Bacillus subtilis Lipase A—Lipase or Esterase?
by
 and
and
Paula Bracco
1, 
Nelleke van Midden
1,
Epifanía Arango
1,
Guzman Torrelo
1,
Valerio Ferrario
2,
Lucia Gardossi
2 and
Ulf Hanefeld
1,*
1
Biokatalyse, Afdeling Biotechnologie, Technische Universiteit Delft, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
2
Dipartimento di Scienze Chimiche e Farmaceutiche, Università degli Studi di Trieste, Via Licio Giorgieri 1, 34127 Trieste, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(3), 308; https://doi.org/10.3390/catal10030308
Submission received: 20 February 2020
/
Revised: 4 March 2020
/
Accepted: 5 March 2020
/
Published: 7 March 2020
(This article belongs to the Special Issue Novel Enzyme and Whole-Cell Biocatalysts)
Abstract
:The question of how to distinguish between lipases and esterases is about as old as the definition of the subclassification is. Many different criteria have been proposed to this end, all indicative but not decisive. Here, the activity of lipases in dry organic solvents as a criterion is probed on a minimal α/β hydrolase fold enzyme, the Bacillus subtilis lipase A (BSLA), and compared to Candida antarctica lipase B (CALB), a proven lipase. Both hydrolases show activity in dry solvents and this proves BSLA to be a lipase. Overall, this demonstrates the value of this additional parameter to distinguish between lipases and esterases. Lipases tend to be active in dry organic solvents, while esterases are not active under these circumstances.
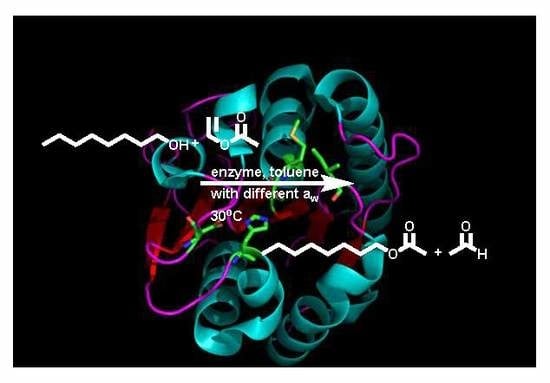
1. Introduction
Lipases and esterases both catalyze the hydrolysis of esters. This has led to the longstanding question: how can we distinguish between a lipase and an esterase? As the simple hydrolysis of an ester does not suffice, a range of different criteria has been suggested [1,2,3,4,5]. (1) The oldest distinction is the kinetic and structural criterion of interfacial activation, which was already described in 1936 [6]. However, several lipases do not fulfill this; in particular, the much-used Candida antarctica lipase B (CALB) does not [7]. (2) Directly linked to the interfacial activation is the lid that covers the active site of many lipases and, via a conformational change, makes the active site more accessible once an interface is present. Again, this is not the case for all lipases [1,2,3,7,8]. (3) Primary sequence data were shown not to be distinctive enough [2]. (4) Substrates and inhibitors, such as Orlistat, can be utilized to distinguish between esterases and lipases but, again, they are not precise. However, the different substrate ranges are indicative. Esterases tend to be capable of the hydrolysis of water-soluble esters and, in general, short and/or branched side chain esters, while lipases hydrolyze triglycerides, apolar esters, substituted with linear side chains, as well as waxes. This is seen as a reliable but not decisive criterion [2,9]. (5) The activity of the enzyme in the presence of (water-miscible) organic solvents has been proposed as a property of lipases, but other enzymes fulfill this criterion, too [2,10,11,12,13,14]. (6) A parameter already investigated some time ago is the activity of lipases in the absence of water, i.e., in modestly polar, water-non-miscible solvents at very low water activities (aw). Out of all enzymes tested, only lipases and the closely related cutinases are active at low aw [1,2,3,4,5,10,15,16,17,18,19]. While not all lipases display this property, it is highly distinctive [20,21].
To probe whether aw is indeed a suitable parameter to distinguish between lipases and esterases and between lipases and other hydrolases in general, we studied the behavior of Bacillus subtilis lipase A (BSLA) [9]. BSLA is a small (181 amino acids, 19 kDa) serine hydrolase (Figure 1). It is neither interfacially activated nor does it have a lid (criteria one and two) [9,22,23,24] and sequence data are not conclusive, but it is a minimal α/β hydrolase fold enzyme [9,23,25]. The substrate range clearly qualifies BSLA as a lipase, as does the stability in the presence of solvents [9,26,27,28,29]. This stability has even been significantly improved in recent mutational studies and BSLA mutants can be very stable in the presence of water-miscible solvents, such as dimethyl sulfoxide (DMSO), dioxane and trifluoroethanol [30,31]. Studies on BSLA in dry organic solvents are, however, missing. As an experimental parameter, we demonstrate the activity of BSLA in dry toluene. Toluene is not water-miscible and has a logP of 2.5 [32]. It is commonly used in organic synthesis and is highly suitable for lipases and also other enzymes with an α/β hydrolase fold. To date, only lipases were shown to be active in toluene with a very low aw [1,3].
