
Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons


Emanuel Institute of Biochemical Physics, Russian Academy of Science, 4 Kosygin Street, 119334 Moscow, Russia
*
Author to whom correspondence should be addressed.
†
Deceased.
Catalysts 2021, 11(9), 1032; https://doi.org/10.3390/catal11091032
Submission received: 5 July 2021
/
Revised: 22 August 2021
/
Accepted: 22 August 2021
/
Published: 26 August 2021
(This article belongs to the Special Issue Catalytic Oxidation of Hydrocarbons)
Abstract
:Earlier, we established that nickel or iron heteroligand complexes, which include PhOH (nickel complexes) or tyrosine residue (nickel or iron complexes), are not only hydrocarbon oxidation catalysts (in the case of PhOH), but also simulate the active centers of enzymes (PhOH, tyrosine). The AFM method established the self-organization of nickel or iron heteroligand complexes, which included tyrosine residue or PhOH, into supramolecular structures on a modified silicon surface. Supramolecular structures were formed as a result of H-bonds and other non-covalent intermolecular interactions and, to a certain extent, reflected the structures involved in the mechanisms of reactions of homogeneous and enzymatic catalysis. Using the AFM method, we obtained evidence at the model level in favor of the involvement of the tyrosine fragment as one of the possible regulatory factors in the functioning of Ni(Fe)ARD dioxygenases or monooxygenases of the family of cytochrome P450. The principles of actions of these oxygenases were used to create highly efficient catalytic systems for the oxidation of hydrocarbons.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The development of industrial processes for the oxidation of hydrocarbons is determined by the ability of researchers to control these processes. An effective method of controlling the rate and mechanism of hydrocarbon oxidation is the use of a catalyst.
Currently, economically justified works in the field of homogeneous catalytic oxidation of hydrocarbons in the world are carried out in two directions: free radical chain oxidation, catalyzed by complexes of transition metals, and catalysis by metal complexes that simulate the action of enzymes [1]. The latter direction has not yet experienced proper development due to a low degree of conversion of hydrocarbons, which is associated with the rapid deactivation of catalysts.
The attention of researchers studying the mechanisms of action of axial modifying ligands that control the activity of metal complex catalysts is, as a rule, focused on the steric and electron-donating properties of ligands. The influences in the outer coordination sphere, the role of hydrogen bonds, and other non-covalent interactions have been studied to a much lesser extent. The formation of supramolecular structures due to hydrogen bonds is characteristic of the catalysis mechanism. In addition, hydrogen bonds are known to play an important role in biology. The facts of the formation of supramolecular structures open up the possibility of assessing the existence of non-covalent interactions [1].
For the first time, we used atomic force microscopy (AFM) to investigate the possibility of assessing H-bonds and the formation of supramolecular nanostructures based on heteroligand complexes of nickel and iron, which are effective catalysts for the oxidation of ethylbenzene to α-phenyl ethyl hydroperoxide and are simultaneously models of Ni(Fe)ARD dioxygenase [2,3].
In this self-review article, we discuss the participation of phenol and supramolecular structures in the mechanisms of selective catalysis of hydrocarbons, as well as, at the model level, the probable role of the Tyr fragment and supramolecular structures in the mechanisms of action of Ni(Fe)ARD dioxygenases and cytochrome P450 monooxygenases using the AFM method. We present for the first time the AFM image of supramolecular structures based on {Hem+PhOH+His} complexes, as well as the design of the structures of {Hem+Tyr+His} complexes based on the obtained AFM data.
The data collected can be considered a step towards understanding regulator activity of Tyr (and His) in the active sites of enzymes Ni(Fe)ARD dioxygenases or the family of cytochrome P450-dependent monooxygenases, the action principles of which were used to create highly efficient catalytic systems.
2. Results
2.1. Triple Systems Ni(acac)2+L2+PhOH That Are the Catalysts of Reaction of Selective Ethylbenzene Oxidation in α-Phenyl Ethyl Hydroperoxide
Some of the most efficient catalytic systems for the oxidation of ethylbenzene to α-phenyl ethyl hydroperoxide (PEH), developed by L.I. Matienko (and also by L.A. Mosolova), are three-component Ni-containing catalytic systems {Ni(acac)2+L2+PhOH} (L2 = N-methyl-2-pirrolidone (NMP), HMPA, MSt (M = Li, Na)) [1,4]. Studies observed the synergistic effect of an increase in the selectivity (S) and conversion (C) of ethylbenzene oxidation into PEH with additions of a third component, phenol PhOH, to the reaction, catalyzed by the {Ni(acac)2+L2} two-component system. This fact indicated the formation of active triple complexes {Ni(acac)2+L2+PhOH} [1,4]. The reaction rate during oxidation practically did not change until the conversion of ethylbenzene C = 20–30% (S = 87–90%) [1]. At that point, unlike the catalysis in the majority of research using binary systems {Ni(acac)2+L2}, in the course of the reaction of ethylbenzene oxidation, catalyzed with triple systems, the byproducts AP (acetophenone) and MPC (methylphenylcarbinol) were formed parallel to PEH throughout the process [1,4]. Researching kinetic data indicated the important role of intra- and intermolecular H-bonds in these catalytic reactions [1,4].
