
Recent Progress of Metal-Oxide-Based Catalysts for Non-Oxidative Coupling of Methane to Ethane and Hydrogen
by
 ,
,
Junbu Wang
1,2,†, 
Zhiqiang Rao
1,2,†,
Zeai Huang
1,2,*,
Yaolin Chen
1,2,
Fang Wang
2 and
Ying Zhou
1,2,* 1
State Key Laboratory of Oil and Gas Reservoir Geology and Exploitation, Southwest Petroleum University, Chengdu 610500, China
2
School of New Energy and Materials, Southwest Petroleum University, Chengdu 610500, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Catalysts 2023, 13(4), 719; https://doi.org/10.3390/catal13040719
Submission received: 14 March 2023
/
Revised: 29 March 2023
/
Accepted: 30 March 2023
/
Published: 10 April 2023
(This article belongs to the Special Issue Recent Progress of Catalysis in “Dual Carbon Targets”)
Abstract
:Methane is the fundamental raw material of the C1 chemical industry, with abundant reserves. Its direct conversion into high-value-added chemicals has great scientific significance and broad commercial potential for the efficient use of methane resources. However, it is difficult to convert methane into more useful hydrocarbons and hydrogen, as the reaction usually requires external energy to overcome thermodynamic limitations. Non-oxidative coupling of methane to produce ethane and hydrogen is a promising supply technology. Catalysts which can be adapted to various energy sources are key to this technology. In recent years, considerable progress has been made in the design and application of these thermal and photocatalysts. This review outlines some typical catalysts, and reviews the progress in the understanding of reaction mechanisms. Finally, suggestions for the development of high-selectivity and high-stability catalysts for the future are presented.
1. Introduction
With the global temperature rising each year, climate anomalies are becoming more and more frequent, greatly impacting the bulk commodity market, especially in energy. Consequently, the utilization of clean energy natural gas is rapidly expanding with the exploitation of shale gas and combustible ice [1]. In the past, natural gas was mainly used as fuel [2], but now, in order to achieve green and sustainable development of the energy system, its industrial utilization is gaining more attention [3].
Natural gas is mainly composed of methane (CH4)-the basic raw gas of the C1 chemical industry. Thus, the conversion of CH4 to high-value chemicals (such as hydrogen (H2), alcohols, alkanes, and olefins) is a promising technology in chemical and energy supply. Traditionally, these chemicals are obtained through the indirect conversion of CH4, that is, CH4 is first converted into syngas (CO and H2), and then processed through the Fischer–Tropsch process. Unfortunately, this method has some disadvantages, such as a complicated process, low utilization rate of CH4, high energy consumption, and high cost [4,5].
The industry has been seeking cost-effective and low-carbon ways to utilize CH4, of which ethene (C2H6) is a highly important commodity chemical for many manufacturing products [6]. Non-oxidative coupling of methane (NOCM) produces C2H6 and H2 (Equation (1)), making it the simplest C-C coupling process and a model reaction for converting CH4 to multi-carbons.
2CH4 → C2H6 + H2 ΔH (298K) = 68.6 kJ mol−1
However, the thermodynamic barrier of this reaction necessitates the use of energy sources to trigger CH4 coupling [2,7], such as thermal catalysis and photocatalysis. Thermal catalysis is the most common method, but carbon deposition occurs easily at relatively high temperatures (>773 K) [8] and complete dehydrogenation of CH4 reduces C2H6 selectivity. Solar energy, on the other hand, is the richest and cleanest renewable energy and has been used to promote NOCM reactions under mild conditions [9,10], improving system safety and reducing reaction temperature.
From an economic standpoint, Gerceker et al. suggested that the selectivity of C2H6 should be at least 25% and coke formation should be less than 20%, making it a viable option for commercial applications [11]. Additionally, CH4 conversion efficiency should be taken into consideration. To this end, this paper reviews a series of catalysts from the perspective of catalytic efficiency and the mechanism of typical thermal catalysis and photocatalysis in NOCM reactions. In thermal catalysis, the catalysts reduce the reaction barrier and regulate the reaction intermediates in the “adsorption-activation-desorption” process. For photocatalysis, catalysts with different electronic properties are excited by solar energy to regulate the C-C coupling process. An overview of the properties and mechanism of the active catalysts is provided, as well as factors that affect CH4 activation and C2H6 selectivity. Designing efficient catalysts for direct NOCM is of great importance for the future.
2. Thermal Catalytic NOCM
2.1. Fundamentals of Thermal Catalysis
Generally, it is assumed that the reaction of Thermal Catalytic NOCM consists mainly of three processes [12]: (1) CH4 is adsorbed onto the active site of the catalyst; (2) the C-H bond is dissociated by the active component and forms adsorbed methyl or H2; (3) the identical adsorbed groups are coupled, then desorbed to generate C2H6 and H2 in the gas phase. However, as the most stable alkane, the CH4 molecule is sp3 hybrid [13] and its C-H bond dissociation energy is up to 439 kJ mol−1 [14,15], making the activation energy of the reaction extremely high, despite the catalyst reducing the energy effectively. The first C-H bond breaking of CH4 is more difficult, whereas the successive dehydrogenation reaction is easy to occur, thus leading to methyl cracking without needing a coupling process.
In thermal catalytic NOCM, the C-H bond of the CH4 molecule is broken through high-temperature thermal energy. The generation of reaction intermediates is regulated by introducing a thermal catalyst, thus avoiding the fracture of the CH3-CH3 skeleton, and effectively improving the catalytic performance. Some typical thermal catalysts are summarized in Table 1.
2.2. Precious Metal Catalysts
Noble metal catalysts can more effectively control the adsorption and desorption of intermediate products in thermal catalytic NOCM, thus enabling the directional conversion of CH4 to C2H6 [19]. In 1991, Belgued et al. [17] explored the effect of temperature on the selectivity of C2H6 over Pt/CeO2 catalyst (Figure 1a,b). With increasing temperature, the adsorption of methyl (-CH3) on the Pt surface decreased, whereas the formation of intermediates such as -CH2, -C2H3, and -C2H2 accelerated, thus reducing the selectivity of C2H6. Moya et al. [18] confirmed that the highly dispersed Pd active sites chemisorbed intermediate carbon species, promoting the desorption balance of methyl (-CH3) and proton hydrogen (H) over a Pd/α-Al2O3 catalyst during the coupled hydrogenolysis of CH4 at low temperature (500–750 K) (Figure 1c). Thermodynamic constraints in the thermal catalytic reaction motivate research into CH4 activation at high temperatures. However, traditional metal cluster catalysts suffer from severe coking and significant deactivation in the high-temperature catalytic process. Xie et al. [21] used a single atom Pt1@CeO2 catalyst to achieve a 40 h catalytic cycle (1173 K). The continuous desorption of intermediates (-CH3 and -CH2) at atomically dispersed Pt sites improves the effective collision of carbon-containing intermediates on the surface of active sites and inhibits carbon deposition, which covers the active sites, thereby aiding the dynamic stability of the catalyst structure. In conclusion, it can be stated that precious metal catalysts can facilitate the adsorption energy of -CH3 on the catalyst surface under appropriate catalytic temperature conditions, while inhibiting the adsorption of H atoms by unsaturated coordination carbon-containing intermediates, thus enhancing the effective collision between adsorbed CH3, and consequently promoting the coupling stability of C2H6. Recently, Pt nanolayers anchored at the hexagonal close-packed sites of the MXene support were able to activate the primary C-H bond of CH4 to create methyl radicals that favor desorption over subsequent dehydrogenation, suppressing coke deposition. At 973 K, the catalyst could operate continuously for 72 h without deactivation and CH4 conversion reached 7% (Figure 1d,e) [19].