Additionally, we extend the structural assignment of the hydrolase character with the GRID-based (Fortran program [33]) Global Positioning System in Biological Space (Bio GPS) investigation [33]. BioGPS utilizes surface shape and polarity as criteria. It is neither based on direct sequence comparison, nor on structure superimposition [33,34]. Earlier studies with this method had placed CALB in both the esterase and lipase group. CALB works extremely well in dry solvents and is, therefore, often applied in reactions that require these conditions, such as dynamic kinetic resolutions [1,3,7,35]. On the other hand, it misses interfacial activation and major conformational changes do not take place when CALB comes into contact with an apolar second phase (see above). As such, BioGPS recognized the ambivalence in the assignment of CALB as a lipase well.
Here, we describe the investigation of BSLA by BioGPS and a comparison to other lipases, in particular CALB. We also probe the lipase character of both BSLA and CALB at different aw. In this manner, new experimental and computational criteria for the esterases and lipases are introduced and investigated.
2. Results
2.1. BioGPS
BioGPS descriptors can be utilized to explore enzyme active site properties and to group them according to their similarities and differences. As such, they can help to explore promiscuous activities. In an earlier study, the character of CALB was investigated in a set of 42 serine hydrolases. The set contained 11 amidases, nine proteases, 11 esterases and 11 lipases, one of them being CALB [33]. Here, we expand this set with BSLA, utilizing the pdb 1R50 with a resolution of 1.4 Å for the structural information (Table S1). Three probes were used to map specific electrostatic and geometrical active site properties. The O-probe evaluates the H-bond donor properties of the enzymes; the N1 probe, on the contrary, evaluates the H-bond acceptor properties and the DRY probe evaluates the hydrophobic interactions [33]. The DRY probe is clearly of special importance for enzymes that accept hydrophobic substrates, as is the case for lipases.
Considering each property separately, the O-probe located BSLA (pdb 1R50) not among the lipases, but in the amidases cluster, together with a number of esterases (Figure S1a). Equally, the N1-probe (Figure S1b) placed BSLA among the amidases. The DRY probe (Figure S1c), again, placed BSLA amongst the amidases and esterases. This is, in all cases, in contrast to CALB (pdb 1TCA) but it should also be noted that Candida rugosa lipase (CRL), a classic lipase with a prominent movement of the lid (criteria one and two) is also always outside the lipase cluster in the different analyses. The previous study ascribed this behavior to the lower hydrophobic nature of the active site of CRL (pdb 1CRL) when compared to the other lipases [8,33].
In the global score, which considers all the mapped properties of the BioGPS together, BSLA can be found firmly among the amidases and esterases (Figure 2), while CALB is in the lipase cluster in the area overlapping with the esterase cluster. CRL, again, is outside the lipase cluster and indeed seems to take a separate position.
BSLA is, according to its substrate range, very clearly a lipase and not an esterase. Amidase activity has to date not been reported for BSLA. While initially surprising, these results also indicate that the study should be extended with an activity assay for amidases.
2.2. Amidase Activity
To probe for amidase activity in BSLA, an amidase activity assay is utilized (Scheme 1). This assay employs benzyl chloroacetamide as standard amide. The released amine reacts with 4-nitro-7-chloro-benzo-2-oxa-1,3-diazole, yielding an adduct that can directly be quantified spectrophotometrically at 475 nm [36].
BSLA showed no activity in this assay. A control experiment with another serine hydrolase, the acyltransferase from Mycobacterium smegmatis (MsAct), was performed. This enzyme is an acyltransferase [37,38] and displays promiscuous amidase activity [39,40]. MsAct exhibited activity in this 24 h assay (> 20% conversion of the 5 mM substrate), showing that even minor, promiscuous activities are detectable. This rules out amidase activity for BSLA and supports the earlier assignment of the enzyme as a lipase.
2.3. BSLA Activity in Dry Organic Solvents
To probe the activity of BSLA at low aw, toluene was used as the solvent and the transesterification of 1-octanol with vinyl acetate was performed as a test reaction (Scheme 2). The use of 1-octanol as a long chain aliphatic compound is a good substrate for lipases [1,2,3,4,5,9] and vinyl acetate is a readily available and widely utilized acyl donor in lipase catalyzed acylation reactions [1,3,41,42]. All reactions were performed with lyophilized BSLA. In parallel, CALB was also tested to ensure direct comparability with one of the most-used lipases. CALB was utilized both as lyophilized enzyme and immobilized as Novozym 435. The latter preparation is most commonly employed, both in the laboratory and on industrial scale [43].
BSLA and CALB were produced by expressing the codon-optimized genes in E. coli BL21 (DE3) within pET22b. Subsequent purification gave both enzymes a good purity (Figure 3). With this expression system, both enzymes are not glycosylated. The CALB Novozym 435 produced and immobilized by Novozymes, however, is expressed in Aspergillus oryzae and it is, therefore, glycosylated [44].
