
Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations


Abstract
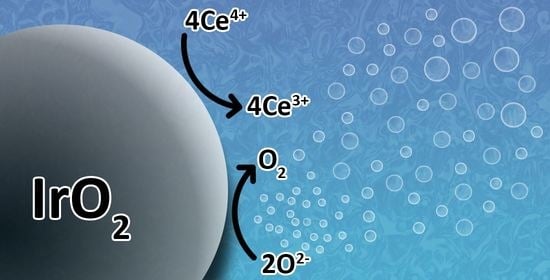
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
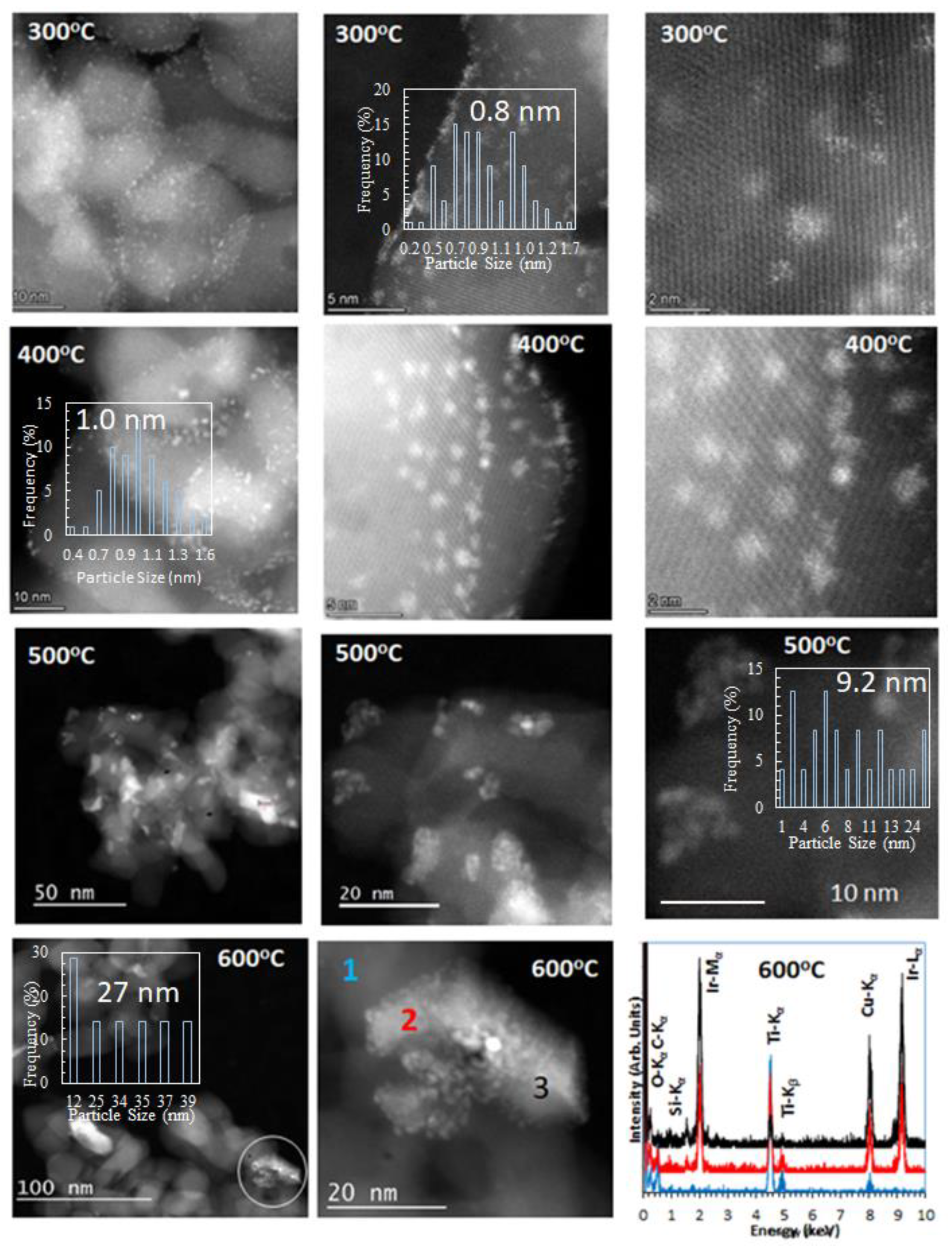
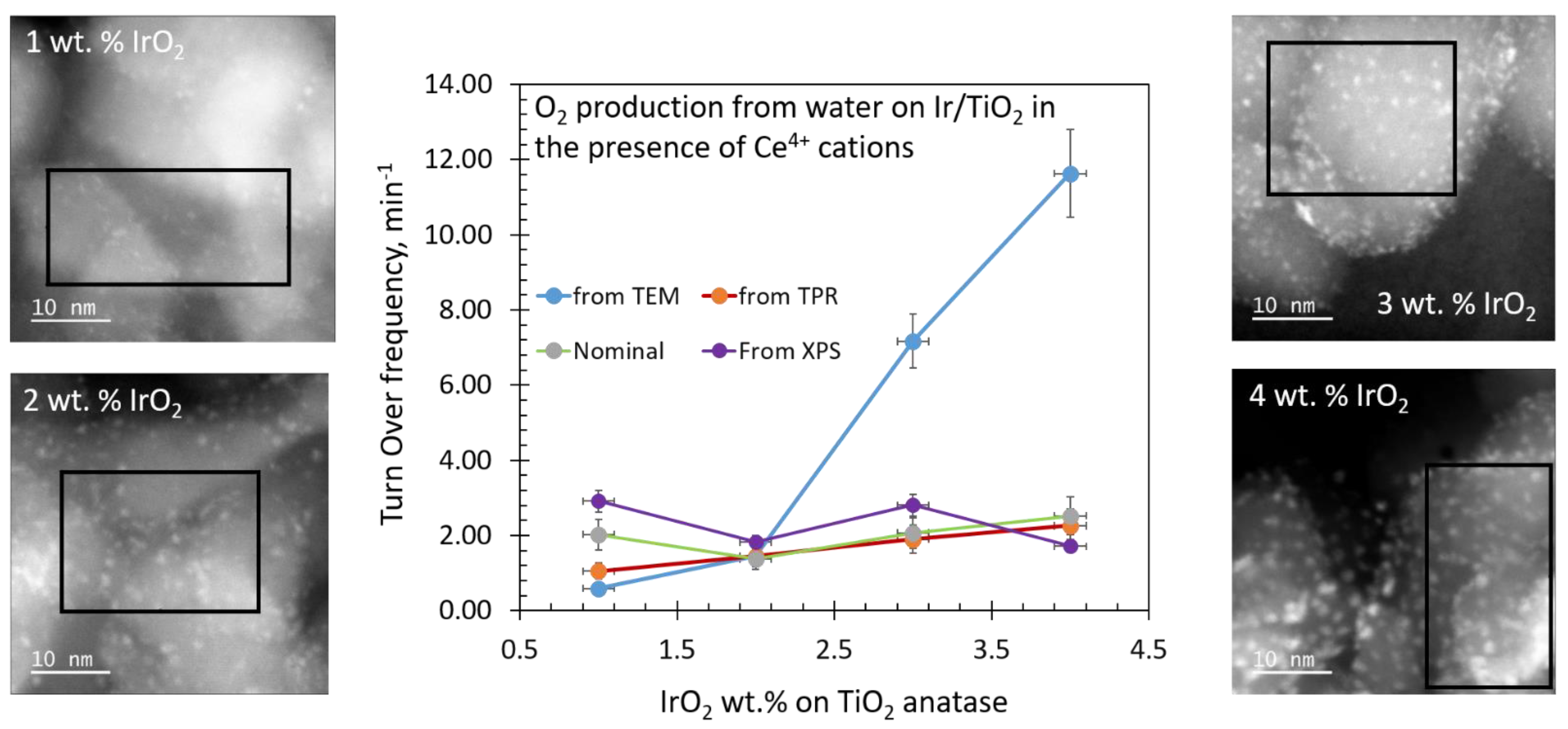
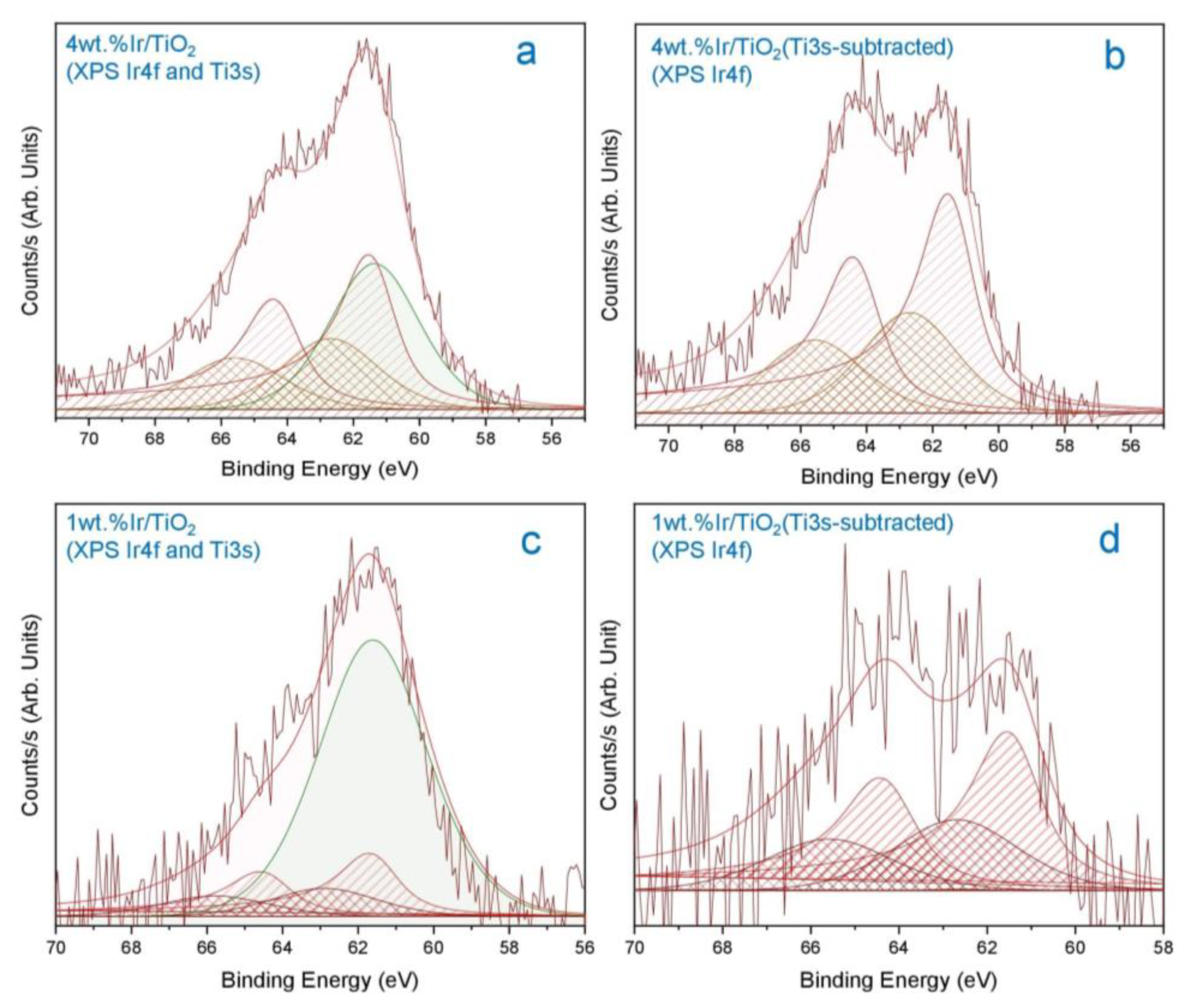
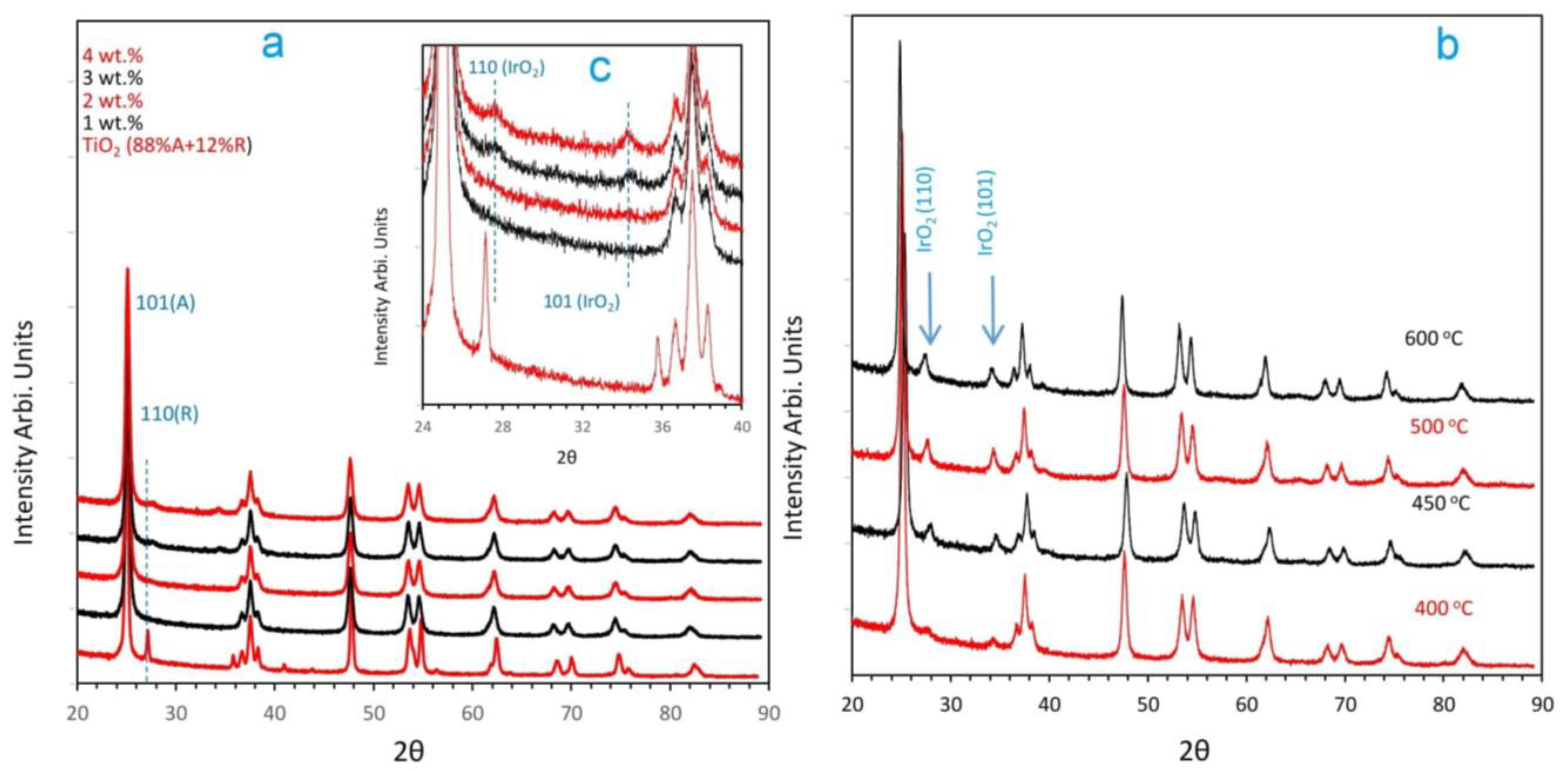
2.1. Catalyst Dispersion and Structure
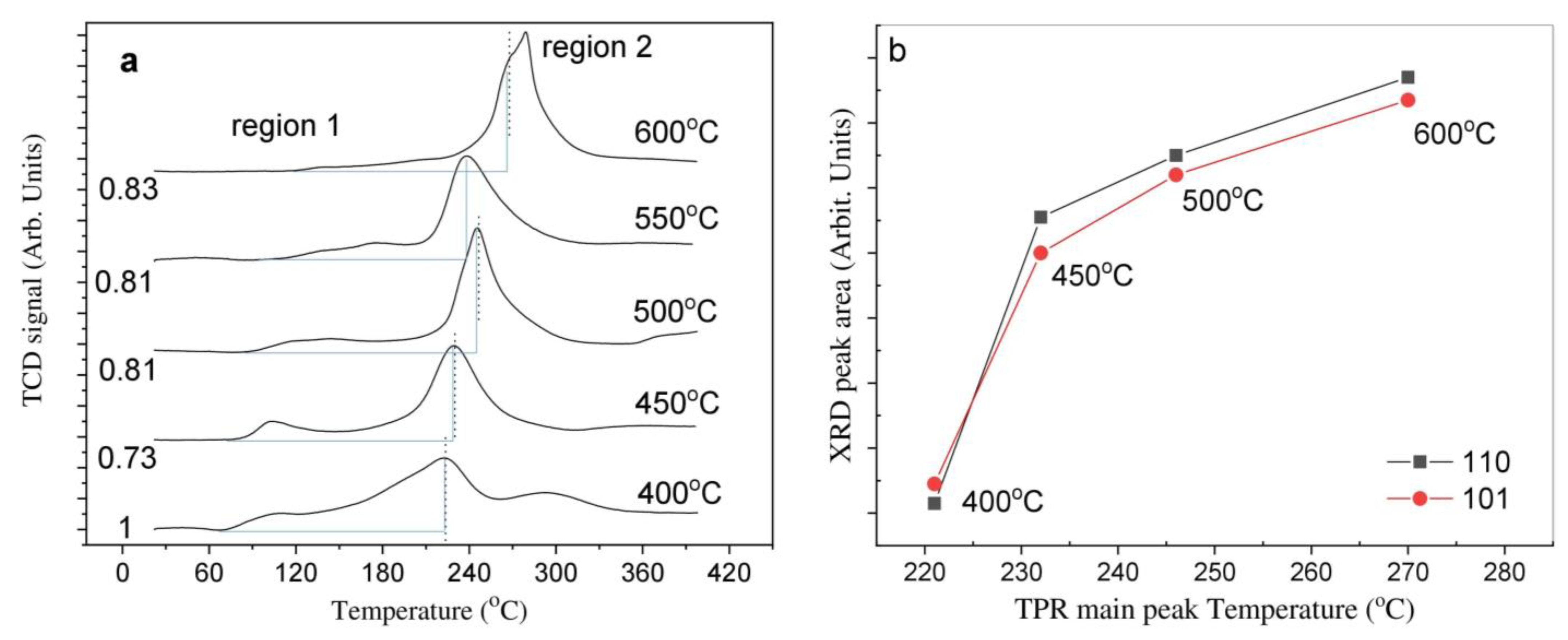
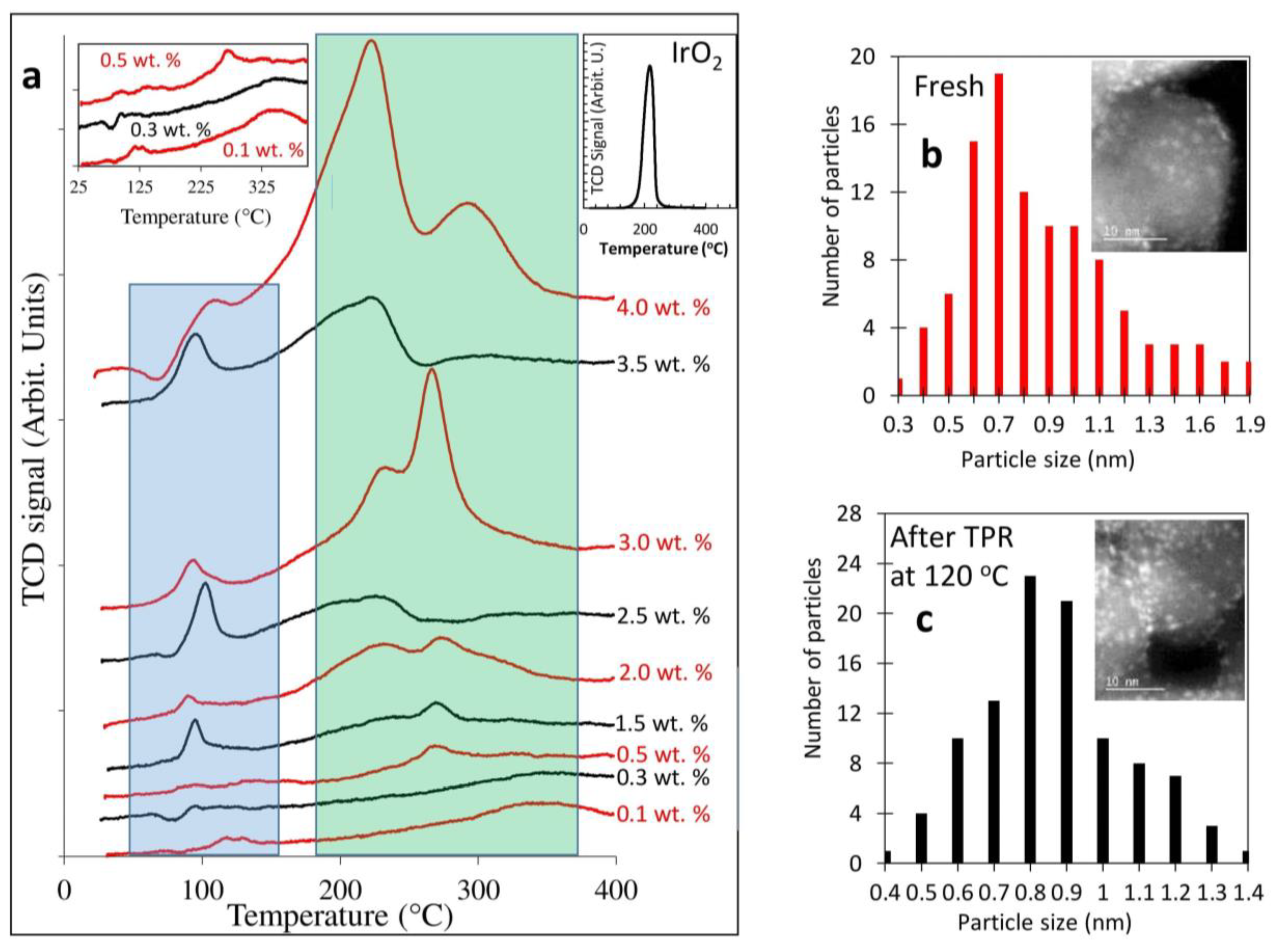
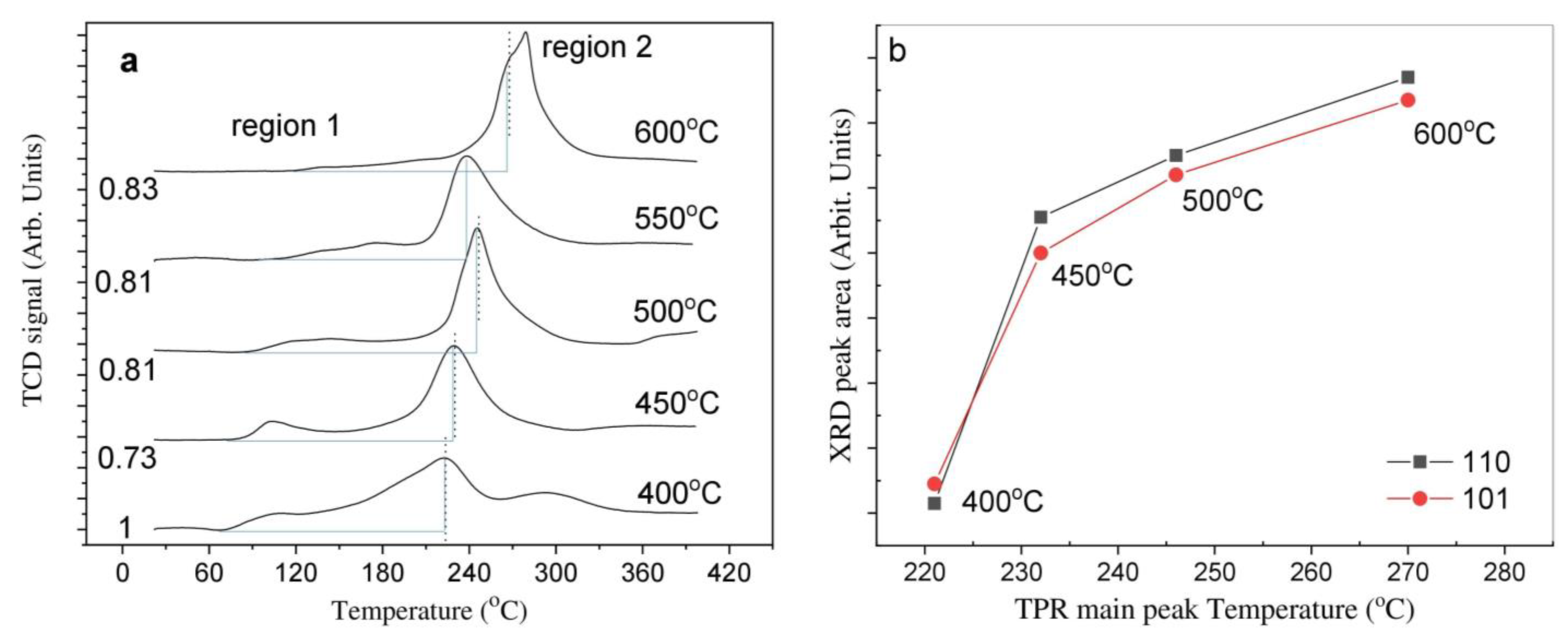
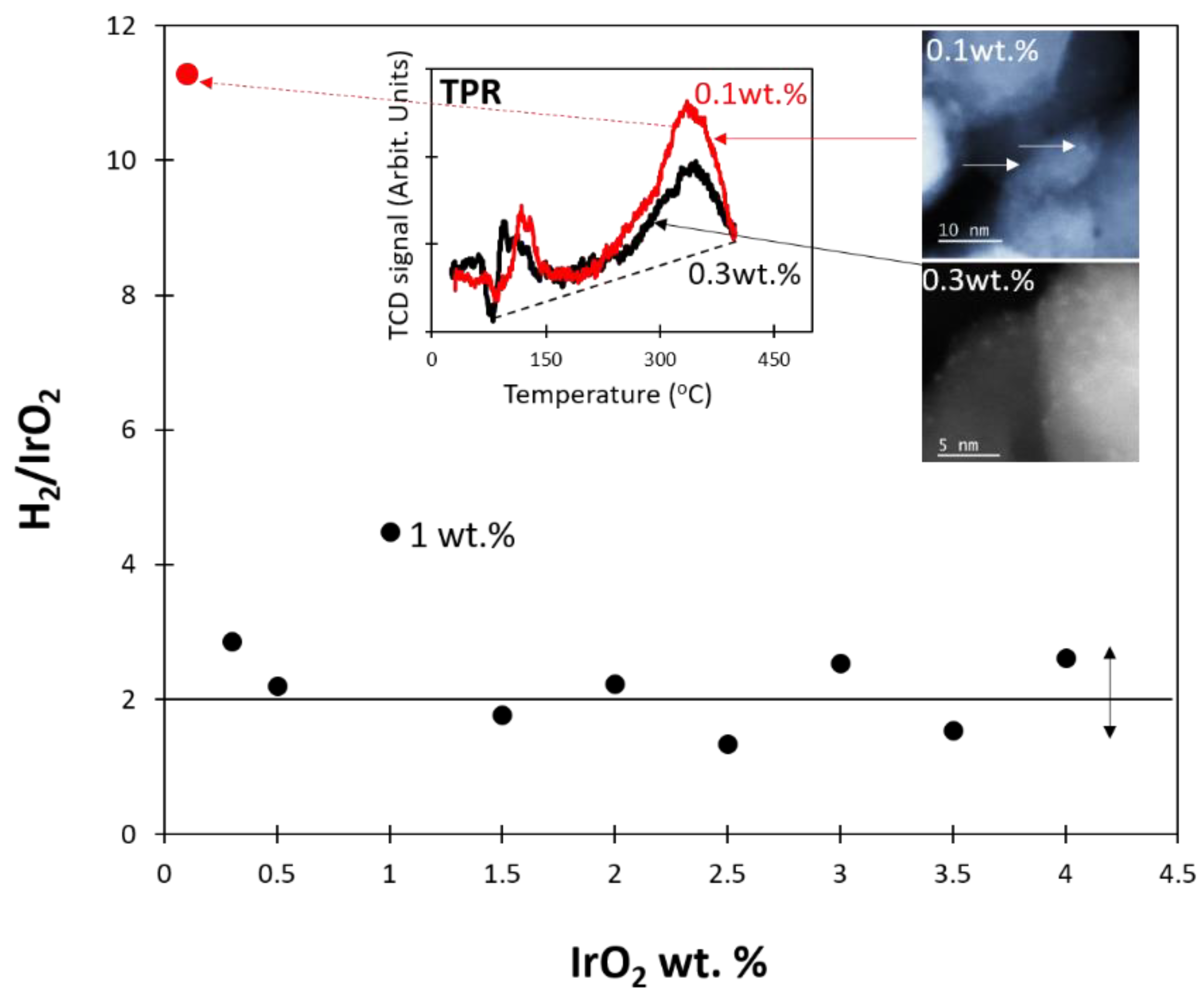
2.2. Temperature-Programmed Reduction and Effect of Calcination Temperature on Catalyst Dispersion
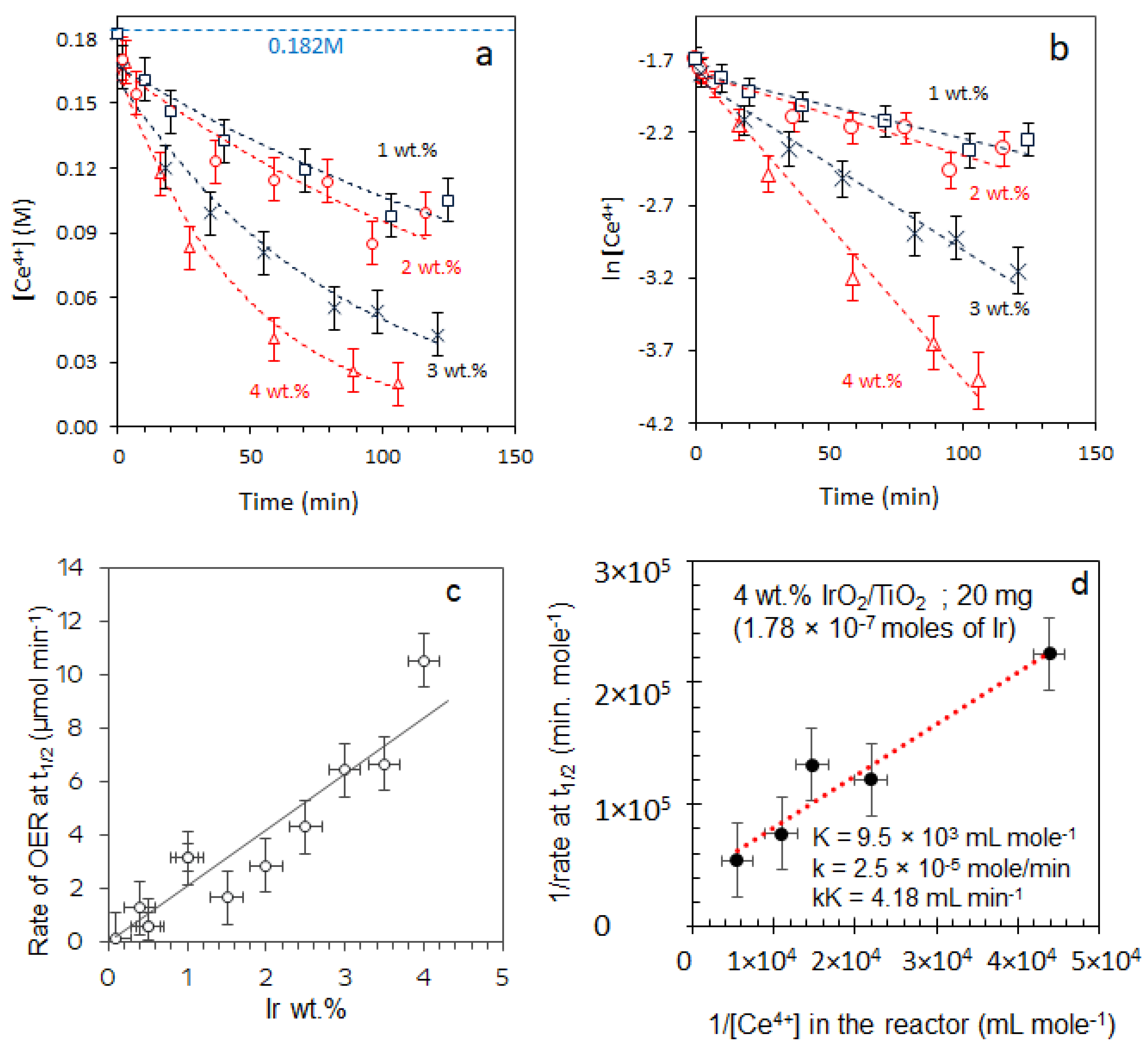
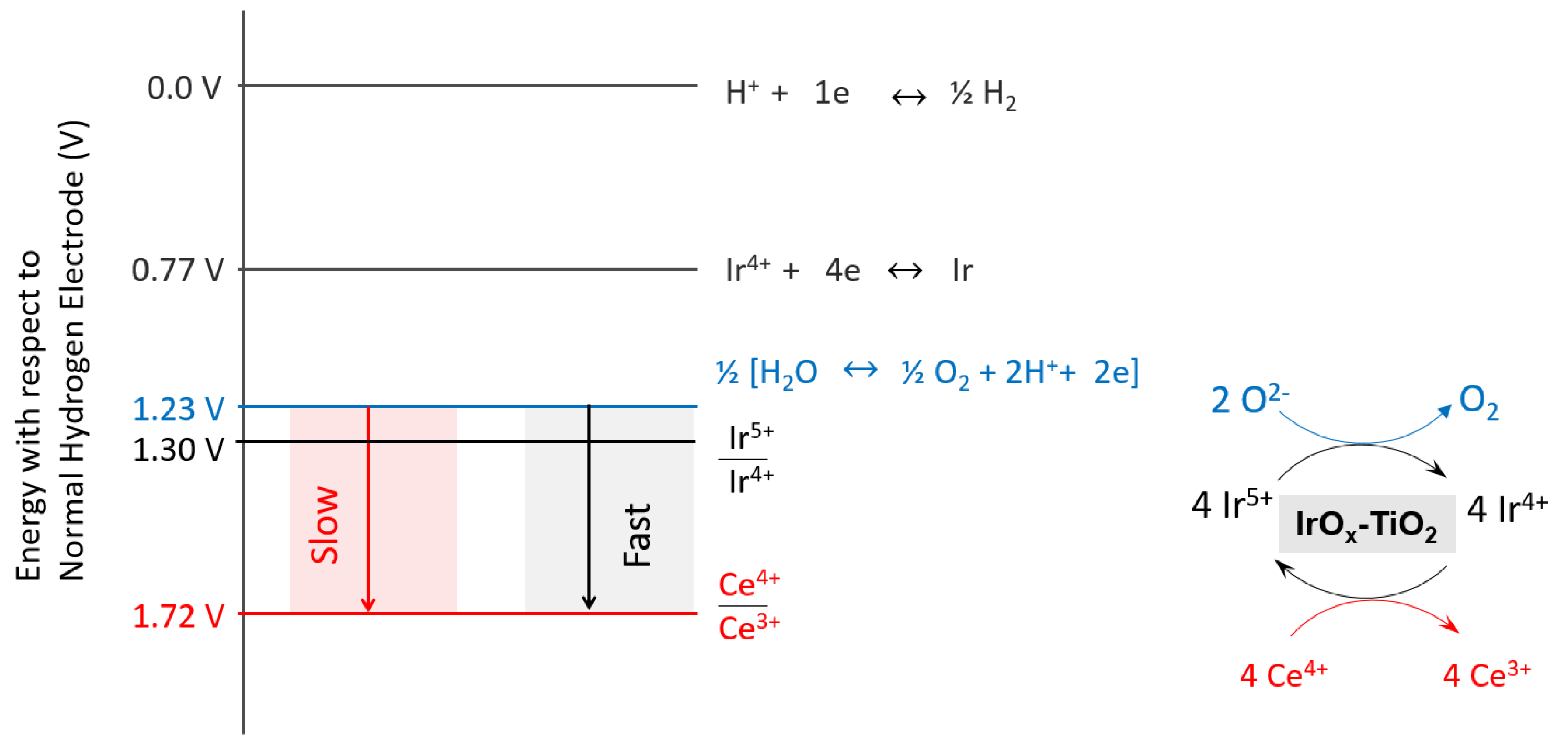
2.3. Oxygen Production from Water in the Presence of Ce4+ Cations
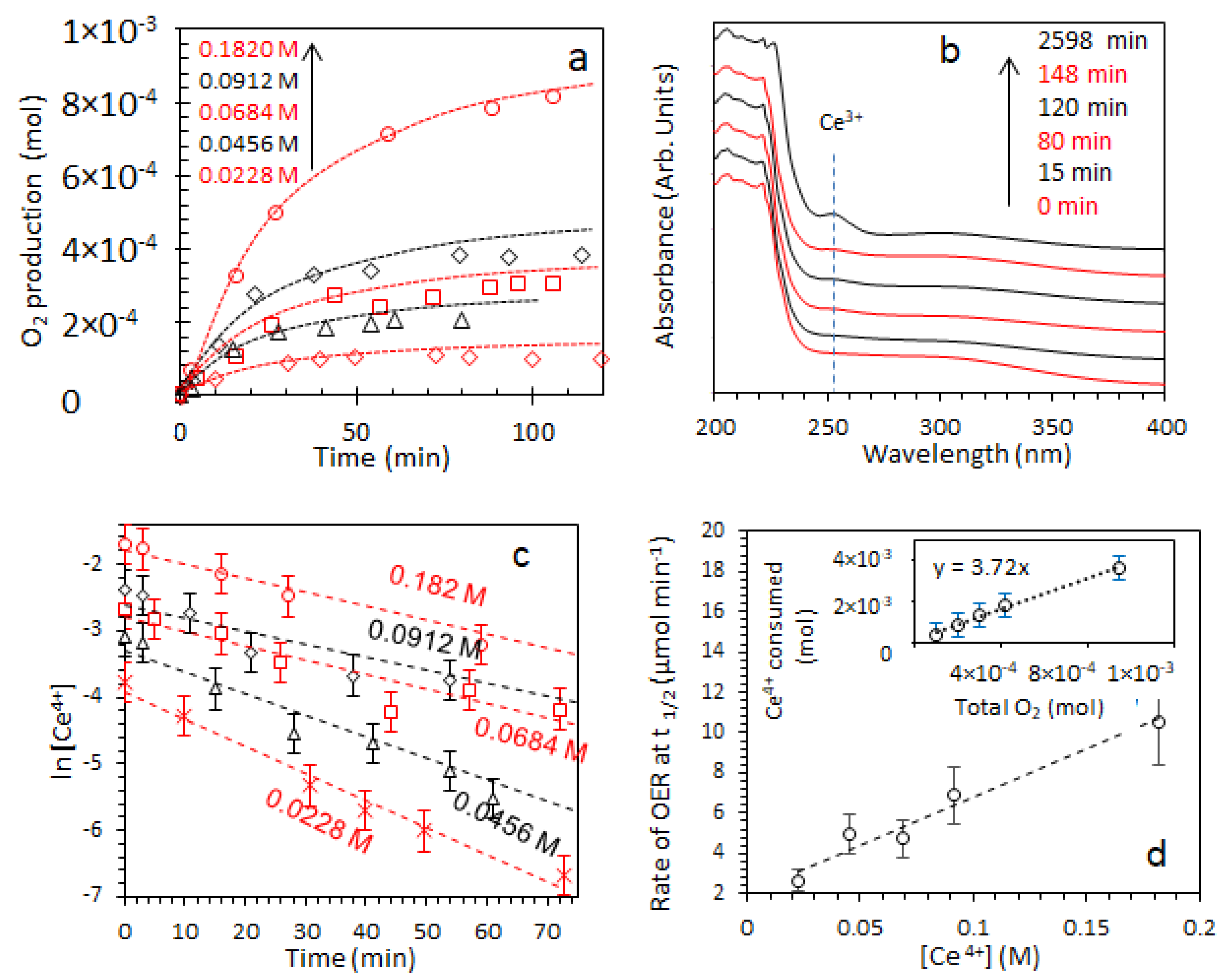
2.3.1. Effect of [Ce4+]
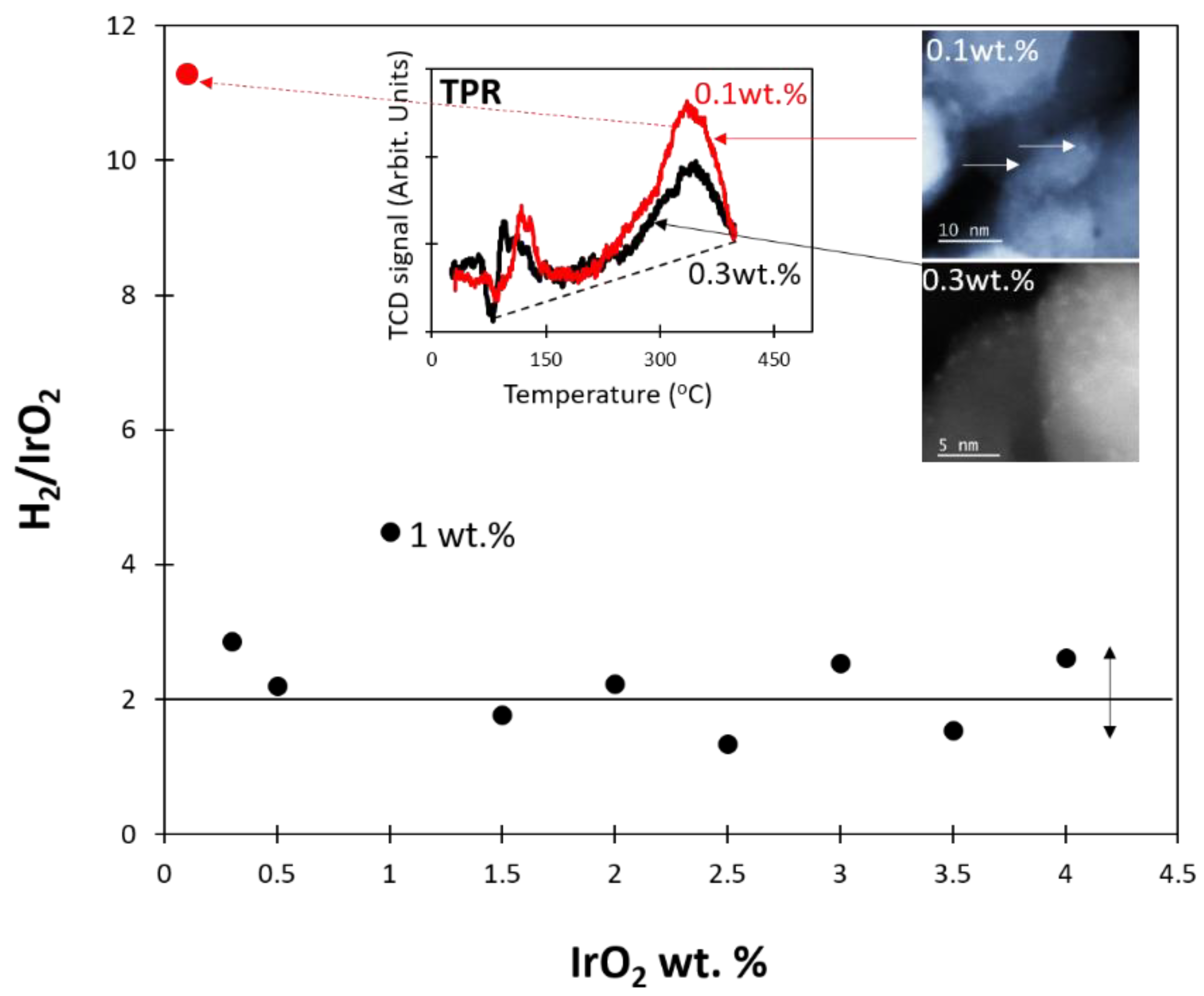
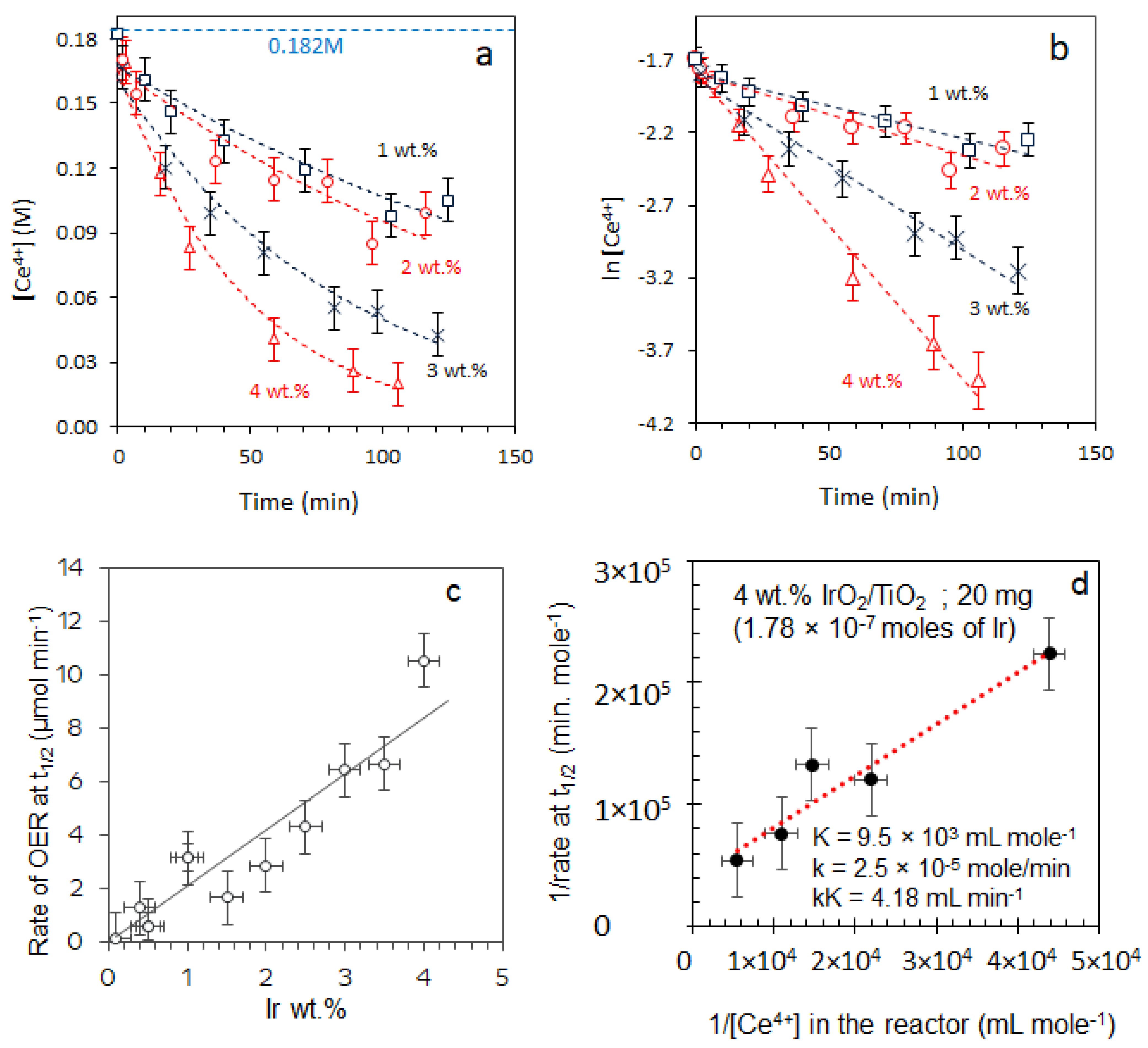
2.3.2. Effect of [IrO2]
2.4. Catalyst Stability and the Location of Deposited Cerium Cations
2.5. Turnover Frequency Calculations
3. Experimental
3.1. Catalyst Preparation
3.2. Reaction Setup
3.3. Catalyst Characterization
3.3.1. UV-Vis Absorbance
3.3.2. X-ray Diffraction (XRD)
3.3.3. X-ray Photoelectron Spectroscopy (XPS)
3.3.4. Temperature-Programmed Reduction (TPR)
3.3.5. Electron Microscopy (EM)