As follows from the kinetic data, the higher efficiency of the {Ni(acac)2∙L2∙PhOH} triple system in comparison with catalysis by the {Ni(acac)2∙L2} binary system is apparently due to the fact that the {Ni(acac)2∙L2∙PhOH} system is more stable and apparently does not undergo oxidative transformation during the oxidation of ethylbenzene. When catalyzed by a two-component system {Ni(acac)2∙L2}, during the ethylbenzene oxidation a selective form of the catalyst was formed as a result of electrophilic inclusion of molecular oxygen to the γ-C atom of one of the acac‒ ligands [1]. In this case, an active selective catalyst was an intermediate form of oxidation of the complex Ni(acac)2∙L2,-Ni2(acac)(OAc)3 L2−•2H2O (“A”) (L2 = NMP, MSt: M = Na, Li). (This is stage II of the oxidation process. In stage I, there were the prime complex Ni(acac)2∙L2 forms.) The decrease in the S of the oxidation process into PEH was associated with the formation of Ni(OAc)2, a product of the complete oxidation of Ni(acac)2∙L2 with molecular oxygen. Ni(OAc)2 was an effective catalyst for the heterolysis of PEH into phenol and acetaldehyde products (stage III of the process) [1]. We established that the conversion of complex Ni(acac)2∙L2 into active form “A” (stage II of the oxidation process) proceeded with a similar mechanism of action for NiARD dioxygenase (see below) [1].
It is known that heteroligand complexes are more active in reactions with electrophiles than homoligand complexes [1]. It is possible that the stability of heteroligand complexes Ni2(OAc)3(acac)NMP•2H2O (“A”) with respect to electrophilic inclusion of O2 can be explained by the formation of supramolecular structures due to intermolecular hydrogen bonds (H2O-MP, H2O-acetate (or acac-) group) [1]. We synthesized complex “A”, and its structure was established using various physicochemical methods: mass spectrometry, electronic and IR spectroscopy, and elemental analysis [1]. It was confirmed by kinetic methods that complexes “A” actually catalyzed the selective oxidation of ethylbenzene in PEH [1]. Figure 1a shows the likely structure of the supramolecular binuclear complex Ni2(OAc)3(acac)NMP•2H2O (“A”).
Using atomic force microscopy (AFM), we showed for the first time the self-organization of supramolecular complexes Ni2(OAc)3(acac)NMP•2H2O due to H-bonds and perhaps other non-covalent interactions (Figure 1b) [2,3].
The stability and efficiency of triple complexes {Ni(acac)2∙L2∙PhOH}, catalyzing the oxidation of ethylbenzene in PEH, could be associated with the formation of supramolecular structures due to intermolecular hydrogen bonds (phenol-carboxylate bonds), as well as other non-covalent interactions (Matienko LI, Mosolova LA: [1,4]). This assumption, based on kinetic data, is confirmed by UV spectroscopy data, which indicate the intra- and outer-sphere coordination of the activating ligands L2 = NMP and L3 (in the case of L3 = L-tyrosine) with Ni(acac)2 [4].
The possibility of the formation of intermolecular bonds (H-bonds and other non-covalent interactions) and supramolecular structures based on triple complexes {Ni(acac)2∙L2∙PhOH} (L2 = N-methyl-2-pyrrolidone (NMP), HMPA, MSt (M = Li, Na)), is evidenced by the data we obtained using the AFM method (Figure 2). The supramolecular structures were formed because of fixation of the {Ni(acac)2∙L2∙PhOH} systems on a specially prepared silicon surface due to H-bonds and subsequent spontaneous self-organization in nanostructures due to intermolecular H-bonds and, possibly, other non-covalent interactions [5].
