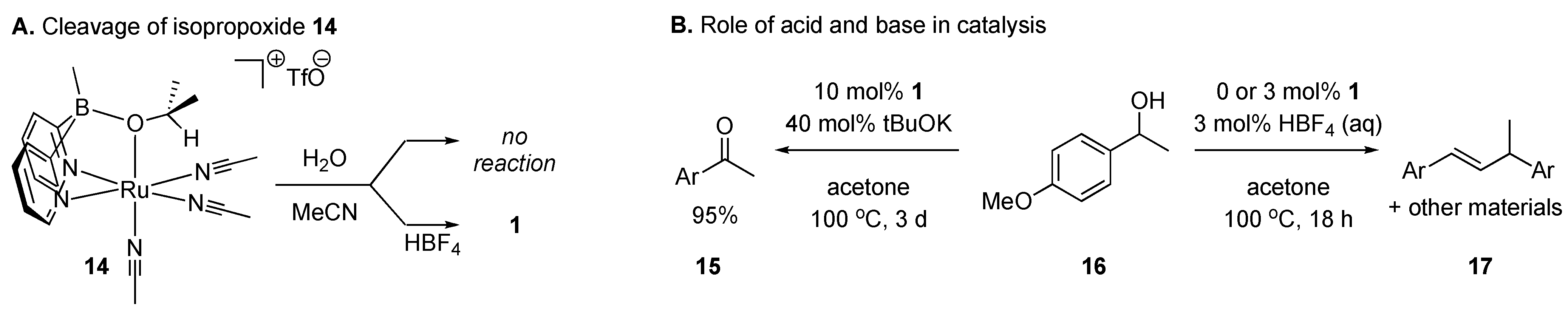
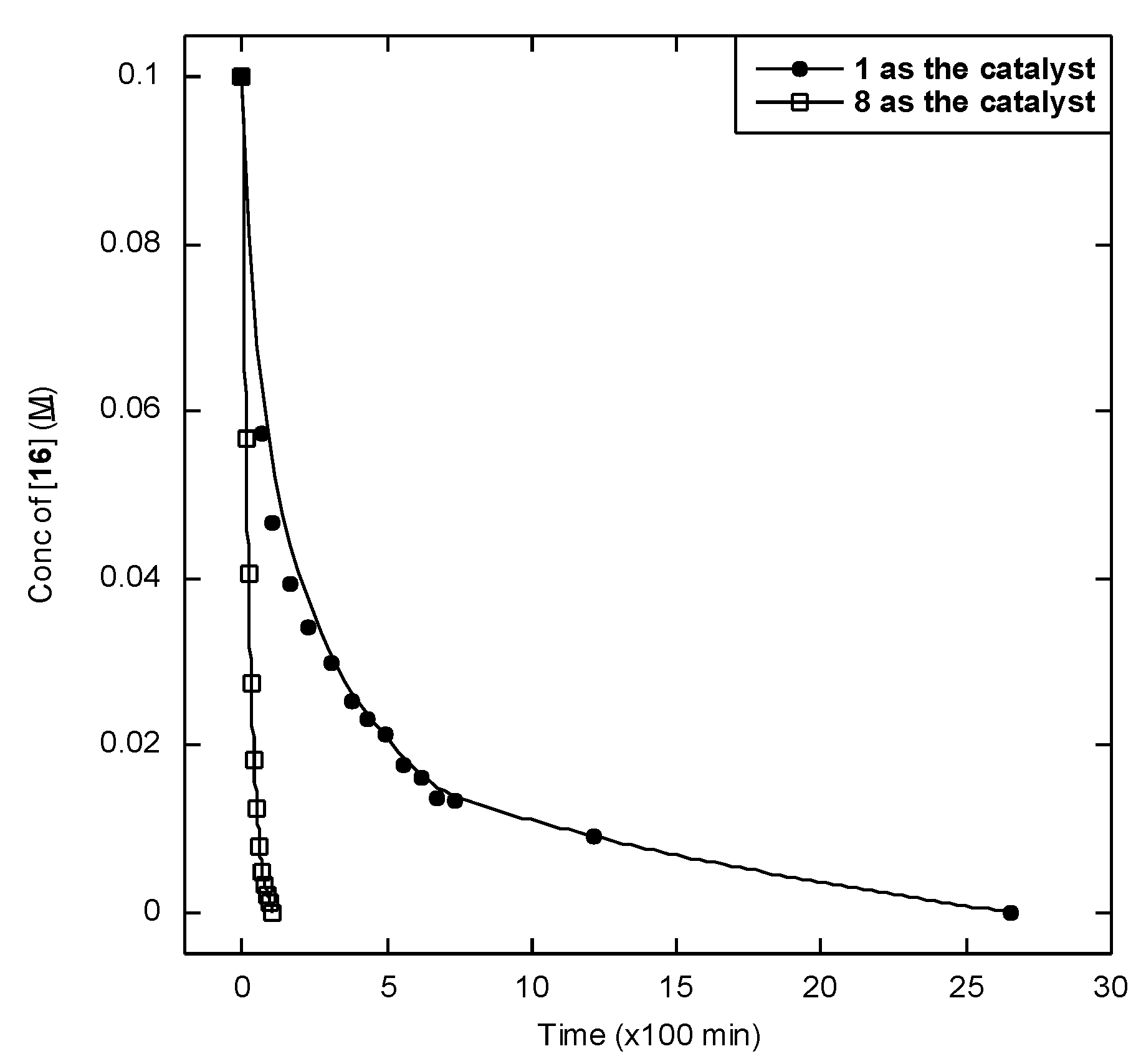
2.2. Catalytic Studies
3. Experimental
3.1. General Procedures
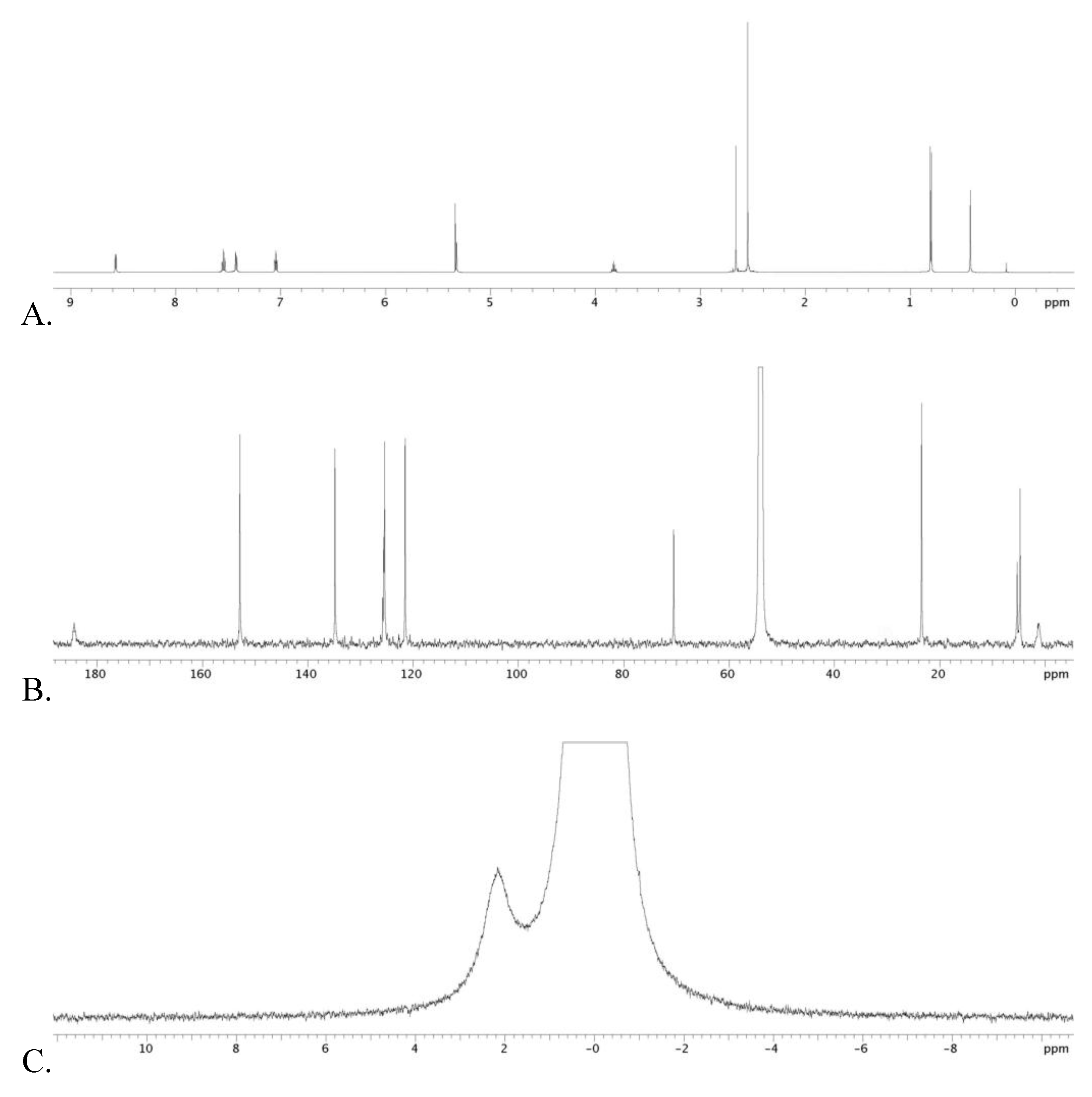
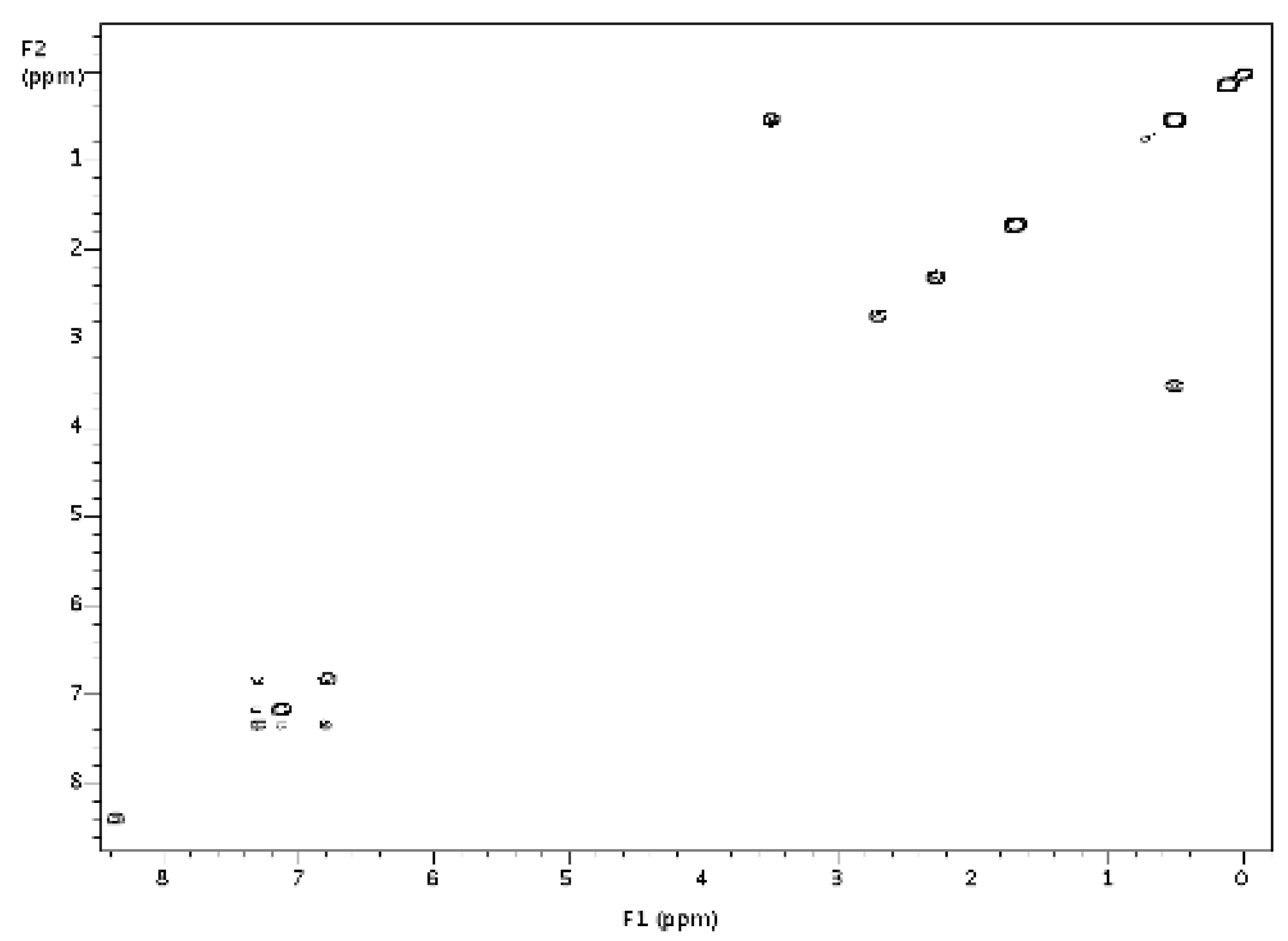
3.2. Trisacetonitrile μ-Isopropoxy Ruthenium Triflate 14
3.3. Diarylbutene 17
4. Conclusions
Acknowledgments
References and Notes
- Shvo, Y.; Czarkie, D.; Rahamim, Y. A new group of ruthenium complexes: structure and catalysis. J. Am. Chem. Soc. 1986, 108, 7400–7402. [Google Scholar] [CrossRef]
- Noyori, S.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 2007, 30, 97–102. [Google Scholar]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Gunanathan, C.; Ben-David, Y.; Milstein, D. Direct synthesis of amides from alcohols and amines with liberation of H2. Science 2007, 317, 790–792. [Google Scholar] [CrossRef]
- Deno, N.C.; Peterson, H.J.; Saines, G.S. The hydride-transfer reaction. Chem. Rev. 1960, 60, 7–14. [Google Scholar] [CrossRef]
- Bäckvall, J.-E. Transition metal hydrides as active intermediates in hydrogen transfer reactions. J. Organomet. Chem. 2002, 652, 105–111. [Google Scholar] [CrossRef]
- Li, Z.; Bohle, D.S.; Li, C.-J. Cu-catalyzed cross-dehydrogenative coupling: a versatile strategy for C–C bond formations via the oxidative activation of sp3 C–H bonds. Proc. Natl. Acad. Sci. USA. 2006, 103, 8928–8933. [Google Scholar] [CrossRef]
- Conley, B.L.; Williams, T.J. Dual site catalysts for hydride manipulations. Comments Inorg. Chem. 2012, 32, 195–218. [Google Scholar]
