A Bifunctional Electroactive Ti4O7-Based Membrane System for Highly Efficient Ammonia Decontamination



Abstract
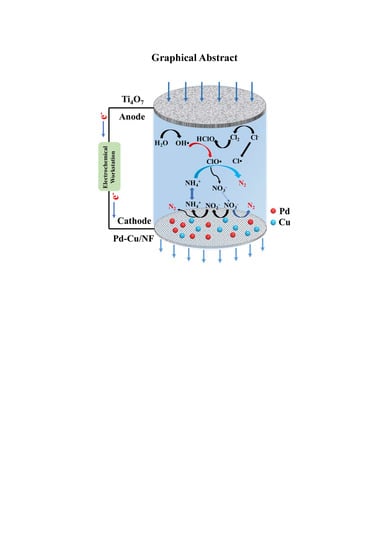
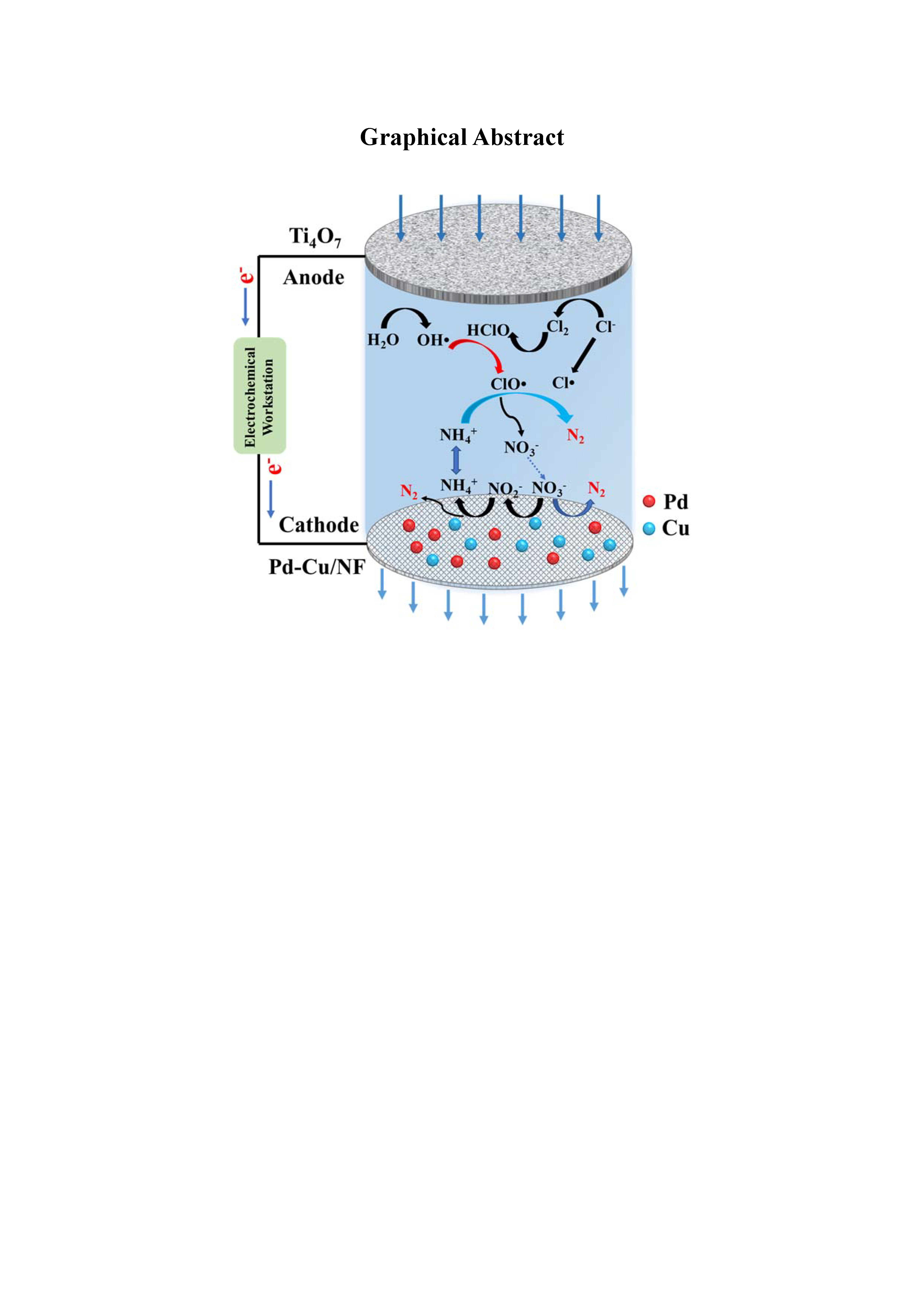
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Characterizations
2.2. Ammonia Conversion
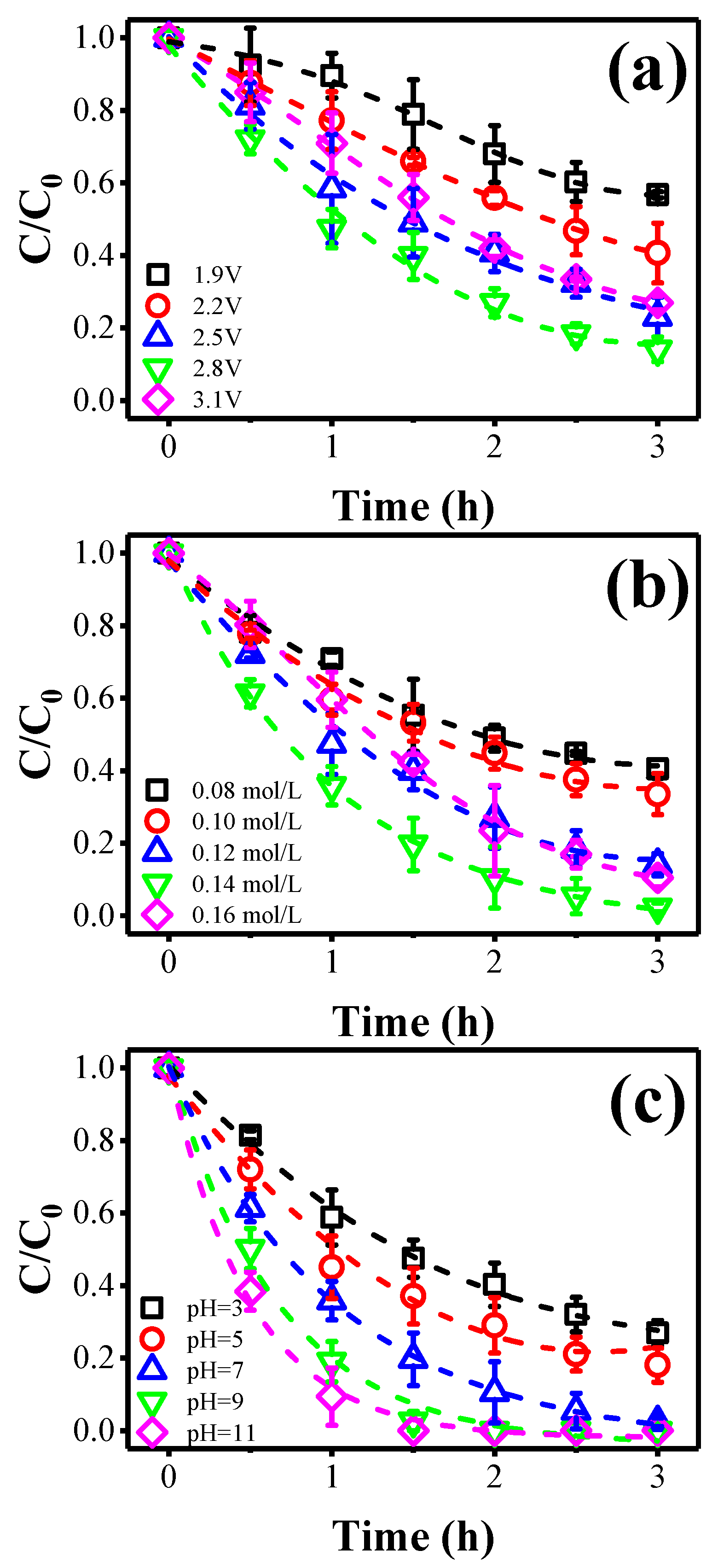
2.3. Impacts of Key Operational Parameters
2.3.1. Effect of Anode Potential
2.3.2. Effect of [Cl−]
2.3.3. Effect of Solution pH
2.3.4. Effect of Flow Rate
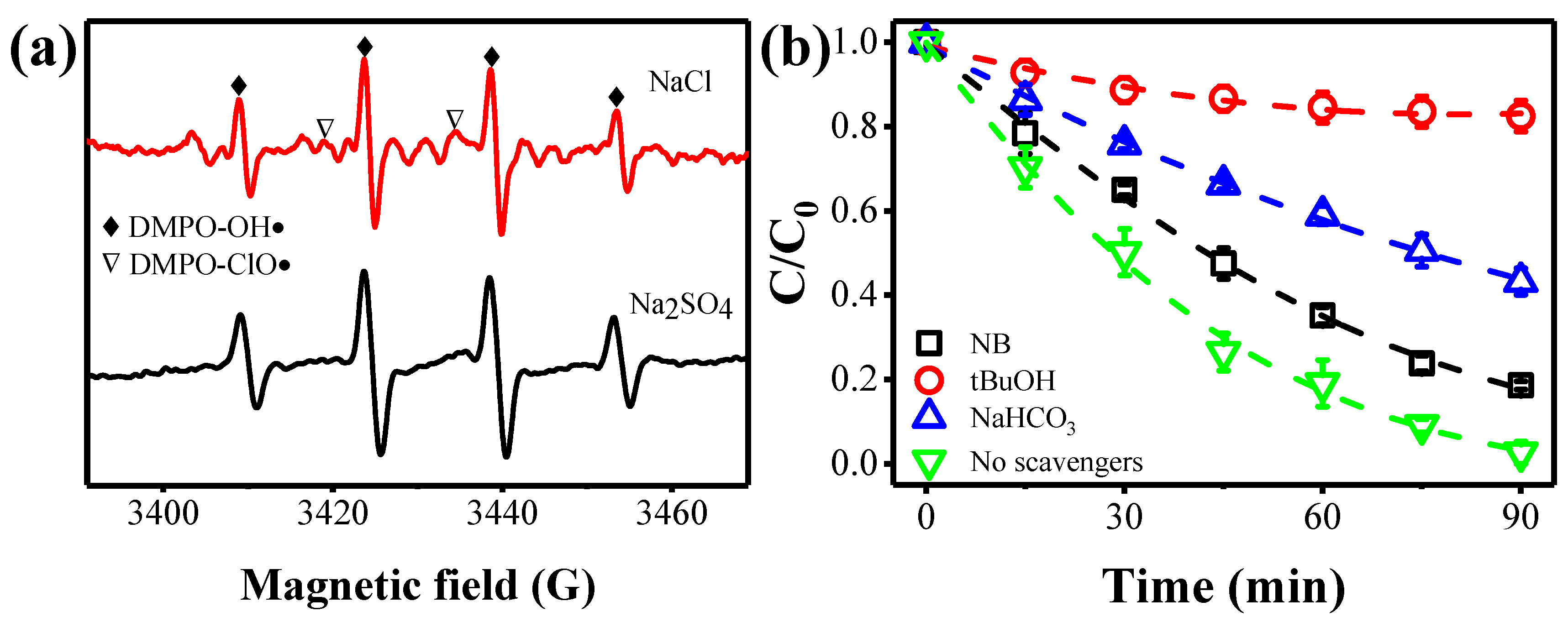
2.4. Ammonia Decontamination Mechanism
2.5. Nitrate Reduction on the Pd-Cu/NF Cathode
2.6. Stability Evaluation of the Ti4O7 Electrode
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Preparation of Electrodes
3.3. Electrochemical Experiments
3.4. Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Weihrauch, D.; Donini, A.; O’Donnell, M.J. Ammonia transport by terrestrial and aquatic insects. J. Insect Physiol. 2012, 58, 473–487. [Google Scholar] [CrossRef]
- Zhang, C.Y.; He, D.; Ma, J.X.; Waite, T.D. Active chlorine mediated ammonia oxidation revisited: Reaction mechanism, kinetic modelling and implications. Water Res. 2018, 145, 220–230. [Google Scholar] [CrossRef]
- Gayen, P.; Chen, C.; Abiade, J.T.; Chaplin, B.P. Electrochemical oxidation of atrazine and clothianidin on Bi-doped SnO2-TinO2n−1 electrocatalytic reactive electrochemical membranes. Environ. Sci. Technol. 2018, 52, 12675–12684. [Google Scholar] [CrossRef]
- Li, M.H.; Liu, Y.B.; Dong, L.M.; Shen, C.S.; Li, F.; Huang, M.H.; Ma, C.Y.; Yang, B.; An, X.Q.; Sand, W. Recent advances on photocatalytic fuel cell for environmental applications-The marriage of photocatalysis and fuel cells. Sci. Total Environ. 2019, 668, 966–978. [Google Scholar] [CrossRef]
- Chaplin, B.P. The prospect of electrochemical technologies advancing worldwide water treatment. Acc. Chem. Res. 2019, 52, 596–604. [Google Scholar] [CrossRef]