2.2. Possible Effect of Tyr Fragment
Hydrogen bonds have importance in catalysis and biology. The variation of H-bond cleavage energies is from~15–40 kcal/mol (the strongest interactions) to ~4 kcal/mol (the weakest ones). Strong hydrogen bonds are covalent in nature, and a larger electrostatic contribution is attributed to weak hydrogen bonds [6,7].
Due to the relatively large phenol-amphipathic side chain, tyrosine residues located in different regions of the protein are involved in the formation of various hydrogen bonds [8] and in conformation and molecular recognition [9].
The role of tyrosine in enzymatic reactions is no less important.
Involvement of tyrosine residues in the mechanism of action of heme oxygenase (HO) was studied. It is known that the hemeless HO enzyme catalyzes the decomposition of protoporphyrin IX (Por)Fe(III) to biliverdin, CO, and Fe(II). In [10], in the mechanism of action of isoform-1 of human hemoxygenase (hHO-1), the role of tyrosyl radicals, which are formed after the reaction of conversion of Fe(III)(Por) to Fe(IV) = O(Por (+)), is discussed.
The Tyr fragment can take part in O2 activation with an iron-containing catalyst. It is assumed that binding through the Tyr fragment can lead to a decrease in the rate of O2 oxidation of the substrate in catalysis by homoprotocatechuate 2.3-dioxygenase [11].
The roles of Tyr and His fragments in the transfer of the methyl group from S-adenosylmethionine (AdoMet) in dopamine are discussed in [12,13]. The QM calculations suggest that carbonyl group of His-293 engaged in substantially stronger CH•••O hydrogen bonding to AdoMet than the tyrosine side chain. It was assumed, based on a model of methyltransferase and a structural study, that methyl CH•••O H-bonding represents a function of AdoMet-dependent methyltransferases as the universal mechanism for methyl transfer [13].
Tyr149 was found in astacin endopeptidases and serralisines. Tyr149, donating a proton, bonded with zinc as the fifth ligand. This switch played an important role, taking part in the stabilization of the transition state when the substrate bound to the enzyme [14].
Mononuclear monooxygenase non-heme iron enzyme, that is tyrosine hydroxylase (an enzyme in the central nervous system), catalyzed the hydroxylation of tyrosine to L-3,4-dihydroxyphenylalanine. This reaction limited the rate of biosynthesis of catecholamine neurotransmitters. Catalysis was conducted with the participation of tyrosine, tetrahydrobiopterin, and O2. The active center of the enzyme included the ferrous ion, coordinated by the facial side chains of 2 histidines and glutamate [15].
Additional protection of enzymes during oxidative transformations can be provided by the presence of chains of nearby tyrosine (Tyr) and tryptophan (Trp) residues. Redox residues Tyr and Trp can carry out the transfer of oxidizing equivalents of cpd I on the surface of proteins. This protective mechanism may be important for the catalysis of P450 [16,17].
2.2.1. Catalysis with Acireductone Dioxygenases Ni(Fe)ARD
Earlier, we established that the conversion of complex Ni(acac)2∙L2 into active form “A” during ethylbenzene oxidation with O2 proceeded with a mechanism similar of action of NiARD dioxygenase mechanism [1]. This, as well as the established phenomenal effect of phenol on the oxidation of ethylbenzene with O2, catalyzed by nickel systems {Ni(acac)2+L2} [1], suggested the involvement of the tyrosine fragment in the action of acireductone dioxygenase NiARD.
Acireductone dioxygenases Ni(Fe)ARD are involved in the methionine salvage pathway (MSP), which is the universal recycling pathway for the conversion of sulfur-containing metabolites to methionine. Ni(Fe)ARD represent an unusual case of catalysis. Depending on the nature of the metal ion in the active site, nickel–iron enzymes differ in their mechanism of action. They catalyze the conversion of the same substrates (1,2-dihydroxy-3-oxo-5 (methylthio), pent-1-ene (β-diketone acireductone), and molecular oxygen) into various products [18]. FeARD catalyzes the penultimate step in the metabolic pathway of acireductone oxidation in formate and 2-keto-4- (thiomethyl) butyrate, which is a precursor of methionine. The NiARD-catalyzed reaction pathway does not produce methionine, but this reaction produces CO, which is a neurotransmitter. CO has been identified as an anti-apoptotic molecule in mammals. Recently, it was found that a human enzyme regulated the activity of matrix metalloproteinase I (MMP-I), which is involved in tumor metastasis, by binding to the cytoplasmic transmembrane tail peptide MMP-I [19].
