

Polymer-Grafted 3D-Printed Material for Enzyme Immobilization—Designing a Smart Enzyme Carrier

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
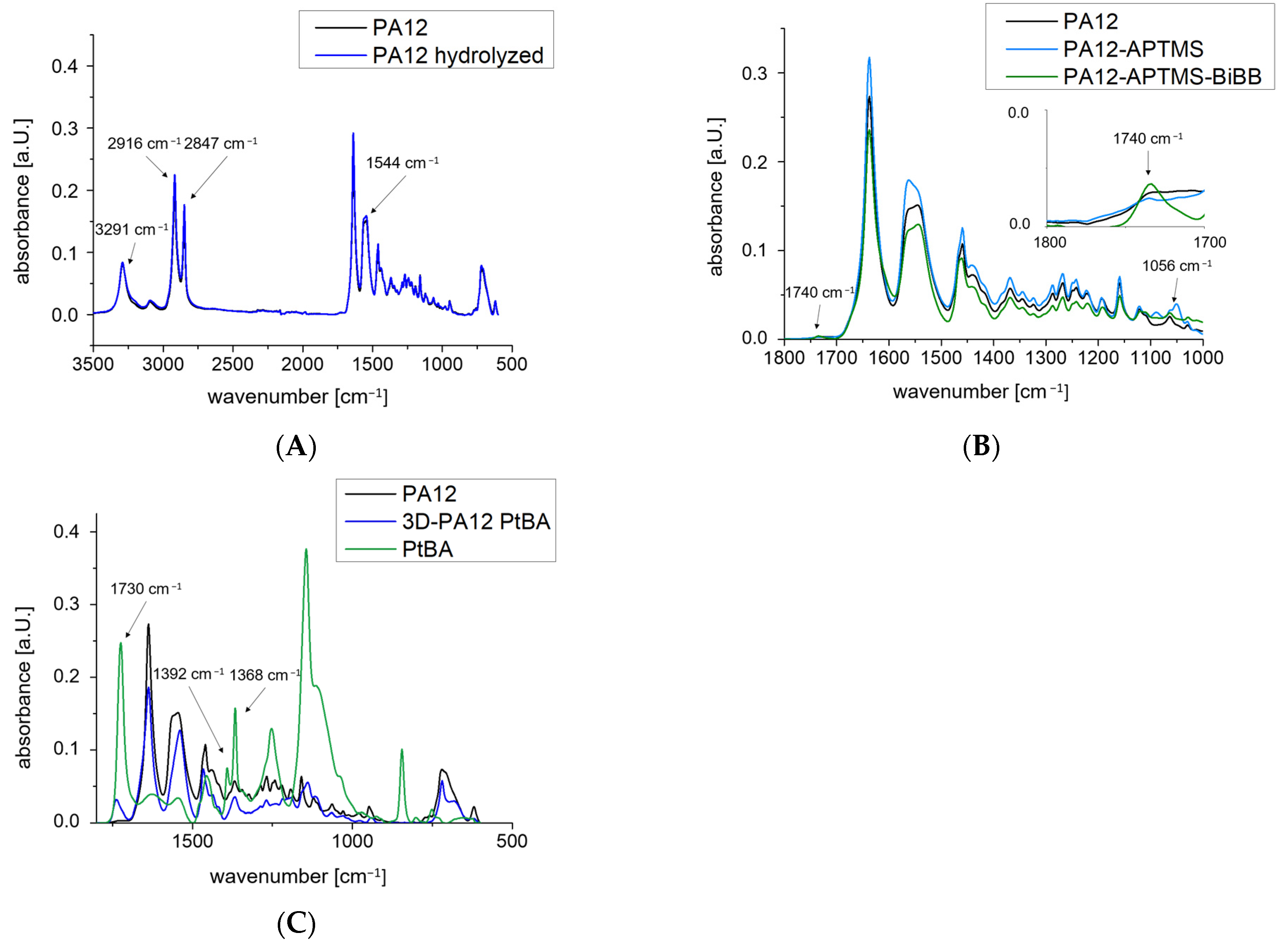
2.1. Surface Functionalization
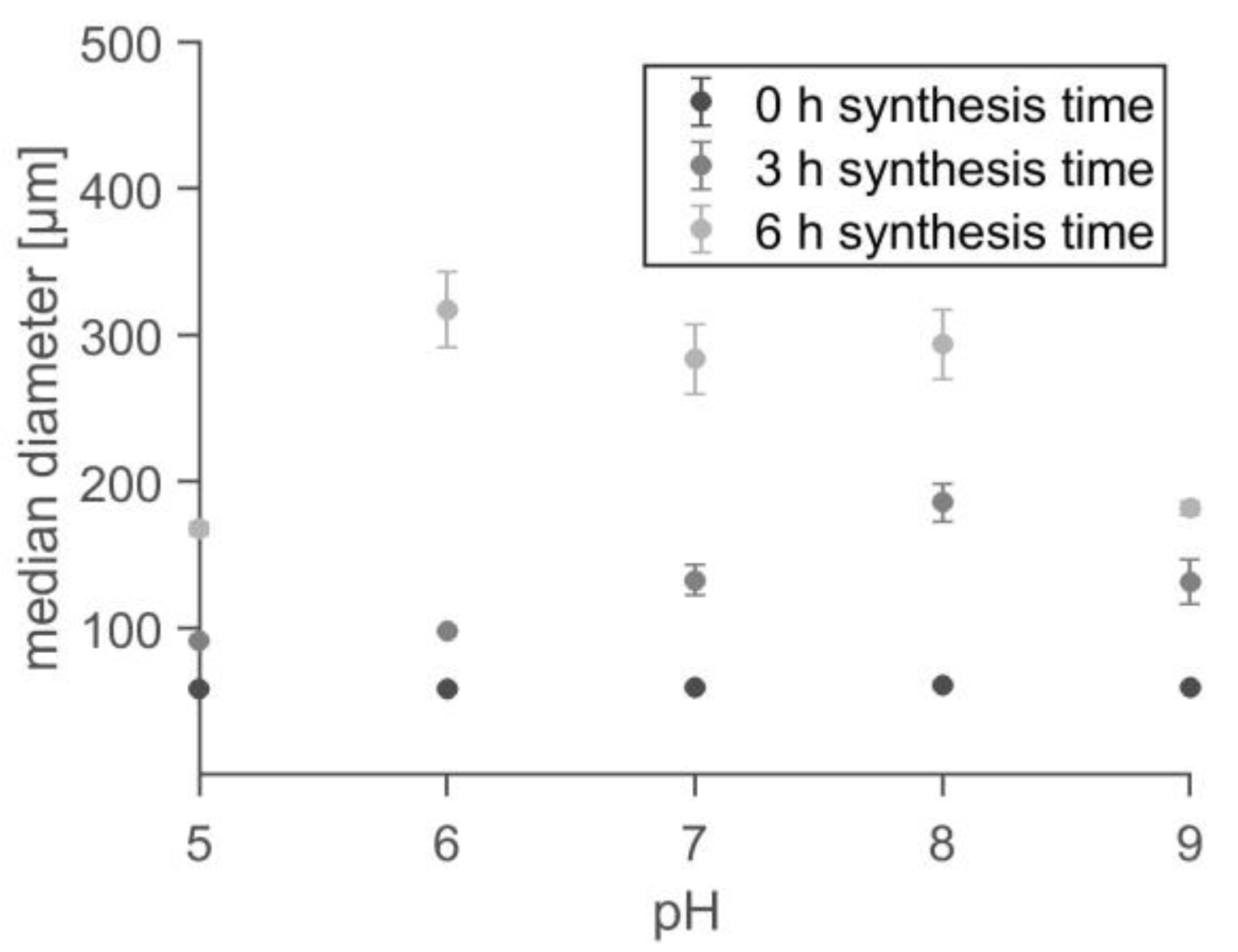
2.2. Polymerization and Topografic Characterization
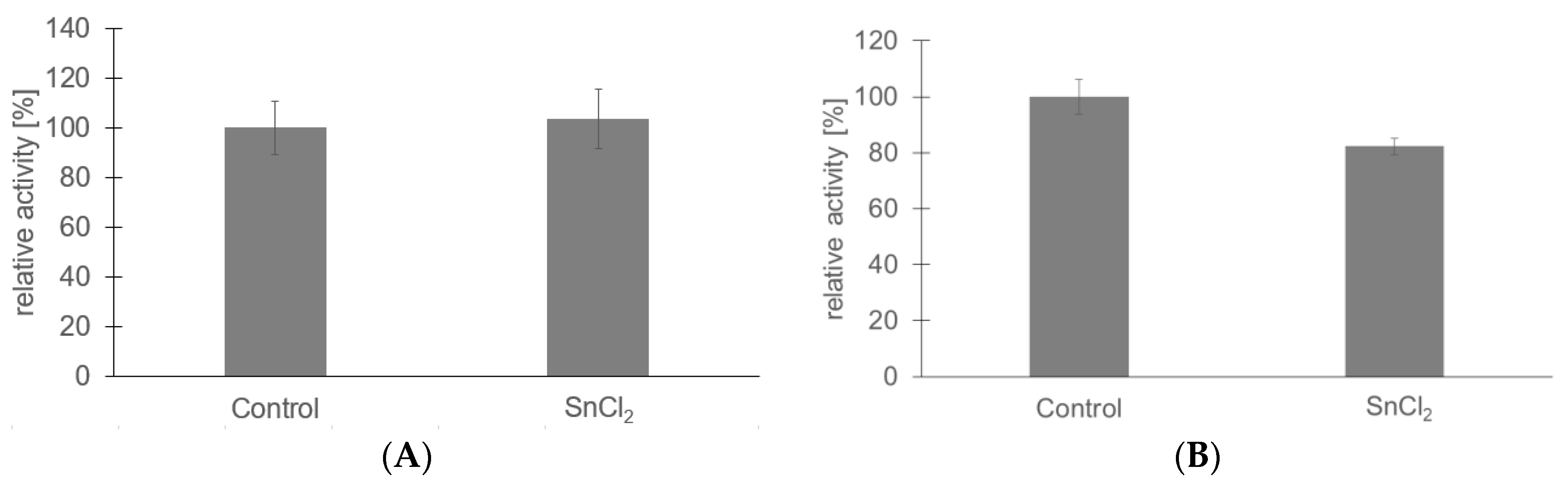
2.3. Influence of Grafted Polymer Layer on Stimuli Responsivity and Enzyme Immobilization
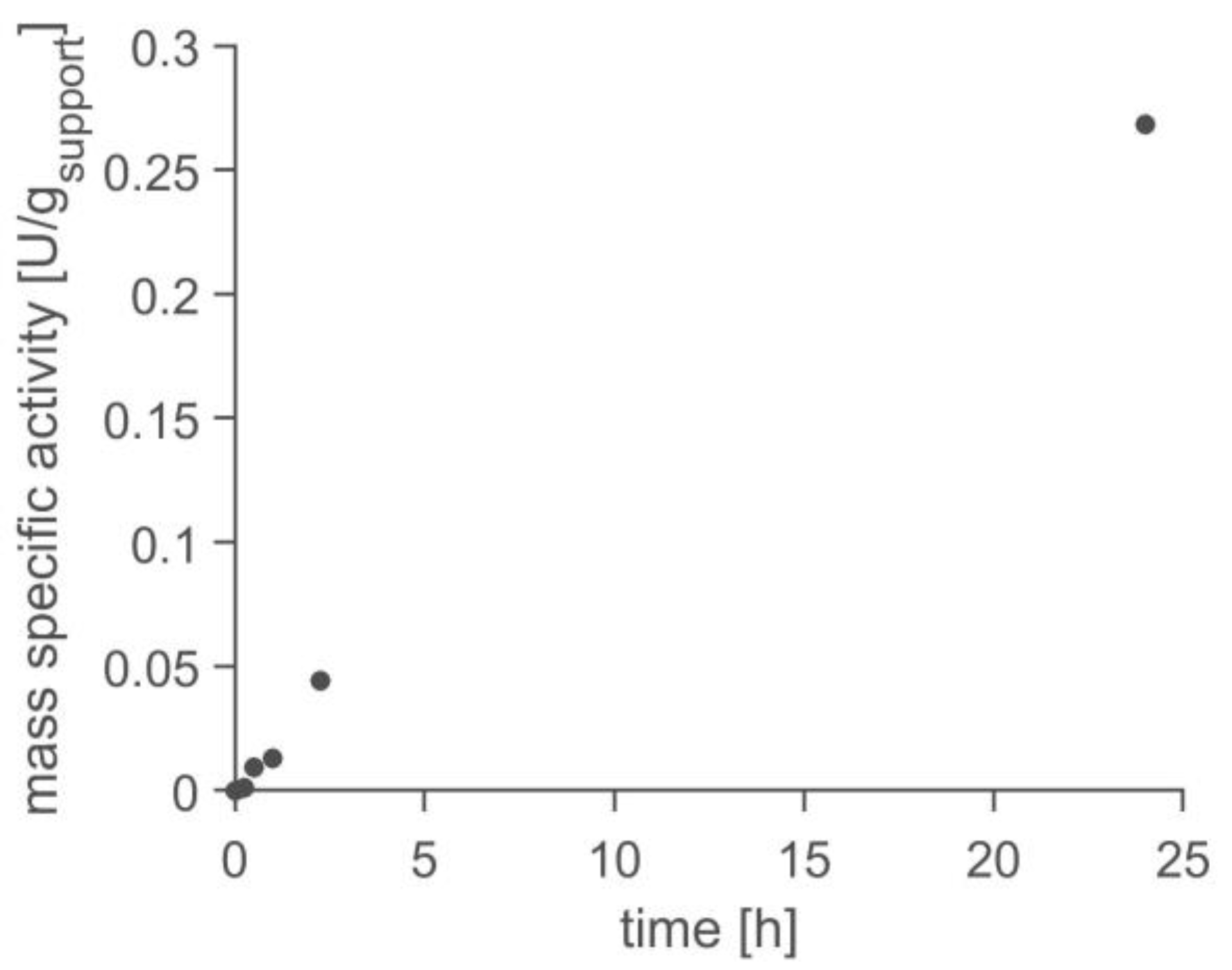
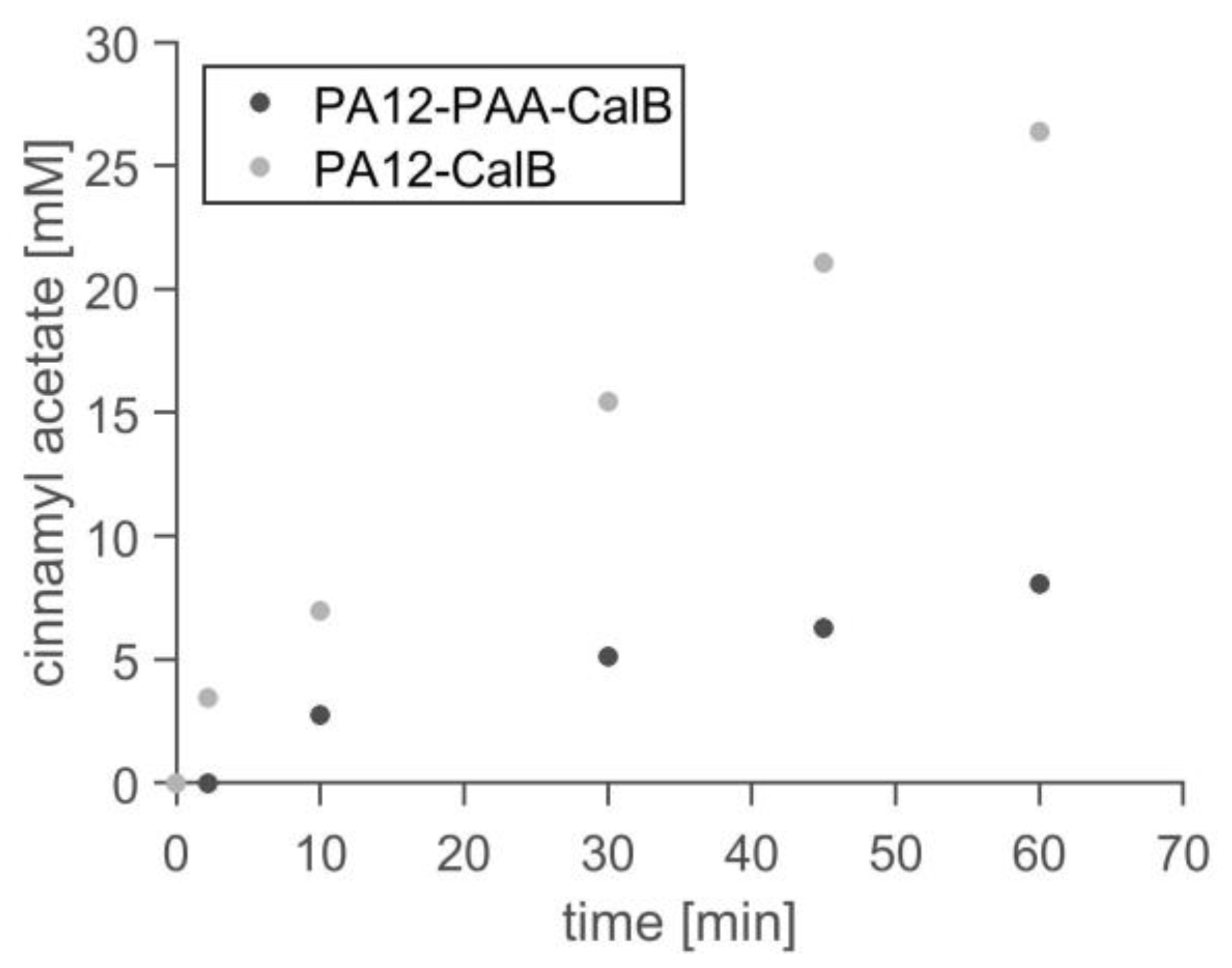
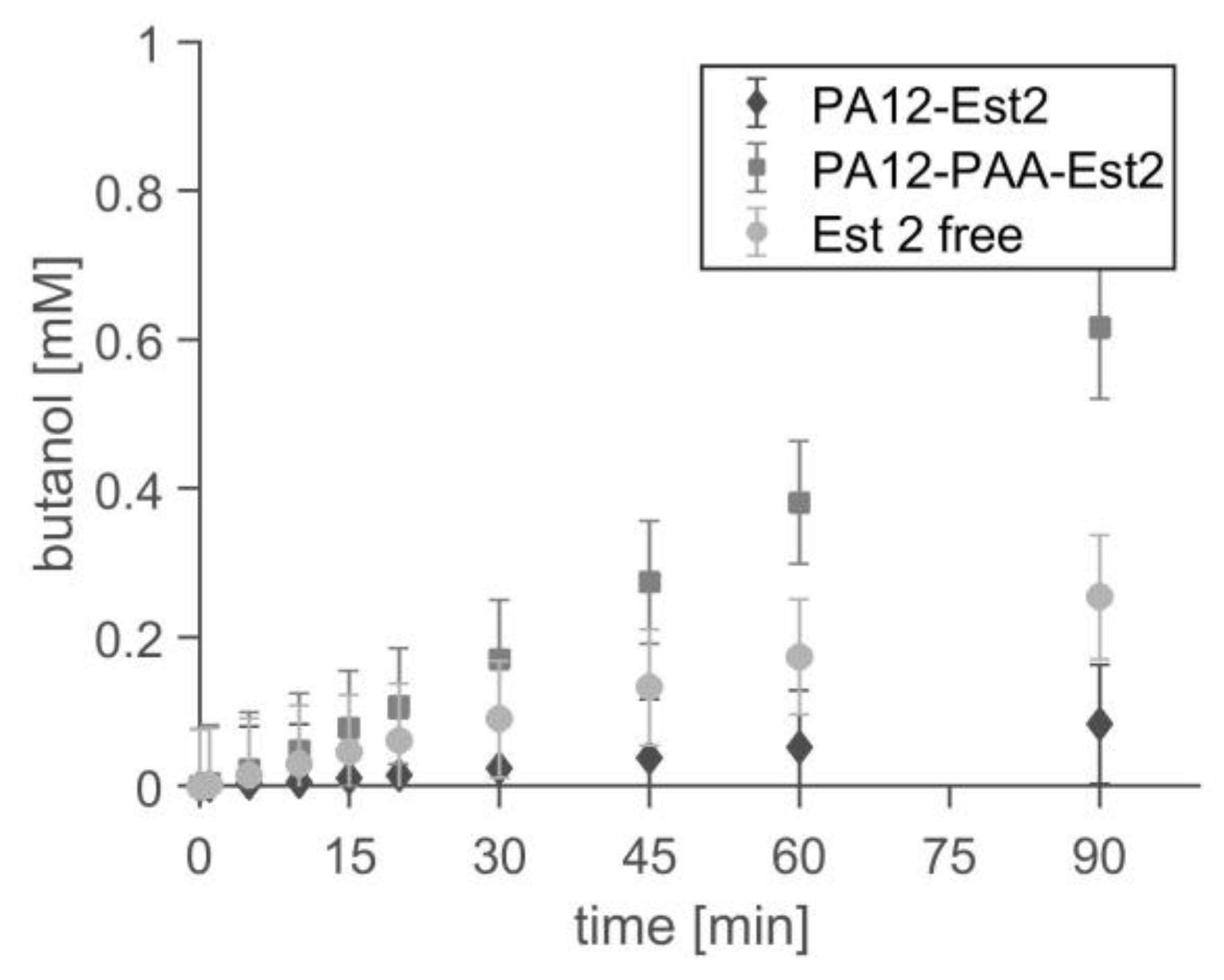
2.4. Application in Batch Reactor
3. Materials and Methods
3.1. 3D Printing of PA12 Structure
3.2. Surface Modification of PA12
3.3. Polymer Synthesis on Modified PA12
3.4. Enzyme Immobilization on Unmodified and PAA-Grafted PA12
3.4.1. Standard Activity Assay
3.4.2. Determination of Protein Concentration
3.4.3. Model Reaction in Aqueous Media
3.4.4. Model Reaction in Organic Media
3.5. Analytical Methods
3.5.1. Proton Nuclear Magnetic Resonance
3.5.2. Fourier-Transform Infrared Spectroscopy
3.5.3. X-ray Photoelectron Spectroscopy
3.5.4. Fluorescence Microscopy
3.5.5. Scanning Electron Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, L. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 2005, 9, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T. Immobilizing enzymes: How to create more suitable biocatalysts. Angew. Chem. Int. Ed. Engl. 2003, 42, 3336–3337. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme Immobilization: The Quest for Optimum Performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Cao, L. Carrier-Bound Immobilized Enzymes: Principles, Applications and Design/Linqiu Cao; Wiley-VCH: Weinheim, Germany; John Wiley: Chichester, UK, 2005; ISBN 978-3-527-31232-0. [Google Scholar]