- Conley, B.L.; Williams, T.J. Thermochemistry and molecular structure of a remarkable agostic interaction in a heterobifunctional ruthenium−boron complex. J. Am. Chem. Soc. 2010, 132, 1764–1765. [Google Scholar] [CrossRef]
- Brookhart, M.; Green, M.L.H.; Parkin, G. Coordination chemistry of saturated molecules special feature: agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. USA. 2007, 104, 6908–6914. [Google Scholar]
- Crabtree, R.H. The organometallic chemistry of alkanes. Chem. Rev. 1985, 85, 245–269. [Google Scholar] [CrossRef]
- Jiang, H.L.; Xu, Q. Catalytic hydrolysis of ammonia borane for chemical hydrogen storage. Catal. Today 2011, 170, 56–63. [Google Scholar] [CrossRef]
- Denney, M.C.; Pons, V.; Hebden, T.J.; Heinekey, D.M.; Goldberg, K.I. Efficient catalysis of ammonia borane dehydrogenation. J. Am. Chem. Soc. 2006, 128, 12048–12049. [Google Scholar] [CrossRef]
- Dietrich, B.L.; Goldberg, K.I.; Heinekey, D.M.; Autrey, T.; Linehan, J.C. Iridium-catalyzed dehydrogenation of substituted amine boranes: kinetics, thermodynamics, and implications for hydrogen storage. Inorg. Chem. 2008, 47, 8583–8585. [Google Scholar]
- Conley, B.L.; Guess, D.; Williams, T.J. A robust, air-stable, reusable ruthenium catalyst for dehydrogenation of ammonia borane. J. Am. Chem. Soc. 2011, 133, 14212–14215. [Google Scholar] [CrossRef]
