
Electrochemical Degradation of Methylene Blue Using a Ni-Co-Oxide Anode



Department of Chemical Engineering, McGill University, 3610 University St., Montreal, QC H3A 0C5, Canada
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(7), 793; https://doi.org/10.3390/catal11070793
Submission received: 4 June 2021
/
Revised: 18 June 2021
/
Accepted: 23 June 2021
/
Published: 29 June 2021
(This article belongs to the Special Issue Advanced Oxidation Treatment of Refractory Polluted Wastewaters)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The potential of using thermally prepared Ni0.6Co0.4-oxide for the electrochemical degradation of organic contaminants was investigated using methylene blue (MB) in an aqueous solution, as a model pollutant. The results of UV spectroscopy obtained during galvanostatic electrolyses at the anode indicated the complete removal of the methylene blue dye. The high removal of chemical oxygen demand (COD) and total organic carbon (TOC) suggested a high level of mineralization of its intermediates. It was found that the electrocatalytic performance of the electrode in the anodic degradation of the organic pollutant was significantly enhanced by the presence of chloride ions in the solution. The improvement in the degradation rate of MB was attributed to the in situ electrogeneration of chlorine active species. The results show that Ni0.6Co0.4-oxide anode can be employed as a stable energy-efficient electrocatalyst in the electrochemical purification of wastewater.
1. Introduction
Industries that utilize dye stuff including the textile and pharmaceutical industries are sources of contaminants prevalent in colored wastewater. These dyes, although in a trace amount, can pose preventable health risks and are toxic potential carcinogens [1,2]. Thus, it is essential to employ a wastewater treatment system in the water cycle to remove these pollutants. The degradation of dye stuff from industrial wastewater is a challenging task that would typically involve multiple steps.
Generally, a conventional wastewater treatment scheme consists of a pre-treatment step followed by primary, secondary, and tertiary treatments that involve physical, chemical, and biological processes, which are selected and designed based on the characteristics of the wastewater and the target contaminants. However, conventional wastewater treatment methods are often ineffective or insufficient for the removal of recalcitrant organic compounds and they may cause the formation of hazardous or even more toxic transformation products [3,4]. To address certain issues associated with conventional wastewater treatment plants, most of the recent research has been focused on the development of advanced oxidation processes (AOPs) [3,4,5,6,7,8,9,10]. These processes are based on the in situ generation of a sufficient quantity of highly reactive oxidizing agents such as ozone or hydroxyl radicals that are efficient in breaking down persistent organic contaminants. AOPs can be utilized as an effective pre-treatment step to increase the biodegradability of recalcitrant organic matters that are difficult to degrade through biological treatment. Furthermore, the advanced treatment procedures used as tertiary treatments can help to reduce the residual total organic load to meet specific effluent requirements. Among the advanced oxidation technologies such as ozonation [9], photocatalytic degradation [10], and Fenton process [11] just to name a few, the electrochemical oxidation method has received wide recognition for its effectiveness in the removal of toxic and bio-refractory organic compounds [3,5,6,12].
The electrochemical oxidation of wastewater offers a plethora of advantages, in comparison to other AOPs, such as its low cost, simplicity, versatility, amenability of automation, environmental compatibility, small footprint and modularity, process safety, and minimum waste production [5,6]. Consequently, there is a greater interest in the utilization of this method for the efficient treatment of certain organics-contaminated wastewaters. In electrochemical oxidation, the choice of anode material is a critical factor as it influences the overall efficiency and cost of the electrocatalytic process and the selectivity of the anodic reaction [13,14]. Studies have shown that the anodic oxidation (removal) of pollutants can follow either the direct or indirect mechanistic pathways, and this is very well documented in the literature [13,15,16,17,18]. In direct oxidation, there is a possibility of electrode fouling leading to the passivation of the electrode surface, which results in a poor chemical decontamination [13,16,17].
Several studies that employed different electrode materials such as boron-doped diamond (BDD) [19,20,21,22] and metal oxides such as PbO2 [23,24], SnO2 [25], Sb-doped SnO2 [26,27], and Sb-doped Sn0.8W0.2-Ox [6] anodes for the electrochemical decontamination of wastewater via indirect anodic oxidation have been reported. BDD has shown to be the best anode material for this purpose, but its commercial use is prohibited by its very high cost. The main issues with the other anode materials cited above is their long-term stability issue and the slow kinetics of the degradation reaction, depending on the pollutant. Thus, there is a need to develop better anode materials for wastewater treatment.
One major characteristic of electrochemical wastewater treatment anodes is that they should offer high overpotential towards the oxygen evolution reaction (OER) which is, in this case, an unwanted parallel reaction that decreases the efficiency and increases the electricity consumption of the wastewater treatment process [5]. In our previous research on the influence of Ni-Co-oxide composition on their electrocatalytic activity in the OER, Ni0.6Co0.4-oxide was identified as exhibiting the largest OER overpotential among the investigated compositions [28]. Therefore, this low-cost, durable, non-toxic binary metal-oxide anode was identified as a promising material for the electrochemical degradation of wastewater contaminants.
Consequently, this original and novel work reports the results on the degradation of a model organic contaminant, methylene blue (MB) dye, using a Ni0.6Co0.4-oxide electrode material. Furthermore, it has been shown in the literature that the decolorization and mineralization of solutions containing model compounds can be achieved faster on metal oxides with chlorine-mediated indirect electrolysis. Hence, the influence of in situ electrogenerated active chlorine species on the electrocatalytic performance of Ni0.6Co0.4-oxide in the electrochemical degradation of MB-containing wastewater was investigated.
2. Results and Discussion
2.1. Electrochemical Degradation of MB in the Absence of Chlorides
The performance of Ni0.6Co0.4-oxide (The topographical and crystallographic characterization of this electrode was presented in our previous manuscript on the development of NiCo-oxide electrodes for oxygen evolution [28]) catalyst in the anodic oxidation of MB was first evaluated by the electrochemical oxidation of 50 mg/L methylene blue dye dissolved in 0.17 M sodium sulfate electrolyte (no chlorides were present). The mechanism of the MB electrochemical oxidation is well established in the literature and is based on the action of anodically electrogenerated active species, which in this case could be the hydroxyl radicals [5,13]; however, some MB could also be degraded through the direct electrochemical oxidation at the anode surface [3]. The disappearance of the blue color of MB during the electrolysis was used to monitor the apparent kinetics of the degradation process by utilizing UV-Vis spectrophotometry. The representative MB absorption spectra depicted in Figure 1 display two prominent peaks in the visible region at ca. 610 and 660 nm, which correspond to the MB dimer (MB+)2 and monomer (MB+), respectively [29,30,31]. The more pronounced absorption peak at 660 nm was used to correlate MB absorbance with its concentration in the solution.
As shown in Figure 1, the MB absorption peaks decreased with time, evidencing the decrease in MB concentration in the electrolyte. It was also visually observed that the color intensity of the blue dye solution diminished with time. The inset to Figure 1 more clearly depicts the gradual decrease in MB peak absorbance with treatment time.