- Liese, A.; Hilterhaus, L. Evaluation of immobilized enzymes for industrial applications. Chem. Soc. Rev. 2013, 42, 6236–6249. [Google Scholar] [CrossRef]
- Hicks, R.E. Pressure Drop in Packed Beds of Spheres. Ind. Eng. Chem. Fund. 1970, 9, 500–502. [Google Scholar] [CrossRef]
- Freund, H.; Zeiser, T.; Huber, F.; Klemm, E.; Brenner, G.; Durst, F.; Emig, G. Numerical simulations of single phase reacting flows in randomly packed fixed-bed reactors and experimental validation. Chem. Eng. Sci. 2003, 58, 903–910. [Google Scholar] [CrossRef]
- Ngo, T.D.; Kashani, A.; Imbalzano, G.; Nguyen, K.T.Q.; Hui, D. Additive manufacturing (3D printing): A review of materials, methods, applications and challenges. Compos. Part B Eng. 2018, 143, 172–196. [Google Scholar] [CrossRef]
- Hübner, E.G.; Lederle, F. Spezielle labortechnische Reaktoren: 3D-gedruckte Reaktoren. In Handbuch Chemische Reaktoren; Reschetilowski, W., Ed.; Springer Reference Naturwissenschaften; Springer Spektrum: Berlin/Heidelberg, Germany, 2020; pp. 1361–1389. ISBN 978-3-662-56434-9. [Google Scholar]
- Wang, G.; Wang, G.; Hill, N.S.; Zhu, D.; Xiao, P.; Coote, M.L.; Stenzel, M.H. Efficient Photoinitiating System Based on Diaminoanthraquinone for 3D Printing of Polymer/Carbon Nanotube Nanocomposites under Visible Light. ACS Appl. Polym. Mater. 2019, 1, 1129–1135. [Google Scholar] [CrossRef]
- Parra-Cabrera, C.; Achille, C.; Kuhn, S.; Ameloot, R. 3D printing in chemical engineering and catalytic technology: Structured catalysts, mixers and reactors. Chem. Soc. Rev. 2018, 47, 209–230. [Google Scholar] [CrossRef]