- Li, F.; Peng, X.; Liu, Y.B.; Mei, J.C.; Sun, L.W.; Shen, C.S.; Ma, C.Y.; Huang, M.H.; Wang, Z.W.; Sand, W.G. A chloride-radical-mediated electrochemical filtration system for rapid and effective transformation of ammonia to nitrogen. Chemosphere 2019, 229, 383–391. [Google Scholar] [CrossRef]
- Liu, Y.B.; Wu, P.; Liu, F.Q.; Li, F.; An, X.Q.; Liu, J.S.; Wang, Z.W.; Shen, C.S.; Sand, W. Electroactive modified carbon nanotube filter for simultaneous detoxification and sequestration of Sb(III). Environ. Sci. Technol. 2019, 53, 1527–1535. [Google Scholar] [CrossRef]
- Zaky, A.M.; Chaplin, B.P. Porous substoichiometric TiO2 anodes as reactive electrochemical membranes for water treatment. Environ. Sci. Technol. 2013, 47, 6554–6563. [Google Scholar] [CrossRef]
- Le, T.X.H.; Haflich, H.; Shah, A.D.; Chaplin, B.P. Energy-efficient electrochemical oxidation of perfluoroalkyl substances using a Ti4O7 reactive electrochemical membrane anode. Environ. Sci. Technol. Lett. 2019, 6, 504–510. [Google Scholar] [CrossRef]
- Chaplin, B.P. Critical review of electrochemical advanced oxidation processes for water treatment applications. Environ. Sci. Process. Impacts 2014, 16, 1182–1203. [Google Scholar] [CrossRef]
- Donaghue, A.; Chaplin, B.P. Effect of select organic compounds on perchlorate formation at boron-doped diamond film anodes. Environ. Sci. Technol. 2013, 47, 12391–12399. [Google Scholar] [CrossRef]
- Liu, Y.B.; Mei, J.C.; Shen, C.S.; Huang, M.H.; Yang, M.; Wang, Z.W.; Sand, W.; Li, F. Rapid and selective electrochemical transformation of ammonia to N2 by substoichiometric TiO2-based electrochemical system. RSC Adv. 2020, 10, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.J.; Wu, Z.H.; Ren, Z.R.; Guo, K.H.; Hou, S.D.; Hua, Z.C.; Li, X.C.; Fang, J.Y. Degradation of lipid regulators by the UV/chlorine process: Radical mechanisms, chlorine oxide radical (ClO•)-mediated transformation pathways and toxicity changes. Water Res. 2018, 137, 242–250. [Google Scholar] [CrossRef]