3.3.6. Metal Dispersion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Zhang, Q.; Feng, X. Support and Interface Effects in Water-Splitting Electrocatalysts. Adv. Mater. 2018, 31, e1808167. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H. Towards large scale hydrogen production from water, what have we learned and what are the main hurdles to cross for commercialization. Energy Technol. 2021, 9, 2000843. [Google Scholar] [CrossRef]
- Alsabban, M.M.; Yang, X.; Wahyudi, W.; Fu, J.-H.; Hedhili, M.N.; Ming, J.; Yang, C.-W.; Nadeem, M.A.; Idriss, H.; Lai, Z.; et al. Design and Mechanistic Study of Highly Durable Carbon-Coated Cobalt Diphosphide Core–Shell Nanostructure Electrocatalysts for the Efficient and Stable Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2019, 11, 20752–20761. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötza, R.; Schmidt, T.J. Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef] [Green Version]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef]
- Yang, C.; Grimaud, A. Factors Controlling the Redox Activity of Oxygen in Perovskites: From Theory to Application for Catalytic Reactions. Catalysts 2017, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Minguzzi, A.; Lugaresi, O.; Achilli, E.; Locatelli, C.; Vertova, A.; Ghigna, P.; Rondinini, S. Observing the oxidation state turnover in heterogeneous iridium-based water oxidation catalysts. Chem. Sci. 2014, 5, 3591–3597. [Google Scholar] [CrossRef] [Green Version]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 14, 4029–4030. [Google Scholar] [CrossRef]