We assumed that the different activities of NiARD and FeARD with respect to the same substrates (acireductone and O2) might be connected to the formation of different macrostructures due to intermolecular H-bonds. We also assumed the role of the Tyr fragment in the second coordination sphere (see Scheme 1 [18]). In the case of catalysis by Ni-dioxygenase, NiARD, the Tyr fragment, participating in the mechanism, could reduce the NiARD activity.
Using the AFM method, we obtained convincing evidence on the model level in favor of the involvement of a tyrosine fragment in the stabilization of primary NiARD complexes as one of the possible regulatory factors in the Ni(Fe)ARD action. For the first time, we observed self-organization of model triple complexes including Tyr and His fragments {Ni(acac)2•NMP•Tyr}, {Ni(acac)2•His•Tyr} (His = L-histidine, Tyr = L-tyrosine) into very stable supramolecular structures due to H-bonds and possibly other non-covalent interactions (Figure 3) [5].
Self-assembly of complexes of iron into supramolecular structures can favor the formation of methionine.
Using the AFM method, we established the self-organization of complexes FeIIIx(acac)y18C6m(H2O)n, and FeIIIx(acac)y(His)m(Tyr)n(H2O)p, which simulated the active center of FeARD, into supramolecular hollow ring-shaped cellular structures (Figure 4a,b), resembling the tubular shape of the tubulin protein. These structures had an average diameter of 20–30 nm, which is approximately the same diameter as that of the tube of the tubulin protein. In the case of the two-component systems {Fe(acac)3+His}, we observed the formation of less distinct structures that were less stable. UV spectroscopy data have revealed the outer-sphere coordination of L-histidine (H-bond) with Fe(acac)3 [20].
The mechanism of action of FeARD, presumably, included the stage of O2-activation of FeII + O2→FeIII − O2−. This oxygen activation mechanism is proposed for FeII-acetylacetone dioxygenase (Dke1) [21]. The oxygen activation, and the subsequent reactions (the regioselective inclusion of activated O2 to the acireductone, and the reactions leading to the formation of methionine) can be facilitated by the formation of structures similar to tubulin tubules. In fact, structures similar to these were formed as a result of self-organization of complexes FeIIIx(acac)y18C6m(H2O)n [1,5], and FeIIIx(acac)y(His)m(Tyr)n(H2O)p (Figure 4) on the surface due to H-bonds.
2.2.2. Catalysis with the Family of Cytochrome P450-Dependent Monooxygenases
The original molecular structure of myoglobin (Mb) was determined in 1958. Since then, scientists have used advances in structural biology, biophysics, computational chemistry, and spectroscopy to obtain necessary information about the effect of various structural factors on the reactivity of metal cofactors. These studies have shown that the coordination spheres of the active centers of proteins play a decisive role in determining the properties of metal cofactors [22].
The family of cytochrome P450-dependent monooxygenases (cytochrome P450) are hemoproteins with significantly different functions. Proteins containing heme are widespread and found in almost all organisms, from bacteria to humans. These proteins are involved in many metabolic reactions. They have different mechanisms of action and participate in the oxidation of numerous compounds [23].
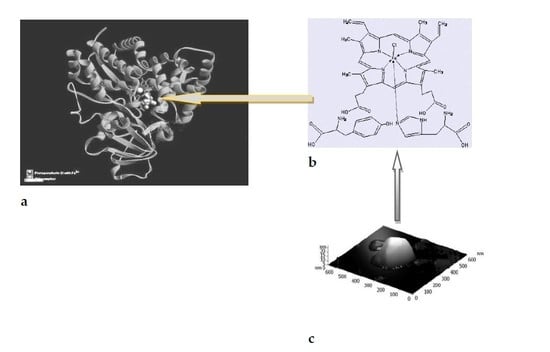
In nature, various types of binding of amino acids to metalloporphyrins are observed. It is known, for example, that Tyr (tyrosine) forms hydrogen bonds with His (histidine) [1,24,25].
For the first time, we used the AFM method to study the formation of supramolecular structures based on metal complexes of porphyrin with amino acids, tyrosine, and histi-dine, which are part of the active sites of enzymes, in particular, cytochrome P450-dependent monooxygenases. The fact of the formation of such structures can be used to analyze non-covalent intermolecular interactions [26] in the mechanisms of enzymatic catalysis [27,28].
Based on published data on the regulatory role of His and Tyr fragments, as well as porphyrins in the functioning of enzymes (heme proteins) of the P450 family [28], self-organization of supramolecular structures based on the models of the P450 family can be expected [29]. The binding of porphyrins through H-bonds is a common intermolecular interaction in nature. The formation of a dimer based on a carboxylic acid group is an example of the simplest artificially self-assembling supramolecular porphyrin system [24].