- Nayak, S.; Chaplin, B.P. Fabrication and characterization of porous, conductive, monolithic Ti4O7 electrodes. Electrochim. Acta 2018, 263, 299–310. [Google Scholar] [CrossRef]
- Liang, S.T.; Lin, H.; Yan, X.F.; Huang, Q.G. Electro-oxidation of tetracycline by a Magneli phase Ti4O7 porous anode: Kinetics, products, and toxicity. Chem. Eng. J. 2018, 332, 628–636. [Google Scholar] [CrossRef]
- Tao, X.Y.; Wang, J.G.; Ying, Z.G.; Cai, Q.X.; Zheng, G.Y.; Gan, Y.P.; Huang, H.; Xia, Y.; Liang, C.; Zhang, W.K.; et al. Strong sulfur binding with conducting Magneli-phase TinO2n−1 nanomaterials for improving lithium-sulfur batteries. Nano Lett. 2014, 14, 5288–5294. [Google Scholar] [CrossRef]
- Boffa, A.B.; Galloway, H.C.; Jacobs, P.W.; Benitez, J.J.; Batteas, J.D.; Salmeron, M.; Bell, A.T.; Somorjai, G.A. The growth and structure of titanium oxide films on Pt(111) investIgated by LEED, XPS, ISS, and STM. Surf. Sci. 1995, 326, 80–92. [Google Scholar] [CrossRef]
- Jiang, X.H.; Xing, Q.J.; Luo, X.B.; Li, F.; Zou, J.P.; Liu, S.S.; Li, X.; Wang, X.K. Simultaneous photoreduction of Uranium(VI) and photooxidation of Arsenic (III) in aqueous solution over g-C3N4/TiO2 heterostructured catalysts under simulated sunlight irradiation. Appl. Catal. B-Environ. 2018, 228, 29–38. [Google Scholar] [CrossRef]
- Zeng, H.B.; Liu, S.S.; Chai, B.Y.; Cao, D.; Wang, Y.; Zhao, X. Enhanced photoelectrocatalytic decomplexation of Cu-EDTA and Cu recovery by persulfate activated by UV and cathodic reduction. Environ. Sci. Technol. 2016, 50, 6459–6466. [Google Scholar] [CrossRef]
- Liu, C.; Hirohara, M.; Maekawa, T.; Chang, R.; Hayashi, T.; Chiang, C.Y. Selective electro-oxidation of glycerol to dihydroxyacetone by a non-precious electrocatalyst-CuO. Appl. Catal. B.-Environ. 2020, 265, 12. [Google Scholar] [CrossRef]