2.3. Non-Precious Metal Catalysts
Compared with expensive precious metals, relatively cheaper non-precious metal catalysts such as Fe have been found to successfully catalyze the direct dehydrogenation of CH4 to generate carbonaceous metal compounds (MeCx) [13,27], resulting in improved stability of the catalyst even under high-temperature conditions. In 2014, Bao et al. [20] used an Fe©SiO2 catalyst to achieve catalytic stability for 60 h continuously, with a selectivity of C2H6 of around 3% at 1223 K (Figure 2a,b). FeOx is confined in the SiO2 lattice, with -CH3 species released from the constrained Fe atom. After CH4 is completely dehydrogenated, the carbon species are directly converted into a FeCx solid solution from the C-Fe=CH2 structure, avoiding carbon deposition. Kim et al. [22] found that the concentration of -CH3 species affects the catalytic process, with a high concentration of -CH3 (logPCH3 > −2) atmosphere being conducive to the formation of C2H6. Conversely, low concentrations of -CH3 (logPCH3 < −3) conditions result in C2H6 continuing to dehydrogenate as a medium for the formation of C2H4 or C2H2 (Figure 2c). It was shown that by regulating the formation energy of -CH3 species over Fe©CRS (CRS:SiO2) catalyst at 1353 K, catalytic stability could be achieved for 50 h, with a selectivity of C2H6 maintained at 7.9%. Mechanism studies confirmed that the energy barrier to be overcome for complete CH4 cracking on the confined Fe3C surface (ΔG = 1.1 eV) is lower than that of the pure Fe3C surface (ΔG = 2.8 eV) (Figure 2d), with the formation energy of -CH3 species on the restricted Fe3C surface site being lower than that of the pure Fe3C site (Figure 2d). Li et al. [28] proposed a quasi “Mars-van Krevelen” surface reaction mechanism, and research showed that the extraction and refilling of surface carbon atoms was the key to the transformation of surface carbon species. The -CH3 species on the surface of Fe1©SiO2 catalyst are difficult to desorb into the gas phase at the single iron atom site, whereas the carbon sites adjacent to the iron atom are more likely to desorb -CH3 and transfer protons H, thereby promoting the cyclic conversion of carbon atoms on the surface. Kashaboina et al. [29] confirmed that the in situ transformation of the unsaturated coordination structure of the In-C bond is a necessary condition for the formation of C2H6 on In/SiO2 catalyst by in situ X-ray absorption spectroscopy. Moya and Rashid et al. [26,30] proved that Ni2+ is conducive to CH4 adsorption, with the CH4 adsorption capacity of the catalyst containing Ni2+ being 13 times that of the catalyst containing Ni0. Dipu et al. [23] found that the selectivity of C2H6 can reach 80% on the variable valence metal Ni-P/SiO2 catalyst at 1098 K (Figure 2e). Lin et al. [31] showed that the divalent metal Ta2+ site is the active center on the Ta/SiO2 catalyst, with the Si-O-Ta bond in (-SiO3)2Ta being broken and the H2-Ta-CH2CH3 structure formed during activation of CH4, which can then be dehydrogenated to Ta-H and C2H6. Soulivong et al. [25] achieved a selectivity of the products of around 50%, with catalytic stability maintained for 200 h over Ta/SiO2 catalyst at 573 K. Therefore, non-precious metal catalysts can promote the C-C coupling of CH4 through a “restriction strategy” or “metal valency change strategy” to obtain the target products of C2H6 and H2.
3. Photocatalytic NOCM
3.1. Fundamentals of Photocatalysis
In the photocatalytic NOCM process involving a semiconductor catalyst, solar energy is utilized to stimulate the catalyst, creating photogenerated carriers (electrons and holes) [15]. The generated holes at the valence band trigger CH4 to enter an excited state, forming methyl radicals (•CH3) and H protons (CH4 + h+ → •CH3 + H+). Subsequently, methyl radicals are combined to yield adsorbed C2H6 (2•CH3 → C2H6), whereas the protons H are reduced by electrons to give adsorbed H2 (2H+ + 2e− → H2) [32]. Finally, C2H6 and H2 are desorbed in the CH4 conversion process at low temperatures [33].
Moreover, several catalysts with no inherent semiconductor characteristics are also applied for photocatalytic NOCM, which are distinguished by dispersing highly active centers on non-photoresponsive insulator supports and localizing on specific sites by taking advantage of the interaction between active components and supports. Some typical photocatalysts that can improve CH4 conversion or C2H6 selectivity are summarized in Table 2.
3.2. Metal-Oxide-Based Catalysts
In 1998, Yoshida et al. [34] first reported that a SiO2-Al2O3 catalyst could be utilized for NOCM, but its C2H6 selectivity was limited to 0.01%. Subsequently, Yoshida et al. found that high-energy ultraviolet light could induce the CH4 molecule to transition directly from its ground state to an excited state, and the phosphorescent excitation sites of SiO2 could desorb hydroxyl (-OH) groups from the surface [49]. Yuliati also confirmed that the dehydroxylation of SiO2 could be achieved by two active sites, namely, the ≡Si–O• (NBOHC, non-bridging oxygen hole center) and the •Si≡ (E’ center, dioxasilirane group), with electrons being obtained at the NBOHC site and holes generated at the E’ center site [35].
To further increase the light absorption capacity of the catalyst, some researchers have introduced the highly dispersed photoactive component TiO2. In 2002, Yoshida et al. [37,38] reported that a ternary oxide catalyst of SiO2-Al2O3-TiO2 achieved a C2H6 selectivity of 2.07%. The mechanism study showed that CH4 was activated by ultraviolet light, and its surface hydroxyl groups desorbed to form (SiO)3Ti-O-Al(OSi)3, which acted as a highly active synergistic site to promote the electron transfer from the oxygen center to the titanium site [38]. In 2021, Zhang et al. [39] proposed an efficient “coupling-desorption mechanism” strategy to regulate the yield of C2H6, with the yield of C2H6 reaching 1.7 μmol g−1 h−1 over the n-type semiconductor Nb-TS (single atom Nb-doped TiO2-SiO2) catalyst (Figure 3). Nb doping replaced the six coordinated Ti6c sites, generating additional active sites to provide additional local electrons which facilitated the directional migration of electrons to local •CH3, thereby accelerating the coupling between CH3-CH3 species and desorbing more C2H6.
Based on the above research, it can be determined that in the photocatalytic NOCM process, metal oxide catalysts mainly utilize high-energy ultraviolet light to induce CH4 to directly transition from the ground state to the excited state, and then adjust the surface electron transfer process through the oxide surface adsorption characteristics, thus improving CH4 conversion and C2H6 selectivity by promoting the formation of adsorbed protons (H) and adsorbed •CH3.
3.3. Metal-Modified Catalysts
The semiconductor catalyst has fixed intrinsic characteristics, making it difficult to meet the needs of photocatalytic reactions. Therefore, it is necessary to adjust the electronic structure of the catalyst by modifying the cocatalyst to optimize its catalytic performance. One strategy is to use metal-modified catalysts to broaden the light response region and improve the conversion efficiency of solar energy. Moreover, metal-modified photocatalysts can generate hot electrons or local electric fields to activate the C-H bond of CH4, promoting the reaction [50]. The metal can also undergo a change from Me1+x to Mex during the reaction, which regulates the adsorption-desorption reaction involving CH4 [50,51].
Yoshida et al. [42,43] found that adjusting the metal loading could achieve metal valence state modulation, thereby modifying the selectivity of C2H6. Ce3+ promotes fast electron migration in the low-loaded Ce(0.01–0.1%)/SiO2 catalyst, resulting in C2H6 selectivity of up to 60%. However, a higher loading of Ce(2–5%)/SiO2 catalyst yields a selectivity of 99% due to Ce4+ widening the band gap and inhibiting CH3 dehydrogenation. Later, Yoshida et al. [40] proposed the “Ag+-Ag0-Ag+ cycle balance charge” mechanism, and a C2H6 selectivity of 4.40% was achieved over Ag-MFI (MFI: SiO2-A12O3) catalyst. The photoexcited Ag+ adsorbs CH4 to form a complex (ZO-(Ag+-CH4), which promotes the adsorption of CH4. During the conversion of Ag+ to Ag0, the C-H bond is broken and •CH3 species accumulate on the catalyst surface, increasing the C-C collision opportunity to form C2H6. In 2020, Yu et al. [2] confirmed the reversibility of “Ag+-Ag0-Ag+” over the Ag-HPW/TiO2 catalyst. The yield of C2H6 was found to reach 150 μmol g−1 h−1, and the optical quantum efficiency was 3.5% (362 nm). It was further demonstrated that during the photocatalytic CH4 coupling reaction, the Lewis acid sites around the low coordination Ag+ are converted into Brønsted acid, and the Ag0 formed in situ is the active center site for the generation of •CH3 (Figure 4a,b).
