Reaction Mechanism for Methane-to-Methanol in Cu-SSZ-13: First-Principles Study of the Z2[Cu2O] and Z2[Cu2OH] Motifs


1
Department of Physics, Chalmers University of Technology, 412 96 Göteborg, Sweden
2
Competence Center for Catalysis, Chalmers University of Technology, 412 96 Göteborg, Sweden
*
Authors to whom correspondence should be addressed.
Catalysts 2021, 11(1), 17; https://doi.org/10.3390/catal11010017
Submission received: 15 November 2020
/
Revised: 9 December 2020
/
Accepted: 15 December 2020
/
Published: 25 December 2020
(This article belongs to the Special Issue Catalytic Oxidation of Hydrocarbons)
Abstract
:As transportation continues to increase world-wide, there is a need for more efficient utilization of fossil fuel. One possibility is direct conversion of the solution gas bi-product CH into an energy-rich, easily usable liquid fuel such as CHOH. However, new catalytic materials to facilitate the methane-to-methanol reaction are needed. Using density functional calculations, the partial oxidation of methane is investigated over the small-pore copper-exchanged zeolite SSZ-13. The reaction pathway is identified and the energy landscape elucidated over the proposed motifs Z[CuO] and Z[CuOH]. It is shown that the Z[CuO] motif has an exergonic reaction path, provided water is added as a solvent for the desorption step. However, a micro-kinetic model shows that neither Z[CuO] nor Z[CuOH] has any notable activity under the reaction conditions. These findings highlight the importance of the detailed structure of the active site and that the most stable motif is not necessarily the most active.
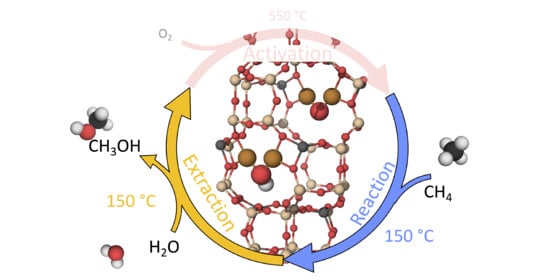
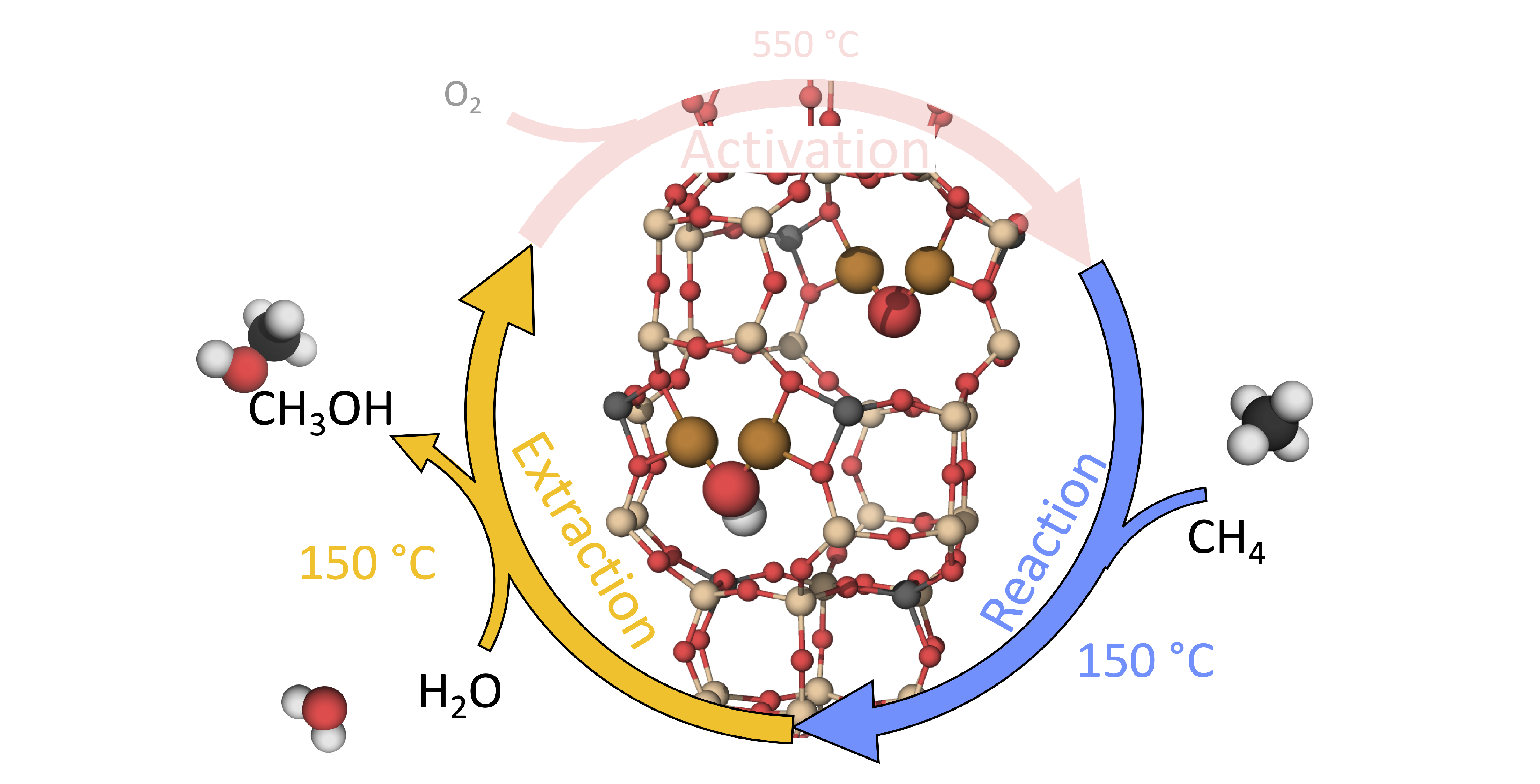
1. Introduction
The growing need for transportation makes a more efficient usage of combustion fuel necessary [1]. Methane, which is extracted together with crude oil, is a not fully utilized bi-product. As methane is a gas at standard conditions with high sunlight absorption properties, methane is often flared into CO instead of being utilized [2]. Such a scenario can be avoided if it were possible to directly convert methane into methanol, which is a liquid at standard conditions. The management and distribution system is already in place to handle liquid fuels. The current method for methane-to-methanol (MTM) conversion is based on a large-scale two step process operating at high temperature and high pressure [3]. A major development would be a catalytic material that can transform methane into methanol in one step (direct methane-to-methanol (DMTM)) on a small scale, at low temperature and standard pressure.