- Büscher, N.; Spille, C.; Kracht, J.K.; Sayoga, G.V.; Dawood, A.W.H.; Maiwald, M.I.; Herzog, D.; Schlüter, M.; Liese, A. Countercurrently Operated Reactive Extractor with an Additively Manufactured Enzyme Carrier Structure. Org. Process Res. Dev. 2020, 24, 1621–1628. [Google Scholar] [CrossRef]
- Spille, C.; Tholan, V.P.; Straiton, B.; Johannsen, M.; Hoffmann, M.; Marashdeh, Q.; Schlüter, M. Electrical Capacitance Volume Tomography (ECVT) for Characterization of Additively Manufactured Lattice Structures (AMLS) in Gas-Liquid Systems. Fluids 2021, 6, 321. [Google Scholar] [CrossRef]
- Büscher, N.; Sayoga, G.V.; Rübsam, K.; Jakob, F.; Schwaneberg, U.; Kara, S.; Liese, A. Biocatalyst Immobilization by Anchor Peptides on an Additively Manufacturable Material. Org. Process Res. Dev. 2019, 23, 1852–1859. [Google Scholar] [CrossRef]
- Ye, J.; Chu, T.; Chu, J.; Gao, B.; He, B. A Versatile Approach for Enzyme Immobilization Using Chemically Modified 3D-Printed Scaffolds. ACS Sustain. Chem. Eng. 2019, 7, 18048–18054. [Google Scholar] [CrossRef]
- Guddati, S.; Kiran, A.S.K.; Leavy, M.; Ramakrishna, S. Recent advancements in additive manufacturing technologies for porous material applications. Int. J. Adv. Manuf. Technol. 2019, 105, 193–215. [Google Scholar] [CrossRef]
- Stoffregen, H.; Fischer, J.; Siedelhofer, C.; Abele, E. Selective Laser Melting of Porous Structures. In Proceedings of the Solid Freeform Fabrication Symposium, Austin, TX, USA, 17 August 2011; pp. 680–695. [Google Scholar]