Li et al. [41] replaced Ga cations on the titanate framework (ETS-10, one-dimensional TiO2 nanowires surrounding SiO2) to obtain C2H6 selectivity of up to 70%. The study showed that CH4 is adsorbed on the Ga site and the holes are captured by the -OH group. Then, hydroxyl radical (•OH) forms by attacking the C-H bond to generate •CH3. Subsequently, the same kind of groups of •CH3 are coupled to form C2H6(Figure 4d). At the same time, Ti4+ captures electrons to form Ti3+, and Ti3+ further transfers its electrons to the lattice oxygen to form the interaction between superoxide radicals (O2−) and H2O (Figure 4c,d). Later, Li et al. [44] designed an electron delocalization state in the Zn2+-ZSM-5− catalyst, achieving a selectivity of C2H6 close to 100%. UV light-excited free electrons transform to the 4s orbital of Zn2+ to form Zn+. Zn+ attracts the H atom of the CH4 molecule, then the anti-bond orbital obtains the 4s orbital electrons of Zn+. This facilitates the desorption of a large number of protons and induces C-C coupling to form C2H6 (Figure 4f–g).
Ga2O3 with a wide band gap can withstand a strong electric field to polarize CH4 molecules, and its cluster center bridges oxygen center radicals to promote the decomposition of CH4 molecules under mild conditions [52]. In 2008, Yuliati et al. [45] achieved a selectivity of 94% for C2H6 on Ga(0.1)/SiO2 catalyst. The tetrahedral coordinated Ga2O3 active site selectively activated CH4 molecules by limiting electrons in the Si-O bond through the Si-O-Ga structure. This is distinct from the electron transfer from O2− to Ga3+ during photoexcitation, which is more conducive to the selective generation of C2H6. Moreover, Pratap Singh et al. [32] found in 2020 that increasing light intensity can not only promote the activation of the C-H bond but also accelerate the photooxidation reaction of •CH3 (•CH3 + 3h+ → C + 3H+)(Figure 5a), which is not beneficial for forming C2H6.
Zhang et al. [46] studied the influence of the size effect of the active metal Pt. The catalytic activity showed a volcanic trend when the crystal size of Pt increased from 1.5 nm to 2.7 nm, and the maximum selectivity of C2H6 reached 90%. Small grain size is favorable for Ptδ+ transfer of photogenerated holes from Ga2O3 to adsorbed CH4 molecules. Though the selectivity of C2H6 on metal-modified Ga2O3 is not low, its CH4 conversion is not high (Figure 5b). Wu et al. [48] reported that Ga doping increased the free electron concentration on the Pt site. More Pt0 can effectively dissociate the C-H bond. Furthermore, the Mott–Schottky heterojunction formed at Pt-TiO2 accelerates the separation of the photogenerated carrier. Thus, the selectivity of C2H6 is 90% and the conversion of CH4 is 28% on the n-type semiconductor Pt/HGTS catalyst (HGTS: Ga doped TiO2-SiO2). Lang et al. [53] found that the lower the interface potential of metal modification, the more difficult the recombination of photogenerated carriers, which is more conducive to enhancing catalytic performance. By comparing a series of TiO2 (P25) catalysts modified by precious metals (Ru, Rh, Pd, Ag, Ir, Pt, and Au), it was found that the interface potential on Au-TiO2 is the lowest, which is more conducive to rapid electron migration to activate CH4 molecules. Consequently, the yield of C2H6 can reach 81.7 μmol g−1 h−1 on the Au-TiO2 catalyst (Figure 5c,d).
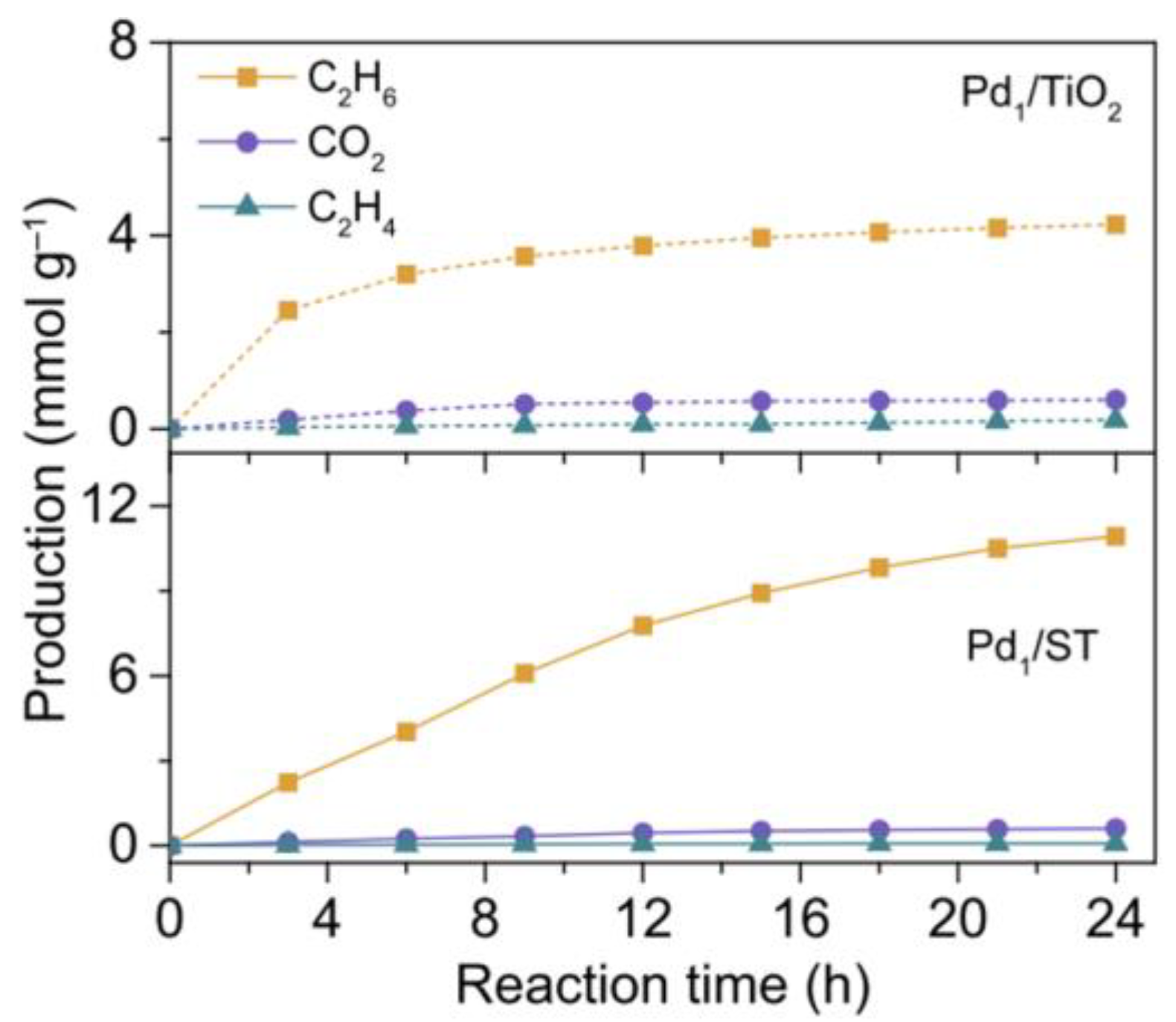
Generally, photogenerated holes used to activate CH4 are mainly concentrated at the lattice oxygen sites of metal oxide semiconductors, which makes CH4 prone to overoxidation by lattice oxygen atoms to produce CO, CO2, and other by-products. Xiong et al. [54] proposed the single-atom coordination method to regulate the valence band electronic structure of the Pd/TiO2 catalyst. This resulted in a selectivity of up to 94.3% for C2H6, a C2H6 yield of 0.91 mmol g−1 h−1, and H2 products with an equal stoichiometric ratio (Figure 6). Photogenerated holes were clustered on the “Pd-O4” coordination structure, thus reducing the contribution of the oxygen site to the valence band and improving the stability of lattice oxygen. This was shown to improve photocatalytic performance and reduce excessive oxidation in NOCM.
In addition, some other studies have found that an increase in CH4 pressure in the reaction system (from 0.27 kPa to 0.49 KPa) can reduce the catalytic activity [36]. Furthermore, the type of light source, light intensity, the reaction device, temperature, and pressure of the reaction system are all important factors affecting the anaerobic coupling of photocatalytic CH4.
4. Conclusions and Outlook
Direct conversion of CH4 to C2H6 and H2 has gained much attention due to its low cost, low carbonization, and other benefits. This review seeks to systematically analyze the development of thermal and photocatalytic NOCM, including the study of material properties and the exploration of reaction mechanisms. In contrast to other CH4 conversions, non-oxidative coupling of CH4 generates many by-products.