- Comas-Vives, A.; Ujague, G.; Lledós, A. Hydrogen transfer to ketones catalyzed by Shvo's ruthenium hydride complex: a mechanistic insight. Organometallics 2007, 26, 4135–4144. [Google Scholar] [CrossRef]
- Romero, I.; Rodríguez, M.; Sens, C.; Mola, J.; Kollipara, M.R.; Francàs, L.; Mas-Marza, E.; Escriche, L.; Llobet, A. Ru complexes that can catalytically oxidize water to molecular dioxygen. Inorg. Chem. 2008, 47, 1824–1834. [Google Scholar]
- Noyori, R.; Sandoval, C.A.; Muñiz, K.; Ohkuma, T. Metal–ligand bifunctional catalysis for asymmetric hydrogenation. Phil. Trans. R. Soc. A 2005, 363, 901–912. [Google Scholar] [CrossRef]
- Conley, B.L.; Pennington-Boggio, M.K.; Boz, E.; Williams, T.J. Discovery, applications, and catalytic mechanisms of Shvo’s catalyst. Chem. Rev. 2010, 110, 2294–2312. [Google Scholar] [CrossRef]
- Karvembu, R.; Prabhakaran, R.; Natarajan, K. Shvo’s diruthenium complex: a robust catalyst. Coord. Chem. Rev. 2005, 249, 911–918. [Google Scholar] [CrossRef]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar]
- Casey, C.P.; Guan, H. Cyclopentadienone iron alcohol complexes: synthesis, reactivity, and implications for the mechanism of iron-catalyzed hydrogenation of aldehydes. J. Am. Chem. Soc. 2009, 131, 2499–2507. [Google Scholar] [CrossRef]
- Blum, Y.; Czarkie, D.; Rahamim, Y.; Shvo, Y. (Cyclopentadienone)ruthenium carbonyl complexes - a new class of homogeneous hydrogenation catalysts. Organometallics 1985, 4, 1459–1461. [Google Scholar] [CrossRef]