- Urban, M.W. Handbook of Stimuli-Responsive Materials; Wiley-VCH: Weinheim, Germany, 2011; ISBN 978-3-527-32700-3. [Google Scholar]
- Cullen, S.P.; Liu, X.; Mandel, I.C.; Himpsel, F.J.; Gopalan, P. Polymeric brushes as functional templates for immobilizing ribonuclease A: Study of binding kinetics and activity. Langmuir 2008, 24, 913–920. [Google Scholar] [CrossRef]
- Ferrand-Drake Del Castillo, G.; Koenig, M.; Müller, M.; Eichhorn, K.-J.; Stamm, M.; Uhlmann, P.; Dahlin, A. Enzyme Immobilization in Polyelectrolyte Brushes: High Loading and Enhanced Activity Compared to Monolayers. Langmuir 2019, 35, 3479–3489. [Google Scholar] [CrossRef]
- Koenig, M.; König, U.; Eichhorn, K.-J.; Müller, M.; Stamm, M.; Uhlmann, P. In-situ-Investigation of Enzyme Immobilization on Polymer Brushes. Front. Chem. 2019, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Swift, T.; Swanson, L.; Geoghegan, M.; Rimmer, S. The pH-responsive behaviour of poly(acrylic acid) in aqueous solution is dependent on molar mass. Soft Matter 2016, 12, 2542–2549. [Google Scholar] [CrossRef] [Green Version]
- Jose, J.; Thomas, S.; Thakur, V.K. Nano Hydrogels; Springer: Singapore, 2021; ISBN 978-981-15-7137-4. [Google Scholar]
- Chen, Q.; Li, S.; Feng, Z.; Wang, M.; Cai, C.; Wang, J.; Zhang, L. Poly(2-(diethylamino)ethyl methacrylate)-based, pH-responsive, copolymeric mixed micelles for targeting anticancer drug control release. Int. J. Nanomed. 2017, 12, 6857–6870. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Cai, X.; Wang, X.; Yang, J. “Paintable” 3D printed structures via a post-ATRP process with antimicrobial function for biomedical applications. J. Mater. Chem. B 2013, 1, 6644–6649. [Google Scholar] [CrossRef]