Three different aw were tested < 0.1 to establish whether BSLA shows the activity in dry solvent only observed for lipases, with aw = 0.23 as a low value at which most enzymes lose all their activity and aw = 0.75, an activity at which most enzymes are active [10,21,45,46]. To rigorously ascertain these values of the solvent and reagents, including the internal standard, decane and the enzyme preparations were equilibrated via the vapor phase with dried molecular sieves (activated at elevated temperatures, 5 Å) for aw < 0.1 [47]. For the other aw, the enzyme preparations and the other components were equilibrated via the gas phase with an oversaturated solution of potassium acetate (aw = 0.23) and sodium chloride (aw = 0.75) [48,49,50,51,52]. For all components, the water content was determined by Karl Fischer titration and equilibrations were considered complete when no changes were observed any more (24–48 h, Table 1). As vinyl acetate was found to negatively affect the Karl Fischer titration, it was freshly distilled and dried with activated molecular sieves for 16 h before use. The activity of the different enzyme preparations was also followed with the tributyrin and p-nitrophenol acetate activity assays [2,5,53,54,55,56] during equilibration, to establish optimal equilibration times. For BSLA, a small loss of activity over time was observed, while both CALB preparations were stable.
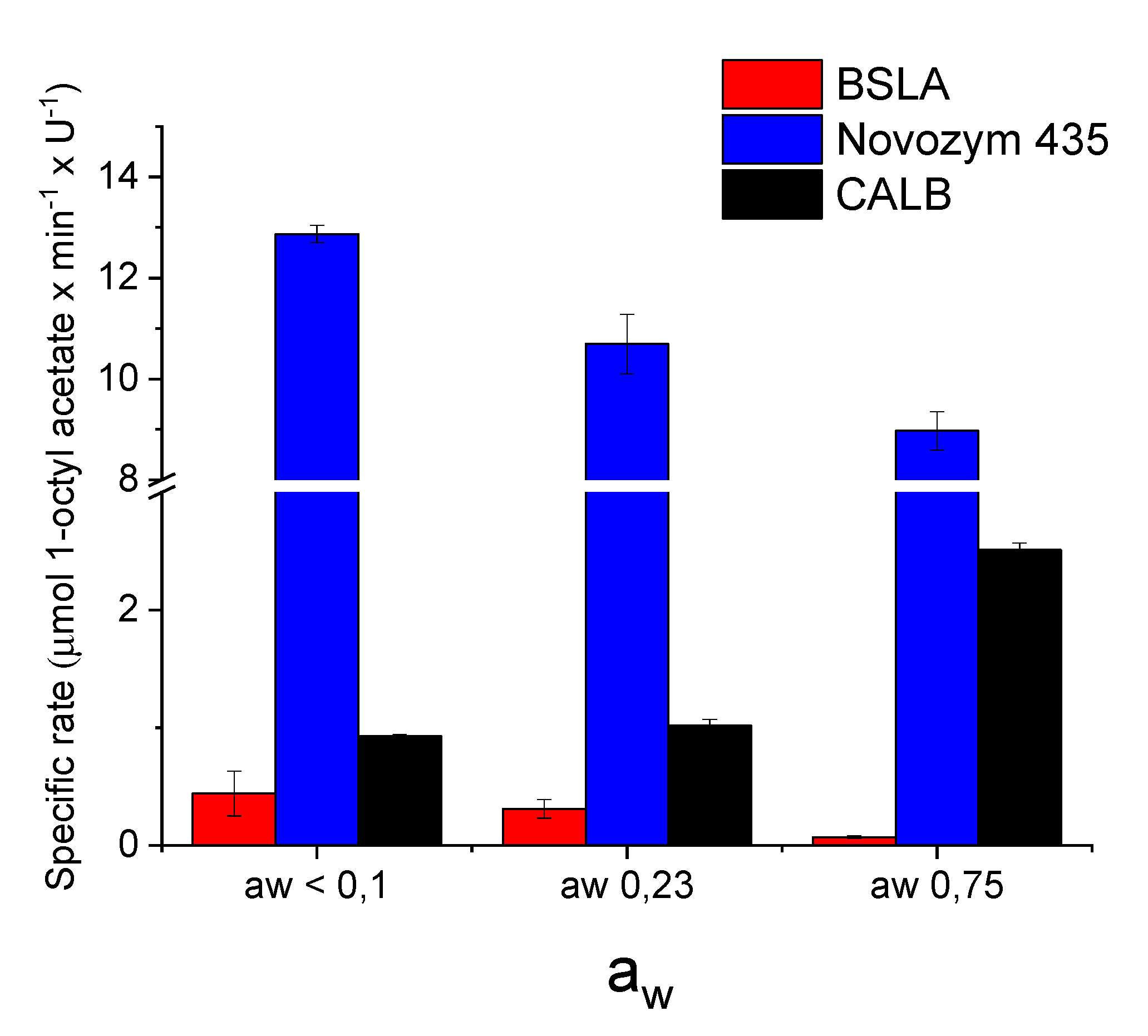
Once reagents and enzymes were equilibrated, the reactions were performed with 100 mM 1-octanol and 500 mM vinyl acetate in previously equilibrated toluene at 30 °C and 1000 rpm (Figure 4). Equal activity of the enzymes (Units) was utilized as determined with the tributyrin activity assay. CALB and, in particular, the well-established commercial preparation of CALB, Novozym 435, performed very well. In both cases, full conversion to 1-octyl acetate was observed. In comparison, BSLA displayed lower conversions (Figure 4). However, the key indicator for a lipase is its activity at low aw. Here, BSLA and Novozym 435 performed best. For the synthesis of 1-octyl acetate, the trend is a reduction in specific rate at higher aw (Figure 5). BSLA is very active in dry solvent, as is Novozym 435. Both display lower activities at higher aw. CALB does not follow this trend.
