
RuNi/MMO Catalysts Derived from a NiAl-NO3-LDH Precursor for CO Selective Methanation in H2-Rich Gases

Guangdong Provincial Key Laboratory of Green Chemical Product Technology, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Catalysts 2023, 13(9), 1245; https://doi.org/10.3390/catal13091245
Submission received: 24 June 2023
/
Revised: 1 August 2023
/
Accepted: 25 August 2023
/
Published: 27 August 2023
(This article belongs to the Special Issue Layered Double Hydroxide-Based Catalysts for Advanced Chemical Technologies)
Abstract
:CO selective methanation (CO-SMET) is a promising method for deep CO removal from H2-rich gases. In this study, a series of RuNi/MMO catalysts are prepared using the support MMO-N derived from NiAl-NO3-LDHs, which was prepared from NiAl-CO3-LDHs via an acid–alcohol ion-exchange reaction. The prepared catalysts were characterized by XRD, SEM, TEM, XPS, H2-TPR, CO-TPD, CO2-TPD, NH3-TPD, and TG. The RuNi/MMO-N catalyst demonstrated excellent CO-SMET performance, successfully reducing the CO to less than 10 ppm with a selectivity greater than 50% in a reaction temperature window ranging from 180 °C to 260 °C. Compared with similar catalysts derived from NiAl-CO3-LDHs, the exceptional CO-SMET capability of the RuNi/MMO-N catalyst is suggested to be associated with a more effective hydrogen spillover, a larger number of electron-rich Ni sites, and a higher density of acid sites on the surface of RuNi/MMO-N, which are conducive to CO adsorption and the inhibition of CO2 methanation.
Keywords:
LDHs; mixed metal oxides; RuNi/MMO catalyst; CO methanation; hydrogen-rich gas; CO removal
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Hydrogen or hydrogen-rich gas used as fuel for proton exchange membrane fuel cells (PEMFCs) is mainly derived from the steam reforming of hydrocarbons. It is vital to remove as much of the carbon monoxide (CO) as possible and obtain concentrations lower than 10 ppm since the presence of CO can poison the Pt electrode of the PEMFCs and degrade the performance [1,2,3]. CO selective methanation (CO-SMET) is regarded as a potential technology for deep CO removal from H2-rich gases because it does not require the addition of extra gas [4,5,6]. Because reforming gas contains approximately 20 vol% carbon dioxide (CO2), CO-SMET is typically accompanied by side reactions of CO2 methanation and reverse water gas shift (RWGS) [7,8,9]. Therefore, a suitable CO-SMET catalyst can remove CO to levels below 10 ppm while inhibiting the methanation of CO2.
Recently, various Ru- and Ni-based catalysts have been reported for CO-SMET [10,11,12]. Many support materials have been investigated, including metal oxides [13,14] and carbon materials [15,16]. Mixed metal oxides (MMOs) [17,18] derived from layered double hydroxides (LDHs) via calcination are widely used as support catalysts in CO-SMET owing to their superior thermal stability, excellent metal dispersion, and large specific surface area [19,20]. However, the presence of numerous basic sites on such catalysts is beneficial for the adsorption and activation of CO2, resulting in numerous CO2 methanation side reactions [21,22].
Zhang et al. [23] reported that the acidity and basicity of the calcined material can be adjusted by changing the interlayer anions of LDHs. Liu et al. [24] showed that anions with a lower charge in LDHs exhibited stronger acidity. Wang et al. [25] found that the interlayer anions of LDHs affect their surface area, morphology, and thermal stability. The performance of LDH-based catalysts is significantly improved by changing the interlayer anion of LDHs [26,27]. It remains a major challenge to improve the performance of CO-SMET catalysts by optimizing the interlayer anions of LDHs to weaken the surface alkaline sites of MMO derived from LDHs, thereby effectively inhibiting the methanation of CO2. However, relevant investigations are currently limited.
In this study, a series of RuNi/MMO catalysts are fabricated by an impregnation method using an MMO-N support derived from a nitrate intercalated LDH (LDHs-N) precursor, which was prepared from a carbonate-intercalated LDH (LDHs-C) acid–alcohol ion-exchange reaction. Compared with similar catalysts in the literature, the RuNi/MMO-N catalyst exhibited a higher activity with effluent CO concentrations of less than 10 ppm in a temperature window ranging from 180 °C to 260 °C for CO-SMET.
2. Results and Discussion
Figure 1 shows the X-ray diffraction (XRD) patterns of the prepared LDH and MMO samples with different concentrations of HNO3 ethanol solutions (0.03 M, 0.06 M, 0.09 M, and 0.12 M) in the acid–alcohol ion-exchange reaction. As shown in Figure 1a, the diffraction peaks of the LDHs-C samples at 11.4°, 23.2°, 35.2°, and 62.1° correspond to the (003), (006), (012), and (110) crystal planes of the LDHs (JCPDS 48-3170), respectively [28]. The typical LDH crystal plane diffraction peaks of the LDHs-N samples were almost identical to those of the LDHs-C samples, indicating that the LDHs-N samples were formed and that the acid–alcohol ion-exchange reaction did not destroy their layered structure. Compared to LDHs-C, the diffraction peak of LDHs-N shifted to a lower angle, possibly due to the expansion of their interlayer distance by the insertion of NO3− anions into the LDHs [24].
The average particle size of the LDHs in the c direction (DLDHs) was calculated from the (003) reflection based on the Scherrer equation and is summarized in Table S1. The d003 of LDHs-C is 0.781 nm, which is close to the 0.75 nm value for that of standard carbonate intercalated LDH [11]. The d003 of LDHs-N (0.06–0.09 M) was 0.831–0.834 nm, which was closest to the 0.833 nm of d003 of nitrate-intercalated LDH previously reported [29,30], indicating that LDH materials containing nitrate intercalation were successfully prepared by ion-exchange reactions. This was further confirmed by the elemental analysis of LDHs-N (0.09 M) (Table S2).
Figure 1b shows that the diffraction peaks of the NiO species appeared for all MMO samples transformed from the LDH precursor by calcination (JCPDS 47-1049) [31]. It is worth noting that the absence of the characteristic diffraction peaks of Al2O3 and spinel species for all MMO samples indicates that no crystalline phase of Al2O3 or nickel aluminate spinel was formed during the calcination of LDHs.
The RuNi/MMO catalyst synthesized using the MMO-N (0.09 M) support, which was transferred from LDHs-N (0.09 M), exhibited a lower hydrogen reduction temperature (Figure S1) and superior catalytic performance than the other RuNi/MMO-N in the CO-SMET reaction (Figure S2). Therefore, unless otherwise specified, in this paper, MMO-N denotes MMO-N (0.09 M) derived from LDHs-N (0.09 M), and LDHs-N denotes LDHs-N (0.09 M) prepared from LDHs-C using acid–alcohol ion exchange in a 0.09 M HNO3 ethanol solution.
Figure S3 is the N2 adsorption–desorption isotherms of RuNi/MMO-C and RuNi/MMO-N catalysts. All catalysts exhibited the typical IV type isotherms with an H1-type hysteresis loop according to the IUPAC classification, which are characteristic of mesoporous materials [32]. The Brunauer−Emmett−Teller (BET) surface area, average pore size, and pore volume of RuNi/MMO-C and RuNi/MMO-N catalysts are summarized in Table S3. Compared with RuNi/MMO-C, the BET surface area of RuNi/MMO-N increased significantly, which facilitates the dispersion of the active metal and thus improves the performance of the catalyst.
The scanning electron microscopy (SEM) images of LDHs-C, LDHs-N, MMO-C, and MMO-N are shown in Figure 2. As shown in Figure 2a,b, both LDHs-C and LDHs-N exhibited LDH nanosheet structures. As shown in Figure 2c,d, the layered structure of the LDHs collapsed after calcination. Numerous agglomerations of blocks or grain granules were observed in MMO-C and MMO-N. Smaller grain granules and LDH nanosheets with a few sheets or a single-layered structure were observed over some MMO-N compared to the MMO-C catalyst, indicating that the precursor of MMO treated with the HNO3 ethanol solution has a good thermal stability.