Figure 5a,b show that the AFM images of stable nanostructures of model systems {Hem+Tyr+His} had the form of triangular prisms. These structures, apparently, were formed due to the coordination of intermolecular interactions of the components of the {Hem•Tyr•His} complex during spontaneous self-organization with the participation of mainly hydrogen bonds and other intermolecular non-covalent interactions.
In the case of the {Hem+PhOH+His} complexes, i.e., substituting PhOH for Tyr, we also observed the formation of supramolecular structures. However, in this case, the structures based on {Hem+PhOH+His} differed in shape from nanostructures based on complexes {Hem•Tyr•His} (compare Figure 5 and Figure 6).
One of the possible schemes for the formation of triangular prisms due to H-bonds NH•••O or N•••HO [25,27] is shown in Figure 7a. The combination of individual triangles into structures (such as Sierpinski triangular motifs [30], Figure 7b) due to H-bonds and π–π- intermolecular interplanar interactions can lead to the formation of triangular prisms.
3. Discussion
We used the AFM method to study the role of self-organized supramolecular nanostructures and phenol in mechanisms of catalysis of selective hydrocarbon oxidation and also the possible role of supramolecular nanostructures and tyrosine in mechanisms of enzymatic catalysis, in actions of dioxygenases Ni(Fe)ARD, and of enzymes of the P450 cytochrome family. The fact of the formation of self-organized supramolecular structures can be used to analyze non-covalent molecular interactions [27]. Using the AFM method, we obtained convincing evidence on the model level in favor of the involvement of a tyrosine fragment in the stabilization of primary NiARD complexes as one of the possible regulatory factors in the functioning of Ni(Fe)ARD. Self-assembly of iron complexes into supramolecular structures, such as tubulin tubules, can facilitate reactions leading to the formation of methionine (FeARD catalysis). We used the AFM method to study the formation of supramolecular structures, based on porphyrin metal complexes with amino acids, tyrosine, and histidine, which are part of the active centers of enzymes, in particular, the family of cytochrome P450-dependent monooxygenases. The nanostructures, based on the model {Hem•Tyr•His} complex in the form of triangular prisms, apparently, were formed through spontaneous self-organization due to the coordination of intermolecular interactions of the components of the complex {Hem•Tyr•His} with the participation of mainly hydrogen bonds and other intermolecular non-covalent interactions. We did not observe such structures in the case of complexes {Hem•PhOH•His}, i.e., when replacing Tyr with PhOH. A possible scheme for the formation of nanostructures based on the {Hem•Tyr•His} complex is proposed.
The data obtained can be considered a step towards understanding the regulatory activity of Tyr (and His) with respect to the active centers of the enzymes Ni(Fe)ARD dioxygenases or the family of cytochrome P450-dependent monooxygenases, the principles of which were used to create highly efficient catalytic systems.
4. Materials and Methods
For the AFM study, we used the scanning probe microscope SOLVER P47 SMENA (Adm. Distr. Zelenograd of Moscow, 124490, Russia) at a frequency of 150 kHz using an NSG30 cantilever with a radius of curvature of 10 nm. We also used an NSG30_SS cantilever (NanosensorsTM Advanced TecTM AFM probes, CH-2000 Neuchatel, Switzerland) with a radius of curvature of 2 nm, a resonance frequency of 300 kHz, and a force constant of 22–100 N/m in tapping mode was used for AFM research of supramolecular structures. Sampling was carried out using a spin coating process, from a CHCl3 solution (Ni(acac)2∙L2∙PhOH complexes), an aqueous solution ({Fe(acac)3+His+Tyr} mixture), or a water-alcohol solution of the mixture {Hem+Tyr+His} (Hem = hemin, Tyr = L-tyrosine, His = L-histidine, molar ratio of 1:1:1). The measurement was conducted on air-dried samples. In the course of scanning the studied samples, it was found that the structures were fixed on the surface rather strongly, perhaps due to the H-bonds. In fact, we registered the self-assembly of the three components of the system into nanostructures with the participation, we believe, of hydrogen bonds and perhaps other non-covalent interactions. The spontaneous process of self-organization of the nanostructures, based on researched systems on a specially prepared modified silicon surface, was due to the balance between molecular–surface and intermolecular interactions.