- Casey, C.P.; Beetner, S.E.; Johnson, J.B. Spectroscopic determination of hydrogenation rates and intermediates during carbonyl hydrogenation catalyzed by Shvo’s hydroxycyclopentadienyl diruthenium hydride agrees with kinetic modeling based on independently measured rates of elementary reactions. J. Am. Chem. Soc. 2008, 130, 2285–2295. [Google Scholar]
- Casey, C.P.; Singer, S.W.; Powell, D.R.; Hayashi, R.K.; Kavana, M. Hydrogen transfer to carbonyls and imines from a hydroxycyclopentadienyl ruthenium hydride: Evidence for concerted hydride and proton transfer. J. Am. Chem. Soc. 2001, 123, 1090–1100. [Google Scholar] [CrossRef]
- Almeida, M.L.S.; Beller, M.; Wang, G.-Z.; Bäckvall, J.-E. Ruthenium(II)-catalyzed oppenauer-type oxidation of secondary alcohols. Chem. Eur. J. 1996, 2, 1533–1536. [Google Scholar] [CrossRef]
- Johnson, J.B.; Bäckvall, J.-E. Mechanism of ruthenium-catalyzed hydrogen transfer reactions. concerted transfer of OH and CH hydrogens from an alcohol to a (cyclopentadienone)ruthenium complex. J. Org. Chem. 2003, 68, 7681–7684. [Google Scholar] [CrossRef]
- Thorson, M.K.; Klinkel, K.L.; Wang, J.; Williams, T.J. Mechanism of hydride abstraction by cyclopentadienone-ligated carbonylmetal complexes (M = Ru, Fe). Eur. J. Inorg. Chem. 2009, 295–302. [Google Scholar]
- Kaneda, K.; Mori, K.; Hara, T.; Mizugaki, T.; Ebitani, K. Design of hydroxyapatite-bound transition metal catalysts for environmentally-benign organic syntheses. Catal. Surv. Asia 2004, 8, 231–239. [Google Scholar] [CrossRef]