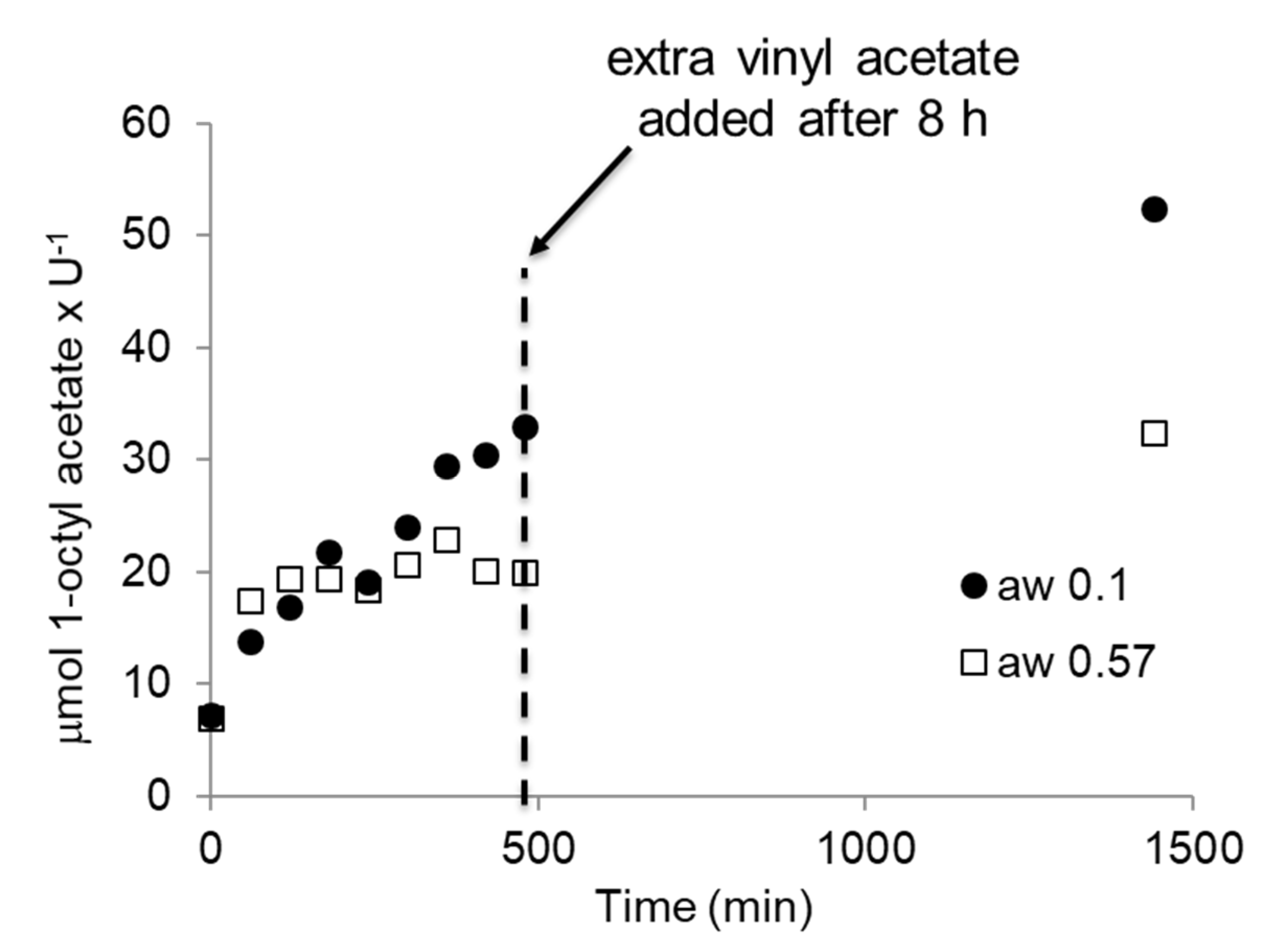
In an earlier study, it had been demonstrated, for different CALB preparations, that this change in activity in the synthesis reaction to 1-octyl acetate can be due to the hydrolysis of the acyl donor vinyl acetate [47]. Therefore, the synthesis reaction at aw < 0.1 was repeated for BSLA with a 1-octanol to vinyl acetate ratio of 1:1 (Figure 6). Almost the same rate and conversion was observed as with the 1:5 ratio, indicating that, at this low aw, essentially no hydrolysis occurred, as was the case for Novozym 435, as reported earlier. Overall, these differences in performance at altered aw can be ascribed to several influences [47,57,58]. Novozyme 435 is an immobilized enzyme and its high activity can be linked to the dispersion of the enzyme on a large surface, promoting its mass transfer and preventing particle aggregation. In contrast, the lyophilized enzymes have a reduced accessibility of the individual enzymes in the preparation. Furthermore, it is well established that immobilized enzymes are better protected against the acetaldehyde that is a side product of the acylation reaction [59]. A difference in susceptibility to acetaldehyde induced deactivation might also cause the alterations in rate between the two pure enzymes. However, similarly, ionization and water clustering can influence the activity [60,61], leading to these alterations. To demonstrate that the observed effect is general, the experiments were repeated, but this time with BSLA that was dried by co-lyophilization with a salt to establish the desired aw [62,63]. The enzyme is now in a different environment and two different aw were established, < 0.1 and 0.57. At < 0.1, very similar results were obtained. Equally, at higher aw, the ester formation slowed down as before, but could be restarted by adding additional vinyl acetate (Figure 7).