In traditional thermal catalytic NOCM, the C-H bond of CH4 is broken through the application of high-temperature thermal energy. The range of 1053–1123 K is more conducive to the selective generation of C2H6, since it is an optimal balance between breaking the C-H bond and the CH3-CH3 coupling progress. However, with increasing temperature, the adsorption of methyl (-CH3) on the precious metal catalysts surface weakens and the formation of intermediates such as -CH2, -C2H3, and -C2H2 accelerates, thereby reducing the selectivity of C2H6. Non-precious metal catalysts, on the other hand, can promote the C-C coupling of CH4 through the “restriction strategy” or “metal price change strategy” to obtain the target products of C2H6 and H2.
Photochemical reactions driving NOCM can break the restriction of thermodynamic equilibrium at mild temperatures. Metal-oxide-based catalysts which do not have the inherent characteristics of semiconductors can be applied for photocatalytic NOCM, whereby CH4 is directly converted from the ground state to the excited state under solar energy, thereby promoting the formation of adsorbed protons (H) and adsorbed •CH3. This is achieved by dispersing highly active centers on non-photoresponsive insulator supports and localizing them on specific sites through the interaction between active components and supports. Additionally, it is useful for metal-modified catalysts to broaden the light response region and improve the conversion efficiency of solar energy. This can generate hot electrons or local electric fields to activate the C-H bond of CH4. The metal, as an electron donor, undergoes the change from Me1+x to Mex during the reaction. Furthermore, hydroxyl radical (•OH) can be generated to attack the C-H bond to generate •CH3 on metal-modified catalysts.
There are many by-products in the NOCM process, and the conversion efficiency of CH4 is still relatively low. Thus, further research is warranted. In view of this, we propose the following methods for creating a photocatalyst for the NOCM reaction: (1) Developing in-situ technologies, such as X-ray Absorption Fine Structure (XAFS), X-ray Photoelectron Spectroscopy (XPS), and Diffuse Reflectance Fourier Transform Infrared Spectroscopy (DRIFTS), to investigate the structural changes of intermediates and catalysts in an ultra-short time scale, which could help to distinguish between thermal and photocatalytic effects. (2) The laboratory typically utilizes Xe lamps as the solar energy source, limiting the use of sunlight of different wavebands. To improve the efficiency of solar energy conversion and utilization, researchers could consider using solar simulators for indoor testing, or solar concentrators to directly convert and utilize solar energy. (3) More research should focus on bimetallic catalysts to improve C2H6 selectivity by exploiting the synergistic effects of different metals. It is important to further elucidate the reaction mechanism of cocatalysts.
Author Contributions
J.W. and Z.R. contributed equally. Editing and review, J.W. and Z.R.; Review, Y.C.; Supervision, F.W., Z.H. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been financially supported by the National Natural Science Foundation of China (No. 202209136), the Sichuan Provincial International Cooperation Project (No. 22ZDYF3690, 2021YFH0055 and 2022YFH0084), the Cheung Kong Scholars Program of China, the Science Project of SWPU (No. 2021JBGS10), Central Government Funds of Guiding Local Scientific and Technological Development for Sichuan Province (No. 2021ZYD0035).
Data Availability Statement
Data sharing is not applicable to this article. No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kerr, R.A. Natural Gas From Shale Bursts Onto the Scene. Science 2010, 328, 1624–1626. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zholobenko, V.L.; Moldovan, S.; Hu, D.; Wu, D.; Ordomsky, V.V.; Khodakov, A.Y. Stoichiometric methane conversion to ethane using photochemical looping at ambient temperature. Nat. Energy 2020, 5, 511–519. [Google Scholar] [CrossRef]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, S.; Chen, G. High-temperature steam reforming of methanol over ZnO–Al2O3 catalysts. Appl. Catal. B 2011, 101, 409–416. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Najari, S.; Saeidi, S.; Concepcion, P.; Dionysiou, D.D.; Bhargava, S.K.; Lee, A.F.; Wilson, K. Oxidative dehydrogenation of ethane: Catalytic and mechanistic aspects and future trends. Chem. Soc. Rev. 2021, 50, 4564–4605. [Google Scholar] [CrossRef]
- Xiao, Y.; Varma, A. Highly Selective Nonoxidative Coupling of Methane over Pt-Bi Bimetallic Catalysts. ACS Catal. 2018, 8, 2735–2740. [Google Scholar] [CrossRef]
- Julian, I.; Ramirez, H.; Hueso, J.L.; Mallada, R.; Santamaria, J. Non-oxidative methane conversion in microwave-assisted structured reactors. Chem. Eng. J. 2019, 377, 119764. [Google Scholar] [CrossRef]
- Wang, G.; Mu, X.; Li, J.; Zhan, Q.; Qian, Y.; Mu, X.; Li, L. Light-induced Nonoxidative Coupling of Methane using Stable Solid-Solutions. Angew. Chem. Int. Ed. Engl. 2021, 13, 20760–20764. [Google Scholar] [CrossRef]
- Meng, L.; Chen, Z.; Ma, Z.; He, S.; Hou, Y.; Li, H.-H.; Yuan, R.; Huang, X.-H.; Wang, X.; Wang, X.; et al. Gold plasmon-induced photocatalytic dehydrogenative coupling of methane to ethane on polar oxide surfaces. Energy Environ. Sci. 2018, 11, 294–298. [Google Scholar] [CrossRef]
- Huang, K.; Miller, J.B.; Huber, G.W.; Dumesic, J.A.; Maravelias, C.T. A General Framework for the Evaluation of Direct Nonoxidative Methane Conversion Strategies. Joule 2018, 2, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Bajec, D.; Kostyniuk, A.; Pohar, A.; Likozar, B. Nonoxidative methane activation, coupling, and conversion to ethane, ethylene, and hydrogen over Fe/HZSM-5, Mo/HZSM-5, and Fe–Mo/HZSM-5 catalysts in packed bed reactor. Int. J. Energy Res. 2019, 43, 6852–6868. [Google Scholar] [CrossRef]
- Schwach, P.; Pan, X.L.; Bao, X.H. Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef] [PubMed]
- Amos, R.D. An accurateab initiostudy of the multipole moments and polarizabilities of methane. Mol. Phys. 1979, 38, 33–45. [Google Scholar] [CrossRef]
- Reddy, P.V.L.; Kim, K.-H.; Song, H. Emerging green chemical technologies for the conversion of CH4 to value added products. Renew. Sustain. Energy Rev. 2013, 24, 578–585. [Google Scholar] [CrossRef]
- Belgued, M.; Pareja, P.; Amariglio, A.; Amariglio, H. Conversion of Methane into Higher Hydrocarbons on Platinum. Nature 1991, 352, 789–790. [Google Scholar] [CrossRef]
- Bajec, D.; Kostyniuk, A.; Pohar, A.; Likozar, B. Micro-kinetics of non-oxidative methane coupling to ethylene over Pt/CeO2 catalyst. Chem. Eng. J. 2020, 396, 125182. [Google Scholar] [CrossRef]
- Moya, S.F.; Martins, R.L.; Ota, A.; Kunkes, E.L.; Behrens, M.; Schmal, M. Nanostructured supported palladium catalysts—Non-oxidative methane coupling. Appl. Catal. A 2012, 411–412, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xiao, Y.; Chowdhury, P.R.; Wu, Z.; Ma, T.; Chen, J.Z.; Wan, G.; Kim, T.-H.; Jing, D.; He, P.; et al. Direct methane activation by atomically thin platinum nanolayers on two-dimensional metal carbides. Nat. Catal. 2021, 4, 882–891. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Ma, C.; Wu, X.; Deng, D.; Wei, M.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Xie, P.; Pu, T.; Nie, A.; Hwang, S.; Purdy, S.C.; Yu, W.; Su, D.; Miller, J.T.; Wang, C. Nanoceria-Supported Single-Atom Platinum Catalysts for Direct Methane Conversion. ACS Catal. 2018, 8, 4044–4048. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, H.W.; Han, S.J.; Lee, S.W.; Shin, J.; Kim, Y.T. Mechanistic and microkinetic study of non-oxidative methane coupling on a single-atom iron catalyst. Commun. Chem. 2020, 3, 58. [Google Scholar] [CrossRef] [PubMed]