The search for a catalyst for the partial oxidation of MTM has been the topic of many studies [4,5,6,7]. One line of research has been to mimic the porous structure and ionic metal sites of the naturally occurring enzymatic methane monooxygenases (MMOs) [8], which has been accomplished by metal ion-exchanged zeolites.
The basic building-blocks of zeolites are corner-sharing SiO and AlO tetrahedrons, with Si and Al tetrahedrally coordinated (T-site). A porous framework with pore sizes ranging from two to 12 membered rings (MRs) is formed by connecting tetrahedrons. One important characteristic of the zeolite system is the Si:Al ratio, which is commonly in the range of 5–20:1.
Experimentally, direct conversion of MTM over zeolites has been observed using a quasi-catalytic three step process: activation of the oxidant (O, NO or NO) at high temperature (>350 C); reaction phase at 50–210 C and with the addition of CH; extraction at 25–210 C, where the reactants are flushed out using water/ethanol or some other solvent [9,10]. For instance, studies of the MFI zeolite ZSM-5 have shown catalytic activity for DMTM conversion, with the cation system of Cu-ZSM-5 being the most active for conversion [11]. However, the detailed configuration of the active site is still under debate [12,13,14]. An additional use of Cu-ZSM-5 is as a catalyst for the selective catalytic reduction reaction (SCR) of NO using NH as the reducing agent [15]. However, the NH-SCR reaction is preferably performed over the small-pore chabazite system of Cu-SSZ-13 [16]. It is, thus, interesting to investigate whether Cu-SSZ-13 also would show activity for partial oxidation of methane [17,18,19,20].
Over the CuO motif in Cu-ZSM-5, the reaction mechanism for direct conversion has been shown to occur via a three step reaction path: activation of the C–H bond in CH over the active site and a rotation of the reactant, which leads to the formation of the CHOH-complex [11]. The active site in Cu-SSZ-13 has been suggested to be CuOH [21,22]. The proposed reaction mechanism [21] for CuOH is more complex than for CuO in MFI. Direct insertion of a methyl radical can be done either with a possible two step reaction path or a five (alternatively six) step reaction path. The longer path shows an energy profile with low barriers. Furthermore, including water in the reaction path makes methanol formation energetically favourable [21]. However, the presence of the CuOH motif has also been explained as it is a possible precursor for the formation of either CuO or CuO [23]. Larger Cu clusters (Cu > 2), namely CuO [24,25], have also been suggested as the active motif. The trimer structure has been shown to decrease the energy barrier for the activation of the C–H bond when compared with the smaller Cu monomer and dimer [21]. However, only large pore zeolites such as MFI and mordenite (MOR), are believed to contain these large clusters; in small pore zeolites, they have been shown to be unstable [22].
It has been shown that the active site motif is dynamic and depends sensitively on the operating conditions, such as the temperature and partial pressure of relevant gases [26,27]. Previous work [27] reports that for a quasi-catalytic reaction cycle in the zeolite Cu-SSZ-13 (with Si:Al = 10:2 and Al:Cu = 2:2, in the 12 T-site unit cell), the CuOH motif, as seen in Figure 1a, is the energetically preferred structure for low temperature and low partial pressure of oxygen, while for high temperature and high partial pressure of oxygen, the CuO motif, as can be seen in Figure 1b, is preferred. However, the reaction mechanism and character of the intermediates are not yet known.
In this study, we use first-principles calculations to determine the reaction mechanism for the partial methane oxidation over the active site motifs of CuO and CuOH in the small-pore zeolite SSZ-13. As water is often included as a solvent in the experimental procedure, its influence on the reaction mechanism is investigated in detail, and a micro-kinetic modelling is used to explore the activity of the considered sites.
2. Results and Discussion
The reaction mechanism is elucidated for the reaction and the extraction phase of the DMTM reaction. This is experimentally done at 448 K and flow reactor conditions with CH and CHOH with respect to atmospheric pressure [10]. Under these conditions, previous work has identified Z[CuO] and Z[CuOH], with the active sites situated in the 8MR in the chabazite (CHA) (see Figure 1a,b), as the most stable active site motifs [27].
In the description of the intermediate steps in the reaction mechanism, the two copper atoms of the active site are denoted as *. Hence, *X,Y implies that reaction intermediate X is coordinated to both Cu atoms, forming the current state of the active site, while reaction intermediate Y is either adsorbed on the active site or free in the zeolite.