- Vijayasri, K.; Rajaram, J.; Kuriacose, J.C. Ruthenium(III)-catalysed oxidation of secondary alcohols by N-methylmorpholine N-oxide (NMO). J. Mol. Cat. 1987, 333, 203–217. [Google Scholar]
- Casey, C.P.; Johnson, J.B.; Singer, S.W.; Cui, Q. Hydrogen elimination from a hydroxycyclopentadienyl ruthenium(II) hydride: Study of hydrogen activation in a ligand−metal bifunctional hydrogenation catalyst. J. Am. Chem. Soc. 2005, 127, 3100–3109. [Google Scholar] [CrossRef]
- Koren-Selfridge, L.; Query, I.P.; Hanson, J.A.; Isley, N.A.; Guzei, I.A.; Clark, T.B. Synthesis of ruthenium boryl analogues of the Shvo metal−ligand bifunctional catalyst. Organometallics 2010, 29, 3896–3900. [Google Scholar] [CrossRef]
- Koren-Selfridge, L.; Londino, H.N.; Vellucci, J.K.; Simmons, B.L.; Casey, C.P.; Clark, T.B. A boron-substituted analogue of the Shvo hydrogenation catalyst: catalytic hydroboration of aldehydes, imines, and ketones. Organometallics 2009, 28, 2085–2090. [Google Scholar]
- CCDC 738031 and 738030 contain supplementary crystallographic data for compounds 8 and 1 respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif.
- Martín-Mature, B.; Åberg, J.B.; Edin, M.; Bäckvall, J.-E. Racemization of secondary alcohols catalyzed by cyclopentadienylruthenium complexes: Evidence for an alkoxide pathway by fast β-hydride elimination–readdition. Chem. Eur. J. 2007, 13, 6063–6072. [Google Scholar] [CrossRef]
- Takao, I.; Masakatsu, S. Bifunctional Molecular Catalysis, 1st ed; Springer: Berlin, Germany, 2011; Volume 37. [Google Scholar]