- Dipu, A.L.; Ohbuchi, S.; Nishikawa, Y.; Iguchi, S.; Ogihara, H.; Yamanaka, I. Direct Nonoxidative Conversion of Methane to Higher Hydrocarbons over Silica-Supported Nickel Phosphide Catalyst. ACS Catal. 2019, 10, 375–379. [Google Scholar] [CrossRef]
- Han, S.J.; Lee, S.W.; Kim, H.W.; Kim, S.K.; Kim, Y.T. Nonoxidative Direct Conversion of Methane on Silica-Based Iron Catalysts: Effect of Catalytic Surface. ACS Catal. 2019, 9, 7984–7997. [Google Scholar] [CrossRef]
- Soulivong, D.; Norsic, S.; Taoufik, M.; Cope’ret, C.; Thivolle-Cazat, J.; Chakka, S.; Basset, J.-M. Non-Oxidative Coupling Reaction of Methane to Ethane and Hydrogen Catalyzed by the Silica-Supported Tantalum Hydride: (tSiO)2Ta-H. J. Am. Chem. Soc. 2008, 130, 5044–5045. [Google Scholar] [CrossRef]
- Moya, S.F.; Martins, R.L.; Schmal, M. Monodispersed and nanostructrured Ni/SiO2 catalyst and its activity for non oxidative methane activation. Appl. Catal. A 2011, 396, 159–169. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, J.; Pupucevski, M.; Impeng, S.; Yang, B.; Chen, G.; Kuboon, S.; Zhong, Q.; Faungnawakij, K.; Zheng, L.; et al. High-Performance Binary Mo–Ni Catalysts for Efficient Carbon Removal during Carbon Dioxide Reforming of Methane. ACS Catal. 2021, 11, 12087–12095. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.C.; Li, T.H.; Duan, Z.H.; Zhang, T.Y.; Yan, M.; Li, W.L.; Xiao, H.; Wang, Y.G.; Chang, C.R.; et al. Unravelling the Enigma of Nonoxidative Conversion of Methane on Iron Single-Atom Catalysts. Angew. Chem. Int. Ed. Engl. 2020, 59, 18586–18590. [Google Scholar] [CrossRef]
- Kashaboina, U.; Nishikawa, Y.; Wakisaka, Y.; Sirisit, N.; Nagamatsu, S.-I.; Bao, D.; Ariga-Miwa, H.; Takakusagi, S.; Inami, Y.; Kuriyama, F.; et al. Metamorphosis-like Transformation during Activation of In/SiO2 Catalyst for Non-oxidative Coupling of Methane: In Situ X-ray Absorption Fine Structure Analysis. Chem. Lett. 2019, 48, 1145–1147. [Google Scholar] [CrossRef]
- Al Rashid, M.H.; Dipu, A.; Nishikawa, Y.; Ogihara, H.; Inami, Y.; Obuchi, S.; Yamanaka, I.; Nagamatsu, S.; Kido, D.; Asakura, K. Active Phase Structure of the SiO2-supported Nickel Phosphide Catalysts for Non-oxidative Coupling of Methane (NOCM) Reactions. E-J. Surf. Sci. Nanotechnol. 2020, 18, 24–27. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Xi, Y.; Zhang, G.; Phillips, D.L.; Guo, W. A Reaction Mechanism of Methane Coupling on a Silica-Supported Single-Site Tantalum Catalyst. Organometallics 2014, 33, 2172–2181. [Google Scholar] [CrossRef]
- Singh, S.P.; Anzai, A.; Kawaharasaki, S.; Yamamoto, A.; Yoshida, H. Non-oxidative coupling of methane over Pd-loaded gallium oxide photocatalysts in a flow reactor. Catal. Today 2020, 375, 264–272. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Jansen, I.; Schuttlefield Christus, J.D. Renewable energy based catalytic CH4 conversion to fuels. Catal. Sci. Technol. 2014, 4, 2397–2411. [Google Scholar] [CrossRef]
- Kato, Y.; Yoshida, H.; Hattorib, T. Photoinduced non-oxidative coupling of methane over silica-alumina and alumina around room temperature. Chem. Commun. 1998, 21, 2389–2390. [Google Scholar] [CrossRef]
- Yuliati, L.; Tsubota, M.; Satsuma, A.; Itoh, H.; Yoshida, H. Photoactive sites on pure silica materials for nonoxidative direct methane coupling. J. Catal. 2006, 238, 214–220. [Google Scholar] [CrossRef]
- Yoshida, H.; Chaskar, M.G.; Kato, Y.; Hattori, T. Active sites on silica-supported zirconium oxide for photoinduced direct methane conversion and photoluminescence. J. Photochem. Photobiol. A 2003, 160, 47–53. [Google Scholar] [CrossRef]
- Kato, Y.; Matsushita, N.; Yoshida, H.; Hattori, T. Highly active silica–alumina–titania catalyst for photoinduced non-oxidative methane coupling. Catal. Commun. 2002, 3, 99–103. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsushita, N.; Kato, Y.; Hattori, T. Synergistic Active Sites on SiO2-Al2O3-TiO2 Photocatalysts for Direct Methane Coupling. J. Phys. Chem. B 2003, 107, 8355–8362. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, S.; Ma, J.; Mine, S.; Toyao, T.; Matsuoka, M.; Wang, L.; Zhang, J. Non-oxidative Coupling of Methane: N-type Doping of Niobium Single Atoms in TiO(2)-SiO(2) Induces Electron Localization. Angew. Chem. Int. Ed. Engl. 2021, 60, 11901–11909. [Google Scholar] [CrossRef]
- Yoshida, H.; Hamajima, T.; Kato, Y.; Shibata, J.; Satsuma, A.; Hattori, T. Active Ag species in MFI zeolite for direct methane conversion in the light and dark. Res. Chem. Intermed. 2003, 29, 7–9. [Google Scholar] [CrossRef]
- Li, L.; Cai, Y.Y.; Li, G.D.; Mu, X.Y.; Wang, K.X.; Chen, J.S. Synergistic effect on the photoactivation of the methane C-H bond over Ga(3+)-modified ETS-10. Angew. Chem. Int. Ed. Engl. 2012, 51, 4702–4706. [Google Scholar] [CrossRef] [PubMed]
- Yuliati, L.; Hamajima, T.; Hattori, T.; Yoshida, H. Highly dispersed Ce(III) species on silica and alumina as new photocatalysts for non-oxidative direct methane coupling. Chem. Commun. (Camb) 2005, 38, 4824–4826. [Google Scholar] [CrossRef] [PubMed]
- Yuliati, L.; Hamajima, T.; Hattori, T.; Yoshida, H. Nonoxidative Coupling of Methane over Supported Ceria Photocatalysts. J. Phys. Chem. C 2008, 112, 7223–7232. [Google Scholar] [CrossRef]
- Li, L.; Li, G.D.; Yan, C.; Mu, X.Y.; Pan, X.L.; Zou, X.X.; Wang, K.X.; Chen, J.S. Efficient sunlight-driven dehydrogenative coupling of methane to ethane over a Zn(+)-modified zeolite. Angew. Chem. Int. Ed. Engl. 2011, 50, 8299–8303. [Google Scholar] [CrossRef]
- Yuliati, L.; Hattori, T.; Itoh, H.; Yoshida, H. Photocatalytic nonoxidative coupling of methane on gallium oxide and silica-supported gallium oxide. J. Catal. 2008, 257, 396–402. [Google Scholar] [CrossRef]
- Ma, J.; Tan, X.; Zhang, Q.; Wang, Y.; Zhang, J.; Wang, L. Exploring the Size Effect of Pt Nanoparticles on the Photocatalytic Nonoxidative Coupling of Methane. ACS Catal. 2021, 11, 3352–3360. [Google Scholar] [CrossRef]
- Singh, S.P.; Yamamoto, A.; Fudo, E.; Tanaka, A.; Kominami, H.; Yoshida, H. A Pd-Bi Dual-Cocatalyst-Loaded Gallium Oxide Photocatalyst for Selective and Stable Nonoxidative Coupling of Methane. ACS Catal. 2021, 11, 13768–13781. [Google Scholar] [CrossRef]
- Wu, S.; Tan, X.; Lei, J.; Chen, H.; Wang, L.; Zhang, J. Ga-Doped and Pt-Loaded Porous TiO2-SiO2 for Photocatalytic Nonoxidative Coupling of Methane. J. Am. Chem. Soc. 2019, 141, 6592–6600. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsushita, N.; Kato, Y.; Hattori, T. Active sites in sol–gel prepared silica-alumina for photoinduced non-oxidative methane coupling. Phys. Chem. Chem. Phys. 2002, 4, 2459–2465. [Google Scholar] [CrossRef]
- Talebi, P.; Kistanov, A.A.; Rani, E.; Singh, H.; Pankratov, V.; Pankratova, V.; King, G.; Huttula, M.; Cao, W. Unveiling the role of carbonate in nickel-based plasmonic core@shell hybrid nanostructure for photocatalytic water splitting. Appl. Energy 2022, 322, 119461. [Google Scholar] [CrossRef]
- Song, H.; Meng, X.; Wang, Z.-J.; Liu, H.; Ye, J. Solar-Energy-Mediated Methane Conversion. Joule 2019, 3, 1606–1636. [Google Scholar] [CrossRef]
- Schwarz, H.; Shaik, S.; Li, J. Electronic Effects on Room-Temperature, Gas-Phase C-H Bond Activations by Cluster Oxides and Metal Carbides: The Methane Challenge. J. Am. Chem. Soc. 2017, 139, 17201–17212. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.; Ma, Y.; Wu, X.; Jiang, Y.; Hu, Y.H. Highly efficient light-driven methane coupling under ambient conditions based on an integrated design of a photocatalytic system. Green Chem. 2020, 22, 4669–4675. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, C.; Low, J.; Duan, D.; Ma, J.; Jiang, W.; Chen, Y.; Liu, H.; Qi, Z.; Long, R.; et al. High-performance photocatalytic nonoxidative conversion of methane to ethane and hydrogen by heteroatoms-engineered TiO2. Nat. Commun. 2022, 13, 2806. [Google Scholar] [CrossRef]
Figure 1.