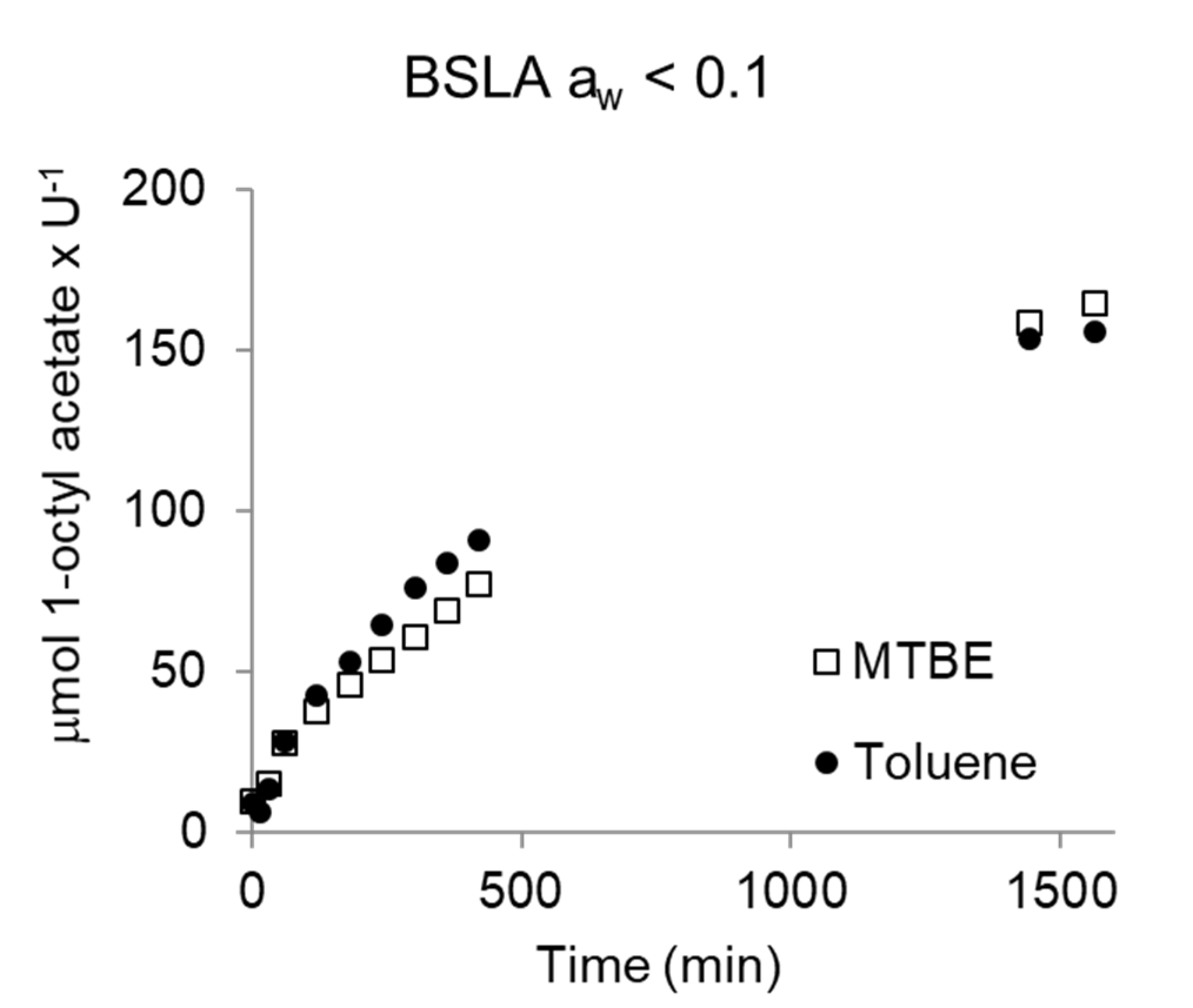
To confirm this activity of BSLA (equilibrated via the gas phase) in dry toluene as a general property, the reaction was repeated in dry methyl-t-butyl ether (MTBE) at the same low aw < 0.1. Enzymes display the same activity in organic solvents when these have the same aw [64]. Indeed, the BSLA-catalyzed esterification displayed a very similar reaction progress in MTBE and toluene (Figure 8). This confirms the activity of BSLA at low aw, in line with the earlier observed catalytic activity of CALB at low aw [47] and of Rhizomucor miehei lipase at very low aw [65].