It is common knowledge that the kinetics of electrochemical oxidation of organic pollutants, and thus also that of MB, depends on the current passing through the electrochemical cell. Consequently, we investigated the influence of applied current density on the kinetics of MB degradation, and the results are presented in Figure 2A. It can be observed that the MB degradation kinetics increases with an increase in current density, as expected. After two hours, 70% of the MB initially present was degraded at 10 mA/cm2, while at 60 mA/cm2, all of the MB was degraded during that same period of time. The percentage of MB that degraded was directly proportional to the charge passed through the anode, and Figure 2B shows that this dependence is linear. This indicates good faradaic performance of the anode in MB degradation. Namely, the anodic MB degradation reaction is always paralleled by the oxygen evolution reaction (the same is true in the case of anodic electrochemical degradation of any organic compound in an aqueous solution). If the Ni0.6Co0.4-oxide anode was a poor MB-degradation electrocatalyst, with an increase in current density, the proportion of current going to MB degradation would decrease on account of the OER current increase. Figure 2B shows that the kinetics of MB degradation is proportional to the current density (i.e., charge), evidencing that the Ni0.6Co0.4-oxide is indeed a suitable MB-degradation anode. Furthermore, during the electrolysis, the solution pH remained stable at 5.8 ± 0.1, also indicating that the predominant anodic reaction was MB degradation. The pH was not adjusted prior to the experiments and further research should investigate the effect of pH on the mechanisms and kinetics of MB degradation on the anode material studied here.
The kinetic analyses of the data in Figure 2A confirm that the degradation of MB under the experimental conditions employed follows the first-order kinetics with respect to MB. The corresponding apparent reaction rate constants, kapp are listed in Table S1 in the Supplementary Material. The dependence between these rate constants and the applied current density was found to be linear, as shown in Figure S1.
As mentioned earlier, electrochemical oxidation of organic pollutant can proceed through the direct pathway, where the pollutant is directly oxidized on the electrode surface, or through the indirect (mediated) pathway involving electrochemical formation of OH-radicals, which then oxidize the pollutant. To investigate a plausible mechanistic pathway for MB degradation on the Ni0.6Co0.4-oxide electrode in 0.17 M Na2SO4 aqueous solution, methanol, which is a well-known scavenger of hydroxyl radicals, was added into the MB solution [14,32]. The result of this test is presented in Figure 2C. It was observed that the addition of 0.2 M methanol to the MB-containing solution did not have any effect on the apparent kinetics of degradation of MB. This confirms that the MB degradation on the Ni0.6Co0.4-oxide surface occurs mainly through direct oxidation of the molecule, rather than through the OH-radical mediated oxidation.
2.2. Influence of Chloride-Ion Concentration on the Kinetics of MB Degradation
The results presented above demonstrate that the Ni0.6Co0.4-oxide anode is capable of degrading MB with a mechanism driven by direct oxidation at the electrode surface, rather than by hydroxyl-radical mediated oxidation. The apparent kinetics of degradation was increased by increasing the degradation current; however, another approach could also be to introduce specific ions into the electrolyte in order to produce additional active species that are strong oxidants and that would contribute to the electrochemical oxidation of the pollutant and possibly enhance the degradation kinetics. One such species is the chloride ion, which is either already present in certain wastewaters or could be added to the wastewater. The dissolved chloride ion can be oxidized at the anode to active chlorine species (such as Cl2, HOCl, ClO−), which are strong oxidants and, unlike hydroxyl radicals, are long-lived species that can contribute to the oxidation of MB [5]. This approach would be suitable for applications allowing the discharge of the treated water (which could be in the range of 0.25–2 g/L chlorides, as used in this work) into receiving water bodies that can tolerate the presence of chlorides (e.g., seawater).
Figure 3A shows the degradation of MB at 20 mA/cm2 in the presence of chlorides in the electrolyte as a function of current and time.
Figure 3A clearly reports a faster degradation of MB in the presence of the chloride ion and with its increasing concentration, which could be attributed to the strong oxidative action of electrogenerated active chlorine species. The kinetic analysis of the data in Figure 3A confirmed that the degradation of MB in the presence of chlorides in the electrolyte and under the experimental conditions employed follows the first-order kinetics with respect to MB. The corresponding apparent reaction rate constants, kapp,Cl are listed in Table S2 in the Supplementary Material. The dependence between these rate constants and the concentration of dissolved chlorides was found to be linear up to the chloride concentration of 1 g/L (Figure S2), while the apparent rate constant obtained at 2 g/L of dissolved chlorides was only slightly higher than that at the concentration of 1 g/L, indicating that a further increase in the chloride concentration would not significantly influence the MB apparent degradation kinetics.
To confirm that active chlorine species were responsible for the improvement in the MB degradation kinetics, 40 mM dopamine hydrochloride (DHCl), a known active chlorine species scavenger [33], was added into the degrading dye solution, and the scavenging effect was studied by performing three experiments using different conditions. The results are presented in Figure 3B. The control experiments consisted of running the treatment for 20 min, switching off the power supply and then monitoring the degradation of MB over a period of 40 min post-treatment (blue line/symbols) in the absence of the scavenger. For the “DHCl with power off” experiment, the same experiment as the control one was run, but DHCl was added into the solution after the 20 min of treatment at which point the power was switched off (red line/symbols). For the “DHCl with power on” experiment, DHCl was also added into the solution after 20 min of treatment but this time the current supplied to the cell was maintained (green line/symbols).
In the control experiment (blue line/symbols), the MB continued to degrade even after the power supply to the cell was turned off at the 20th minute. This continuation in MB degradation can be explained by the presence of active chlorine species generated during the first 20 min of the experiment, which continued degrading MB after the power was switched off. This confirms that active chlorine species persisted in the electrolyte for a longer time (at least for another 40 min). However, after adding DHCl into the solution at 20 min and turning off the power (red line/symbols), the degradation of MB stopped, which is due to the scavenging of active chlorine species by DHCl. On the other hand, when the power was maintained after the addition of DHCl (green line/symbols), the degradation of MB was initially stopped for 10–20 min, but the MB degradation resumed afterward and continued until the end of the monitoring period. This restart was due to the decrease in DHCl concentration in the electrolyte due to its electrochemical degradation occurring in parallel with MB degradation, enabling again the formation of active chlorine species that continued degrading MB.
2.3. Influence of Current Density on the Kinetics of MB Degradation in the Presence of Chlorides
Although the level of MB degradation at the applied current density of 20 mA/cm2 increased significantly with increasing concentration of Cl– ion (Figure 3A, this effect was not pronounced for [NaCl] ≥ 1 g/L. Consequently, the effect of current density on the kinetics of MB degradation was further investigated at a fixed chloride concentration of 2 g/L, and the results are presented in Figure 4. Results indicate that the MB oxidation rate increased with an increase in current density. Thus, it took ca. 50 min for the concentration of MB to drop to zero at 10 mA/cm2, while at 60 mA/cm2 this occurred within less than 15 min from the start of the process. During the electrochemical degradation, the temperature of the electrolyte was observed to change only slightly, by ca. 1.8 ± 0.6K. Hence, the observed improvement in the oxidation rate with current density is negligibly affected by the change in electrolyte temperature.