The structural properties of gas phase HO, CH and CHOH are in good agreement with the experimental values [28]; for details, see Table S1.
2.1. Reaction Mechanism over Z[CuO]
The identified reaction mechanisms for DMTM at 448 K over Z[CuO] in SSZ-13 are presented in Figure 2, where the difference in the Gibbs free energy of two different mechanisms, with and without water, is compared (total energies for the mechanism can be found in Figure S2).
Starting with an empty zeolite, the free energy cost of moving one methane molecule into the zeolite framework is 0.28 eV. The first transition state (TS) is CH dissociation, suggested to occur via a CH radical [29]. Here, the methyl structure was found by constrained geometry optimizations and proved to be a saddle point through vibrational analysis and is shown as TS1 in Figure 2.
Passing through the methyl radical, with a barrier of 1.12 eV, the first intermediate step, i.e., *OH, CH is an OH-group coordinated to both Cu atoms at a distance of 1.86/1.87 Å and a planar CH at a distance of 2.08/2.07 Å. This configuration is 0.25 eV more stable than if CH attaches to the framework. The second TS shows the carbon atom coordinating to one Cu atom at a distance of 2.15 Å and the OH-group lying in the plane of the 8MR. Under dry conditions (p = 2%, p = 10%), the barrier to form an attached *CHOH is 0.84 eV, where the oxygen in the methanol bridges both Cu atoms at a distance of 1.98/1.99 Å. The subsequent desorption energy of the CHOH molecule is 0.73 eV. The empty site after desorption is a Z[Cu] structure with an interatomic distance of 2.46 Å, which is 0.07 Å larger than the Cu–Cu distance in the Z[CuO] structure. The energy is 1.84 eV higher than the most stable Z[Cu] structure, where the copper atoms are situated in the 6MRs. This is clearly seen in the reactivity of the final structure. For instance, the adsorption energy of oxygen in the final structure, with the copper atoms residing in the 8MR as a reference, is −2.55 eV, as compared to −0.71 eV if using the most stable configuration as the reference.
The complete reaction under dry conditions, as marked by red squares in Figure 2, is exergonic by −1.02 eV. If water is added to the reaction mechanism after TS2, the methanol desorption becomes exergonic by −1.81 eV. This confirms the need for a solvent during the extraction phase [9,30]. The most exergonic reaction pathway ends in a Z[CuHO] structure with a Cu–Cu distance of 2.43 Å, Cu–O distances of 1.96/1.97 Å and a Cu–O–Cu angle of 76.3. This is stretched and more acute as compared to the Z[CuO] structure of the original active site, where the distances are 2.39 Å and 1.74/1.74 Å and the angle 86, respectively. In the energetic landscape, the relative energies of the reaction path show a decrease in energy when water is added (see Figure S2).
2.2. Reaction Mechanism over Z[CuOH]
The Gibbs free energy along the reaction path under dry conditions, over the Z [CuOH]-site, is reduced to a one step reaction, marked by purple diamonds in Figure 3. The addition of water (blue crosses in Figure 3) to the mechanism adds one intermediate step and makes the reaction less exergonic. Total energies for the reaction path can be found in Figure S5.
Starting with an empty zeolite, *OH, the introduction of methane into the zeolite is exergonic at −0.38 eV (treating the entropy of methane in the harmonic approximation increases the cost to 0.08 eV; see Figure S7).As in the Z[CuO] system, the methyl radical is found to have the lowest TS energy for CH dissociation with a barrier of 2.65 eV.
Following the methyl radical along the dry path, the second intermediate (*H,CHOH) is 1.66 eV higher in energy than the first intermediate (*OH,CH). Methanol desorption is 2.30 eV lower in energy than the CHOH formation step in the reaction, and as a whole, the reaction is exergonic with 1.02 eV. The empty site after CHOH desorption is a Cu–H–Cu complex with a Cu–Cu distance of 2.34 Å, Cu–H bonds of 1.60/1.59 Å and a Cu–H–Cuangle of 95, displaying shorter bonds and a more obtuse angle when compared to the Cu–OH–Cu structure, where the distances are 2.50 Å and 1.82/1.82 Å and the angle 86.
All intermediates in the reaction are found to be doublets. The energy difference between the lowest and second lowest spin states is found in Table 1. A Bader analysis shows a charge of approximately 0.88/0.90 on the Cu atoms for all but the final state, *H, where the charges instead are calculated to be 0.95/0.95.
There is an additional path to the direct formation of CHOH on the Z[CuOH] structure. After the methyl TS1, an intermediate with a 0.18 eV lower formation energy than the direct formation of methanol, *CHOH,H, is found (see Figure S5).This is a CH–Cu–HO structure with a 0.69 eV higher free energy of formation (blue cross in Figure 3). To form methanol via this intermediate, water is included in the reaction, and the subsequent CHOH formation occurs at no cost. The CHOH molecule is attached to the Cu–H–Cu cluster on one side at a distance of 1.99 Å, and the HO molecule is on the other side at a distance of 2.08 Å. The most exergonic final state is that of Z[CuH] for completely dry conditions, i.e., including no water molecules. When water is included, the final state is a Z[CuH(HO)] structure with Cu–H bond of 1.59 and 1.58 Å and Cu–HO bonds of 2.17 and 2.14 Å, 0.83 eV higher in energy. The inclusion of water in the mechanism binds methanol stronger to the framework, hindering the desorption of the products. This is undesired and in contrast to established experimental procedure, where a solvent is introduced in order to extract the produced methanol [31]. Since the zeolite contains water [26,27], our results indicate that the participation of the Z[CuOH] site in the reaction is limited. In the energy landscape, the relative energies of the reaction path show a decrease in energy when water is added; see Figure S5.