- Das, R.N.; Sarma, K; Pathak, M.G.; Goswami, A. Silica-supported KHSO4: an efficient system for activation of aromatic terminal olefins. Synlett 2010, 2908–2912. [Google Scholar]
- Conley, B.L.; Williams, T.J. Dehydrogenation of ammonia-borane by Shvo’s catalyst. Chem. Commun. 2010, 4815–4817. [Google Scholar]
- Lu, Y.; Conley, B.L.; Williams, T.J. A three-Stage mechanistic model for ammonia–borane dehydrogenation by Shvo’s catalyst. Organometallics. 2012, 31. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, Z.; Malinoski, B.; Flores, A.V.; Conley, B.L.; Guess, D.; Williams, T.J. Alcohol Dehydrogenation with a Dual Site Ruthenium, Boron Catalyst Occurs at Ruthenium. Catalysts 2012, 2, 412-421. https://doi.org/10.3390/catal2040412
Lu Z, Malinoski B, Flores AV, Conley BL, Guess D, Williams TJ. Alcohol Dehydrogenation with a Dual Site Ruthenium, Boron Catalyst Occurs at Ruthenium. Catalysts. 2012; 2(4):412-421. https://doi.org/10.3390/catal2040412
Chicago/Turabian StyleLu, Zhiyao, Brock Malinoski, Ana V. Flores, Brian L. Conley, Denver Guess, and Travis J. Williams. 2012. "Alcohol Dehydrogenation with a Dual Site Ruthenium, Boron Catalyst Occurs at Ruthenium" Catalysts 2, no. 4: 412-421. https://doi.org/10.3390/catal2040412
APA StyleLu, Z., Malinoski, B., Flores, A. V., Conley, B. L., Guess, D., & Williams, T. J. (2012). Alcohol Dehydrogenation with a Dual Site Ruthenium, Boron Catalyst Occurs at Ruthenium. Catalysts, 2(4), 412-421. https://doi.org/10.3390/catal2040412