The analysis of the trends in Figure 4 confirmed the first-order kinetics with respect to MB at all current densities, and the corresponding apparent reaction rate constants, kapp,Cl,j, are listed in Table S3 in the Supplementary Material. Similar to the findings in the chloride-free electrolyte (Figure 2A and Figure S1), the dependence between these rate constants and the applied current density was found to be linear (Figure S3). However, comparing the apparent rate constant values obtained in the chloride-containing solution (Table S3) to those obtained in the chloride-free solution (Table S1), it is evident that the ratio of the two increases with current density, indicating that this increase in current density has a larger effect on the MB degradation kinetics in the presence of dissolved chlorides.
One of the negative consequences of the increased kinetics of organic pollutant oxidation at higher current densities is the decrease in faradaic efficiency of the process, which is due to the increase in kinetics of the parallel (unwanted) oxygen evolution reaction. To determine if this also occurred during the MB degradation experiments presented in Figure 4, the charge required to decrease the MB concentration to zero was determined at each current density employed; the average value was 38.7 ± 2.1 C/cm2, and the narrow distribution range indicates that the current that passed through the cell was predominantly invested in MB degradation (direct and active-chloride-species mediated), rather than in oxygen evolution. Furthermore, this small relative standard deviation also indicates that most of the chloride ions oxidized at the anode into active chlorine species to degrade MB; otherwise, the charge required to degrade MB to zero concentration would result in a larger increase with an increase in current density.
2.4. Chemical Oxygen Demand, Total Organic Carbon and Color Removal Efficiency
Although it is useful to determine the kinetics of MB oxidation by monitoring the concentration of MB by UV-Vis spectrophotometry, these measurements do not provide information on the extent of MB mineralization, but only on the extent of MB oxidation to a range of possible transformation products. In order to estimate the degree of mineralization of MB during treatment, chemical oxygen demand and the total organic carbon measurements were performed. The level of TOC and COD as a function of treatment time, representing an estimation of the mineralization, are presented in Figure 5, along with the removal of color corresponding to the removal of the parent compound MB. For the initial concentration of MB of 50 mg/L, the initial (time zero) COD and TOC values were ca. 63 mg/L and 26.5 mg/L, respectively. The figure shows that the COD and TOC removal increased over time, leading to the complete mineralization of the methylene blue dye. It took ca. 30 min to decolorize the MB-containing solution, while ca. 90% and ca. 88% of the initial COD and TOC were, respectively, removed during the same period of time. The complete abatement of COD and TOC was achieved after 50 and 60 min, respectively. The COD and TOC results in Figure 5 are indicative of the formation of intermediate products that may be less rapidly converted to carbon dioxide than the disappearance of the methylene blue [24].
The apparent kinetics of the removal of COD and TOC showed that the COD trend follows the kinetics characterized by the 1.89th order with respect to MB (Figure S4), yielding a rate constant of tCOD = 7.23 min−1 mg/L−0.89, while the TOC trend is of the 1.87th order with respect to MB (Figure S5), with the rate constant of tTOC = 8.35 min−1 mg/L−0.87.
These results further support Ni0.6Co0.4-oxide as an effective material for the electrochemical degradation of the MB. Notably, at 20 mA/cm2 current density and 2 g/L NaCl concentration, the NiCo-oxide material showed the MB degradation kinetics comparable to TiRuO2, BDD, PbO2/Ti, G/β-PbO2, which are among some of the best electrochemical anodes for MB degradation reported in the literature [3,34,35]. However, in terms of cost, the NiCo-oxide anode is much cheaper than the Ti/TiRuO2 and especially BDD anodes [3], and it is also significantly more environmentally friendly than the PbO2/Ti [34] and G/β-PbO2 anodes [35].
2.5. Stability of the Anode Material
Considering that anodes must be electrochemically stable over a longer period of time to minimize replacement costs, a preliminary evaluation of the stability of the anode was performed. One way of estimating the stability of an anode is to monitor its potential (or cell voltage) over time at a constant current density. Figure 6 shows the cell voltage measured when a constant current density of 20 mA/cm2 was applied between the Ni0.6Co0.4-oxide anode and the stainless steel cathode over a period of 50 h of electrolysis of 1000 mg/L MB in 0.17 M Na2SO4 aqueous solution (pH = 4.2) which is more aggressive than the regular MB test concentration (50 mg/L; pH = 5.8). Results indicate that the anode material is relatively stable over the monitored time interval. Furthermore, energy-dispersive X-ray spectroscopic (EDX) analysis of the residual solutions after evaporation of the solvent revealed no appreciable dissolution of the active electrode metals (Ni and/or Co) during the electrochemical degradation measurements. Nevertheless, it should be noted that the 50 h stability test in Figure 6 does not represent a result that can be used to reliably estimate the long-term service life/stability of the anode material when used at the industrial scale, which should be in up to thousands of hours. However, the electrode stability can be addressed through improvement of the coating-production method, which was not in the scope of the research reported in this manuscript.
2.6. Energy Consumption Determination
Although it may be beneficial to achieve fast degradation rates at higher current densities, this may lead to increased energy consumption [14]. Consequently, to appreciate the cost-effectiveness of the electrochemical oxidation treatment technique, it is important to determine the energy efficiency of the process. Typically, the energy consumption in electrochemical treatment is dependent on key parameters such as reactor configuration, electrode material, concentration of treated contaminant, to name a few [10]. In the present work, the overall energy consumed was primarily estimated as the electrical energy consumption per volume of treated solution (in kWh/m3) as described in the literature [10].
Figure 7 presents the total energy consumption for the complete oxidation of 50 mg/L methylene blue in 0.17 M Na2SO4 containing 2 g/L of NaCl (blue circles, EEC_A), and also the energy required to achieve a 70% degree of degradation in the absence of chloride ions (black triangles, EEC_B) for different current densities. In both cases, a linear relationship was found to exist between the total input energy and the applied current density. The results indicate that although the degradation process is faster when operating at higher current densities, this leads to a corresponding increase in operational cost. As previously discussed, the charge required to degrade MB, either in the absence or presence of chloride ions, was constant with current density. Thus, the increase in power consumption seen in Figure 7 results from the increase in cell potential (potential difference between the anode and cathode) required to “push” the required current through the electrochemical cell.
The main observation in Figure 7 is that the power needed to completely degrade MB is significantly lower when chloride ions are present in the electrolyte, with a value even lower than the power required for a non-complete (70%) degradation of MB in the absence of chlorides. The power requirement could be further reduced by the optimization of the design of the electrochemical treatment reactor; however, this was not within the scope of this project.
It is useful to determine the specific energy consumption and the corresponding current efficiency based on COD, since this outlines the amount of energy necessary for the complete mineralization of the MB. Hence, the instantaneous current efficiency (ICE, Equation (1)), which is the ratio of the charge employed in the anodic oxidation of each MB compound to the total charge passed during the electrolysis, was evaluated from COD values [2,36]. The specific energy consumption, in kWh/kgCOD, which is the energy needed to remove one kg of COD from the wastewater was also calculated from Equation (2) [37]:
where CODt and CODΔt are the COD (gO2/L) at time t and t + Δt, respectively, the number “8” represents the oxygen equivalent mass (g/eq), F is the Faraday constant (96486 C/mol), V is the potential difference between the electrodes (V), I is the applied current (A), Δt is the electrolysis time (in seconds for Equation (1) and in hours for Equation (2)), ΔCOD is the COD reduction in g/L during the time Δt and ν is the volume of treated solution in litres.