3. Discussion
Interestingly, the BioGPS analysis seems to identify features of the BSLA active site which are shared by other amidase enzymes. In particular, BSLA seems to share similar H-bond capabilities with amidases, as evidenced by the single-probe clustering. The possible promiscuous amidase activity of BSLA was probed with an amidase activity assay (Scheme 1) [36]. This revealed a complete absence of amidase activity. While indicative, this is not conclusive, as this might also be due to substrate specificity. Amidases are characterized by a developed network of H-bond acceptors and donors as described in previous work [33]. The aromatic moiety of the substrate molecule might thus prevent a good interaction with such H-bond/hydrophilic network. More generally, for amidases, the necessity of a hydrogen bond network that stabilizes the NH hydrogen to suppress its deprotonation was reported earlier [66]. Given the very open active site of BSLA, the minimal serine hydrolase with an α/β hydrolase fold, it is not entirely surprising that this type of hydrogen bond network has never been described for this enzyme.
The test of aw as parameter for the assignment of a serine hydrolase as lipase gave conclusive results. BSLA and CALB displayed good activity at low aw. In line with earlier results, the synthetic catalytic activity of CALB varies depending on the preparation. Earlier studies had shown that the observed synthetic catalytic activity competes with the hydrolytic activity—that is to say, the parallel hydrolysis of vinyl acetate [47]. This can lead to an apparent decrease in synthetic activity, as is observed for Novozym 435 and BSLA at higher aw (Figure 5). This trend has already been reported for Novozym 435 [47]. Just like Novozym 435 [47], BSLA displays essentially no hydrolysis of vinyl acetate at low aw < 0.1. This is confirmed in experiments with a ratio of 1:1 of alcohol to vinyl acetate; a similar rate of synthesis was observed (Figure 6 and Figure 7). The fact that free CALB displays higher synthetic rates at higher aw is also in line with the literature [47]. It had earlier been demonstrated for CALB that the ratio of synthesis to hydrolysis depends on the preparation of the enzyme used and that it increases with aw for purified, free CALB [47]. The activity of BSLA at low aw was proven also with a different solvent, MTBE (Figure 8).
BioGPS is a complimentary computational tool to investigate the character of an enzyme and delivers a useful input to help us explore the scope of an enzyme more thoroughly. The parameter aw is an indicative tool to determine whether an enzyme is a lipase or and esterase. Just like the substrate scope, it is not absolute, but is highly indicative. Essentially, a serine hydrolase that is active at low aw is a lipase and not an esterase, while the reverse statement is not valid. Or, as it was recently summarized: “This long-standing and biased question could be compared to the search for differences between humans and mammals, which implicitly means that one does not consider humans as mammals! Obviously, lipases are a special kind of esterases like humans are a special kind of mammals.” [2].
4. Materials and Methods
4.1. Materials
Chemicals and Enzymes
1-propanol, 1-octanol, toluene extra dry, decane, p-nitrophenylbutyrate, p-nitrophenol, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), tributyrin and 2-methyl-2-propanol, butyric acid and caprylic acid were purchased from Sigma-Aldrich (Schnelldorf, Germany) and Acros (Geel, Belgium), and used without previous purification. Vinyl acetate was purchased from Sigma-Aldrich and distilled before use. Novozym 435 (immobilized lipase B from Candida antarctica) was made available by Novozymes (Bagsværd, Denmark). Bovine serum albumin protein and lysozyme from chicken egg whites were purchased from Sigma Aldrich. Bradford reagent was purchased from Biorad (Hercules, C.A., USA). Medium and buffer components were purchased from BD, Merck (Darmstadt, Germany) or J.T. Baker (Geel, Belgium).
Strains and Plasmids
Strains Escherichia coli (E. coli) HB2151 and E. coli HB2151 pCANTAB 5E bsla were kindly provided by Prof. Bauke Dijkstra and Prof. Wim Quax, University of Groningen, the Netherlands. Strains Escherichia coli (E. coli) BL21 (DE3), E. coli TOP10 and plasmid pET22b(+) were utilized for all further work.
4.2. Methods
Cloning pET22bbsla and pET22bcalb
The gene of BSLA (as confirmed by sequencing ID: CP011115.1, range from 292296 to 292841, protein AKCA5803.1) was amplified by PCR from vector pCANTAB 5E BSLA using primers BSLA F: 5′-CCTTTCTATGCGGCCCAGC–3′ and BSLA + XhoI R: 5′-CCGCTCGAGCGCCTTCGTATTCTGG-3′. Thereby, restriction site XhoI was introduced for subsequent cloning of BSLA (NcoI, XhoI) into vector pET22b+ (in frame with pelB and His-tag signals). The resulting vector was named pET22bBSLA. The wild type CALB was synthesized by BaseClear (Leiden, The Netherlands). The codon-optimized genes were cloned into pET22b(+) using the NcoI and NotI restriction sites as previously described [67], in order to be in frame with the pelB sequence and a C-terminal His-tag of the plasmid.