(a) Reaction mechanism of Pt/CeO2 catalyst [17], (b) CH4 conversion and C2 selectivity on Pt/CeO2 catalyst [17], copyright 2020, Elsevier. (c) Conversion of CH4 and C-based selectivity to C2 products [18], copyright 2011, Elsevier. (d) Catalytic performance of Pt/Mo2TiC2Tx for non-oxidative coupling of CH4, and (e) a proposed catalytic circle of NOCM over Pt/Mo2TiC2Tx catalyst [19]. Copyright 2021, Springer Nature.
Figure 1.
(a) Reaction mechanism of Pt/CeO2 catalyst [17], (b) CH4 conversion and C2 selectivity on Pt/CeO2 catalyst [17], copyright 2020, Elsevier. (c) Conversion of CH4 and C-based selectivity to C2 products [18], copyright 2011, Elsevier. (d) Catalytic performance of Pt/Mo2TiC2Tx for non-oxidative coupling of CH4, and (e) a proposed catalytic circle of NOCM over Pt/Mo2TiC2Tx catalyst [19]. Copyright 2021, Springer Nature.

Figure 2.
(a) Comparison of different catalysts at 1223 K [20], (b) effect of reaction temperatures and space velocities on the 0.5% Fe©SiO2 catalyst. Blue circles denote CH4 conversion, whereas bars represent product selectivity, CH4 conversion, and C2 hydrocarbon selectivity over Fe©SiO2 catalyst [20], copyright 2014, The American Association for the Advancement of Science. (c) C2H6, C2H4, C2H2, and C6H6 selectivity as functions of catalyst contact time and gas-phase residence time. Experimental NOCM results using Fe@CRS catalyst in a fixed-bed reactor [22], copyright 2020, Springer Nature. (d) The activation energy barrier of CH4 on SiO2, Fe3C, and Fe©SiO2 catalysts [24], and (e) product distribution and CH4 conversion on Ni-P/SiO2 catalyst as a function of temperature [23]. Copyright 2020, American Chemical Society.
Figure 2.
(a) Comparison of different catalysts at 1223 K [20], (b) effect of reaction temperatures and space velocities on the 0.5% Fe©SiO2 catalyst. Blue circles denote CH4 conversion, whereas bars represent product selectivity, CH4 conversion, and C2 hydrocarbon selectivity over Fe©SiO2 catalyst [20], copyright 2014, The American Association for the Advancement of Science. (c) C2H6, C2H4, C2H2, and C6H6 selectivity as functions of catalyst contact time and gas-phase residence time. Experimental NOCM results using Fe@CRS catalyst in a fixed-bed reactor [22], copyright 2020, Springer Nature. (d) The activation energy barrier of CH4 on SiO2, Fe3C, and Fe©SiO2 catalysts [24], and (e) product distribution and CH4 conversion on Ni-P/SiO2 catalyst as a function of temperature [23]. Copyright 2020, American Chemical Society.
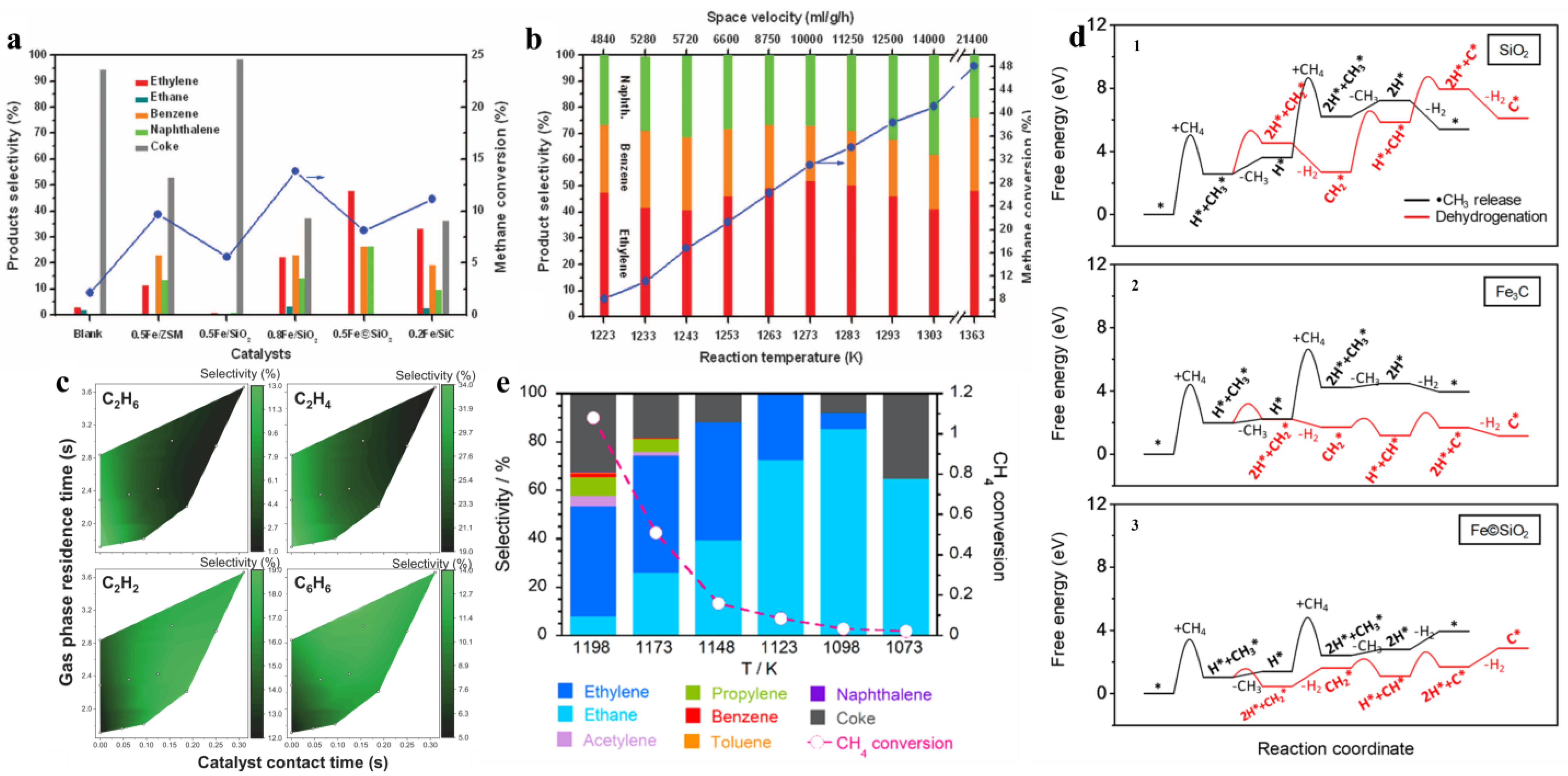
Figure 3.
The hydrocarbon products, H2 yields, and the CH4 conversion rate over Nb-T [39]. Copyright 2021, John Wiley and Sons.