The complete electrochemical mineralization of MB at 20 mA/cm2 in the chloride-containing electrolyte (Figure 5) yielded a current efficiency of 26.4% which is close to the value obtained with a Ti/TiRuO2 anode (26%) [3]. The results presented in Figure 5 also show that the SEC needed to completely mineralize MB is 101.6 kWh/kgCOD, whereas to achieve ca. 70% degradation of 50 mg/L MB at SEC value of 12.9 kWh/kgCOD was needed, which is substantially lower than 82.4 kWh/kgCOD required to remove 20mg/L MB on the Pb/PbO2 anode in another study [2].
3. Materials and Methods
3.1. Anode Preparation
The synthesis of the anode material involved several preparation steps that ranged from the pre-treatment of the underlying 50 mm × 50 mm × 2 mm Ti substrate to the formation of the Ni0.6Co0.4-oxide film coating on the Ti substrate, as described in our earlier work [28]. The titanium substrate support (99% pure, McMaster Carr, Chicago, IL, USA) was first pretreated via wet-polishing using a 600-grit SiC sandpaper. The substrate was then rinsed in deionized water of resistivity 18.2 MΩcm and afterwards sonicated in ethanol for 30 min to remove any remaining polishing residues. The polished sample was next etched in a boiling equivolume mixture of deionized water and HCl (36.5 wt.%) for 30 min and then dried with argon gas (purity 99.998 wt.%, MEGS Specialty Gases Inc., St-Laurent, QC, Canada) [38].
A coating precursor solution of 0.5 M concentration was prepared by dissolving appropriate amounts of NiCl2·6H2O (ReagentPlus, 100%, Sigma Aldrich, St. Louis, MO, USA) and Co(NO3)2·6H2O (99% pure, ACROS Organics, Bridgewater, NJ, USA) in an equivolume mixture of isopropanol (Fisher Scientific, Nepean, ON, Canada) and deionized water to yield a coating of relative Ni/Co atomic ratio of 3:2. The surface characterization of the oxide film by energy-dispersive X-ray spectroscopic (EDX) analysis showed that the oxide is composed of 60.8 ± 0.8 atomic percentage of Ni and the rest is Co, which is very close to the nominal composition of the oxide [28]. Additionally, EDX was used to analyze the elemental composition of the dried residue obtained from the leftover electrolyte after degradation testing. The Ni-Co-oxide coating was formed by brush-painting the precursor salt solution onto one side of the pretreated Ti substrate. This was followed by a 5 min drying in an oven at 383 K to vaporize the solvent and then calcination for 15 min in a furnace at 773 K. The coated surface was cooled down to room temperature in 20 min. Subsequently, the coating process was repeated six times to form a seven-layered coating on the Ti surface. Finally, the sample was annealed for 2 h at 773 K to form a uniform binary metal oxide coating. X-ray diffraction (XRD) analysis was used to characterize the coatings’ crystalline oxide phases, as presented in our previous work [28]. The uncoated side of the Ni0.6Co0.4-oxide/Ti sample was covered with a waterproof, electrically insulating film (3M Electrical Tape, Ruban isolant, St. Paul, MN, USA) while the oxide-covered side served as the working electrode. All experiments presented in the manuscript were run at least in duplicate to ensure the reliability of the data.
3.2. Electrochemical Degradation of Methylene Blue
The methylene blue degradation studies were conducted in a one-compartment, two-electrode electrochemical cell at atmospheric pressure and temperature of 295 ± 2 K. The electrochemical cell comprised of the Ni0.6Co0.4-oxide working electrode (anode), described above, and a 50 mm × 53 mm × 2 mm flat stainless steel (316L) counter electrode (cathode). The vertically suspended electrodes were kept ca. 1 cm apart and connected to a power supply that operated in a constant current mode during the experiment. The electrolyte was a 50 mg/L methylene blue (MB) solution in 0.17 M sodium sulfate (≥99% pure, Fisher Scientific, Nepean, ON, Canada) of pH 5.8, prepared from a stock solution of 500 mg/L MB (Certified biological stain, Fisher Chemical, Nepean, ON, Canada). A total of 25 cm2 geometric area of the working electrode was exposed to the 250 mL of electrolyte. The solution was stirred by a magnetic stirring bar at 300 rpm to ensure adequate mixing. Aliquots of 500 μL were taken from the treated solution at selected time intervals for analysis. At each sampling time, the corresponding potential difference between the electrodes was recorded, and the electrolyte temperature at the start and end of the experiment was also recorded. The collected aliquots were then diluted in a cuvette with a solution of 0.17 M sodium sulfate by a requisite factor for analysis. It should be noted that the determination of MB concentration in kinetic experiments was carried out immediately (1 min) after taking the aliquot.
The observed electrolyte color removal, i.e., the MB degradation rate, was monitored over time by measuring the reduction in the peak absorbance of MB at 660 nm wavelength employing UV/Vis spectrophotometry (ThermoScientific Evolution 300, Nepean, ON, Canada), and the scanning range was from 380–780 nm. The measured absorbance value was correlated to the concentration of MB using a linear calibration curve (R2 = 0.996). The electrochemical degradation of the methylene blue was studied at different constant current densities of 10, 20, 40, and 60 mA/cm2.
The effect of chloride ion concentration on the electrochemical degradation performance of Ni0.6Co0.4-oxide for the decontamination of MB (50 mg/L MB in 0.17 M Na2SO4) was investigated at a current density of 20 mA/cm2 and at NaCl concentrations of 0.25, 0.5, 1.0, and 2.0 g/L. Note that the relatively low concentrations of chlorides selected is comparatively lower than the concentration of chlorides in seawater (35 g/L). To confirm the role of electrochemically generated oxidants such as hydroxyl radical and active chlorine species during the MB degradation process, methanol (Fisher Scientific, Nepean, ON, Canada) and dopamine chloride (100% pure, PubChem) were added to the reaction vessel as scavengers, respectively.
Chemical oxygen demand (COD) and total organic carbon (TOC) measurements were also performed to determine the level of mineralization obtained during the electrochemical degradation of MB. The COD measurements were performed using a low range (0–150 mg/L) COD digestion vials (K-7355, CHEMetrics, Calverton, VA, USA), COD reactor (HACH, DRB 200) and a HACH spectrophotometer (DR/2500). The HACH method used is based on the ASTM D 1252-95 standard. The TOC test was performed using a Rosemount DC-80 Total Organic Carbon Analyzer and the EPA Method 415.2. All the measurements were carried out in duplicates and the presented data are the average values with the accompanying mean standard deviations.