Expression and Purification of BSLA
This protocol was adapted from [24]. A freshly grown colony of E. coli HB 2151 pCANTAB 5E BSLA was used to inoculate a 1 L shake flask containing 100 mL of 2xTY medium (1.6% w/v bactotryptone, 1% w/v bacto yeast extract and 0.5% w/v sodium chloride), ampicillin (100 µg/mL final concentration) and isopropyl-β-d-galactopyranoside (IPTG, 1 mM final concentration). After 16 h at 28 °C and 150 rpm (Innova Incubator, Hamburg, Germany) the cells were harvested and washed with 10 mM Tris Buffer pH 7.4 and stored at −20 °C. The periplasm isolation protocol was adapted from [67] and consisted of the resuspension of the overexpressed cells in 1 mL of 10 mM Tris buffer pH 8.0 containing sucrose (25 % w/v), EDTA (2 mM) and lysozyme (0.5 mg/mL). After incubation on ice for 20 min, 250 µL of 10 mM Tris buffer pH 8.0 containing sucrose (20% w/v) and MgCl2 (125 mM) was added. The suspension was centrifuged and the supernatant containing the periplasmic fraction was desalted using a PD10 column (GE, Healthcare, New York, N.Y., USA) to 100 mM potassium phosphate buffer pH 7.4. Afterwards, the solution was shock-frozen with liquid nitrogen and stored at −20 °C for future biocatalysis applications. BSLA was purified mainly from the media. First, proteins were precipitated by adding 50% v/v saturated ammonium sulphate (2.8 M final concentration) for 5 h at 4 °C. After centrifugation, the solid fraction was dissolved in 100 mL of 100 mM potassium phosphate buffer pH 7.4, filtered through a 0.45 μm filter and loaded into a 5 mL His-Trap previously equilibrated column (GE Healthcare) using a NGC chromatography system (BIORAD, Hercules, C.A., USA). The loaded proteins were washed with equilibration buffer potassium phosphate (100 mM, pH 7.4) containing 500 mM NaCl and 20 mM imidazole. The His-tagged BSLA was eluted with a linear gradient from 0–100% potassium phosphate (100 mM, pH 7.4) containing 500 mM NaCl and 500 mM imidazole. The progress of the purification was monitored at 280 nm. Fractions containing the target protein (as confirmed by SDS-PAGE and activity assay, 50–60% of the gradient) were combined, concentrated and desalted with a PD-10 column (GE Healthcare) to potassium phosphate buffer (100 mM, pH 7.4). The purified enzyme (68–148 μg/L medium) was aliquoted (2.5 U/vial, units determined by tributyrin assay) and freeze dried for 16 h, −80 °C and stored at −20 °C under nitrogen atmosphere.
Protein Sequence of BSLA-His (AKCA5803.1):
MAAEHNPVVMVHGIGGASFNFAGIKSYLVSQGWSRDKLYAVDFWDKTGTNYNNGPVLSRFVQKVLDETGAKKVDIVAHSMGGANTLYYIKNLDGGNKVANVVTLGGANRLTTGKALPGTDPNQKILYTSIYSSADMIVMNYLSRLDGARNVQIHGVGHIGLLYSSQVNSLIKEGLNGGGQNTKALEHHHHHH
Expression and Purification of CALB
An LB-Amp plate (100 µg/mL) was used to freshly grow E. coli BL21 (DE3) pET22bCALB from a −80 °C DMSO stock. After incubation at 37 °C for 16 h, a single colony was used to inoculate a 5 mL LB-Amp (100 µg/mL) preculture and grown for 8 h at the same temperature. Large-scale expressions were carried out in 0.5 L of ZYM-5052 media (placed in 2 L shake flask), 2% v/v of the preculture was used for inoculation. After 17 h expression at 22 °C and 170 rpm, an optical density (600 nm) of approximately 3 was obtained in all cases. Afterwards, cells were spun down, washed with 10 mM potassium phosphate buffer pH 7.4 and stored at −20 °C. ZYM-5052 medium [68]: The main cultures were grown in ZYM-5052 medium containing 50 mL 50xM (Na2HPO4·12H2O 448 g/L, KH2PO4 170 g/L, NH4Cl 134 g/L, Na2SO4 35.5 g/L), 20 mL 50x5052 (100 g/L α-D-lactose, 250 g/L glycerol and 25 g/L glucose dissolved in ddH2O) and 2 mL of MgSO4 solution (1 M in ddH2O) and filled to 1 L with ZY medium (casamino acids 10 g/L—tryptone in this case—yeast extract 5 g/L). Additionally, 0.2 mL of trace element solution was added to the media. The purification of His-tagged CALB was performed from the periplasmic fraction, as described for BSLA in the previous sections.
Protein Sequence of CALB-His (Sequence ID: 4K6G_A):
MALPSGSDPAFSQPKSVLDAGLTCQGASPSSVSKPILLVPGTGTTGPQSFDSNWIPLSTQLGYTPCWISPPPFMLNDTQVNTEYMVNAITALYAGSGNNKLPVLTWSQGGLVAQWGLTFFPSIRSKVDRLMAFAPDYKGTVLAGPLDALAVSAPSVWQQTTGSALTTALRNAGGLTQIVPTTNLYSATDEIVQPQVSNSPLDSSYLFNGKNVQAQAVCGPLFVIDHAGSLTSQFSYVVGRSALRSTTGQARSADYGITDCNPLPANDLTPEQKVAAAALLAPAAAAIVAGPKQNCEPDLMPYARPFAVGKRTCSGIVTPAAALEHHHHHH
Bradford Assay
Total protein concentration was determined using Bradford reagent in a microtiter plate (MTP) reader format (96 well plates) [69]. Properly diluted samples were mixed with Bradford reagent (5x), incubated at room temperature (RT) for 5 min and the absorbance measured at 595 nm (in triplicate). The calibration curve was carried out using bovine serum albumin protein, as is standard.