2.3. Micro-Kinetic Model
With the help of a micro-kinetic model using the reaction paths shown in Figure 2 and Figure 3, the performance of the active sites is analysed in Figure 4. The dry reactions over the Z[CuO] and Z[CuOH] sites in SSZ-13 are here compared to Z[CuO]HO in SSZ-13, as well as previous results for the same reaction over the Z[CuO] site in the large pore zeolite ZSM-5 [11]. In Figure 4a, the fraction of empty sites is plotted over time at a fixed temperature of 523 K, and Figure 4b shows the half-time of the sites as a function of temperature. The poor activity of the dry reactions over the sites in SSZ-13, as indicated in the free energies of the reaction landscape, is here confirmed. Additionally, due to the change in spin for TS1 in Z[CuO] SSZ-13 (as shown in Table 2), the rate is an upper limit.
The time needed to convert the active sites into CHOH is shown in Figure 4a. The MFI framework converted all active sites after 2 s. Second fastest is the CuO site in CHA containing water (blue stars) at 9000 s; the CuO site without water does not start the conversion until 316 years have passed, and the CuOH site in CHA still has done no conversion at s. Treating the reactants according to the harmonic approximation instead of in the gas phase has no significant effect on the activity of the sites (see Figures S8 and S9).
The half-time of the sites as a function of temperature is shown in Figure 4b. The poorly performing systems show a clear increase in activity when the temperature is increased. Over the temperature range, the half-time for the Z[CuOH] SSZ-13 site is reduced from ∼10 s to ∼10 s. The Z[CuO] SSZ-13 is equally sensitive to a change in temperature, falling from ∼10 s to ∼10 s. The two most active systems, Z[CuO]HO SSZ-13 and Z[CuO] ZSM-5, remain largely constant over the given temperature range. Here, it should be noted that the chabazite framework is only stable for temperatures below ∼800 K [32]. Overall, the result shown in Figure 4 implies that neither the Z[CuO] nor Z[CuOH] site can be considered active sites for DMTM. However, the addition of HO to the Z[CuO] in SSZ-13 leads to a significant increase in the activity.
The reaction mechanism over Z[CuO] ZSM-5 is very similar to that over Z[CuO] SSZ-13, passing through a radical TS1 and then forming CHOH on the site [11]. However, as can be seen in Figure S10, it has a flatter energy landscape than that over Z[CuO] SSZ-13; all barriers are lower in ZSM-5, as well as the final desorption step. Thus, comparing the activity of the two different zeolite frameworks, the Z[CuO] site in ZSM-5 is clearly more active than the sites in SSZ-13. In the transient kinetics in Figure 4, the Z[CuO] ZSM-5 sites are all consumed after ∼0.6 s, and the site shows a half-time of – s. This higher activity corresponds well with experiments where large pore zeolites (such as ZSM-5 or mordenite) show higher turnovers for DMTM than small-pore zeolites (such as SSZ-13) [19].
3. Materials and Methods
Density functional calculations were performed using the Vienna Ab initio Simulation Package (VASP, Vienna, Austria) [33,34,35,36,37,38]. The projector augmented wave (PAW) method [39,40] is used to describe the interaction between the valence electrons and the core. PAW potentials were used with the valence states H(1s), O(2s2p), C(2s2p), Al(3s3p), Si(3s3p) and Cu(3d4s). The exchange-correlation interaction was treated using the vdW-DF-cx functional [41,42,43], which includes van der Waals interactions in the exchange-correlation by taking non-local screening into account. In zeolites, the van der Waals interaction has been found to be important to include in the functional. Moreover, the Cu–O bond has shown sensitivity to the localization of the 3d-electrons [44]. The effect of using a +U correction of U = 6 eV is shown in Figures S3 and S6 for Z[CuO] and Z[CuOH], respectively. Overall, the adsorption energies of the reactants increases when the correction is applied.
The Kohn-Sham orbitals were represented using a plane wave basis set with 480 eV as the cut-off energy. A Gaussian smearing of 0.05 eV was applied to the Fermi-level discontinuity. The electronic energies were converged to eV in the self-consistent loop, and ionic positions were considered to be relaxed when the largest atomic force in the system was smaller than 0.01 eV/Å. A Monkhorst-Pack grid with 2 × 2 × 2 -centred k-points was used to sample the Brillouin zone. The gas phase molecules were treated in a cubic box with sides of 10 Å. All calculations were done using spin polarization. All intermediate states were analysed for charge distribution using the Bader analysis method developed by the Henkelman group [45,46,47,48].
Vibrational energies were calculated by constructing the Hessian matrix using atomic forces generated by 0.01 Å displacements of the considered atoms. Only the extra-framework atoms, i.e., active site and reactants, were included in the vibration analysis. All low-lying normal wave numbers of the adsorbed reaction intermediate were set to 100 cm (the included vibrations can be found in Tables S2 and S3). The translations and rotations of water, methane and methanol were calculated using the ideal gas approximation, whereas all vibrations were calculated using the harmonic approximation as implemented in the Themochemistry module in the Atomic Simulation Environment (ASE) [49,50]. Rotations were treated by the rigid motor model [51]. The transition states (TSs) were characterized using the climbing nudged elastic band (NEB) method [52,53,54], during which the spin of the structure was allowed to change.