4. Conclusions
The Ni0.6Co0.4-oxide was shown to be a promising anode material for the electrochemical degradation of aqueous organic contaminants, represented here by methylene blue (MB). The MB removal followed a first-order kinetics while the mineralization conformed to an approximate 1.9th order kinetics. The kinetics of MB degradation was found to be largely dependent on the current density passing through the cell and was found to increase with an increase in current density. It was also observed that the increase in the current density was invested predominantly in improving the kinetics of the MB degradation reaction, rather than in the competing oxygen evolution reaction, making the Ni0.6Co0.4-oxide a suitable candidate for the degradation of organic compounds in aqueous solutions. The presence of chloride ions in the electrolyte was found to significantly improve the kinetics of MB degradation, which was due to the formation of electrogenerated active chlorine species. Although the results of the present study are promising, they were based on the use of a laboratory-scale batch electrochemical reactor and a simple wastewater matrix containing only one organic compound (methylene blue). Further studies are needed to examine the performance of the anode material in larger-scale electrochemical wastewater treatment reactors (batch and flow-through) and using more complex wastewater matrices such as synthetic and real wastewaters.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/catal11070793/s1, Table S1: Apparent reaction rate constants, Table S2: Apparent reaction rate constants, Figure S1: Dependence of the rate constant on the current density, Figure S2: Dependence of the rate constant on the concentration, Table S3: Apparent reaction rate constants, Figure S3: Dependence of the rate constant on the current density, Figure S4: Dependence of COD on time, Figure S5: Dependence of TOC on time.
Author Contributions
Conceptualization, E.O.N. and S.O.; investigation, E.O.N., X.L. and E.P.; methodology, E.O.N.; resources, V.Y. and S.O.; supervision, S.O.; validation, E.O.N. and X.L.; visualization, E.O.N., V.Y. and S.O.; writing—original draft, E.O.N. and X.L.; writing—review and editing, E.O.N., E.P., V.Y. and S.O. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are indebted to the Nigerian Petroleum Technology Development Fund (PTDF), Natural Sciences and Engineering Research Council of Canada (NSERC), and the McGill Engineering Doctoral Award (MEDA) for the financial support.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Acknowledgments
Our immense gratitude also goes to Ranjan Roy and Andrew Golsztajn for their technical input for the TOC and UV/Vis spectrophotometric measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mathur, N.; Bhatnagar, P.; Bakre, P. Assessing mutagenicity of textile dyes from Pali(Rajasthan) using Ames bioassay. Appl. Ecol. Environ. Res. 2006, 4, 111–118. [Google Scholar] [CrossRef]
- Indu, M.; Gupta, A.; Sahoo, C. Electrochemical oxidation of methylene blue using lead acid battery anode. APCBEE Procedia 2014, 9, 70–74. [Google Scholar] [CrossRef]
- Panizza, M.; Barbucci, A.; Ricotti, R.; Cerisola, G. Electrochemical degradation of methylene blue. Sep. Purif. Technol. 2007, 54, 382–387. [Google Scholar] [CrossRef]
- Kraft, A. Electrochemical water disinfection: A short review. Platin. Metals Rev. 2008, 52, 177–185. [Google Scholar] [CrossRef]
- Anglada, A.; Urtiaga, A.; Ortiz, I. Contributions of electrochemical oxidation to waste-water treatment: Fundamentals and review of applications. J. Chem. Technol. Biotechnol. 2009, 84, 1747–1755. [Google Scholar] [CrossRef]
- Ghasemian, S.; Omanovic, S. Fabrication and characterization of photoelectrochemically-active Sb-doped Snx-W(100-x)%-oxide anodes: Towards the removal of organic pollutants from wastewater. Appl. Surf. Sci. 2017, 416, 318–328. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, R. Advanced oxidation processes (AOPs) in wastewater treatment. Curr. Pollut. Rep. 2015, 1, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Dutta, K.; Mukhopadhyay, S.; Bhattacharjee, S.; Chaudhuri, B. Chemical oxidation of methylene blue using a Fenton-like reaction. J. Hazard. Mater. 2001, 84, 57–71. [Google Scholar] [CrossRef]
- Gottschalk, C.; Libra, J.A.; Saupe, A. Ozonation of Water and Waste Water: A Practical Guide to Understanding Ozone and Its Applications, 2nd ed.; John Wiley & Sons GmbH: Weinheim, Germany, 2010; pp. 46–55. [Google Scholar]
- Ghasemian, S.; Nasuhoglu, D.; Omanovic, S.; Yargeau, V. Photoelectrocatalytic degradation of pharmaceutical carbamazepine using Sb-doped Sn80%-W20%-oxide electrodes. Sep. Purif. Technol. 2017, 188, 52–59. [Google Scholar] [CrossRef]
- Lei, J.; Duan, P.; Liu, W.; Sun, Z.; Hu, X. Degradation of aqueous cefotaxime in electro-oxidation—electro-Fenton—persulfate system with Ti/CNT/SnO2–Sb–Er anode and Ni@ NCNT cathode. Chemosphere 2020, 250, 126163. [Google Scholar] [CrossRef]
- Zhao, W.; Xing, J.; Chen, D.; Jin, D.; Shen, J. Electrochemical degradation of Musk ketone in aqueous solutions using a novel porous Ti/SnO2-Sb2O3/PbO2 electrodes. J. Electroanal. Chem. 2016, 775, 179–188. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, L.; Liu, J.; Logan, B.E. Electrochemical technologies for wastewater treatment and resource reclamation. Environ. Sci. Water Res. Technol. 2016, 2, 800–831. [Google Scholar] [CrossRef]
- Ghasemian, S.; Asadishad, B.; Omanovic, S.; Tufenkji, N. Electrochemical disinfection of bacteria-laden water using antimony-doped tin-tungsten-oxide electrodes. Water Res. 2017, 126, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Lo, S.L.; Srivastava, V.C.; Hiwarkar, A.D. Comparative study of electrochemical oxidation for dye degradation: Parametric optimization and mechanism identification. J. Environ. Chem. Eng. 2016, 4, 2911–2921. [Google Scholar] [CrossRef]
- Gattrell, M.; Kirk, D. A study of the oxidation of phenol at platinum and preoxidized platinum surfaces. J. Electrochem. Soc. 1993, 140, 1534. [Google Scholar] [CrossRef]