The structures and compositions of the RuNi/MMO-N and RuNi/MMO-C samples were measured by transmission electron microscopy (TEM), as shown in Figure 3. The average sizes of the Ni and Ru nanoparticles for RuNi/MMO-C and RuNi/MMO-N catalysts are around 4.8 nm and 2.7 nm, respectively. In RuNi/MMO-C (Figure 3a,b), the Ni and Ru nanoparticles exhibited some agglomeration on MMO. Their fringe spacings were 0.204 and 0.234 nm, respectively, consistent with the Ni (111) (JCPDS 04-0850) and Ru (100) (JCPDS 06-0663) planes, and no Ni-Ru alloy nanoparticles were observed. For RuNi/MMO-N (Figure 3c,d), the Ni and Ru nanoparticles were uniformly dispersed on the support without significant agglomeration, as confirmed by the XRD analyses. As shown in Figure S3, the XRD pattern of the RuNi/MMO-N catalyst exhibits weak peaks related to the NiO species in comparison with RuNi/MMO-C, corresponding to the lower crystallinity and smaller grain size of NiO. In addition, the RuNi/MMO-N sample exhibited the same fringe spacing for Ni and Ru nanoparticles as RuNi/MMO-C. The appearance of small particles with an interplanar spacing of 0.210 nm indicates the formation of Ni-Ru alloy nanoparticles [33,34], which can suppress CO2 methanation and further enhances CO hydrogenation to CH4 [35].
The X-ray photoelectron spectroscopy (XPS) spectra of Ni 2p and Ru 3p for RuNi/MMO-C and RuNi/MMO-N are shown in Figure 4. For RuNi/MMO-C, the Ni 2p spectrum displays two peaks at the binding energies of 852.93 eV and 856.15 eV, corresponding to Ni0 species and Ni2+ species, respectively, and the peaks at 861.90 eV are assigned to the oscillating satellite peaks [36,37,38]. The Ru 3d3/2 peak centered at approximately 284.80 eV congenitally overlaps with the C1s signal [39], and the signals at 281.17 eV and 285.66 eV were assigned to Ru 3d5/2 and Ru 3d3/2, respectively [40,41]. On the other hand, only one signal located at 462.33 eV emerges from RuNi/MMO-C, which matches the Ru 3p spectrum of metallic Ru0 [42], indicating that the Ru3+ species was reduced completely. Compared to RuNi/MMO-C, the Ni 2p spectrum in RuNi/MMO-N shifted toward a lower binding energy, whereas the Ru 3d and Ru 3p spectra move toward a higher binding energy, implying that the electron is transferred from Ru to Ni. This indicates the formation of more electron-rich Ni sites, thus promoting the dissociation of CO in CO-SMET [43,44].
The H2 temperature-programmed reduction (H2-TPR) profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts with three reduction peaks are shown in Figure 5. The metallic Ru is characterized by the weak peak of the reduction of Ru3+ around 100–200 °C. The peaks at 429 °C and 605 °C are attributed to the reduction of α-type (surface amorphous NiO or bulk NiO) and γ-type (strongly interacting with MMO or called Al-rich phase) NiO species, respectively [45,46]. It has been reported that abundant acid sites enhance the hydrogen spillover effect [47,48,49]. Since the acid sites of the RuNi/MMO-N catalyst is more abundant than those of the RuNi/MMO-C sample, as shown in the NH3 temperature-programmed desorption experiments, the RuNi/MMO-N catalyst enhanced the hydrogen spillover effect. This promotes the reduction of NiO in the catalyst [41], thus improving the activity of the catalyst. In addition, the RuNi/MMO-N catalyst improved the dispersion of metallic Ni and Ru particles compared to the RuNi/MMO-C catalyst, as verified by TEM (Figure 3), resulting in a decreased reduction temperature and better catalytic activity [15].
The results of CO temperature-programmed desorption (CO-TPD) analyses are shown in Figure 6. Two TPD peaks are observed for the RuNi/MMO-C and RuNi/MMO-N catalysts. The low-temperature (50–300 °C) peak is ascribed to CO adsorbed weakly onto the catalyst surfaces [50]. The high-temperature (300–600 °C) peak is assigned to the bridge-bonded adsorptive CO [28]; it is generally believed that bridged chemical adsorption contributes more to the formation of CH4 than single-site chemisorption [51]. In contrast to the RuNi/MMO-C catalyst, the peak position of bridge-adsorbed CO shifted from 436 °C to 449 °C for RuNi/MMO-N, indicating a stronger interaction between the active sites of RuNi/MMO-N and CO [6]. The enhanced CO adsorption capacity of the RuNi/MMO-N catalyst, which was confirmed by the enhanced peak area of CO-TPD (Figure 6), provides strong evidence of its superior activity and selectivity in the CO-SMET reaction.
The CO2 temperature-programmed desorption (CO2-TPD) profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts possessing three CO2 desorption peaks are shown in Figure 7. In this case, the quantity of CO2 adsorbed indicates the total number of basic sites, and the CO2 desorption temperature demonstrates the strength of the basic sites. The peak below 150 °C corresponds to weak basic sites due to CO2 interaction with surface hydroxyl groups. The peaks observed at 150–300 °C and over 300 °C correspond to moderate and strong basic sites, resulting from CO2 interacting with Lewis acid–base pairings and isolated/surface O2− anions [52]. As shown in Figure 7, the CO2 desorption temperature of RuNi/MMO-N was lower than that of RuNi/MMO-C, indicating that the CO2 adsorption ability of RuNi/MMO-N was lower than that of RuNi/MMO-C. Thus, it is not conducive to the activation of CO2, inhibits the methanation of CO2 [22], and improves the selectivity of the CO methanation reaction.
The peak areas of CO2-TPD for the RuNi/MMO-C and RuNi/MMO-N catalysts are listed in Table S4. The areas of the three basic sites over RuNi/MMO-N were clearly lower than those of RuNi/MMO-C, indicating that the ion exchange of the LDH precursor weakened the basicity of the catalyst’s surface, which reduced the adsorption capacity of CO2 and effectively inhibited the methanation of CO2 [53].
To further corroborate this conclusion, the NH3 temperature-programmed desorption (NH3-TPD) measurements were conducted on the RuNi/MMO-C and RuNi/MMO-N catalysts, and the results are shown in Figure 8. Similar to CO2-TPD, the NH3 desorption temperature represents the strength of the acid sites and the area of the desorption peak indicates the total number of acid sites. The low-temperature peaks at 100–300 °C correspond to weak Lewis acid sites, while the high-temperature peaks at 300–550 °C represent strong Lewis acid sites [54]. The NH3 desorption temperature of RuNi/MMO-N was higher than that of RuNi/MMO-C in terms of strong acid sites, indicating the enhanced acidity of the RuNi/MMO-N samples. Compared with the RuNi/MMO-C sample, the increase in the peak area of NH3-TPD at the two acid sites on RuNi/MMO-N indicates an increase in the number of acid sites. This promotes the adsorption of CO molecules and inhibits the dissociation of CO2 [55,56], thus facilitating the CO-SMET reaction.
The performances of the RuNi/MMO-C and RuNi/MMO-N catalysts in the CO-SMET are shown in Figure 9. In Figure 9a, it was additionally observed that the CO outlet concentrations of CO-SMET initially decreased to below 10 ppm over the RuNi/MMO-C and RuNi/MMO-N catalysts and then increased with increasing reaction temperature owing to competitive CO2 methanation in the reaction system [11]. As shown in Figure 9b, the reaction selectivity for all catalyst samples remained near 100% at low temperatures and then decreased rapidly as the reaction temperature increased. This is because CO methanation mainly produces CH4 at low temperatures, while CO2 also participates in competitive methanation reactions to produce large amounts of CH4 at higher temperatures, resulting in a reduced selectivity for CO-SMET [11].
Compared to RuNi/MMO-C, a drastic improvement in the CO-SMET activity was observed owing to the more favorable hydrogen spillover effect [49], better dispersion of Ni and Ru particles [15], and increased electron-rich Ni sites [43] on the RuNi/MMO-N catalyst. In addition, the low density of the basic sites and high density of the acid sites present on the surface of RuNi/MMO-N are conducive to CO adsorption and inhibition of CO2 methanation [22,55], thus improving the performance of the CO-SMET reaction.
In this study, the reaction temperature window, with a CO concentration in the effluent of less than 10 ppm and a CO selectivity greater than 50%, was considered as the suitable working temperature window of the catalyst for the CO-SMET reaction [13]. As shown in Figure 9c, the RuNi/MMO-N catalyst displays excellent catalytic performance under the suitable working temperature window ranging from 180 °C to 260 °C compared with RuNi/MMO-C. In addition, the CO-SMET performance of the RuNi/MMO-N catalyst is superior to the majority of those previously reported for Ru- and Ni-based catalysts (Table S5).
The stability of the RuNi/MMO-N catalyst was examined by long-term durability tests at 210 °C, as shown in Figure 9d. The effluent CO concentration was relatively stable at 2 ppm over the reaction time of 120 h. Notably, the CO selectivity of the RuNi/MMO-N catalyst was maintained above 70%, yet decreased slightly as the reaction progressed, which may be due to the sintering of nickel and ruthenium particles and carbon deposition during the long-term test [57].