- Yuan, H.K.; Kusema, B.T.; Yan, Z.; Streiff, S.; Shi, F. Highly selective synthesis of 2,5-bis(aminomethyl)furan via catalytic amination of 5-(hydroxymethyl)furfural with NH3 over a bifunctional catalyst. RSC Adv. 2019, 9, 38877–38881. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.Z.; Bai, J.; Li, J.H.; Luo, T.; Qiao, L.; Zeng, Q.Y.; Zhou, B.X. Highly selective transformation of ammonia nitrogen to N2 based on a novel solar-driven photoelectrocatalytic-chlorine radical reactions system. Water Res. 2017, 125, 512–519. [Google Scholar] [CrossRef]
- Deng, Y.; Englehardt, J.D. Electrochemical oxidation for landfill leachate treatment. Waste Manag. 2007, 27, 380–388. [Google Scholar] [CrossRef]
- Kim, K.W.; Kim, Y.J.; Kim, I.T.; Park, G.I.; Lee, E.H. The electrolytic decomposition mechanism of ammonia to nitrogen at an IrO2 anode. Electrochim. Acta 2005, 50, 4356–4364. [Google Scholar] [CrossRef]
- Bunce, N.J.; Bejan, D. Mechanism of electrochemical oxidation of ammonia. Electrochim. Acta 2011, 56, 8085–8093. [Google Scholar] [CrossRef]
- Candido, L.; Gomes, J. Evaluation of anode materials for the electro-oxidation of ammonia and ammonium ions. Mater. Chem. Phys. 2011, 129, 1146–1151. [Google Scholar] [CrossRef]
- Kim, K.W.; Kim, Y.J.; Kim, I.T.; Park, G.I.; Lee, E.H. Electrochemical conversion characteristics of ammonia to nitrogen. Water Res. 2006, 40, 1431–1441. [Google Scholar] [CrossRef]
- Hao, R.L.; Mao, X.Z.; Wang, Z.; Zhao, Y.; Wang, T.H.; Sun, Z.H.; Yuan, B.; Li, Y.K. A novel method of ultraviolet/NaClO2-NH4OH for NO removal: Mechanism and kinetics. J. Hazard. Mater. 2019, 368, 234–242. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.H.; Bai, J.; Li, L.S.; Chen, S.; Zhou, T.S.; Wang, J.C.; Xia, L.G.; Xu, Q.J.; Zhou, B.X. Extremely efficient decomposition of ammonia N to N2 using ClO• from reactions of HO• and HOCl generated in situ on a novel bifacial photoelectroanode. Environ. Sci. Technol. 2019, 53, 6945–6953. [Google Scholar] [CrossRef]