- Li, J.-J.; Zhou, Y.-N.; Luo, Z.-H. Polymeric materials with switchable superwettability for controllable oil/water separation: A comprehensive review. Prog. Polym. Sci. 2018, 87, 1–33. [Google Scholar] [CrossRef]
- Jiao, Y.; Niu, L.-N.; Ma, S.; Li, J.; Tay, F.R.; Chen, J.-H. Quaternary ammonium-based biomedical materials: State-of-the-art, toxicological aspects and antimicrobial resistance. Prog. Polym. Sci. 2017, 71, 53–90. [Google Scholar] [CrossRef]
- Wan, F.; Ye, Q.; Yu, B.; Pei, X.; Zhou, F. Multiscale hairy surfaces for nearly perfect marine antibiofouling. J. Mater. Chem. B 2013, 1, 3599–3606. [Google Scholar] [CrossRef] [PubMed]
- Touris, A.; Turcios, A.; Mintz, E.; Pulugurtha, S.R.; Thor, P.; Jolly, M.; Jalgaonkar, U. Effect of molecular weight and hydration on the tensile properties of polyamide 12. Results Mater. 2020, 8, 100149. [Google Scholar] [CrossRef]
- Fabb-It pro3D Additive Group. Material-Details: PA12 Datasheet. Available online: http://www.fabb-it.com/material/details?material=nylon-pa12 (accessed on 11 August 2022).
- Beamson, G.; Briggs, D. High Resolution XPS of Organic Polymers: The Scienta ESCA300 Database; Wiley: Chichester, UK, 1992; ISBN 9780471935926. [Google Scholar]
- Ma, N.; Liu, W.; Ma, L.; He, S.; Liu, H.; Zhang, Z.; Sun, A.; Huang, M.; Zhu, C. Crystal transition and thermal behavior of Nylon 12. e-Polymers 2020, 20, 346–352. [Google Scholar] [CrossRef]
- Launer, P.J.; Arkles, B. Infrared Analysis of Organosilicon Compounds: Spectra-Structure Relationships. In Silicon Compounds: Silanes and Silicones, 2nd ed.; Gelest, Inc.: Morrisville, PA, USA, 2008; pp. 223–226. [Google Scholar]