- Chatzisymeon, E.; Dimou, A.; Mantzavinos, D.; Katsaounis, A. Electrochemical oxidation of model compounds and olive mill wastewater over DSA electrodes: 1. The case of Ti/IrO2 anode. J. Hazard. Mater. 2009, 167, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Comninellis, C. Electrocatalysis in the electrochemical conversion/combustion of organic pollutants for waste water treatment. Electrochim. Acta 1994, 39, 1857–1862. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Application of diamond electrodes to electrochemical processes. Electrochim. Acta 2005, 51, 191–199. [Google Scholar] [CrossRef]
- Lacasa, E.; Tsolaki, E.; Sbokou, Z.; Rodrigo, M.A.; Mantzavinos, D.; Diamadopoulos, E. Electrochemical disinfection of simulated ballast water on conductive diamond electrodes. Chem. Eng. J. 2013, 223, 516–523. [Google Scholar] [CrossRef]
- Katsoni, A.; Mantzavinos, D.; Diamadopoulos, E. Sequential treatment of diluted olive pomace leachate by digestion in a pilot scale UASB reactor and BDD electrochemical oxidation. Water Res. 2014, 57, 76–86. [Google Scholar] [CrossRef]
- Solano, A.M.S.; de Araújo, C.K.C.; de Melo, J.V.; Peralta-Hernandez, J.M.; da Silva, D.R.; Martínez-Huitle, C.A. Decontamination of real textile industrial effluent by strong oxidant species electrogenerated on diamond electrode: Viability and disadvantages of this electrochemical technology. Appl. Catal. B Environ. 2013, 130, 112–120. [Google Scholar] [CrossRef]
- Zhou, M.; Dai, Q.; Lei, L.; Ma, C.A.; Wang, D. Long life modified lead dioxide anode for organic wastewater treatment: Electrochemical characteristics and degradation mechanism. Environ. Sci. Technol. 2005, 39, 363–370. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, T.; Su, Q.; Tang, Y.; Xu, X.; Akram, M.; Jiang, B. Highly efficient and mild electrochemical degradation of bentazon by nano-diamond doped PbO2 anode with reduced Ti nanotube as the interlayer. J. Colloid Interface Sci. 2020, 575, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Polcaro, A.; Palmas, S.; Renoldi, F.; Mascia, M. On the performance of Ti/SnO2 and Ti/PbO2 anodesin electrochemical degradation of 2-chlorophenolfor wastewater treatment. J. Appl. Electrochem. 1999, 29, 147–151. [Google Scholar] [CrossRef]
- Wu, T.; Zhao, G.; Lei, Y.; Li, P. Distinctive tin dioxide anode fabricated by pulse electrodeposition: High oxygen evolution potential and efficient electrochemical degradation of fluorobenzene. J. Phys. Chem. C 2011, 115, 3888–3898. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, X.; Miao, J.; Zhang, R.; Zhang, J.; Zhou, M.; Lu, W. Enhanced performance of an Al-doped SnO2 anode for the electrocatalytic oxidation of organic pollutants in water. Mater. Today Commun. 2020, 24, 101164. [Google Scholar] [CrossRef]
- Nwanebu, E.O.; Omanovic, S. The influence of NixCo1-x-oxide composition on its electrocatalytic activity in the oxygen evolution reaction. Mater. Chem. Phys. 2019, 228, 80–88. [Google Scholar] [CrossRef]
- Morgounova, E.; Shao, Q.; Hackel, B.J.; Thomas, D.D.; Ashkenazi, S. Photoacoustic lifetime contrast between methylene blue monomers and self-quenched dimers as a model for dual-labeled activatable probes. J. Biomed. Opt. 2013, 18, 056004. [Google Scholar] [CrossRef] [Green Version]
- Usacheva, M.N.; Teichert, M.C.; Biel, M.A. The role of the methylene blue and toluidine blue monomers and dimers in the photoinactivation of bacteria. J. Photochem. Photobiol. B Biol. 2003, 71, 87–98. [Google Scholar] [CrossRef]
- Spencer, W.; Sutter, J.R. Kinetic study of the monomer-dimer equilibrium of methylene blue in aqueous solution. J. Phys. Chem. 1979, 83, 1573–1576. [Google Scholar] [CrossRef]
- Chung, S.-K.; Toshihiko, O. Hydroxyl radical scavengers from white mustard (Sinapis alba). Food Sci. Biotechnol. 1998, 7, 209–213. [Google Scholar]
- Mishra, O.P.; Popov, A.V.; Pietrofesa, R.A.; Christofidou-Solomidou, M. Gamma-irradiation produces active chlorine species (ACS) in physiological solutions: Secoisolariciresinol diglucoside (SDG) scavenges ACS-A novel mechanism of DNA radioprotection. Biochim. Biophys. Acta 2016, 1860, 1884–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Ghalwa, N.M.; Zaggout, F.R. Electrodegradation of methylene blue dye in water and wastewater using lead oxide/titanium modified electrode. J. Environ. Sci. Health A 2006, 41, 2271–2282. [Google Scholar] [CrossRef]
- Samarghandi, M.R.; Dargahi, A.; Shabanloo, A.; Nasab, H.Z.; Vaziri, Y.; Ansari, A. Electrochemical degradation of methylene blue dye using a graphite doped PbO2 anode: Optimization of operational parameters, degradation pathway and improving the biodegradability of textile wastewater. Arab. J. Chem. 2020, 13, 6847–6864. [Google Scholar] [CrossRef]
- Comninellis, C.; Pulgarin, C. Electrochemical oxidation of phenol for wastewater treatment using SnO2 anodes. J. Appl. Electrochem. 1993, 23, 108–112. [Google Scholar] [CrossRef]
- Zeng, L.; Wei, X.; Miao, J.; Zhang, R.; Zhang, J.; Zhou, M.; Lu, W. Preparation and investigation of a Ni–B-assisted SnO2–Sb anode for electrooxidation of phenol. J. Solid State Electrochem. 2021, 25, 1541–1553. [Google Scholar] [CrossRef]
- Nwanebu, E.O.; Yao, Y.; Omanovic, S. The Influence of Ir Content in (Ni0.4Co0.6)1-xIrx-oxide Anodes on their Electrocatalytic Activity in Oxygen Evolution by Acidic and Alkaline Water Electrolysis. J. Electroanal. Chem. 2020, 865, 114122. [Google Scholar] [CrossRef]
Figure 1.
The variation of absorbance of MB during its electrochemical degradation on the Ni0.6Co0.4-oxide anode electrode in 0.17 M Na2SO4 aqueous solution containing 50 mg/L of MB at current density of 20 mA/cm2. The inset shows the change of 660 nm peak intensity with treatment time.
Figure 1.
The variation of absorbance of MB during its electrochemical degradation on the Ni0.6Co0.4-oxide anode electrode in 0.17 M Na2SO4 aqueous solution containing 50 mg/L of MB at current density of 20 mA/cm2. The inset shows the change of 660 nm peak intensity with treatment time.