The stability of the reaction intermediate states was compared using the change in Gibbs free energy:
T is temperature, and is the change in chemical potential between 0 K and the condition of interest for the relevant gases. Here, , unless water is added into the mechanism, in which case, HO is treated in an analogous way to CH in Equations (2) and (4) and .
is the difference in entropy between the adsorbed and non-adsorbed state,
where Z[CuOHC] and Z[CuOH] are the active sites inside the zeolite framework. When gas phase molecules are present in the zeolite cage, their entropy contribution is affected by the confinement of the zeolite structure and is calculated according to [55,56]:
Here, and are the entropy contributions from the gas phase translations and rotations of the molecule, and is the entropy contribution from the vibrational modes of the molecule inside the zeolite. In Equation (1), the change in enthalpy is approximated as the difference in total energy [57] given by:
3.1. Micro-Kinetic Modelling
The reaction pathways were explored using a mean-field micro-kinetic model for the DMTM reaction. The rate constants of the activated conversion of adsorbed reactants were expressed using the conventional transitions-state theory (TST) [51],
where is the energy difference between the initial and transition state, Z is the partition function for the initial state, is the partition function for the transition state with the reaction coordinate excluded, is Boltzmann’s constant and h Planck’s constant. The adsorption/desorption of methane and methanol was considered to be barrierless, and the rate constant for adsorption is calculated as:
where p is the partial pressure of the molecule in the gas phase, A is the cross-sectional area of the pore and m the mass of the molecule. The corresponding desorption rate constants were obtained through the equilibrium constant:
3.2. Zeolite Framework
In this study, the chabazite (CHA) framework SSZ-13 was used for catalysing the reaction. Considering the small 12 T-site unit cell of the CHA framework, an Si:Al ratio of 10:2 was used and denoted Z. Comparisons were made with systems of a Si:Al ratio of 11:1, denoted Z. The cation introduced to compensate the 2+ charge in the Z zeolite system was a Cu-dimer motif, which formed the active site. The reaction was performed over Z[CuO] and Z[CuOH]; in a previous study identified to be the most stable motifs of the Cu-dimer under the reaction conditions [27].
4. Conclusions
The reaction mechanisms and the activity of DMTM conversion over Z[CuO] and Z[CuOH] in Cu-SSZ-13 are explored by first-principles calculations and micro-kinetic modelling. Over both systems, methane dissociation occurs via a methyl radical state, which is responsible for the highest barrier of the reaction in both systems. During dry reaction conditions: T = 448 K, p = 2% and p = 10% (with respect to atmospheric pressure); methanol desorption is exergonic over both structures. When water is added to the mechanism over Z[CuO], desorption becomes more exergonic; while in Z[CuOH], the addition of any water increases the cost of desorption.
The results show that neither of the dry sites (Z[CuO] and Z[CuOH]) are responsible for any DMTM activity in SSZ-13 under the investigated conditions. Thus, for SSZ-13 to be considered as a candidate for the DMTM reaction, the conditions must be chosen with care; for instance by using a higher partial pressure of methane and including water. Another strategy would be to utilize other candidates for the active site that has a lower transition barrier for methane activation.
It should be noted that the identified reaction mechanism represents a quasi-catalytic cycle. The initial activation and subsequent reactivation of the active site are not taken into account. To close the cycle and make it a truly catalytic process remain as future challenges.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/11/1/17/s1, Table S1: Molecular vibrations, Table S2: Vibrational frequencies of reaction intermediates in Z2[Cu2O], Table S3: Vibrational frequencies of reaction intermediates in Z2[Cu2OH], Figure S1: Unit cell and larger scale structure of SSZ-13, Figure S2: Reaction mechanism over Z2[Cu2O] in relative energies, Figure S3: Reaction mechanism over Z2[Cu2O] showing the effect of using +U, Figure S4: Reaction mechanism over Z2[Cu2O] showing the change in free energy when using the harmonic approximation for reaction species, Figure S5: Reaction mechanism over Z2[Cu2OH] in relative energies, Figure S6: Reaction mechanism over Z2[Cu2OH] showing the effect of using +U, Figure S7: Reaction mechanism over Z2[Cu2OH] showing the change in free energy when using the harmonic approximation for reaction species, Figure S8: Micro-kinetic model, Figure S9: Coverages, Figure S10: Reaction mechanism over Cu-ZSM-5 in relative energy.
Author Contributions
Conceptualization, H.G. and A.H.; methodology, U.E. and A.H.; software, U.E. and A.A.A.; validation, U.E., A.A.A. and A.H.; formal analysis, U.E. and A.A.A.; investigation, U.E. and A.A.A.; resources, H.G. and A.H.; data curation, U.E.; writing, original draft preparation, U.E.; writing, review and editing, U.E., A.A.A., H.G. and A.H.; visualization, U.E.; supervision, A.H.; project administration, H.G.; funding acquisition, H.G. All authors read and agreed to the published version of the manuscript.
Funding
Financial support is gratefully acknowledged from the Knut and Alice Wallenberg Foundation through the project “Atomistic design of catalysts” (No: KAW 2015.0058) and the Competence Centre for Catalysis (KCK) at Chalmers University of Technology. K.C.K. is financially supported by Chalmers University of Technology, the Swedish Energy Agency and the member companies: AB Volvo, ECAPS AB, Johnson Matthey AB, Preem AB, Scania CV AB and Umicore Denmark ApS. Calculations performed at C3SE at Chalmers, with CPU time provided through an SNIC grant.