- Treat, N.D.; Ayres, N.; Boyes, S.G.; Brittain, W.J. A Facile Route to Poly(acrylic acid) Brushes Using Atom Transfer Radical Polymerization. Macromolecules 2006, 39, 26–29. [Google Scholar] [CrossRef]
- Wang, W.; Tang, J.; Jia, Z.; Li, X.; Xiao, Z. Grafting of amphiphilic polymers containing quaternary ammonium group on SiO2 surface via surface-initiated ATRP. J. Polym. Res. 2012, 19, 9804. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Swanson, L. Optical Properties of Polyelectrolytes. In Photochemistry and Photophysics of Polymer Materials; Allen, N.S., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 41–91. ISBN 9780470594179. [Google Scholar]
- Li, B.; Xu, L.; Wu, Q.; Chen, T.; Sun, P.; Jin, Q.; Ding, D.; Wang, X.; Xue, G.; Shi, A.-C. Various Types of Hydrogen Bonds, Their Temperature Dependence and Water-Polymer Interaction in Hydrated Poly(Acrylic Acid) as Revealed by 1H Solid-State NMR Spectroscopy. Macromolecules 2007, 40, 5776–5786. [Google Scholar] [CrossRef]
- Jabbari, E.; Nozari, S. Swelling behavior of acrylic acid hydrogels prepared by γ-radiation crosslinking of polyacrylic acid in aqueous solution. Eur. Polym. J. 2000, 36, 2685–2692. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, G. Periodic swelling and collapse of polyelectrolyte brushes driven by chemical oscillation. J. Phys. Chem. B 2008, 112, 10137–10141. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.; Howitz, S.; Kuckling, D.; Arndt, K.-F. Influence of volume phase transition phenomena on the behavior of hydrogel-based valves. Sens. Actuators B Chem. 2004, 99, 451–458. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef] [Green Version]