- McDaniel, N.D.; Coughlin, F.J.; Tinker, L.L.; Bernhard, S. Cyclometalated Iridium(III) Aquo Complexes: Efficient and Tunable Catalysts for the Homogeneous Oxidation of Water. J. Am. Chem. Soc. 2008, 130, 210–217. [Google Scholar] [CrossRef]
- Kiwi, J.; Grätzel, M. Oxygen Evolution from Water via Redox Catalysis. Angew. Chem. Intern. Ed. 1978, 17, 860–861. [Google Scholar] [CrossRef]
- Beni, G.; Schiavone, L.M.; Shay, J.L.; Dautremont-Smith, W.C.; Schneider, B.S. Electrocatalytic oxygen evolution on reactively sputtered electrochromic iridium oxide films. Nature 1979, 282, 281–283. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis in the anodic evolution of oxygen and chlorine. Electrochim. Acta 1984, 29, 1503–1512. [Google Scholar] [CrossRef]
- Stacy, J.; Regmi, Y.; Leonard, B.; Fan, M. The recent progress and future of oxygen reduction reaction catalysis: A review. Renew. Sustain. Energy Rev. 2017, 69, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Rossmeisl, J.; Qu, Z.-W.; Zhu, H.; Kroes, G.-J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Bucci, A.; Rodriguez, G.M.; Bellachioma, G.; Zuccaccia, C.; Poater, A.; Cavallo, L.; Macchioni, A. An Alternative Reaction Pathway for Iridium-Catalyzed Water Oxidation Driven by Cerium Ammonium Nitrate (CAN). ACS Catal. 2016, 6, 4559–4563. [Google Scholar] [CrossRef] [Green Version]
- Shilpa, N.; Manna, J.; Rajput, P.; Rana, R.K. Water Oxidation Catalyst via Heterogenization of Iridium Oxides on Silica: A Polyamine-Mediated Route to Achieve Activity and Stability. ACS Catal. 2016, 6, 5699–5705. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Brown, D.B.; Martin, J.K.; Marelius, D.C.; Abadjian, M.-C.; Tran, H.N.; Kalyuzhny, G.; Vecchio, K.S.; Specht, Z.G.; Cortes-Llamas, S.A.; et al. Evolution of Iridium-Based Molecular Catalysts during Water Oxidation with Ceric Ammonium Nitrate. J. Am. Chem. Soc. 2011, 133, 19024–19027. [Google Scholar] [CrossRef]
- Hong, D.; Murakami, M.; Yamada, Y.; Fukuzumi, S. Efficient water oxidation by cerium ammonium nitrate with [Ir III (Cp*)(4,4′-bishydroxy-2,2′-bipyridine)(H2O)]2+ as a precatalyst. Energy Environ. Sci. 2012, 5, 5708–5716. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Verho, O.; Johnston, E.V.; Åkermark, B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef]
- Boudart, M.; Djega-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Boudart, M. Turnover Rates in Heterogeneous Catalysis. Chem. Rev. 1995, 95, 661–666. [Google Scholar] [CrossRef]
- English, B.; Min, W.; van Oijen, A.; Lee, K.T.; Luo, G.; Sun, H.; Cherayil, B.J.; Kou, S.C.; Xie, X.S. Ever-fluctuating single enzyme molecules: Michaelis-Menten equation revisited. Nat. Chem. Biol. 2005, 2, 87–94. [Google Scholar] [CrossRef]
- Katsiev, K.; Harrison, G.; Al-Salik, Y.; Thornton, G.; Idriss, H. Gold Cluster Coverage Effect on H2 Production over Rutile TiO2(110). ACS Catal. 2019, 9, 8294–8305. [Google Scholar] [CrossRef]
- Jones, A.J.; Carr, R.T.; Zones, S.I.; Iglesia, E. Acid strength and solvation in catalysis by MFI zeolites and effects of the identity, concentration and location of framework heteroatoms. J. Catal. 2014, 312, 58–68. [Google Scholar] [CrossRef]