Conflicts of Interest
The authors declare no conflict of interest.
References
- U.S. Energy Information Administration (EIA). International Energy Outlook 2019. Available online: https://www.eia.gov/outlooks/ieo/pdf/ieo2019.pdf (accessed on 9 December 2020).
- U.S. Energy Information Administration (EIA). Natural Gas Explained-Natural Gas and the Environment. Available online: https://www.eia.gov/energyexplained/naturalgas/natural-gas-and-the-environment.php (accessed on 9 December 2019).
- Letcher, T. Future Energy: Improved, Sustainable and Clean Options for our Planet; Elsevier Science: Oxford, UK, 2008. [Google Scholar]
- Foster, N.R. Direct catalytic oxidation of methane to methanol—A review. Appl. Catal. 1985, 19, 1–11. [Google Scholar] [CrossRef]
- Chun, J.W.; Anthony, R.G. Catalytic oxidations of methane to methanol. Ind. Eng. Chem. Res. 1993, 32, 259–263. [Google Scholar] [CrossRef]
- Palkovits, R.; Antonietti, M.; Kuhn, P.; Thomas, A.; Schüth, F. Solid Catalysts for the Selective Low-Temperature Oxidation of Methane to Methanol. Angew. Chem. Int. Ed. 2009, 48, 6909–6912. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Yang, Y.; Xu, Y.; Etim, U.; Qiao, K.; Xu, B.; Yan, Z. A review of the direct oxidation of methane to methanol. Chin. J. Catal. 2016, 37, 1206–1215. [Google Scholar] [CrossRef]
- Sirajuddin, S.; Rosenzweig, A.C. Enzymatic Oxidation of Methane. Biochemistry 2015, 54, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Groothaert, M.H.; Smeets, P.J.; Sels, B.F.; Jacobs, P.A.; Schoonheydt, R.A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. [Google Scholar] [CrossRef]
- Wang, X.; Martin, N.; Nilsson, J.; Carlson, S.; Gustafson, J.; Skoglundh, M.; Carlsson, P.A. Copper-Modified Zeolites and Silica for Conversion of Methane to Methanol. Catalysts 2018, 8, 545. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, A.A.; Zhdanov, V.P.; Carlsson, P.A.; Grönbeck, H.; Hellman, A. Metal dimer sites in ZSM-5 zeolite for methane-to-methanol conversion from first-principles kinetic modelling: Is the [Cu–O–Cu]2+ motif relevant for Ni, Co, Fe, Ag, and Au? Catal. Sci. Technol. 2017, 7, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Woertink, J.S.; Smeets, P.J.; Groothaert, M.H.; Vance, M.A.; Sels, B.F.; Schoonheydt, R.A.; Solomon, E.I. A [Cu2O]2+ core in Cu-ZSM-5, the active site in the oxidation of methane to methanol. Proc. Natl. Acad. Sci. USA 2009, 106, 18908–18913. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Vassilev, P.; Sanchez-Sanchez, M.; Lercher, J.A.; Hensen, E.J.; Pidko, E.A. Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol. J. Catal. 2016, 338, 305–312. [Google Scholar] [CrossRef]
- Hammond, C.; Forde, M.M.; AbRahim, M.H.; Thetford, A.; He, Q.; Jenkins, R.L.; Dimitratos, N.; Lopez-Sanchez, J.A.; Dummer, N.F.; Murphy, D.M.; et al. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by using Copper-Promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef] [PubMed]
- Sjövall, H.; Blint, R.J.; Olsson, L. Detailed kinetic modelling of NH3 SCR over Cu-ZSM-5. Appl. Catal. Environ. 2009, 92, 138–153. [Google Scholar] [CrossRef]
- Kwak, J.H.; Tonkyn, R.G.; Kim, D.H.; Szanyi, J.; Peden, C.H. Excellent activity and selectivity of Cu-SSZ-13 in the selective catalytic reduction of NOx with NH3. J. Catal. 2010, 275, 187–190. [Google Scholar] [CrossRef]
- Oord, R.; Schmidt, J.E.; Weckhuysen, B.M. Methane-to-methanol conversion over zeolite Cu-SSZ-13, and its comparison with the selective catalytic reduction of NOx with NH3. Catal. Sci. Technol. 2018, 8, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Newton, M.A.; Knorpp, A.J.; Pinar, A.B.; Sushkevich, V.L.; Palagin, D.; van Bokhoven, J.A. On the Mechanism Underlying the Direct Conversion of Methane to Methanol by Copper Hosted in Zeolites; Braiding Cu K-Edge XANES and Reactivity Studies. J. Am. Chem. Soc. 2018, 140, 10090–10093. [Google Scholar] [CrossRef] [PubMed]
- Borfecchia, E.; Pappas, D.K.; Dyballa, M.; Lomachenko, K.A.; Negri, C.; Signorile, M.; Berlier, G. Evolution of active sites during selective oxidation of methane to methanol over Cu-CHA and Cu-MOR zeolites as monitored by operando XAS. Catal. Today 2019, 333, 17–27. [Google Scholar] [CrossRef]
- Dinh, K.T.; Sullivan, M.M.; Narsimhan, K.; Serna, P.; Meyer, R.J.; Dincă, M.; Román-Leshkov, Y. Continuous Partial Oxidation of Methane to Methanol Catalyzed by Diffusion-Paired Copper Dimers in Copper-Exchanged Zeolites. J. Am. Chem. Soc. 2019, 141, 11641–11650. [Google Scholar] [CrossRef]
- Kulkarni, A.R.; Zhao, Z.J.; Siahrostami, S.; Nørskov, J.K.; Studt, F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016, 6, 6531–6536. [Google Scholar] [CrossRef]
- Yu Mao, P.H. Identification of the active sites and mechanism for partial methane oxidation to methanol over copper-exchanged CHA zeolites. Sci. China Chem. 2020, 63, 850–859. [Google Scholar] [CrossRef]