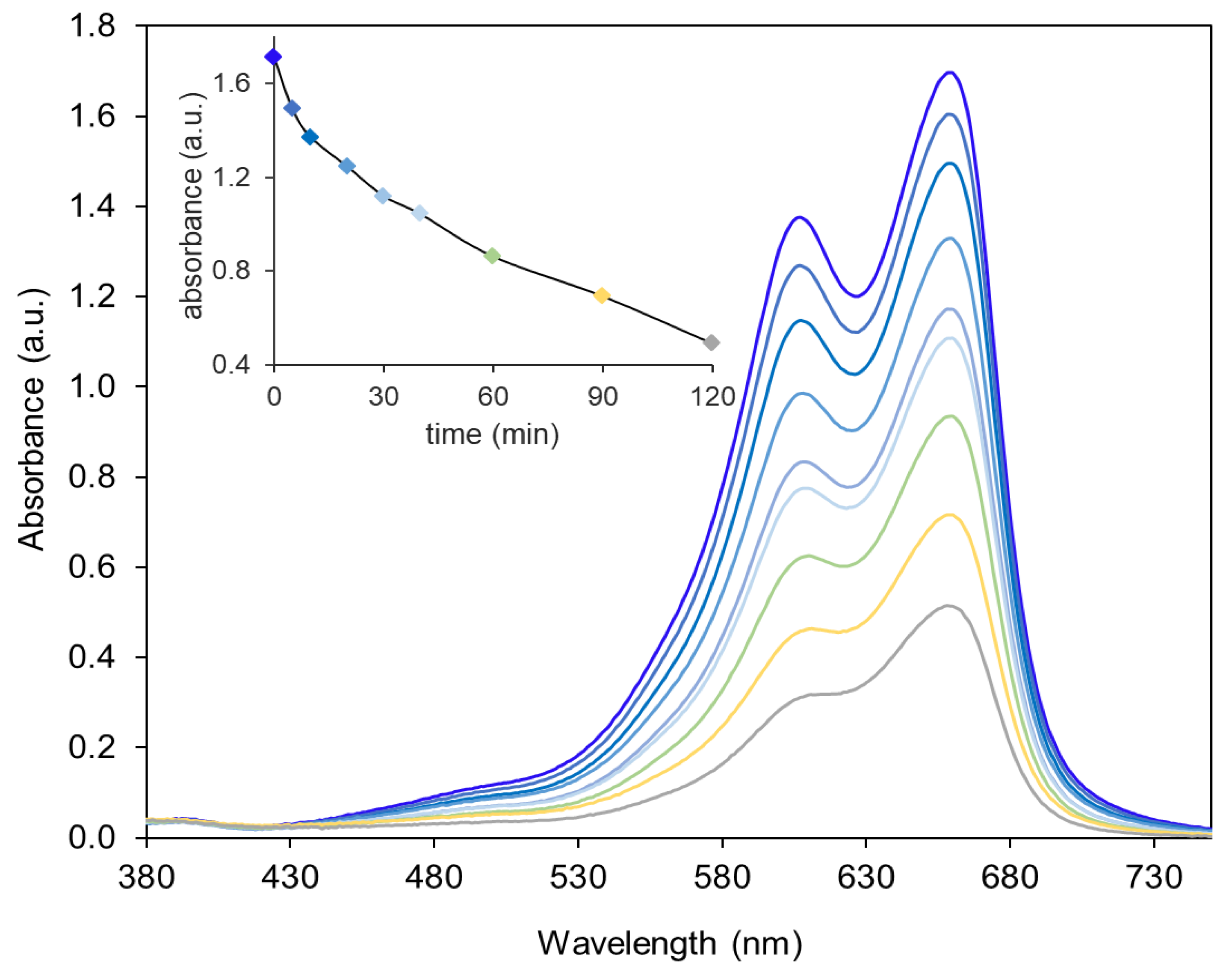
Figure 2.
(A) Electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode recorded at different current densities and as a function of time; (B) Fraction of the initial MB degraded as a function of charge passed through the anode after (●) 60, (■) 90 and (♦) 120 min of degradation. Electrolyte: 0.17 M Na2SO4 aqueous solution; (C) Effect of the presence of 0.2 M methanol in the electrolyte on the degradation of MB at 20 mA/cm2.
Figure 2.
(A) Electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode recorded at different current densities and as a function of time; (B) Fraction of the initial MB degraded as a function of charge passed through the anode after (●) 60, (■) 90 and (♦) 120 min of degradation. Electrolyte: 0.17 M Na2SO4 aqueous solution; (C) Effect of the presence of 0.2 M methanol in the electrolyte on the degradation of MB at 20 mA/cm2.
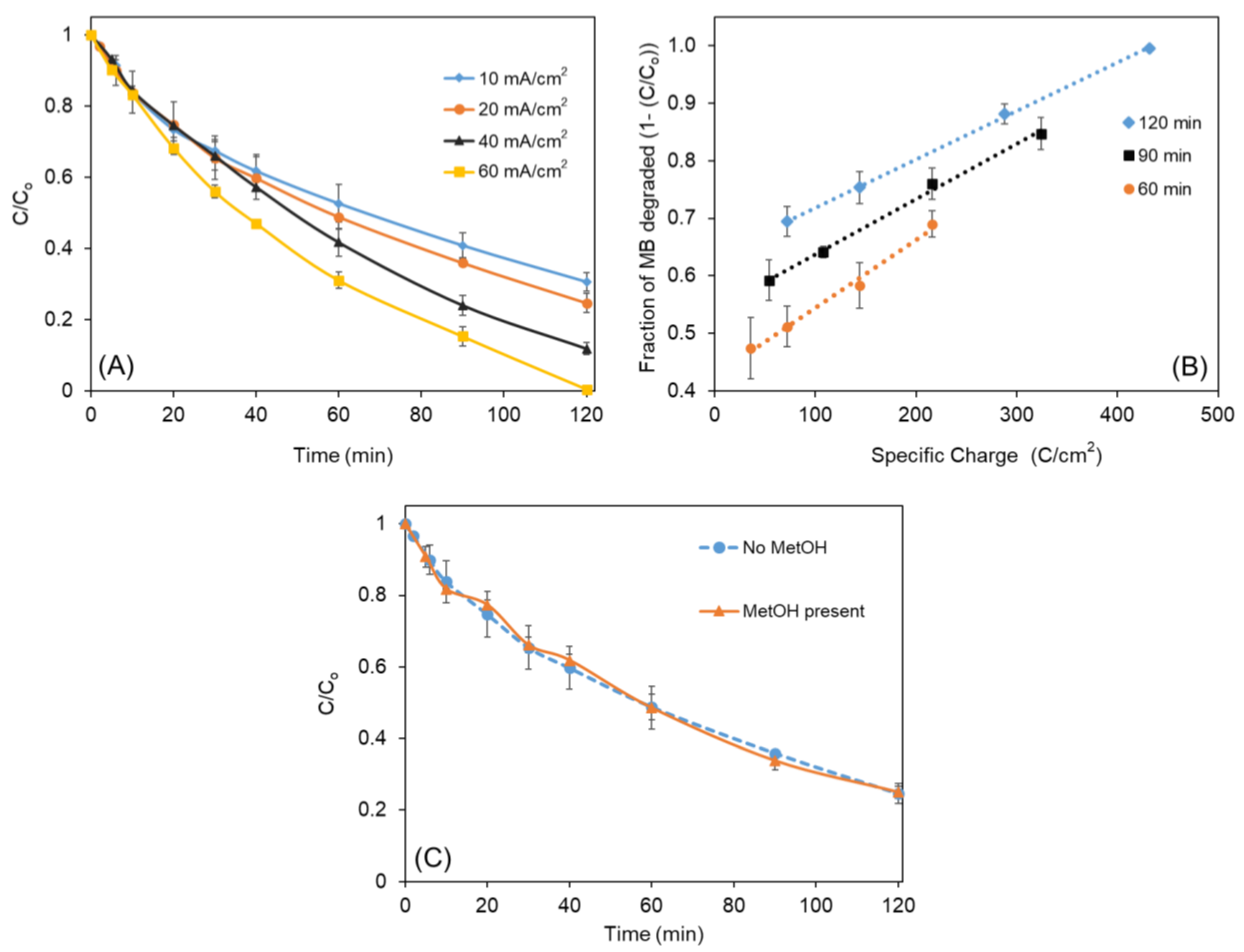
Figure 3.
(A) Influence of NaCl concentration in 0.17 M Na2SO4 aqueous solution on the electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode at 20 mA/cm2; (B) Effect of addition of 40 mM dopamine hydrochloride into the electrolyte at the 20th minute on the kinetics of degradation of MB at 10 mA/cm2 in the case when the current supply to the cell was and was not terminated. In the control experiment, no dopamine hydrochloride was present in the electrolyte, but the current supply to the cell was terminated at the 20th minute.