5. Conclusions
Using the AFM method, we registered the self-assembly of three components of the system ({Ni(acac)2+L2+PhOH(Tyr)}, {Fe(acac)3+His+Tyr}, and {Hem+Tyr+His}) into nanostructures with the participation, we believe, of hydrogen bonds, as well as, possibly, other non-covalent interactions. The images that we see in the examples of complexes, which are models of active sites of enzymes, tell us that such an assembly was taking place. Apparently, this type of interaction in the active center of the enzyme cannot be ignored either.
With the AFM method, we obtained convincing evidence at the model level in favor of the involvement of tyrosine (and histidine) fragments as the possible regulatory factors in the functioning of di- or monooxygenases, namely, Ni(Fe)ARD dioxygenases or enzymes in the family of the cytochrome P450-dependent monooxygenases. The principles of actions of these oxygenases were used to create highly efficient catalytic systems for the oxidation of hydrocarbons.
Author Contributions
Data curation, L.M., A.G.; Methodology, V.B., E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Russian Academy of Sciences and Presidium of the Russian Academy of Sciences RAS 14P, State Register Number: AAAA-A17-117121920169-0.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Acknowledgments
To A.V. Lobanov for the reagents provided and participation in the discussions.
Conflicts of Interest
The authors declare no conflict of interest, financial or otherwise.
Abbreviations
| AFM method | Atomic Force Microscopy method |
| (acac)− | Acetylacetonate ion |
| ARD | Acireductone ligand |
| 18C6 | 18-crown-6 |
| CO | Carbon monoxide |
| Dke1 | Fe-acetylacetone dioxygenase |
| HMPA | Hexamethylphosphorotriamide |
| Hacac | Acetylacetone |
| Hem | Hemin |
| His | L-histidine |
| L2 | Electron-donating mono-, or multidentate ligand |
| MSt | Stearates of Na, Li |
| NMP | N-methyl-2-pirrolidone |
| Ni(Fe)ARD | Ni(Fe) acireductone dioxygenases |
| (OAc)− | Acetate ion |
| PhOH | Phenol |
| Tyr | L-tyrosine |
| UV | Ultraviolet-spectroscopic data |
References
- Matienko, L.I.; Mosolova, L.A.; Zaikov, G.E. Selective Catalytic Hydrocarbons Oxidation. New Perspectives; Nova Science Publ. Inc.: New York, NY, USA, 2010; 150p. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. The new approach to research of mechanism catalysis with nickel complexes in alkylarens oxidation. In Polymer Yearbook 2011; Nova Science Publishers: New York, NY, USA, 2012; pp. 221–230. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. Supramolecular Nanostructures on the Basis of Catalytic Active Heteroligand Nickel Complexes and their Possible Roles in Chemical and Biological Systems. J. Biol. Res. 2012, 1, 37–44. [Google Scholar]
- Matienko, L.I.; Mosolova, L.A.; Binyukov, V.I.; Mil, E.M.; Zaikov, G.E. Triple systems, based on Ni(acac)2, introduced ligands- modifiers HMPA, N-methylpirrolidone-2, PhOH, or Tyrosine, as effective catalysts in selective ethylbenzene oxidation with dioxygen, as models of Ni-ARD Dioxygenase. Oxid. Commun. 2017, 40, 569–580. [Google Scholar]
- Matienko, L.I.; Mil, E.M.; Binyukov, V.I. AFM Research of Supramolecular Structures. Russ. J. Phys. Chem. B 2020, 14, 559–563. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The Cation−π Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.D.; Buytendyk, A.M.; Wang, D.; Bowen, K.H.; Collins, K.D. Strong, Low-Barrier Hydrogen Bonds May Be Available to Enzymes. Biochemistry 2014, 53, 344–349. [Google Scholar] [CrossRef]
- Radi, R. Protein Tyrosine Nitration; Biochemical Mechanisms and Structure Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, S.; Sidhu, S.S. The importance of being tyrosine: Lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem. Biol. 2009, 4, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, V.V.; Roth, J.P. Tyrosine oxidation in heme oxygenase: Examination of long-range proton-coupled electron transfer. J. Biol. Inorg. Chem. 2014, 19, 1137–1148. [Google Scholar] [CrossRef]