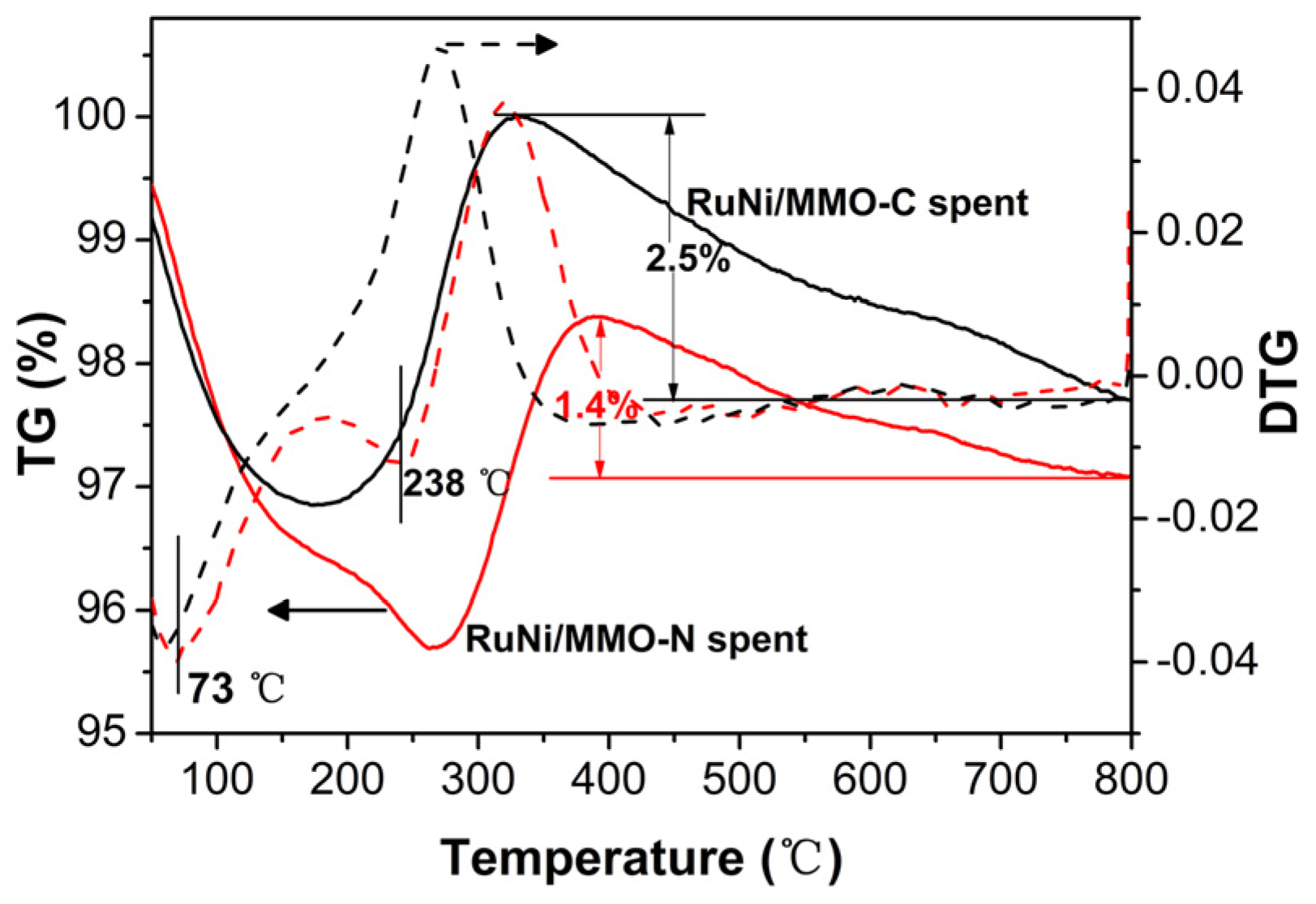
Figure 10 shows the thermogravimetry (TG) and derivative thermogravimetry (DTG) curves of the spent RuNi/MMO-C and RuNi/MMO-N catalysts. The mass loss at temperatures below 250 °C was attributed to the removal of adsorbed water. The weight increase in the temperature window of 200 °C to 350 °C was ascribed to the oxidation of metallic Ni. The weight loss above 350 °C is ascribed to the oxidation of the deposited carbon produced from the cracking reactions of CH4 [58,59]. The weight loss rate of the spent RuNi/MMO-N (1.4%) was lower than that of the spent RuNi/MMO-C (2.5%), indicating that the RuNi/MMO-N catalysts have strong resistance to carbon deposition and, therefore, a good stability during the CO-SMET reaction.
3. Materials and Methods
3.1. Materials
All chemicals and reagents, including RuCl3·nH2O, Ni(NO3)2·6H2O, Al(NO3)3·9H2O, NaOH, and Na2CO3, HNO3, and C2H5OH, were commercially available and directly used as received.
3.2. Preparation of the Catalysts
NiAl-CO3-LDHs were prepared using the co-precipitation technique [11]. A total of 10.91 g Ni(NO3)2·6H2O and 4.69 g Al(NO3)3·9H2O were initially dissolved in 50 mL of deionized water to obtain a homogeneous solution (solution A). A total of 1.00 g of NaOH and 2.65 g of Na2CO3 were dissolved in 50 mL of deionized water to obtain another homogeneous solution (solution B). Solution B was then added dropwise to Solution A until the pH of the mixed solution reached 9. Finally, the precipitates were aged for 18 h at 80 °C, washed with deionized water three times, and dried at 60 °C overnight. The target product was named LDHs-C.
NiAl-NO3-LDHs were prepared by ion exchange [29]. A total of 1.20 g of LDHs-C was added to 200 mL HNO3–ethanol solutions of varying HNO3 concentrations (0.03 M, 0.06 M, 0.09 M, and 0.12 M). The resulting suspension was stirred under N2 flow for 1 h at room temperature for ion exchange. The powders were then filtered, washed with ethanol three times, and dried at 60 °C overnight. The final product NiAl-NO3-LDHs was denoted as LDHs-N (0.03 M, 0.06 M, 0.09 M, and 0.12 M).
The LDHs-C and LDHs-N precursors were calcined in an Ar atmosphere at 450 °C for 1 h to obtain the products MMO-C and MMO-N, respectively.
RuNi/MMO catalysts were synthesized using an incipient wetness impregnation method. A total of 0.30 g of the as-synthesized MMO-C and MMO-N were immersed in a 0.76 mL aqueous solution of 1 wt% RuCl3·nH2O for 24 h, dried at 130 °C overnight, and then reduced at 350 °C in a 50 vol% H2/N2 atmosphere at a flow of 60 mL/min for 1.5 h. The synthesized catalysts were named RuNi/MMO-C and RuNi/MMO-N, respectively. The Ru loading in the RuNi/MMO catalyst was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES), as shown in Table S3.
3.3. Catalyst Characterization
The XRD patterns of the samples were recorded using a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 0.154 nm), operating at a current of 40 mA and a voltage of 40 kV. The scanning range was set between 5 and 80°. Prior to the test, 50 mg powder samples were smeared uniformly onto a sample holder to ensure a flat upper surface.
SEM images were obtained using a Hitachi SU8220 (Tokyo, Japan) scanning electron microscope, set at the working acceleration voltage of 10 kV. Firstly, a conductive adhesive was stuck on the sample table, followed by evenly spreading 10 mg powder samples onto the conductive adhesive. Subsequently, an ear wash ball was used to blow off the non-adhered powder. Prior to the test, the powder samples were sprayed with gold in the vacuum coating apparatus to enhance conductivity.
The morphology and elemental composition of the catalysts were identified by TEM using a JEOL JEM-2100F Transmission Electron Microscope (Tokyo, Japan), operating at the acceleration voltage of 200 kV. Prior to the test, 5 mg powder samples were uniformly dispersed in anhydrous ethanol for 0.5 h using ultrasound, and then a small amount of suspension was transferred onto a copper mesh using a pipette. The samples were subsequently subjected to a drying treatment.
The XPS analysis was conducted using a Kratos Axis Ultra DLD Multifunctional photoelectron spectrometer (Kratos Analytical Ltd., Manchester, UK). An excitation source of Al Kα (1486.6 eV) was used. Specifically, 20 mg powder samples were securely fixed on the sample stage using conductive adhesive. The non-adhered powder was removed by gently blowing with an ear wash ball. The binding energy of carbon C 1s (284.8 eV) on the sample surface served as the internal standard.
The TG analysis of the catalysts was performed using a Thermo Gravimetric Analyzer (NETZSCH TG 209 F3 Tarsus, Germany). Typically, 8 mg catalysts were tested and heated under atmospheric conditions. The heating rate was set at 10 °C/min, and the temperature range for heating was from 50 °C to 800 °C.