Figure 3.
The hydrocarbon products, H2 yields, and the CH4 conversion rate over Nb-T [39]. Copyright 2021, John Wiley and Sons.
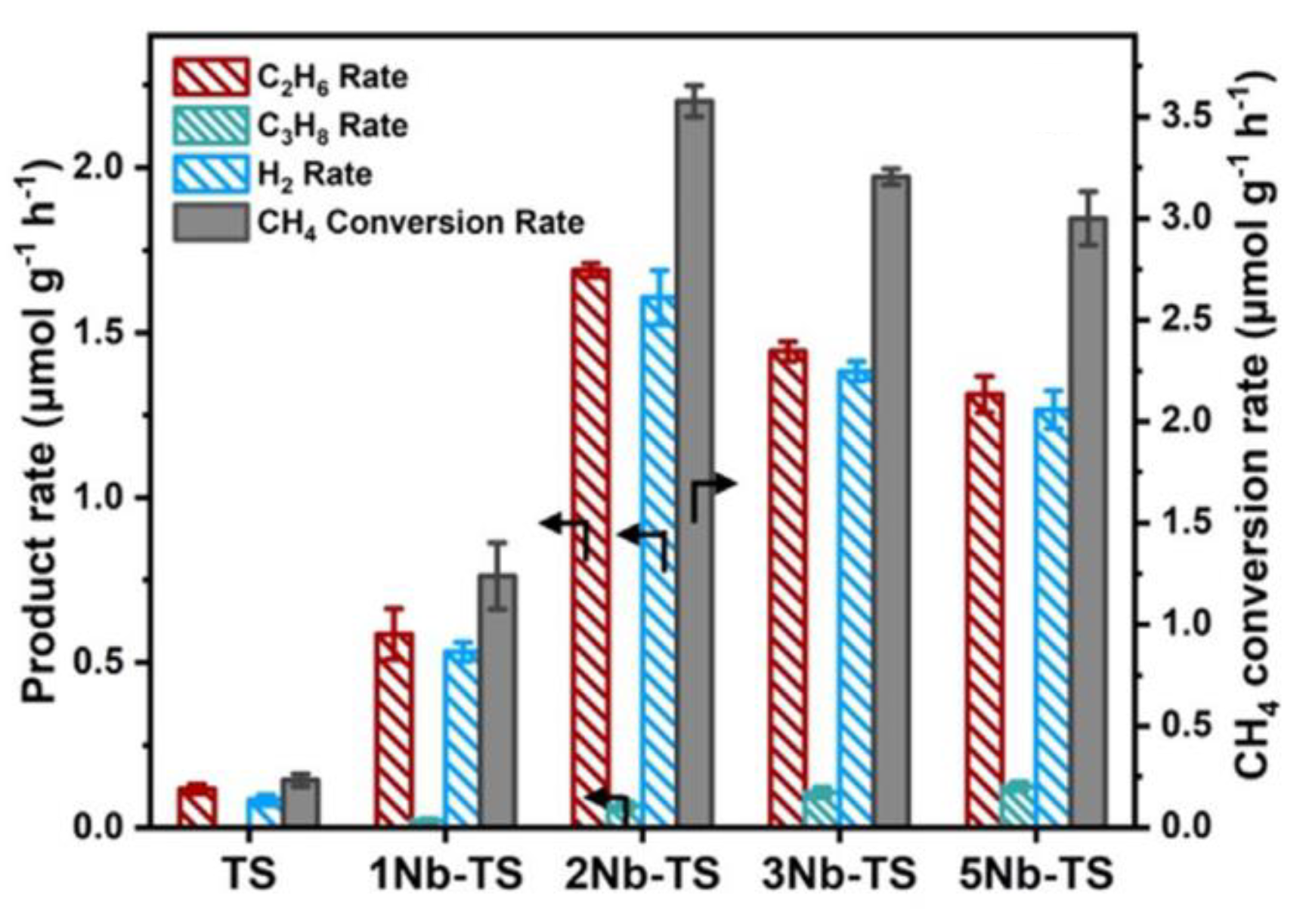
Figure 4.
(a,b) Production and mechanism of Ag–HPW/TiO2 catalyst in photocatalytic NOCM [2], copyright 2020, Springer Nature. (c,d) CH4 conversion and product distribution were obtained in the photo-driven CH4 activation reaction over various substrates under direct irradiation. From left to right: Na, K-ETS-10-0.2, Ga-ETS-10-0.2, Al-ETS-10-0.2, Zn-ETS-10-0.2, Fe-ETS-10-0.2, and Cu-ETS-10-0.2 [41], (e) EPR results of Ga-ETS-10-0.2 [41], copyright 2012, John Wiley and Sons. (f) CH4 conversion and C2H6 selectivity of Zn2+-ZSM-5− catalyst [44], and (g) mechanism of Zn2+-ZSM-5− catalyst in photocatalytic NOCM [44]. Copyright 2011, John Wiley and Sons.
Figure 4.
(a,b) Production and mechanism of Ag–HPW/TiO2 catalyst in photocatalytic NOCM [2], copyright 2020, Springer Nature. (c,d) CH4 conversion and product distribution were obtained in the photo-driven CH4 activation reaction over various substrates under direct irradiation. From left to right: Na, K-ETS-10-0.2, Ga-ETS-10-0.2, Al-ETS-10-0.2, Zn-ETS-10-0.2, Fe-ETS-10-0.2, and Cu-ETS-10-0.2 [41], (e) EPR results of Ga-ETS-10-0.2 [41], copyright 2012, John Wiley and Sons. (f) CH4 conversion and C2H6 selectivity of Zn2+-ZSM-5− catalyst [44], and (g) mechanism of Zn2+-ZSM-5− catalyst in photocatalytic NOCM [44]. Copyright 2011, John Wiley and Sons.

Figure 5.
(a) Production rates on Pd/Ga2O3 with different light intensities [32], copyright 2011, Elsevier. (b) Photocatalytic reaction conversion and selectivity of Pt/Ga2O3 [46], copyright 2021, American Chemical Society. (c) C2H6 yield over TiO2 catalysts supported by different noble metals in NOCM [53], and (d) IR spectra over Au/TiO2 and P25 [53]. Copyright 2020, Royal Society of Chemistry.
Figure 5.
(a) Production rates on Pd/Ga2O3 with different light intensities [32], copyright 2011, Elsevier. (b) Photocatalytic reaction conversion and selectivity of Pt/Ga2O3 [46], copyright 2021, American Chemical Society. (c) C2H6 yield over TiO2 catalysts supported by different noble metals in NOCM [53], and (d) IR spectra over Au/TiO2 and P25 [53]. Copyright 2020, Royal Society of Chemistry.