- Tang, W.J.; Zhang, Y.; Bai, J.; Li, J.H.; Wang, J.C.; Li, L.S.; Zhou, T.S.; Chen, S.; Rahim, M.; Zhou, B.X. Efficient denitrification and removal of natural organic matter, emerging pollutants simultaneously for RO concentrate based on photoelectrocatalytic radical reaction. Sep. Purif. Technol. 2020, 234, 116032. [Google Scholar] [CrossRef]
- Cheng, X.W.; Liu, H.L.; Chen, Q.H.; Li, J.J.; Wang, P. Preparation and characterization of palladium nano-crystallite decorated TiO2 nano-tubes photoelectrode and its enhanced photocatalytic efficiency for degradation of diclofenac. J. Hazard. Mater. 2013, 254, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.H.; Wu, Z.H.; Shang, C.; Yao, B.; Hou, S.D.; Yang, X.; Song, W.H.; Fang, J.Y. Radical chemistry and structural relationships of PPCP degradation by UV/chlorine treatment in simulated drinking water. Environ. Sci. Technol. 2017, 51, 10431–10439. [Google Scholar] [CrossRef]
- Hua, Z.C.; Guo, K.H.; Kong, X.J.; Lin, S.K.; Wu, Z.H.; Wang, L.P.; Huang, H.; Fang, J.Y. PPCP degradation and DBP formation in the solar/free chlorine system: Effects of pH and dissolved oxygen. Water Res. 2019, 150, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.H.; Guo, K.H.; Fang, J.Y.; Yang, X.Q.; Xiao, H.; Hou, S.D.; Kong, X.J.; Shang, C.; Yang, X.; Meng, F.A.; et al. Factors affecting the roles of reactive species in the degradation of micropollutants by the UV/chlorine process. Water Res. 2017, 126, 351–360. [Google Scholar] [CrossRef]
- De Laat, J.; Boudiaf, N.; Dossier-Berne, F. Effect of dissolved oxygen on the photodecomposition of monochloramine and dichloramine in aqueous solution by UV irradiation at 253.7 nm. Water Res. 2010, 44, 3261–3269. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, Y.; Akita, T.; Mikami, I.; Okuhara, T. Hydrogenation of nitrate in water to nitrogen over Pd-Cu supported on active carbon. J. Catal. 2002, 207, 37–45. [Google Scholar] [CrossRef]