- Mbughuni, M.M.; Meier, K.K.; Münck, E.; Lipscomb, J.D. Substrate-Mediated Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by a Tyrosine 257 Variant. Biochemistry 2012, 51, 8743–8754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Klinman, J.P. Enzymatic Methyl Transfer: Role of an Active Site Residue in Generating Active Site Compaction That Correlates with Catalytic Efficiency. J. Am. Chem. Soc. 2011, 133, 17134–17137. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and Functional Importance of Carbon–Oxygen Hydrogen Bonding in AdoMet-Dependent Methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.S.; Beynon, R.J. The Astacin Family of Metalloendpeptidases. Protein Sci. 1995, 4, 1247–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, J.; Eser Bekir, E.; Mannikko, D.; Krzyaniak Matthew, D.; Fitzpatrick Paul, F. HYSCORE Analysis of the Effects of Substrates on Coordination of Water to the Active Site Iron in Tyrosine Hydroxylase. Biochemistry 2015, 54, 3759–3771. [Google Scholar] [CrossRef]
- Gray, H.B.; Winkler, J.R. Hole Hopping Through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. USA 2015, 112, 10920−10925. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Groves John, T. Oxygen Activation and Radical Transformations in Heme Protein and Metalloporphyrins. Chem. Rev. 2018, 118, 2491−2553. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.C.; Dang, T.; Ju, M.; Goldsmith, R.B.; Maroney, M.J.; Pochapsky, T.C. Characterization of Metal Binding in the Active Sites of Acireductone Dioxygenase Isoforms from Klebsiella ATCC 8724. Biochemistry 2008, 47, 2428–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Garber, A.; Ryan, J.; Deshpande, A.; Ringe, D.; Pochapsky, T.C. A Model for the Solution Structure of Human Fe(II)-Bound Acireductone Dioxygenase and Interactions with the Regulatory Domain of Matrix Metalloproteinase I (MMP-I). Biochemistry 2020, 59, 4238–4249. [Google Scholar] [CrossRef] [PubMed]
- Matienko, L.I.; Binykov, V.I.; Mil, E.M.; Zaikov, G.E. Role of Supramolecular Structures in Mechanisms of Catalytic Oxidation, and Action of Ni(Fe)ARD Dioxygenases on Model Systems. Chem. Chem. Tech. 2020, 14, 304–311. [Google Scholar] [CrossRef]
- Leitgeb, S.; Straganz, G.D.; Nidetzky, B. Functional characterization of an orphan cupin protein from Burkholderia xenovorans reveals a mononuclear nonheme Fe2+-dependent oxygenase that cleaves β-diketones. FEBS J. 2009, 276, 5983–5997. [Google Scholar] [CrossRef]
- Cook Sarah, A.; Hill Ethan, A.; Borovik, A.S. Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167−4180. [Google Scholar]
- Savino, C.; Montemiglio, L.C.; Giuliano, S.; Miele Adriana, E.; Kendrew Jemth Per Steven, G.; Gianni, S. Investigating the Structural Plasticity of a Cytochrome P450. Three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 2009, 284, 29170–29179. [Google Scholar] [CrossRef] [Green Version]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.R.; Stem, C. Supramolecular Chemistry of Metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.T.; Tysko, R. Side Chain Hydrogen-Bonding Interactions within Amiloid-like Fibrils Formed by the Low-Complexity Domain of FUS: Evidence from Solid State Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2020, 59, 304–378. [Google Scholar]
- Matienko, L.I.; Binyukov, V.I.; Mil, E.M.; Mosolova, L.A.; Zaikov, G.E. Application of the AFM method to studying the role of Supramolecular structures and Tyr-fragment in the mechanism of Ni(Fe)ARD action on model systems. Oxid. Commun. 2018, 41, 429–440. [Google Scholar]
- Biedermann, F.; Schneider, H.-J. Experimental Binding Energies in Supramolecular Complexes. Chem. Rev. 2016, 116, 5216–5300. [Google Scholar] [CrossRef]
- Basom, E.J.; Bryce, A.M.; Thielges, M.C. Conformational Heterogeneity and the Affinity of Substrate Molecular Recognition by Cytochrome P450cam. Biochemistry 2017, 56, 3248–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, R. Molecular level structural studies of metalloproteins/metalloenzymes by scanning tunneling microscopy; Scopes and promises. Curr. Sci. 2003, 84, 1202–1210. [Google Scholar]
- Sarkar, R.; Xie, T.-Z.; Endres, K.J.; Wang, Z.; Moorefield, C.N.; Saunders, M.J.; Ghorai, S.; Patri, A.K.; Wesdemiotis, C.; Dobrynin, A.V.; et al. Sierpinski Pyramids by Molecular Entanglement. J. Am. Chem. Soc. 2020, 142, 5528–5530. [Google Scholar] [CrossRef]
Figure 1.