- Davis, R. Turnover rates on complex heterogeneous catalysts. AIChE J. 2018, 64, 3778–3785. [Google Scholar] [CrossRef]
- Chan, H.C.; Chen, T.; Xie, L.; Shu, Y.; Gao, Q. Enhancing formaldehyde oxidation on iridium catalysts using hydrogenated TiO2 supports. New J. Chem. 2018, 42, 18381–18387. [Google Scholar] [CrossRef]
- Zhao, S.; Stocks, A.; Rasimick, B.; More, K.; Xu, H. Highly Active, Durable Dispersed Iridium Nanocatalysts for PEM Water Electrolyzers. J. Electrochem. Soc. 2018, 165, F82–F89. [Google Scholar] [CrossRef]
- Kasian, O.; Li, T.; Mingers, A.M.; Schweinar, K.; Savan, A.; Ludwig, A.; Mayrhofer, K. Stabilization of an iridium oxygen evolution catalyst by titanium oxides. J. Phys. Energy 2021, 3, 034006. [Google Scholar] [CrossRef]
- Reyes, P.; Salinas, D.; Campos, C.; Oportus, M.; Murcia, J.; Rojas, H.; Borda, G.; Fierro, J.L.G. Selective hydrogenation of furfural on Ir/TiO2 catalysts. Quím. Nova 2010, 33, 777–780. [Google Scholar] [CrossRef] [Green Version]
- Okumura, M.; Masuyama, N.; Konishi, E.; Ichikawa, S.; Akita, T. CO Oxidation below Room Temperature over Ir/TiO2 Catalyst Prepared by Deposition Precipitation Method. J. Catal. 2002, 208, 485–489. [Google Scholar] [CrossRef]
- Pennycook, S.; Jesson, D. High-resolution Z-contrast imaging of crystals. Ultramicroscopy 1991, 37, 14–38. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Huo, M.; Chen, T.; Li, Q.; Sun, Z.; Song, L. The properties of Irn (n = 2–10) clusters and their nucleation on c-Al2O3 and MgO surfaces: From ab initio studies. Phys. Chem. Chem. Phys. 2015, 17, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Hegmann, T. Determining the composition of gold nanoparticles: A compilation of shapes, sizes, and calculations using geometric considerations. J. Nanopart. Res. 2016, 18, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, V.; Jones, T.E.; Vélez, J.J.V.; Massué, C.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Greiner, M.T.; Allan, J.; et al. The electronic structure of iridium and its oxides. Surf. Interface Anal. 2016, 48, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Freakley, S.; Esquius, J.R.; Morgan, D.J. The X-ray photoelectron spectra of Ir, IrO2 and IrCl3 revisited. Surf. Interface Anal. 2017, 49, 794–799. [Google Scholar] [CrossRef]
- Sakurai, K.; Mizusawa, M. X-ray Diffraction Imaging of Anatase and Rutile. Anal. Chem. 2010, 82, 3519–3522. [Google Scholar] [CrossRef]
- Hanaor, D.A.H.; Sorrell, C.C. Review of the anatase to rutile phase transformation. J. Mater. Sci. 2010, 46, 855–874. [Google Scholar] [CrossRef] [Green Version]
- Burrows, J.P.; Tyndall, G.S.; Moortgat, G.K. Absorption spectrum of NO3 and kinetics of the reactions of NO3 with NO2, Cl, and several stable atmospheric species at 298 K. J. Phys. Chem. 1985, 89, 4848–4856. [Google Scholar] [CrossRef]
- Henderson, M.A.; Perkins, C.L.; Engelhard, M.H.; Thevuthasan, S.; Peden, C.H.F. Redox properties of water on the oxidized and reduced surfaces of CeO2(111). Surf. Sci. 2003, 526, 1–18. [Google Scholar] [CrossRef]