- Gao, W.L.; Guan, N.J.; Chen, J.X.; Guan, X.X.; Jin, R.C.; Zeng, H.S.; Liu, Z.G.; Zhang, F.X. Titania supported Pd-Cu bimetallic catalyst for the reduction of nitrate in drinking water. Appl. Catal. B-Environ. 2003, 46, 341–351. [Google Scholar] [CrossRef]
- Panizza, M.; Cerisola, G. Direct and mediated anodic oxidation of organic pollutants. Chem. Rev. 2009, 109, 6541–6569. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.H.; Bai, J.; Shen, Z.X.; Li, L.S.; Xia, L.G.; Chen, S.; Zhou, B.X. Exhaustive conversion of inorganic nitrogen to nitrogen gas based on a photoelectro-chlorine cycle reaction and a highly selective nitrogen gas generation cathode. Environ. Sci. Technol. 2018, 52, 1413–1420. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, W.; Mei, J.; Liu, Y.; Yang, B.; Li, F.; Fang, X.; Huang, M.; Sand, W. A Bifunctional Electroactive Ti4O7-Based Membrane System for Highly Efficient Ammonia Decontamination. Catalysts 2020, 10, 383. https://doi.org/10.3390/catal10040383
Zhao W, Mei J, Liu Y, Yang B, Li F, Fang X, Huang M, Sand W. A Bifunctional Electroactive Ti4O7-Based Membrane System for Highly Efficient Ammonia Decontamination. Catalysts. 2020; 10(4):383. https://doi.org/10.3390/catal10040383
Chicago/Turabian StyleZhao, Wenchang, Jiancheng Mei, Yanbiao Liu, Bo Yang, Fang Li, Xiaofeng Fang, Manhong Huang, and Wolfgang Sand. 2020. "A Bifunctional Electroactive Ti4O7-Based Membrane System for Highly Efficient Ammonia Decontamination" Catalysts 10, no. 4: 383. https://doi.org/10.3390/catal10040383
APA StyleZhao, W., Mei, J., Liu, Y., Yang, B., Li, F., Fang, X., Huang, M., & Sand, W. (2020). A Bifunctional Electroactive Ti4O7-Based Membrane System for Highly Efficient Ammonia Decontamination. Catalysts, 10(4), 383. https://doi.org/10.3390/catal10040383