H2-TPR analysis was conducted using a Micromeritics AutoChem II 2920 instrument (Micromeritics Instrument Corporation, Norcross, GA, USA). Typically, 200 mg catalysts were placed in a reaction tube. Initially, the catalysts were pretreated in a 10 vol% He atmosphere at the 30 mL/min flow rate at 200 °C for 0.5 h to purify their surface. After cooling down to room temperature, the He flow was switched to a 10 vol% H2/Ar atmosphere at a flow rate of 30 mL/min and heating at a temperature ranging from 80 °C to 800 °C with a heating rate of 10 °C/min.
CO-TPD, CO2-TPD, and NH3-TPD analyses were conducted using Autochem II 2950 (Micromeritics, USA). The effluent gas was detected by a mass spectrometer (MKS Cirrus 2). The concentrations of CO, CO2, and NH3 were determined by the intensities of m/z at 18, 28, and 18, respectively. Briefly, 200 mg catalysts were placed in a reaction tube. Initially, the catalysts were pre-reduced in a 50 vol% H2/N2 atmosphere with a flow rate of 60 mL/min at 350 °C for 1.5 h. After cooling to room temperature with an N2 flow, a CO or CO2 or NH3 atmosphere with a flow rate of 30 mL/min was introduced for 1 h until saturation. Subsequently, the inlet gas was switched to a He flow (30 mL/min) for 1 h to remove the physically adsorbed CO or CO2 or NH3 on the surface of the catalysts. Finally, the temperature was increased to 700 °C (CO) or 600 °C (CO2, NH3) under a He flow (30 mL/min) with a heating rate of 10 °C/min for desorption.
The N2 adsorption−desorption isotherms were examined on an automatic adsorption machine (Micromeritics ASAP2460) at −196 °C. The samples were degassed at 200 °C for 8 h under vacuum before starting N2 adsorption. The specific surface areas of the catalysts were determined according to the BET method. The pore size distributions and pore volumes were derived according to the Barrett–Joyner–Halenda (BJH) method using the desorption branch of the isotherms.
The contents of Ni and Ru in the RuNi/MMO-C and RuNi/MMO-N catalysts were measured on Agilent 5110 ICP-OES (USA). Prior to measurements, the samples were digested in an HF aqueous solution.
3.4. Catalyst Evaluation
The CO-SMET reaction was conducted in a quartz tubular fixed-bed reactor with a diameter of 6 mm at atmospheric pressure. Prior to the measurement, 0.20 g samples were loaded into the center of a quartz tube and fixed with quartz wool, and then, the catalysts were pre-reduced at 350 °C in a 50 vol% H2/N2 atmosphere at the flow of 60 mL/min for 1.5 h. The reaction gas (1 vol% CO, 20 vol% CO2, and 79 vol% H2), which simulated the dry-basis reformate gas, was introduced into the reactor by the mass flow controller (Sevenstar D07-7) at the flow rate of 20 mL/min. The reaction temperature was controlled and measured by the temperature controller (Yudian AI-708P). The compositions of feedstock and effluent gases were examined on-line by a gas chromatograph (Agilent 7820A) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The operating temperatures of TCD, FID, and packed column (ATEO, TDX-01, 3 m × 3 mm) were 200 °C, 250 °C, and 140 °C, respectively. Standard gases containing H2, CO, CO2, and CH4 were purchased from Guangzhou Shengying Gas Company (China). The CO-SMET reaction was performed at temperatures between 150 °C and 350 °C. The catalytic performance was evaluated based on the CO concentration in the effluent [11]. CO selectivity (SCO) was calculated using the following equation:
where is the flow rate of CO in the feedstock, mmol/min; is the flow rate of CO in the effluent, mmol/min; and is the flow rate of CH4 in the effluent, mmol/min.
4. Conclusions
In summary, RuNi/MMO catalysts derived from the LDHs precursor via nitrate intercalation were fabricated for CO selective methanation. Compared to RuNi/MMO-C, the prepared RuNi/MMO-N catalyst demonstrated a higher activity and selectivity for CO-SMET at reaction temperatures ranging from 180 °C to 260 °C. The excellent catalytic performance of the RuNi/MMO-N catalyst was attributed to (1) the better surface dispersion of Ni and Ru particles; (2) enhanced hydrogen overflow effect due to the abundance of acid sites in the catalyst; (3) an increase in electron-rich Ni sites that promoted CO dissociation; and (4) a higher density of acid sites that promoted CO adsorption and inhibited CO2 dissociation. Furthermore, this catalyst demonstrated long-term catalytic stability and carbon resistance.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13091245/s1. Table S1: Structural characteristics of the prepared LDHs; Table S2: C and N chemical compositions of LDHs; Table S3: The physicochemical properties of the RuNi/MMO-C and RuNi/MMO-N catalysts; Table S4: The peak areas of CO2-TPD for the RuNi/MMO-C and RuNi/MMO-N catalysts; Table S5: Summary of the CO-SMET performances for the Ru- and Ni-based catalysts developed in recent years; Figure S1: H2-TPR profiles of different HNO3–ethanol solutions concentrations (0.03 M, 0.06 M, 0.09 M, and 0.12 M) of the RuNi/MMO catalysts; Figure S2: (a) The CO outlet concentration and (b) the selectivity of CO methanation over the different RuNi/MMO-N catalysts; Figure S3: (a) N2 adsorption–desorption isotherms and (b) BJH pore size distributions of the RuNi/MMO-C and RuNi/MMO-N catalysts; Figure S4: XRD patterns of the RuNi/MMO-C and RuNi/MMO-N catalysts. References [15,20,57,60,61,62,63,64,65,66,67] are cited in the Supplementary Materials.
Author Contributions
Methodology, Z.L. and X.Z.; formal analysis, Z.L.; resources, X.Z.; data curation, Z.L.; writing—original draft preparation, Z.L. and X.Z.; writing—review and editing, Z.L. and X.D.; investigation, Z.L., X.Z. and J.M.; supervision, X.D.; funding acquisition, X.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 21978098) and the Natural Science Foundation of Guangdong Province of China (No. 2020A1515010488).
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lan, Y.; Lu, J.; Mu, L.; Wang, S.; Zhai, H. Waste heat recovery from exhausted gas of a proton exchange membrane fuel cell to produce hydrogen using thermoelectric generator. Appl. Energy 2023, 334, 120687. [Google Scholar] [CrossRef]
- Carrette, L.P.L.; Friedrich, K.A.; Huber, M.; Stimming, U. Improvement of CO tolerance of proton exchange membrane (PEM) fuel cells by a pulsing technique. Phys. Chem. Chem. Phys. 2001, 3, 320–324. [Google Scholar] [CrossRef]
- Li, X.; Han, Y.; Huang, Y.; Lin, J.; Pan, X.; Zhao, Z.; Zhou, Y.; Wang, H.; Yang, X.; Wang, A.; et al. Hydrogenated TiO2 supported Ru for selective methanation of CO in practical conditions. Appl. Catal. B Environ. 2021, 298, 120597. [Google Scholar] [CrossRef]
- Fujita, S.I.; Takezawa, N. Difference in the selectivity of CO and CO2 methanation reactions. Chem. Eng. J. 1997, 68, 63–68. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Xu, G. Thermally differential methanation—A novel method to realize highly selective removal of CO from H2-rich reformates. Catal. Commun. 2007, 8, 1953–1956. [Google Scholar] [CrossRef]
- Ping, D.; Wan, Y.; Zhao, X.; Geng, J.; Dong, X. Zr-promoted nickel-rich spinel-supported Ni catalysts with enhanced performance for selective CO methanation. Int. J. Energy Res. 2022, 46, 9128–9137. [Google Scholar] [CrossRef]