- Ipek, B.; Wulfers, M.J.; Kim, H.; Göltl, F.; Hermans, I.; Smith, J.P.; Booksh, K.S.; Brown, C.M.; Lobo, R.F. Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39. ACS Catal. 2017, 7, 4291–4303. [Google Scholar] [CrossRef]
- Wang, G.; Chen, W.; Huang, L.; Liu, Z.; Sun, X.; Zheng, A. Reactivity descriptors of diverse copper-oxo species on ZSM-5 zeolite towards methane activation. Catal. Today 2019, 338, 108–116. [Google Scholar] [CrossRef]
- Mahyuddin, M.H.; Tanaka, T.; Shiota, Y.; Staykov, A.; Yoshizawa, K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. [Google Scholar] [CrossRef]
- Paolucci, C.; Parekh, A.A.; Khurana, I.; Di Iorio, J.R.; Li, H.; Albarracin Caballero, J.D.; Shih, A.J.; Anggara, T.; Delgass, W.N.; Miller, J.T.; et al. Catalysis in a Cage: Condition-Dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites. J. Am. Chem. Soc. 2016, 138, 6028–6048. [Google Scholar] [CrossRef] [PubMed]
- Engedahl, U.; Grönbeck, H.; Hellman, A. First-Principles Study of Oxidation State and Coordination of Cu-Dimers in Cu-SSZ-13 during Methane-to-Methanol Reaction Conditions. J. Phys. Chem. C 2019, 123, 26145–26150. [Google Scholar] [CrossRef]
- NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101. Available online: https://cccbdb.nist.gov (accessed on 21 October 2019).
- Latimer, A.; Kulkarni, A.; Aljama, H.; Montoya, J.; Yoo, J.S.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J. Understanding trends in C-H bond activation in heterogeneous catalysis. Nat. Mater. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J.A. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Am. Assoc. Adv. Sci. 2017, 356, 523–527. [Google Scholar] [CrossRef]
- Creci, S.; Wang, X.; Carlsson, P.A.; Martinelli, A.; Skoglundh, M. Methoxy ad-species in MFI zeotypes during methane exposure and methanol desorption followed by in situ IR spectroscopy. Catal. Today 2020. [Google Scholar] [CrossRef]
- Schmieg, S.J.; Oh, S.H.; Kim, C.H.; Brown, D.B.; Lee, J.H.; Peden, C.H.; Kim, D.H. Thermal durability of Cu-CHA NH3-SCR catalysts for diesel NOx reduction. Catal. Today 2012, 184, 252–261. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Norm-conserving and ultrasoft pseudopotentials for first-row and transition elements. J. Phys. Condens. Matter 1994, 6, 8245–8257. [Google Scholar] [CrossRef]
- Klimes, J.; R Bowler, D.; Michaelides, A. Chemical accuracy for the Van der Waals density functional. J. Phys. Condens. Matter 2010, 22, 022201. [Google Scholar] [CrossRef] [PubMed]
- Klimes, J.; Bowler, D.R.; Michaelides, A. Van der Waals density functionals applied to solids. Phys. Rev. B 2011, 83, 195131. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dion, M.; Rydberg, H.; Schröder, E.; Langreth, D.C.; Lundqvist, B.I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Murray, E.D.; Kong, L.; Lundqvist, B.I.; Langreth, D.C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. [Google Scholar] [CrossRef] [Green Version]
- Berland, K.; Hyldgaard, P. Exchange functional that tests the robustness of the plasmon description of the Van der Waals density functional. Phys. Rev. B 2014, 89, 035412. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Janssens, T.V.W.; Grönbeck, H. A comparative test of different density functionals for calculations of NH3-SCR over Cu-Chabazite. Phys. Chem. Chem. Phys. 2019, 21, 10923–10930. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Sanville, E.; Kenny, S.; Smith, R. An Improved Grid-Based Algorithm for Bader Charge Allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Arnaldsson, A.; Jonsson, H. A Fast and Robust Algorithm for Bader Decomposition of Charge Density. Comput. Mater. Sci. Comput. Mater Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Yu, M.; Trinkle, D. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. 2017, 29, 273002. [Google Scholar]
- Bahn, S.R.; Jacobsen, K.W. An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 2002, 4, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; John Wiley & Sons: Weinheim, Germany, 2006. [Google Scholar]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, M.; Chen, L.; Grönbeck, H. Monte Carlo Potential Energy Sampling for Molecular Entropy in Zeolites. J. Phys. Chem. C 2018, 122, 20351–20357. [Google Scholar] [CrossRef]
- Chen, L.; Janssens, T.V.W.; Vennestrøm, P.N.R.; Jansson, J.; Skoglundh, M.; Grönbeck, H. A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA. ACS Catal. 2020, 10, 5646–5656. [Google Scholar] [CrossRef]
- Reuter, K.; Scheffler, M. Composition, structure, and stability of RuO2(110) as a function of oxygen pressure. Phys. Rev. B 2001, 65, 1–11. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The two investigated active site structures and their positions in the zeolite framework are shown. In (a), the CuOH site in SSZ-13 and in (b) the CuO site in SSZ-13.