(a) Structure of the complex Ni2(OAc)3(acac)NMP·2H2O (“A”); (b) the AFM 2D and 3D images of nanoparticles based on Ni2(OAc)3(acac)NMP·2H2O (2 μm × 2 μm).
Figure 1.
(a) Structure of the complex Ni2(OAc)3(acac)NMP·2H2O (“A”); (b) the AFM 2D and 3D images of nanoparticles based on Ni2(OAc)3(acac)NMP·2H2O (2 μm × 2 μm).

Figure 2.
(a) AFM 3D image of structures with h~80–100 nm, formed on a modified silicon surface, based on triple complexes Ni(acac)2∙NMP∙PhOH (3.5 μm × 3.5 μm); (b) AFM 3D image of structures (30 µm × 30 µm) with h~300 nm, based on triple complexes Ni(acac)2∙NaSt∙PhOH.
Figure 2.
(a) AFM 3D image of structures with h~80–100 nm, formed on a modified silicon surface, based on triple complexes Ni(acac)2∙NMP∙PhOH (3.5 μm × 3.5 μm); (b) AFM 3D image of structures (30 µm × 30 µm) with h~300 nm, based on triple complexes Ni(acac)2∙NaSt∙PhOH.

Scheme 1.
The structure of NiARD with Tyr residue in the outer coordination sphere of a nickel ion [18].
Scheme 1.
The structure of NiARD with Tyr residue in the outer coordination sphere of a nickel ion [18].

Figure 3.
(a) 3D AFM image of nanostructures based on systems {Ni(acac)2 His Tyr} (h = 45 nm) (4.5 μm × 4.5 μm); (b) 3D AFM image of nanostructures based on systems {Ni(acac)2⋅NMP⋅Tyr} (h = 25 nm) (2 μm × 2 μm).
Figure 3.
(a) 3D AFM image of nanostructures based on systems {Ni(acac)2 His Tyr} (h = 45 nm) (4.5 μm × 4.5 μm); (b) 3D AFM image of nanostructures based on systems {Ni(acac)2⋅NMP⋅Tyr} (h = 25 nm) (2 μm × 2 μm).

Figure 4.
(a) The AFM 3D image of nanoparticles based on FeIIIx(acac)y18C6m(H2O)n; (b) the AFM 3D image of nanoparticles based on triple system FeIIIx(acac)y(His)m(Tyr)n(H2O)p.
Figure 4.
(a) The AFM 3D image of nanoparticles based on FeIIIx(acac)y18C6m(H2O)n; (b) the AFM 3D image of nanoparticles based on triple system FeIIIx(acac)y(His)m(Tyr)n(H2O)p.

Figure 5.
AFM two- (a) and three-dimensional (b) image of stable nanostructures based on model {Hem•Tyr•His} complexes.
Figure 5.
AFM two- (a) and three-dimensional (b) image of stable nanostructures based on model {Hem•Tyr•His} complexes.

Figure 6.
(a) AFM two-dimensional image of supramolecular structures based on {Hem+PhOH+His} complexes and a two-dimensional image profile of these structures; (b) AFM three-dimensional image of supramolecular structures, based on {Hem+PhOH+His} complexes.
Figure 6.
(a) AFM two-dimensional image of supramolecular structures based on {Hem+PhOH+His} complexes and a two-dimensional image profile of these structures; (b) AFM three-dimensional image of supramolecular structures, based on {Hem+PhOH+His} complexes.

Figure 7.
The possible triangular structure of {Hem•Tyr•His} complex that is formed due to H-bonds NH•••O or N•••HO (a). The combination of individual triangles into structures such as Sierpinski triangular motifs (b).
Figure 7.
The possible triangular structure of {Hem•Tyr•His} complex that is formed due to H-bonds NH•••O or N•••HO (a). The combination of individual triangles into structures such as Sierpinski triangular motifs (b).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Matienko, L.; Binyukov, V.; Mil, E.; Goloshchapov, A. Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts 2021, 11, 1032. https://doi.org/10.3390/catal11091032
AMA Style
Matienko L, Binyukov V, Mil E, Goloshchapov A. Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons. Catalysts. 2021; 11(9):1032. https://doi.org/10.3390/catal11091032
Chicago/Turabian StyleMatienko, Ludmila, Vladimir Binyukov, Elena Mil, and Alexander Goloshchapov. 2021. "Role of PhOH and Tyrosine in Selective Oxidation of Hydrocarbons" Catalysts 11, no. 9: 1032. https://doi.org/10.3390/catal11091032
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.