Figure 3.
(A) Influence of NaCl concentration in 0.17 M Na2SO4 aqueous solution on the electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode at 20 mA/cm2; (B) Effect of addition of 40 mM dopamine hydrochloride into the electrolyte at the 20th minute on the kinetics of degradation of MB at 10 mA/cm2 in the case when the current supply to the cell was and was not terminated. In the control experiment, no dopamine hydrochloride was present in the electrolyte, but the current supply to the cell was terminated at the 20th minute.

Figure 4.
Kinetics of degradation of MB (50 mg/L) in 0.17 M Na2SO4 aqueous electrolyte containing 2 g/L NaCl at various current densities.
Figure 4.
Kinetics of degradation of MB (50 mg/L) in 0.17 M Na2SO4 aqueous electrolyte containing 2 g/L NaCl at various current densities.

Figure 5.
Variation of COD, TOC and color removal as a function of time during the electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode at 20 mA/cm2 in a 0.17 M Na2SO4 aqueous solution containing 2 g/L NaCl salt. The initial (time zero) COD and TOC values were ca. 63 mg/L and 26.5 mg/L, respectively. The curves presented are the representative plots of the corresponding duplicate experiments.
Figure 5.
Variation of COD, TOC and color removal as a function of time during the electrochemical degradation of MB (50 mg/L) on the Ni0.6Co0.4-oxide anode at 20 mA/cm2 in a 0.17 M Na2SO4 aqueous solution containing 2 g/L NaCl salt. The initial (time zero) COD and TOC values were ca. 63 mg/L and 26.5 mg/L, respectively. The curves presented are the representative plots of the corresponding duplicate experiments.

Figure 6.
Voltage difference between the Ni0.6Co0.4-oxide anode and stainless steel cathode during the electrochemical degradation of 1g/L MB in 0.17 M Na2SO4 at a current density of 20 mA/cm2.
Figure 6.
Voltage difference between the Ni0.6Co0.4-oxide anode and stainless steel cathode during the electrochemical degradation of 1g/L MB in 0.17 M Na2SO4 at a current density of 20 mA/cm2.
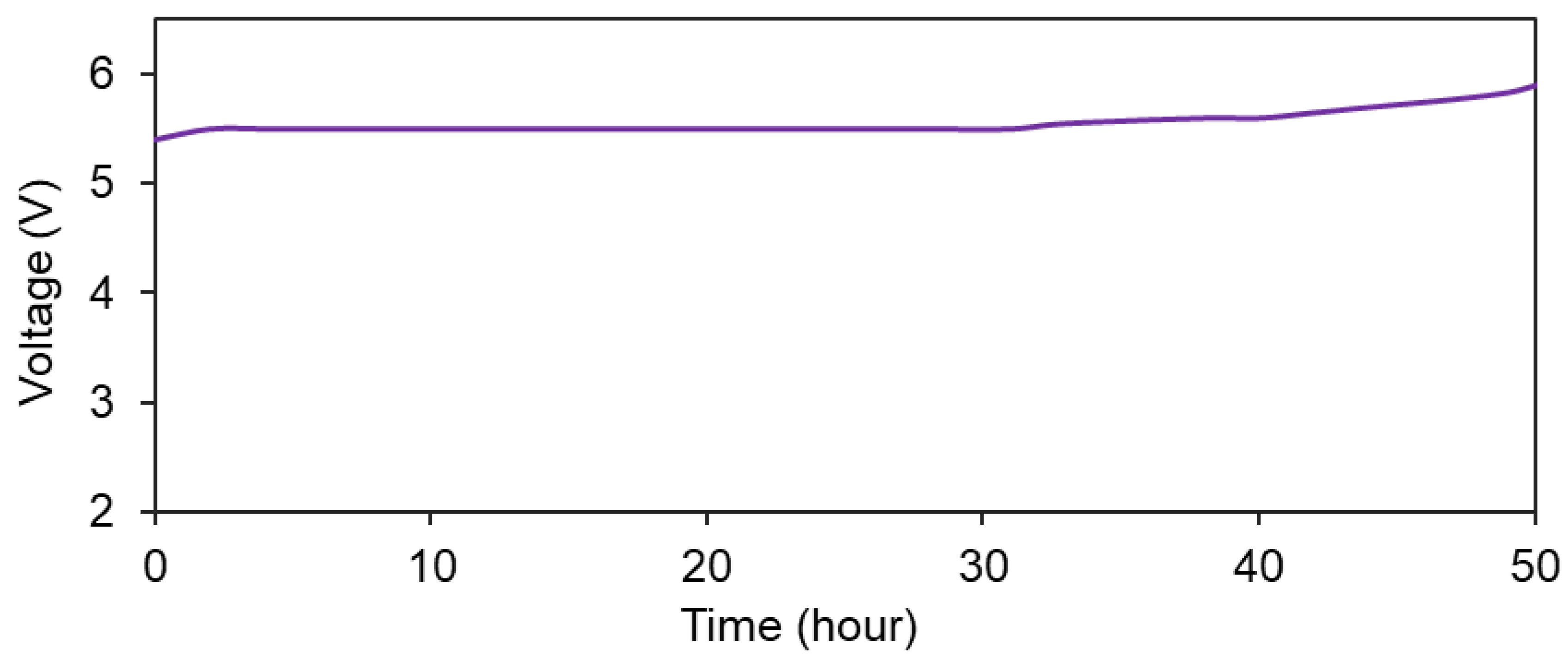
Figure 7.
Energy consumption required for 100% MB (50 mg/L) degradation in 0.17 M Na2SO4 + 2 g/L NaCl (EEC_A) and for 70% MB (50 mg/L) degradation in 0.17 M Na2SO4 in the absence of Cl– ions (EEC_B) in a current density range of 10–60 mA/cm2.
Figure 7.
Energy consumption required for 100% MB (50 mg/L) degradation in 0.17 M Na2SO4 + 2 g/L NaCl (EEC_A) and for 70% MB (50 mg/L) degradation in 0.17 M Na2SO4 in the absence of Cl– ions (EEC_B) in a current density range of 10–60 mA/cm2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nwanebu, E.O.; Liu, X.; Pajootan, E.; Yargeau, V.; Omanovic, S. Electrochemical Degradation of Methylene Blue Using a Ni-Co-Oxide Anode. Catalysts 2021, 11, 793. https://doi.org/10.3390/catal11070793
AMA Style
Nwanebu EO, Liu X, Pajootan E, Yargeau V, Omanovic S. Electrochemical Degradation of Methylene Blue Using a Ni-Co-Oxide Anode. Catalysts. 2021; 11(7):793. https://doi.org/10.3390/catal11070793
Chicago/Turabian StyleNwanebu, Emmanuel Onyekachi, Xiaocheng Liu, Elmira Pajootan, Viviane Yargeau, and Sasha Omanovic. 2021. "Electrochemical Degradation of Methylene Blue Using a Ni-Co-Oxide Anode" Catalysts 11, no. 7: 793. https://doi.org/10.3390/catal11070793
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.