- Shuai, W.; Das, R.K.; Naghdi, M.; Brar, S.K.; Verma, M. A review on the important aspects of lipase immobilization on nanomaterials. Biotechnol. Appl. Biochem. 2017, 64, 496–508. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Cabrera, Z.; Godoy, C.; Fernandez-Lafuente, R.; Palomo, J.M.; Guisan, J.M. Interfacially activated lipases against hydrophobic supports: Effect of the support nature on the biocatalytic properties. Process Biochem. 2008, 43, 1061–1067. [Google Scholar] [CrossRef]
- Yagonia, C.F.J.; Park, K.; Yoo, Y.J. Immobilization of Candida antarctica lipase B on the surface of modified sol–gel matrix. J. Sol-Gel Sci. Technol. 2014, 69, 564–570. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Swinyard, B. Location of three critical phase boundaries in poly(acrylic acid)-dioxane solutions. Polymer 1990, 31, 1507–1513. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Berger, S.; Synytska, A.; Ionov, L.; Eichhorn, K.-J.; Stamm, M. Stimuli-Responsive Bicomponent Polymer Janus Particles by “Grafting from”/“Grafting to” Approaches. Macromolecules 2008, 41, 9669–9676. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase Activities of Human Carbonic Anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Manco, G.; Adinolfi, E.; Pisani, F.M.; Ottolina, G.; Carrea, G.; Rossi, M. Overexpression and properties of a new thermophilic and thermostable esterase from Bacillus acidocaldarius with sequence similarity to hormone sensitive lipase subfamily. Biochem. J. 1998, 332, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | O 1s (at%) | C 1s (at%) | N 1s (at%) | Si 2p (at%) | Br 3d (at%) | Sn 3d (at%) |