- Takenaka, S.; Shimizu, T.; Otsuka, K. Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts. Int. J. Hydrogen Energy 2004, 29, 1065–1073. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R. Preparation of Ru nanoparticles on TiO2 using selective deposition method and their application to selective CO methanation. Catal. Sci. Technol. 2014, 4, 26–29. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R. Mechanistic study and catalyst development for selective carbon monoxide methanation. Catal. Sci. Technol. 2015, 5, 3061–3070. [Google Scholar] [CrossRef]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Ping, D.; Dong, X.; Zang, Y.; Feng, X. Highly efficient Ru/TiO2-NiAl mixed oxide catalysts for CO selective methanation in hydrogen-rich gas. Int. J. Energy Res. 2017, 41, 2308–2317. [Google Scholar] [CrossRef]
- Krämer, M.; Stöwe, K.; Duisberg, M.; Müller, F.; Reiser, M.; Sticher, S.; Maier, W.F. The impact of dopants on the activity and selectivity of a Ni-based methanation catalyst. Appl. Catal. A Gen. 2009, 369, 42–52. [Google Scholar] [CrossRef]
- Chen, A.; Miyao, T.; Higashiyama, K.; Yamashita, H.; Watanabe, M. High Catalytic Performance of Ruthenium-Doped Mesoporous Nickel–Aluminum Oxides for Selective CO Methanation. Angew. Chew. Int. Ed. 2010, 122, 10091–10094. [Google Scholar] [CrossRef]
- Zhang, L.; Xiao, M.; Gao, Z.; Ma, H.; Bao, L.; Li, Z. CO removal via selective methanation over the catalysts Ni/ZrO2 prepared with reduction by the wet H2-rich gas. Int. J. Hydrogen Energy 2018, 43, 15985–15994. [Google Scholar] [CrossRef]
- Ping, D.; Dong, C.; Zhao, H.; Dong, X. A Novel Hierarchical RuNi/Al2O3–Carbon Nanotubes/Ni Foam Catalyst for Selective Removal of CO in H2-Rich Fuels. Ind. Eng. Chem. Res. 2018, 57, 5558–5567. [Google Scholar] [CrossRef]
- Kumi, D.O.; Dlamini, M.W.; Phaahlamohlaka, T.N.; Mhlanga, S.D.; Coville, N.J.; Scurrell, M.S. Selective CO Methanation Over Ru Supported on Carbon Spheres: The Effect of Carbon Functionalization on the Reverse Water Gas Shift Reaction. Catal. Lett. 2018, 148, 3502–3513. [Google Scholar] [CrossRef]
- Meng, X.; Wang, L.; Chen, L.; Xu, M.; Liu, N.; Zhang, J.; Yang, Y.; Wei, M. Charge-separated metal-couple-site in NiZn alloy catalysts towards furfural hydrodeoxygenation reaction. J. Catal. 2020, 392, 69–79. [Google Scholar] [CrossRef]
- Li, S.; Wang, D.; Wu, X.; Chen, Y. Recent advance on VOCs oxidation over layered double hydroxides derived mixed metal oxides. Chin. J. Catal. 2020, 41, 550–560. [Google Scholar] [CrossRef]
- Rosset, M.; Féris, L.A.; Perez-Lopez, O.W. Biogas dry reforming using Ni–Al-LDH catalysts reconstructed with Mg and Zn. Int. J. Hydrogen Energy 2021, 46, 20359–20376. [Google Scholar] [CrossRef]
- Mohaideen, K.K.; Kim, W.; Koo, K.Y.; Yoon, W.L. Highly dispersed Ni particles on Ru/NiAl catalyst derived from layered double hydroxide for selective CO methanation. Catal. Commun. 2015, 60, 8–13. [Google Scholar] [CrossRef]
- He, L.; Lin, Q.; Liu, Y.; Huang, Y. Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticles and a basic support. J. Energy Chem. 2014, 23, 587–592. [Google Scholar] [CrossRef]
- Lima, D.d.S.; Dias, Y.R.; Perez-Lopez, O.W. CO2 methanation over Ni–Al and Co–Al LDH-derived catalysts: The role of basicity. Sustain. Energy Fuels 2020, 4, 5747–5756. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, J.; Jiang, X.; Evans, D.G.; Li, F. Influence of nature of precursors on the formation and structure of Cu–Ni–Cr mixed oxides from layered double hydroxides. J. Phys. Chem. Solids 2006, 67, 1678–1686. [Google Scholar] [CrossRef]
- Liu, H.-M.; Zhao, X.-J.; Zhu, Y.-Q.; Yan, H. DFT study on MgAl-layered double hydroxides with different interlayer anions: Structure, anion exchange, host-guest interaction and basic sites. Phys. Chem. Chem. Phys. 2020, 22, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, Z.; Tay, H.H.; Chen, L.; Liu, Y.; Chang, J.; Zhong, Z.; Luo, J.; Borgna, A. High temperature adsorption of CO2 on Mg–Al hydrotalcite: Effect of the charge compensating anions and the synthesis pH. Catal. Today 2011, 164, 198–203. [Google Scholar] [CrossRef]
- Meyer, O.; Roessner, F.; Rakoczy, R.A.; Fischer, R.W. Impact of Organic Interlayer Anions in Hydrotalcite Precursor on the Catalytic Activity of Hydrotalcite-Derived Mixed Oxides. ChemCatChem 2010, 2, 314–321. [Google Scholar] [CrossRef]
- Du, Y.; Feng, Y.; Zou, C.; Wu, X.; Huang, W. Kinetics and mechanism of acetalisation of furfural to furfural diethyl acetal with Ni–Al layered double hydroxides containing Lewis acid sites. Prog. React. Kinet. Mech. 2018, 43, 21–29. [Google Scholar] [CrossRef]
- Shi, Z.; Feng, J.; Dong, X. Ru–Ni/GA-MMO composites as highly active catalysts for CO selective methanation in H2-rich gases. Int. J. Hydrogen Energy 2023, 48, 24640–24651. [Google Scholar] [CrossRef]
- Iyi, N.; Yamada, H.; Sasaki, T. Deintercalation of carbonate ions from carbonate-type layered double hydroxides (LDHs) using acid–alcohol mixed solutions. Appl. Clay Sci. 2011, 54, 132–137. [Google Scholar] [CrossRef]
- Iyi, N.; Fujii, K.; Okamoto, K.; Sasaki, T. Factors influencing the hydration of layered double hydroxides (LDHs) and the appearance of an intermediate second staging phase. Appl. Clay Sci. 2007, 35, 218–227. [Google Scholar] [CrossRef]
- Liu, M.; Chang, J.; Sun, J.; Gao, L. A facile preparation of NiO/Ni composites as high-performance pseudocapacitor materials. RSC Adv. 2013, 3, 8003–8008. [Google Scholar] [CrossRef]
- Muttakin, M.; Mitra, S.; Thu, K.; Ito, K.; Saha, B. Theoretical framework to evaluate minimum desorption temperature for IUPAC classified adsorption isotherms. Int. J. Heat Mass Transf. 2018, 122, 795–805. [Google Scholar] [CrossRef]
- Zhu, L.; Cao, M.; Li, L.; Sun, H.; Tang, Y.; Zhang, N.; Zheng, J.; Zhou, H.; Li, Y.; Yang, L.; et al. Synthesis of Different Ruthenium Nickel Bimetallic Nanostructures and an Investigation of the Structure–Activity Relationship for Benzene Hydrogenation to Cyclohexane. ChemCatChem 2014, 6, 2039–2046. [Google Scholar] [CrossRef]
- Qin, Y.; Bai, X. Hydrogenation of N-ethylcarbazole over Ni-Ru alloy nanoparticles loaded on graphitized carbon prepared by carbothermal reduction. Fuel 2022, 307, 121921. [Google Scholar] [CrossRef]
- Wang, K.; Men, Y.; Liu, W.; Zhang, J. Recent progress in catalytical CO purification of H2-rich reformate for proton exchange membrane fuel cells. Int. J. Hydrogen Energy 2023, 48, 25100–25118. [Google Scholar] [CrossRef]
- Braos-García, P.; García-Sancho, C.; Infantes-Molina, A.; Rodríguez-Castellón, E.; Jiménez-López, A. Bimetallic Ru/Ni supported catalysts for the gas phase hydrogenation of acetonitrile. Appl. Catal. A Gen. 2010, 381, 132–144. [Google Scholar] [CrossRef]
- Zheng, Z.; Du, X.; Wang, Y.; Li, C.M.; Qi, T. Efficient and Stable NiCo2O4/VN Nanoparticle Catalyst for Electrochemical Water Oxidation. ACS Sustain. Chem. Eng. 2018, 6, 11473–11479. [Google Scholar] [CrossRef]