Figure 1.
The two investigated active site structures and their positions in the zeolite framework are shown. In (a), the CuOH site in SSZ-13 and in (b) the CuO site in SSZ-13.

Figure 2.
Reaction path over Z[CuO]. Gibbs free energies are in eV and relative to that of Z[CuO] with CH in the gas phase. The red squared path shows the reaction for dry conditions. Adding water to the mechanism gives the blue cross path. Both paths are exergonic. Reaction conditions are set to T = 448 K, p = 2%, p = 10% and p = 10%, with respect to atmospheric pressure. TS, transition state.
Figure 2.
Reaction path over Z[CuO]. Gibbs free energies are in eV and relative to that of Z[CuO] with CH in the gas phase. The red squared path shows the reaction for dry conditions. Adding water to the mechanism gives the blue cross path. Both paths are exergonic. Reaction conditions are set to T = 448 K, p = 2%, p = 10% and p = 10%, with respect to atmospheric pressure. TS, transition state.

Figure 3.
Reaction path over Z[CuOH]. Purple diamonds mark the mechanism under dry conditions, blue cross the addition of one HO and grey triangles when two HO are included in the reaction. Gibbs free energies are in eV and relative to that of Z[CuOH] with CH in the gas phase. Reaction conditions are set to T = 448 K, p = 2%, p = 10% and p = 10%, with respect to atmospheric pressure.
Figure 3.
Reaction path over Z[CuOH]. Purple diamonds mark the mechanism under dry conditions, blue cross the addition of one HO and grey triangles when two HO are included in the reaction. Gibbs free energies are in eV and relative to that of Z[CuOH] with CH in the gas phase. Reaction conditions are set to T = 448 K, p = 2%, p = 10% and p = 10%, with respect to atmospheric pressure.

Figure 4.
Coverages calculated for the reaction over four different sites: Z[CuO] in ZSM-5, Z[CuO]HO in SSZ-13, Z[CuO] in SSZ-13 and Z[CuOH] in SSZ-13. (a) shows transient kinetics and the fraction of empty sits as a function of time at a fixed temperature of 523 K. In (b), the half-time of the sites as a function of temperature is shown.
Figure 4.
Coverages calculated for the reaction over four different sites: Z[CuO] in ZSM-5, Z[CuO]HO in SSZ-13, Z[CuO] in SSZ-13 and Z[CuOH] in SSZ-13. (a) shows transient kinetics and the fraction of empty sits as a function of time at a fixed temperature of 523 K. In (b), the half-time of the sites as a function of temperature is shown.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Spin state of the intermediates in the reaction over Z[CuOH] following the reaction mechanisms in fig:pathcu2oh. Energy difference between the two lowest spin states in eV.
Table 1.
Spin state of the intermediates in the reaction over Z[CuOH] following the reaction mechanisms in fig:pathcu2oh. Energy difference between the two lowest spin states in eV.
| Reaction Intermediate | Spin State | Energy Difference (eV) |
|---|---|---|
| *OH | doublet | 3.60 |
| *OH,CH | doublet | 3.60 |
| TS1 | doublet | 1.84 |
| *CHOH,H | doublet | 3.42 |
| *CH,HO | doublet | 3.05 |
| *CHOH,H,HO | doublet | 4.10 |
| *H | doublet | 3.55 |
| *H,HO | doublet | 3.51 |
| *H,2(HO) | doublet | 3.77 |
Table 2.
Spin states of the intermediates in the reaction over Z[CuO] following the reaction mechanisms in Figure 2. The energy difference presented is that between the two lowest spin states in eV.
Table 2.
Spin states of the intermediates in the reaction over Z[CuO] following the reaction mechanisms in Figure 2. The energy difference presented is that between the two lowest spin states in eV.
| Reaction Intermediate | Spin State | Energy Difference (eV) |
|---|---|---|
| *O | singlet | 0.03 |
| *O,CH | singlet | 0.03 |
| TS1 | triplet | 0.45 |
| *OH,CH | singlet | 0.35 |
| TS2 | singlet | 0.08 |
| *CHOH | singlet | 1.71 |
| *CHOH,HO | singlet | 2.01 |
| * | singlet | 0.26 |
| *HO | singlet | 1.69 |
| *HO,HO | singlet | 1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Engedahl, U.; Arvidsson, A.A.; Grönbeck, H.; Hellman, A. Reaction Mechanism for Methane-to-Methanol in Cu-SSZ-13: First-Principles Study of the Z2[Cu2O] and Z2[Cu2OH] Motifs. Catalysts 2021, 11, 17. https://doi.org/10.3390/catal11010017
AMA Style
Engedahl U, Arvidsson AA, Grönbeck H, Hellman A. Reaction Mechanism for Methane-to-Methanol in Cu-SSZ-13: First-Principles Study of the Z2[Cu2O] and Z2[Cu2OH] Motifs. Catalysts. 2021; 11(1):17. https://doi.org/10.3390/catal11010017
Chicago/Turabian StyleEngedahl, Unni, Adam A. Arvidsson, Henrik Grönbeck, and Anders Hellman. 2021. "Reaction Mechanism for Methane-to-Methanol in Cu-SSZ-13: First-Principles Study of the Z2[Cu2O] and Z2[Cu2OH] Motifs" Catalysts 11, no. 1: 17. https://doi.org/10.3390/catal11010017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.