- Mao, Z.; Lustemberg, P.G.; Rumptz, J.R.; Ganduglia-Pirovano, M.V.; Campbell, C.T. Ni Nanoparticles on CeO2(111): Energetics, Electron Transfer, and Structure by Ni Adsorption Calorimetry, Spectroscopies, and Density Functional Theory. ACS Catal. 2020, 10, 5101–5114. [Google Scholar] [CrossRef] [Green Version]
- Ševčíková, K.; Nehasil, V.; Vorokhta, M.; Haviar, S.; Matolín, V.; Matolínová, I.; Mašek, K.; Píš, I.; Kobayashi, K.; Kobata, M.; et al. Altering properties of cerium oxide thin films by Rh doping. Mater. Res. Bull. 2015, 67, 5–13. [Google Scholar] [CrossRef]
- Sheng, P.Y.; Chiu, W.W.; Yee, A.; Morrison, S.J.; Idriss, H. Hydrogen production from ethanol over bimetallic Rh-M/CeO2 (M=Pd or Pt). Catal. Today 2007, 129, 313–321. [Google Scholar] [CrossRef]
- Škoda, M.; Libra, J.; Šutara, F.; Tsud, N.; Skála, T.; Sedláček, L.; Cháb, V.; Prince, K.C.; Matolín, V. A resonant photoemission study of the Ce and Ce-oxide/Pd(111) interfaces. Surf. Sci. 2007, 601, 4958–4965. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron transfer reactions in chemsitry: Theory and experiments. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Silverstein, T.P. Marcus Theory: Thermodynamics CAN Control the Kinetics of Electron Transfer Reactions. J. Chem. Educ. 2012, 89, 1159–1167. [Google Scholar] [CrossRef]