- Sakai, M.; Imagawa, H.; Baba, N. Layered-double-hydroxide-based Ni catalyst for CO2 capture and methanation. Appl. Catal. A Gen. 2022, 647, 118904. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.; Ji, Y.; Yang, R.; Zhang, Z.; Wang, X.; Liu, H. Hybrid nanostructures of pit-rich TiO2 nanocrystals with Ru loading and N doping for enhanced solar water splitting. Chem. Commun. 2019, 55, 2781–2784. [Google Scholar] [CrossRef]
- Ramos-Fernández, E.V.; Silvestre-Albero, J.; Sepúlveda-Escribano, A.; Rodríguez-Reinoso, F. Effect of the metal precursor on the properties of Ru/ZnO catalysts. Appl. Catal. A Gen. 2010, 374, 221–227. [Google Scholar] [CrossRef]
- Luo, Z.; Zheng, Z.; Li, L.; Cui, Y.-T.; Zhao, C. Bimetallic Ru–Ni Catalyzed Aqueous-Phase Guaiacol Hydrogenolysis at Low H2 Pressures. ACS Catal. 2017, 7, 8304–8313. [Google Scholar] [CrossRef]
- Yang, M.; Feng, T.; Chen, Y.; Liu, J.; Zhao, X.; Yang, B. Synchronously integration of Co, Fe dual-metal doping in Ru@C and CDs for boosted water splitting performances in alkaline media. Appl. Catal. B Environ. 2020, 267, 118657. [Google Scholar] [CrossRef]
- Zhu, C.; Cao, J.-P.; Zhao, X.-Y.; Xie, T.; Zhao, M.; Wei, X.-Y. Bimetallic effects in the catalytic hydrogenolysis of lignin and its model compounds on Nickel-Ruthenium catalysts. Fuel Process. Technol. 2019, 194, 106126. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, Z.; Meng, X.; Lv, Y.; Tao, M. Effect of MoO3 on structures and properties of Ni-SiO2 methanation catalysts prepared by the hydrothermal synthesis method. Ind. Eng. Chem. Res. 2013, 52, 14533–14544. [Google Scholar] [CrossRef]
- Zhao, A.; Ying, W.; Zhang, H.; Ma, H.; Fang, D. Ni/Al2O3 catalysts for syngas methanation: Effect of Mn promoter. J. Nat. Gas Chem. 2012, 21, 170–177. [Google Scholar] [CrossRef]
- Chang, F.-W.; Kuo, M.-S.; Tsay, M.-T.; Hsieh, M.-C. Effect of calcination temperature on catalyst reducibility and hydrogenation reactivity in rice husk ash-alumina supported nickel systems. J. Chem. Technol. Biotechnol. 2004, 79, 691–699. [Google Scholar] [CrossRef]
- Liu, N.; Zhao, J.; Dong, Q.; Zhao, Y.; Fan, B. Supported noble metal catalyst with a core-shell structure for enhancing hydro-genation performance. Mol. Catal. 2021, 506, 111543. [Google Scholar] [CrossRef]
- Shun, K.; Mori, K.; Masuda, S.; Hashimoto, N.; Hinuma, Y.; Kobayashi, H.; Yamashita, H. Revealing hydrogen spillover pathways in reducible metal oxides. Chem. Sci. 2022, 13, 8137–8147. [Google Scholar] [CrossRef]
- Li, M.; Yin, W.; Pan, J.; Zhu, Y.; Sun, N.; Zhang, X.; Wan, Y.; Luo, Z.; Yi, L.; Wang, L. Hydrogen spillover as a promising strategy for boosting heterogeneous catalysis and hydrogen storage. Chem. Eng. J. 2023, 471, 144691. [Google Scholar] [CrossRef]
- Santos, D.C.R.M.; Lisboa, J.S.; Passos, F.B.; Noronha, F.B. Characterization of steam-reforming catalysts. Braz. J. Chem. Eng. 2004, 21, 203–209. [Google Scholar] [CrossRef]
- Alstrup, I. On the Kinetics of Co Methanation on Nickel Surfaces. J. Catal. 1995, 151, 216–225. [Google Scholar] [CrossRef]
- Grad, O.; Kasza, A.M.; Turza, A.; Dan, M.; Barbu-Tudoran, L.; Lazar, M.D.; Mihet, M. Facile and efficient synthesis of ordered mesoporous MIL-53(Al)-derived Ni catalysts with improved activity in CO2 methanation. J. Environ. Chem. Eng. 2023, 11, 109456. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, R.; Liu, N.; Dai, C.; Yu, G.; Wang, N.; Chen, B. In situ Ce-doped catalyst derived from NiCeAl-LDHs with enhanced low-temperature performance for CO2 methanation. Appl. Surf. Sci. 2022, 579, 152204. [Google Scholar] [CrossRef]
- Barthos, R.; Lønyi, F.; Onyestyak, G.; Valyon, J. An NH3-TPD and -FR study on the acidity of sulfated zirconia. Solid State Ion. 2001, 141–142, 253–258. [Google Scholar] [CrossRef]
- Huang, L.; Shi, Y.; Xiong, W.; Ding, Y.; Zhang, Y. Facile design of highly effective Fe-modified bimetallic Fex–Ni1−x-MOFs catalysts with rodlike structures for low-temperature NO reduction by CO. J. Mater. Sci. 2021, 56, 9914–9928. [Google Scholar] [CrossRef]
- Al-Mubaddel, F.S.; Kumar, R.; Sofiu, M.L.; Frusteri, F.; Ibrahim, A.A.; Srivastava, V.K.; Kasim, S.O.; Fakeeha, A.H.; Abasaeed, A.E.; Osman, A.I.; et al. Optimizing acido-basic profile of support in Ni supported La2O3+Al2O3 catalyst for dry reforming of methane. Int. J. Hydrogen Energy 2021, 46, 14225–14235. [Google Scholar] [CrossRef]
- Tada, S.; Kikuchi, R.; Wada, K.; Osada, K.; Akiyama, K.; Satokawa, S.; Kawashima, Y. Long-term durability of Ni/TiO2 and Ru–Ni/TiO2 catalysts for selective CO methanation. J. Power Sources 2014, 264, 59–66. [Google Scholar] [CrossRef]
- Sengupta, S.; Ray, K.; Deo, G. Effects of modifying Ni/Al2O3 catalyst with cobalt on the reforming of CH4 with CO2 and cracking of CH4 reactions. Int. J. Hydrogen Energy 2014, 39, 11462–11472. [Google Scholar] [CrossRef]
- Liu, Q.; Zhong, Z.; Gu, F.; Wang, X.; Lu, X.; Li, H.; Xu, G.; Su, F. CO methanation on ordered mesoporous Ni–Cr–Al catalysts: Effects of the catalyst structure and Cr promoter on the catalytic properties. J. Catal. 2016, 337, 221–232. [Google Scholar] [CrossRef]
- Gao, Z.; Cui, L.; Ma, H. Selective methanation of CO over Ni/Al2O3 catalyst: Effects of preparation method and Ru addition. Int. J. Hydrogen Energy 2016, 41, 5484–5493. [Google Scholar] [CrossRef]
- Dai, X.; Liang, J.; Ma, D.; Zhang, X.; Zhao, H.; Zhao, B.; Guo, Z.; Kleitz, F.; Qiao, S. Large-pore mesoporous RuNi-doped TiO2–Al2O3 nanocomposites for highly efficient selective CO methanation in hydrogen-rich reformate gases. Appl. Catal. B Environ. 2015, 165, 752–762. [Google Scholar] [CrossRef]
- Wang, C.; Ping, D.; Dong, X.; Dong, Y.; Zang, Y. Construction of Ru/Ni-Al-oxide/Ni-foam monolithic catalyst for deep-removing CO in hydrogen-rich gas via selective methanation. Fuel Process. Technol. 2016, 148, 367–371. [Google Scholar] [CrossRef]
- Tada, S.; Shoji, D.; Urasaki, K.; Shimoda, N.; Satokawa, S. Physical mixing of TiO2 with sponge nickel creates new active sites for selective CO methanation. Catal. Sci. Technol. 2016, 6, 3713–3717. [Google Scholar] [CrossRef]
- Guo, Q.; Li, S.; Li, J.; Hu, Y.; Li, Y. Effect of SiO2 on the CO Selective Methanation over SiO2/Ni-ZrO2 Catalysts. ChemCatChem 2022, 14, e202101281. [Google Scholar] [CrossRef]
- Feng, X.; Dong, C.; Ping, D.; Geng, J.; Zhang, J.; Dong, X. Zr-Modified SBA-15 Supported Ni Catalysts with Excellent Catalytic Performance of CO Selective Methanation in H2-Rich Fuels. Catal. Lett. 2018, 148, 2967–2973. [Google Scholar] [CrossRef]
- Zyryanova, M.M.; Snytnikov, P.V.; Gulyaev, R.V.; Amosov, Y.I.; Boronin, A.I.; Sobyanin, V.A. Performance of Ni/CeO2 catalysts for selective CO methanation in hydrogen-rich gas. Chem. Eng. J. 2014, 238, 189–197. [Google Scholar] [CrossRef]
- Ping, D.; Zhao, H.; Dong, X. Ni-doped TiO2 nanotubes supported Ru catalysts for CO selective methanation in H2-rich reformate gases. React. Kinet. Mech. Catal. 2018, 124, 619–631. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns of (a) LDHs and (b) the corresponding calcined samples of MMOs.

Figure 2.
SEM images of (a) LDHs-C, (b) LDHs-N, (c) MMO-C, and (d) MMO-N.

Figure 3.
(a) TEM and (b) HRTEM images of RuNi/MMO-C. (c) TEM and (d) HRTEM images of the RuNi/MMO-N.
Figure 3.
(a) TEM and (b) HRTEM images of RuNi/MMO-C. (c) TEM and (d) HRTEM images of the RuNi/MMO-N.