|---|---|---|---|---|---|---|
| PA12 | 11.39 | 82.22 | 4.1 | 2.3 | - | - |
| PA12-HCl | 15.36 | 75.64 | 5.43 | 3.58 | - | - |
| PA12-APTMS | 12.3 | 77.3 | 5.93 | 4.47 | - | - |
| PA12-BiBB | 10.36 | 79.42 | 5.54 | 3.37 | 1.31 | - |
| PA12-PtBA | 29.77 | 47.82 | - | 17.53 | - | 4.88 |
| Pre-Functionalized Particles [g] or 3D-Printed Structure | ca. 1 | |
|---|---|---|
| Tert-butyl acrylate [mL] | 5.0 | 30 mmol |
| Cu(II)Br (catalyst) [g] | 0.0004 | 0.002 mmol |
| N″-pentamethyldiethylenetriamine (ligand) [mL] | 0.0018 | 0.009 mmol |
| ethyl 2-bromoisobutyrate (initiator) [mL] | 0.0009 | 0.006 mmol |
| acetone [mL] | 1.0 | 0.014 mmol |
| tin(II) 2-ethylhexanoate [mL] | 0.3 | 0.926 mmol |
| anisol [mL] | 2.7 | 0.025 mmol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eixenberger, D.; Kumar, A.; Klinger, S.; Scharnagl, N.; Dawood, A.W.H.; Liese, A. Polymer-Grafted 3D-Printed Material for Enzyme Immobilization—Designing a Smart Enzyme Carrier. Catalysts 2023, 13, 1130. https://doi.org/10.3390/catal13071130
Eixenberger D, Kumar A, Klinger S, Scharnagl N, Dawood AWH, Liese A. Polymer-Grafted 3D-Printed Material for Enzyme Immobilization—Designing a Smart Enzyme Carrier. Catalysts. 2023; 13(7):1130. https://doi.org/10.3390/catal13071130
Chicago/Turabian StyleEixenberger, Daniela, Aditya Kumar, Saskia Klinger, Nico Scharnagl, Ayad W. H. Dawood, and Andreas Liese. 2023. "Polymer-Grafted 3D-Printed Material for Enzyme Immobilization—Designing a Smart Enzyme Carrier" Catalysts 13, no. 7: 1130. https://doi.org/10.3390/catal13071130