- Electron Transfer, Chapter 1, Solar Energy Conversion. In Chemical Aspects, 1st ed.; Likhtenshtein, G. (Ed.) Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Abellan, P.; Moser, T.H.; Lucas, I.T.; Grate, J.W.; Evansf, J.E.; Browning, N.D. The Formation of Cerium(III) Hydroxide Nanoparticles by a Radiation Mediated Increase in Local pH. RSC Adv. 2017, 7, 3831–3837. [Google Scholar] [CrossRef] [Green Version]
- Demars, T.J.; Bera, M.K.; Seifert, S.; Antonio, M.R.; Ellis, R.J. Revisiting the Solution Structure of Ceric Ammonium Nitrate. Angew. Chem. Int. Ed. 2015, 54, 7534–7538. [Google Scholar] [CrossRef]
- He, Y.B.; Stierle, A.; Li, W.X.; Farkas, A.; Kasper, N.; Over, H. Oxidation of Ir(111): From O-Ir-O Trilayer to Bulk Oxide Formation. J. Phys. Chem. C 2008, 112, 11946–11953. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrushaid, M.; Nadeem, M.A.; Wahab, K.A.; Idriss, H. Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations. Catalysts 2021, 11, 1030. https://doi.org/10.3390/catal11091030
Alrushaid M, Nadeem MA, Wahab KA, Idriss H. Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations. Catalysts. 2021; 11(9):1030. https://doi.org/10.3390/catal11091030
Chicago/Turabian StyleAlrushaid, Mogbel, Muhammad A. Nadeem, Khaja A. Wahab, and Hicham Idriss. 2021. "Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations" Catalysts 11, no. 9: 1030. https://doi.org/10.3390/catal11091030
APA StyleAlrushaid, M., Nadeem, M. A., Wahab, K. A., & Idriss, H. (2021). Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations. Catalysts, 11(9), 1030. https://doi.org/10.3390/catal11091030