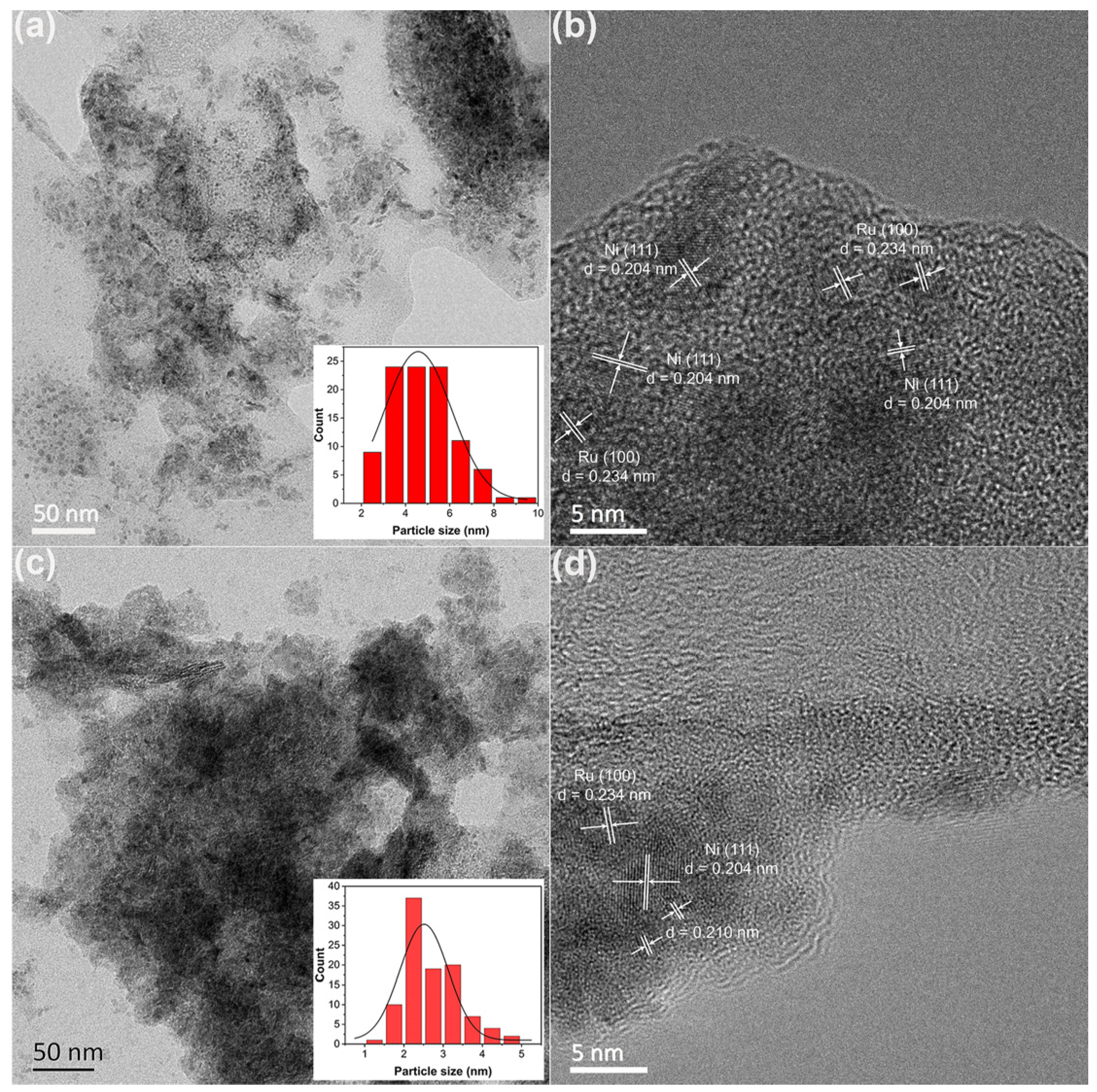
Figure 4.
The (a) Ni 2p and (b) Ru 3d and 3p XPS spectra for the RuNi/MMO-C and RuNi/MMO-N catalysts.
Figure 4.
The (a) Ni 2p and (b) Ru 3d and 3p XPS spectra for the RuNi/MMO-C and RuNi/MMO-N catalysts.

Figure 5.
H2-TPR profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts.

Figure 6.
The CO-TPD profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts.

Figure 7.
The CO2-TPD profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts.
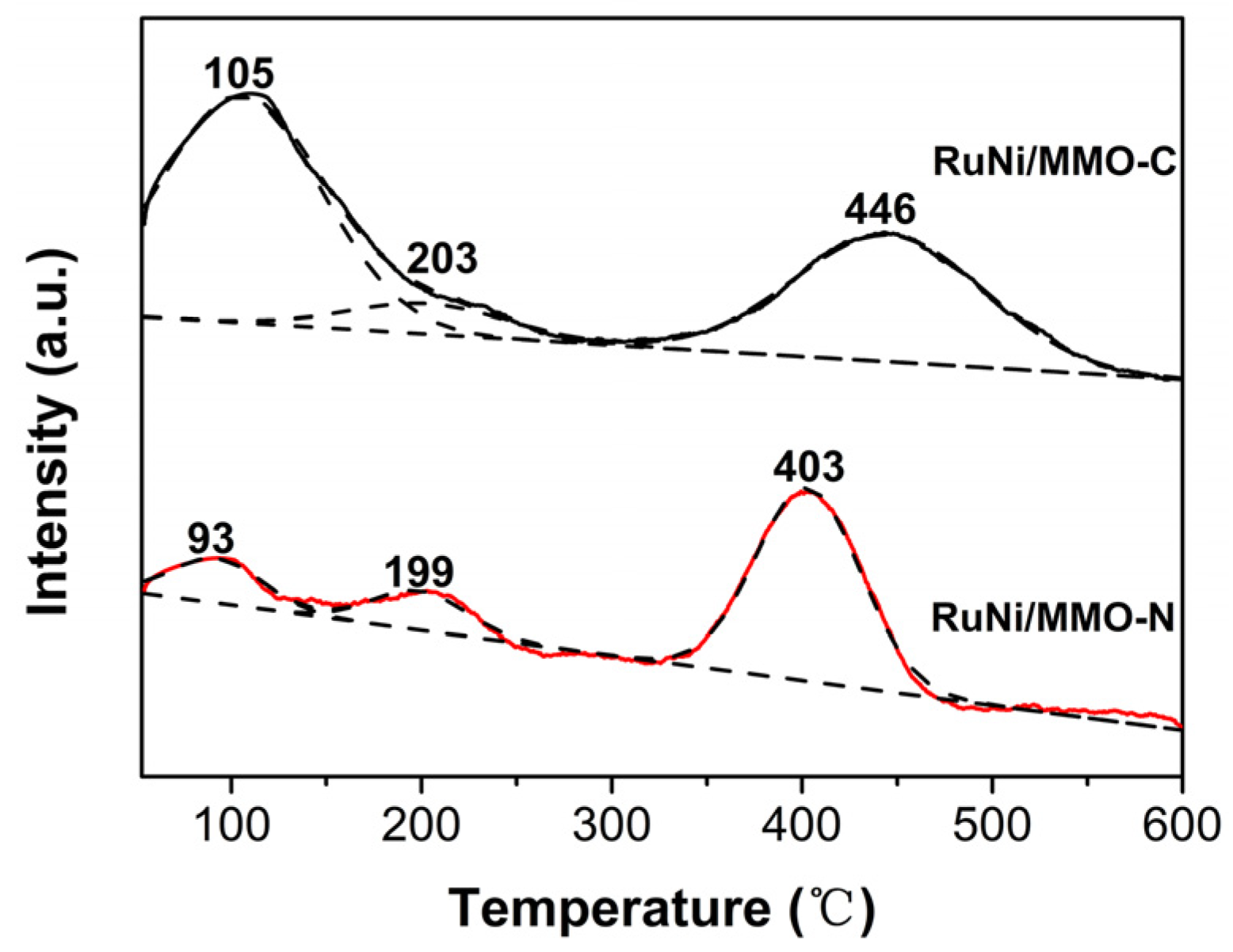
Figure 8.
The NH3-TPD profiles of the RuNi/MMO-C and RuNi/MMO-N catalysts.