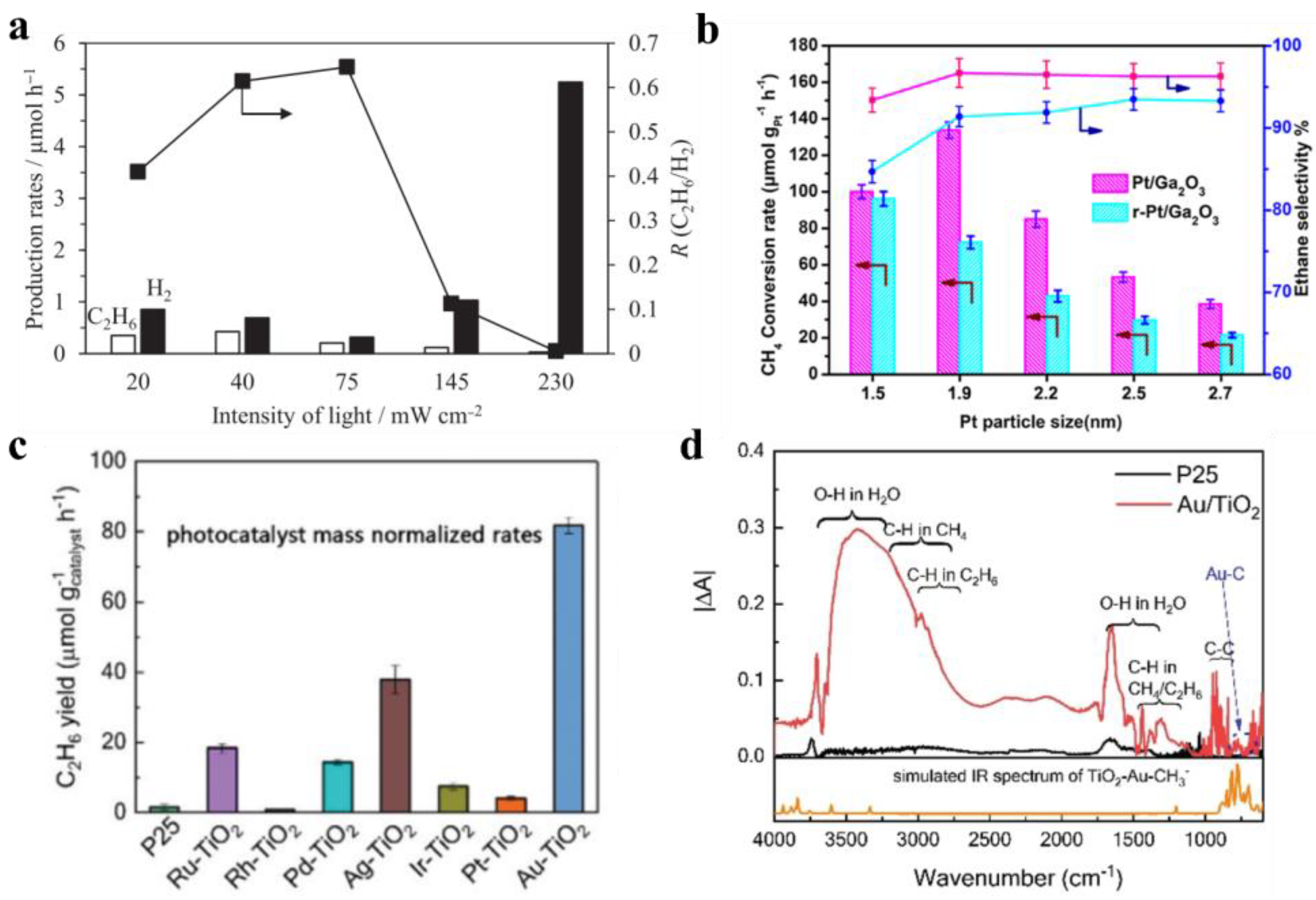
Figure 6.
The comparison of production rate between Pd1/TiO2 and Pd1/ST [54]. Copyright 2022, Springer Nature.
Figure 6.
The comparison of production rate between Pd1/TiO2 and Pd1/ST [54]. Copyright 2022, Springer Nature.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
C2H6 selectivity and CH4 conversion in thermal catalytic NOCM using different catalysts.
| Entry | Catalysts | Temperature (K) | Reaction Conditions | C2H6 Selectivity (%) | CH4 Conversion (%) | Reference |
|---|---|---|---|---|---|---|
| 1 | Pt | 523 | 100 mg, 400 mL min−1 CH4 | - | 19.3 | [16] |
| 2 | Pt/CeO2 | 1053 | 0.36 g, 0.094 h−1 CH4 | 84.8 | 7.92 | [17] |
| 3 | Pd/α-Al2O3 | 550 | 300 mg, 300 mL min−1 CH4 | 4 | - | [18] |
| 4 | Pt/Mo2TiC2 | 1023 | 50 mg, 12.9 GHSV(h−1) | 85 | 6.5 | [19] |
| 5 | Fe©SiO2 | 1223 | 4.84 L gcat−1 h−1 CH4 | 2 | 15 | [20] |
| 6 | Pt1@CeO2 | 1173 | 0.2 g, 20 mL min−1 CH4 | 43.6 | ~6 | [21] |
| 5 | Fe/SiO2 | 1293 | 1.2 g, 2.4 mL gcat−1 s−1 | 13 | 12.2 | [22] |
| 6 | Fe/HZSM-5 | 973 | 0.5 g, 6L gcat−1 h−1 CH4 | 23 | 3 | [12] |
| 7 | Fe©SiO2 | 1098 | 100 mg, 20 mL min−1 CH4 | 80 | 15 | [23] |
| 8 | Fe©SiO2 | 1353 | 0.6 g, 8000 mL gcat−1 h−1 CH4 | 7.9 | 6 | [24] |
| 9 | Ta/SiO2 | 573 | 300 mg, 30 mL min−1 CH4 | 50 | 0.1 | [25] |
| 10 | NiOx/SiO2 | 700 | 300 mg, 300 mL min−1 CH4 | 16 | - | [26] |
Table 2.
C2H6 selectivity and CH4 conversion in photocatalytic NOCM using different catalysts.
| Catalysts | Reaction Conditions | C2H6 Selectivity (% or μmol g−1 h−1) | CH4 Conversion (% or μmol g−1 h−1) | Reference | |
|---|---|---|---|---|---|
| 1 | SiO2-Al2O3 | 250 W Xe lamp, 1.0 g, 100 μmol CH4 | 0.01 | 5.9 | [34] |
| 2 | FSM-16 | 300 W Xe lamp, 0.2 g, 200 μmol CH4 | 1.63 | 5.7 | [35] |
| 3 | ZrO2/SiO2 | 250 W Xe lamp, 0.5 g, 200 μmol CH4 | 0.11 | 0.1 | [36] |
| 4 | SiO2-Al2O3-TiO2 | 250 W Xe lamp, 1.0 g, 200 μmol CH4 | 2.07 | 2.5 | [37] |
| 5 | SiO2-Al2O3-TiO2 | 250 W Xe lamp, 1.0 g, 200 μmol CH4 | 2.07 | 3.7 | [38] |
| 6 | Nb-TiO2-SiO2 | 300 W Xe lamp, 0.1 g, 1 mL CH4 | 1.7 μmol g−1 h−1 | 3.5 μmol g−1 h−1 | [39] |
| 7 | Ag/MFI | 250 W Xe lamp, 0.2 g, 200 μmol CH4 | 4.40 | 4.0 | [40] |
| 8 | Ga-ETS-10 | high-pressure Hg lamp, 0.2 g, 200 μmol CH4 | 70 | 15 | [41] |
| 9 | Ag-HPW/TiO2 | 400 W Xe lamp, 0.1 g, 0.3 MPa CH4 | 150 μmol g−1 h−1 | 4.96 | [2] |
| 10 | Ce/SiO2 | 300 W Xe lamp, 0.2 g, 200 μmol CH4 | 94 | 13 | [42] |
| 11 | Ce/Al2O3 | 300 W Xe lamp, 0.2 g, 200 μmol CH4 | 20 | 15 | [43] |
| 12 | Zn2+-ZSM-5− | high-pressure Hg lamp, 1.0 g, 200 μmol CH4 | 99 | 6 μmol g−1 h−1 | [44] |
| 13 | Ga2O3 | 300 W Xe lamp, 0.2 g, 200 μmol CH4 | 86 | 17 | [45] |
| 14 | Pt/Ga2O3 | 300 W Xe lamp, 0.2 g, 1 mL CH4 | 90 | 140 μmol g−1 h−1 | [46] |
| 15 | Pd/Ga2O3 | 300 W Xe lamp, 0.8 g, 30 mL min−1 CH4 | 0.125 μmol g−1 h−1 | - | [32] |
| 16 | Pd-Bi/Ga2O3 | 300 W Xe lamp, 0.8 g, 450 h−1 CH4 | 97 | 0.03 | [47] |
| 17 | Pt/HGTS | 300 W Xe lamp, 0.2 g, 44.6 μmol CH4 | 3.48 μmol g−1 h−1 | 28 | [48] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, J.; Rao, Z.; Huang, Z.; Chen, Y.; Wang, F.; Zhou, Y. Recent Progress of Metal-Oxide-Based Catalysts for Non-Oxidative Coupling of Methane to Ethane and Hydrogen. Catalysts 2023, 13, 719. https://doi.org/10.3390/catal13040719
AMA Style
Wang J, Rao Z, Huang Z, Chen Y, Wang F, Zhou Y. Recent Progress of Metal-Oxide-Based Catalysts for Non-Oxidative Coupling of Methane to Ethane and Hydrogen. Catalysts. 2023; 13(4):719. https://doi.org/10.3390/catal13040719
Chicago/Turabian StyleWang, Junbu, Zhiqiang Rao, Zeai Huang, Yaolin Chen, Fang Wang, and Ying Zhou. 2023. "Recent Progress of Metal-Oxide-Based Catalysts for Non-Oxidative Coupling of Methane to Ethane and Hydrogen" Catalysts 13, no. 4: 719. https://doi.org/10.3390/catal13040719
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.