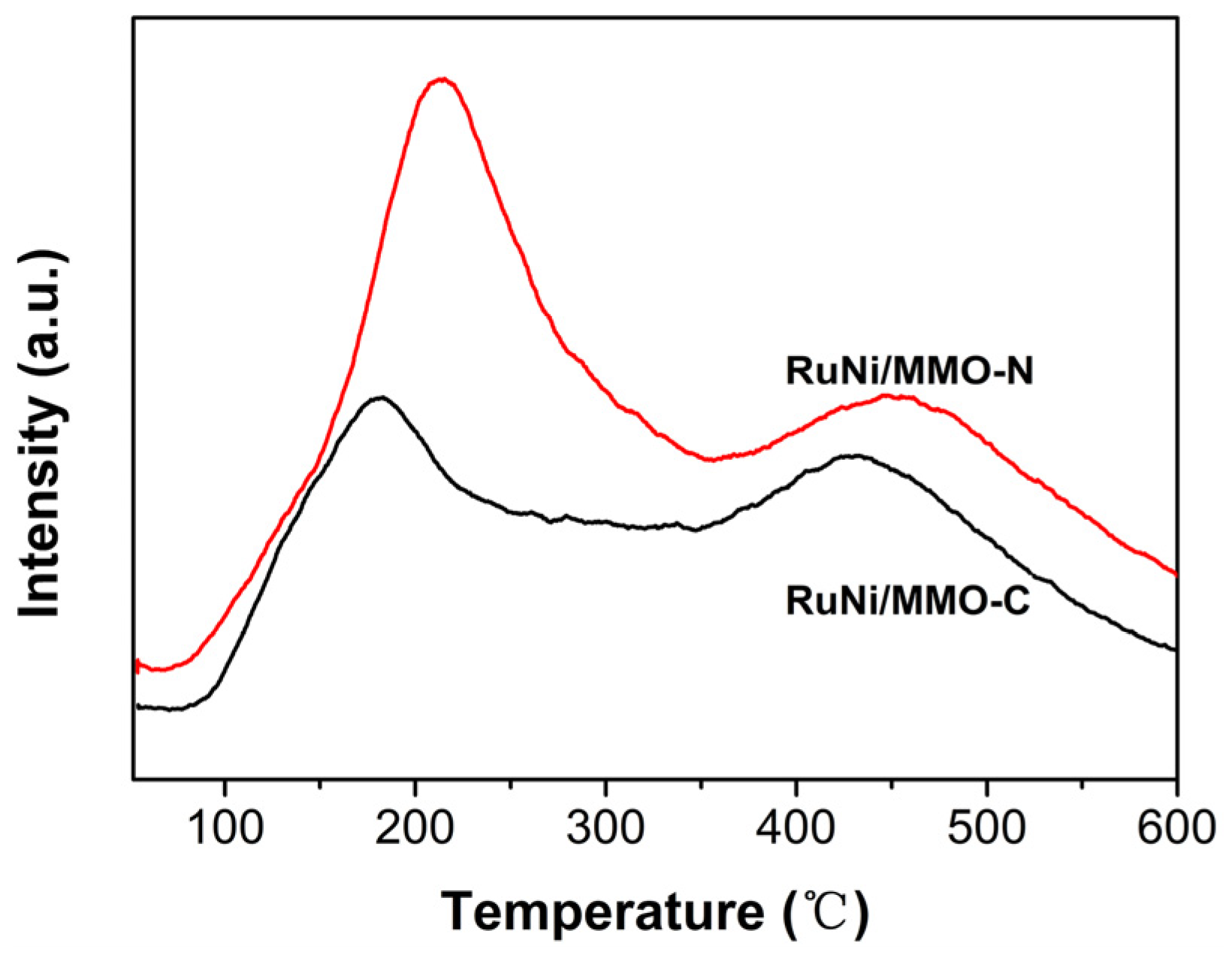
Figure 9.
Catalytic performance of the RuNi/MMO-C and RuNi/MMO-N catalysts for CO-SMET: (a) CO concentration; (b) the selectivity of CO methanation. (c) The suitable working temperature window. (d) The long-term durability test of the RuNi/MMO-N catalyst at 210 °C.
Figure 9.
Catalytic performance of the RuNi/MMO-C and RuNi/MMO-N catalysts for CO-SMET: (a) CO concentration; (b) the selectivity of CO methanation. (c) The suitable working temperature window. (d) The long-term durability test of the RuNi/MMO-N catalyst at 210 °C.
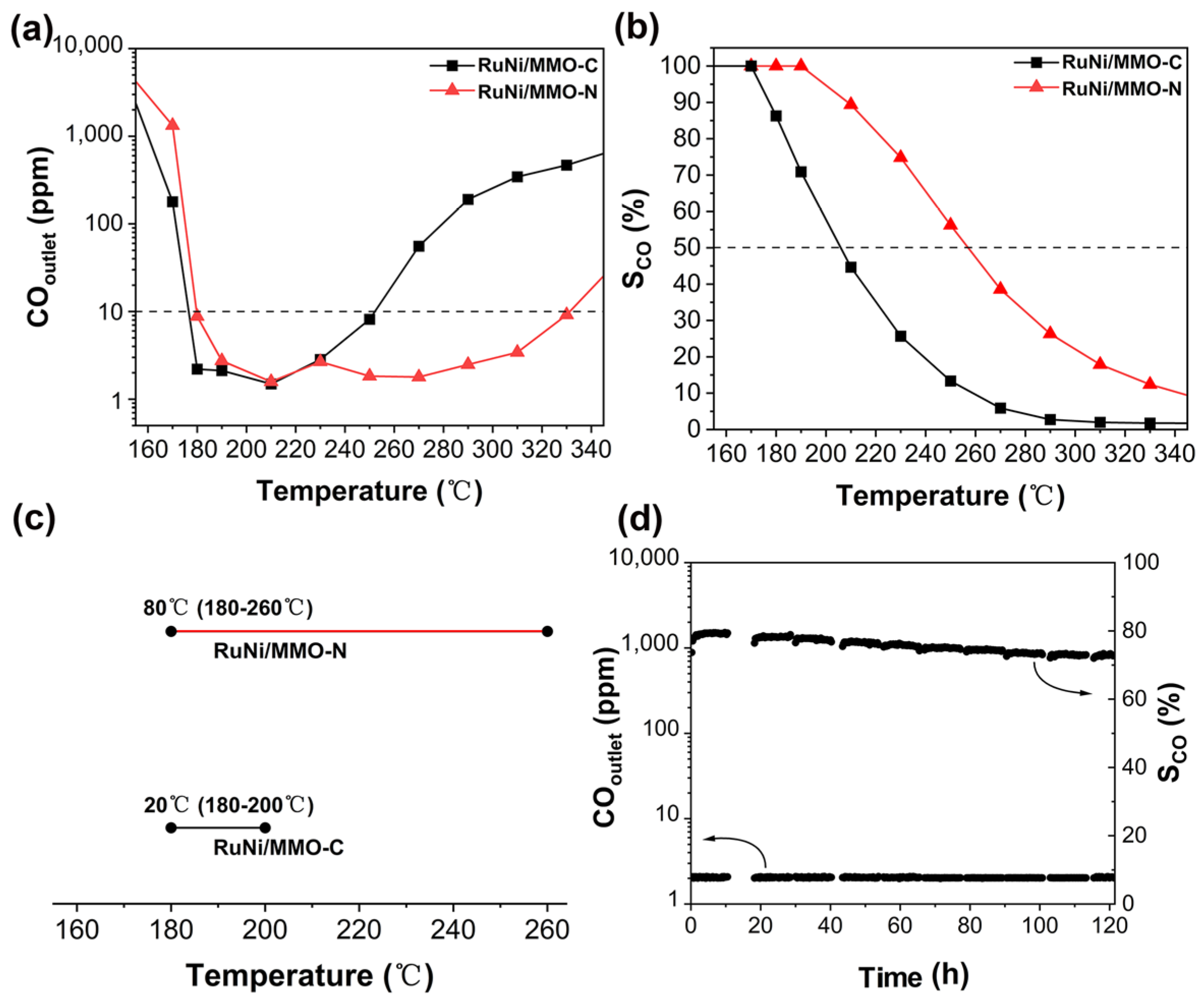
Figure 10.
TG and DTG curves of spent the RuNi/MMO-C and RuNi/MMO-N catalysts.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Z.; Zhao, X.; Ma, J.; Dong, X. RuNi/MMO Catalysts Derived from a NiAl-NO3-LDH Precursor for CO Selective Methanation in H2-Rich Gases. Catalysts 2023, 13, 1245. https://doi.org/10.3390/catal13091245
AMA Style
Li Z, Zhao X, Ma J, Dong X. RuNi/MMO Catalysts Derived from a NiAl-NO3-LDH Precursor for CO Selective Methanation in H2-Rich Gases. Catalysts. 2023; 13(9):1245. https://doi.org/10.3390/catal13091245
Chicago/Turabian StyleLi, Zhihui, Xinyuan Zhao, Jiteng Ma, and Xinfa Dong. 2023. "RuNi/MMO Catalysts Derived from a NiAl-NO3-LDH Precursor for CO Selective Methanation in H2-Rich Gases" Catalysts 13, no. 9: 1245. https://doi.org/10.3390/catal13091245
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.