Lipase Activity: Tributyrin Assay
A tributyrin assay for determining lipase activity was performed according to the literature [54]. The assay is based on pH change by acid formation when tributyrin is hydrolyzed by the enzyme. p-Nitrophenol was used as pH indicator (colorless at pH 5.5 and yellow at pH 7.5) and the acid concentration was determined by a calibration curve with known amounts of butyric acid (from 0 mM to 40 mM). A negative control was performed by adding buffer instead of an enzyme sample. The substrate consumption (0.8 mM initial concentration) was monitored at 410 nm, 30 °C for 15 min, every 38 s by a microtiter plate reader (in 96-well plates, Synergy 2, BioTek, Winooski, V.T., USA). Plates were shaken for 5 s before every read. The different buffers needed for this assay contained 2.5 mM 3-(N-morpholino)propanesulfonic acid (MOPS) (pH 7.2), CHAPS (to dissolve acids) and β-cyclodextrin (to dissolve acids into the solution, to increase the linearity). The activity was determined in U, which is equivalent to μmol acid formed per minute. The assays were done in triplicate. For performing this assay with immobilized enzymes, a larger scale (3 mL) in glass vials with a magnetic stirrer was applied. These were placed on a stirring platform and, for Novozym 435, samples (120 μL) were taken over time and placed in a 96-well plate. If desired, the assay can also be performed with Trioctanoin.
Esterase/Lipase Activity: p-Nitrophenol Assay
This protocol was adapted from [53] to an MTP reader equipped for 96 well plates. The enzymatic hydrolysis of p-nitrophenyl butyrate with the concomitant formation of p-nitrophenol was monitored at 405 nm, 37 °C and recorded for 30 min. For this, a calibration curve of p-nitrophenol in potassium phosphate buffer (100 mM, pH 7.4) was prepared (levels from 0-500 µM, 200 µL total volume, in triplicate) and control reactions without enzyme extracts were performed. Lyophilized cell-free extract or pure enzymes were re-dissolved in potassium phosphate buffer (100 mM, pH 7.4) (approximately 20–30 mg/mL) and proper dilutions were added into a preheated potassium phosphate buffer (100 mM, pH 7.4) solution containing 3 mM p-nitrophenyl butyrate. The esterase activity measured was corrected by subtracting the activity observed in the controls (no enzyme). By definition, one unit of enzyme (U) is equivalent to 1 µmol of p-nitrophenol formed per minute.
Karl Fischer Titration
A Metrohm KF Coulometer Karl Fischer titration setup was used, according to the manufacturer’s instructions, to determine the water content in ppm. Samples (100 μL) were taken from the solvent and injected into the system in duplicate. In general, master mixes of toluene after equilibration with aw < 0.1, aw 0.23 and aw 0.75 contained 20, 120 and 360 ppm respectively. A deviation of 5–10 ppm per sample was observed.
Equilibration of Solvents/Enzymes to the Desired aw
All materials, reagents, enzymes and solvents used for the biocatalytic reactions were carefully dried and kept under nitrogen atmosphere with molecular sieves (5 Å) at all times. In all cases, the water content was monitored by Karl Fischer titration and as standard parameter compounds with a water content below 100 ppm were considered dry and suitable for the reaction.
Vapor Phase Method
Oversaturated salt solutions and activated molecular sieves were used to equilibrate the solvents and enzymes needed in the transesterification reaction with a desired water activity [47,48,49,50]. In the case of working with dry systems, a master mix was prepared including solvent, substrates (without vinyl acetate) and internal standard, all components previously dried with activated molecular sieves achieving aw < 0.1. Lyophilized BSLA, Novozym 435 and CALB were dried over silica in desiccators under a vacuum at room temperature (20–25 °C) for 24, 48 and 24 h, respectively. In order to achieve higher water activities, the master mix and the enzymes were equilibrated over saturated salt solutions of potassium acetate (KAc) and sodium chloride (NaCl) at 30 °C for 48 h, resulting in aw 0.23 and aw 0.75 at 30 °C, respectively [51]. As exceptions, BSLA and free CALB were equilibrated for a shorter period of only 24 h.
Salt Pairs Method
The protocol was adapted from [62]. The enzymes were lyophilized with anhydrous salts (Na2HPO4 or NaAc) in a ratio 1:99 (3 mg pure BSLA or CALB enzyme and 297 mg of the respective salt). For the background reaction (no-enzyme), only lyophilized salts were added. An amount of 10 mg of the co-lyophilized enzyme was added under a nitrogen atmosphere to the previously dried reaction components (except vinyl acetate) and a specific amount of water was introduced under the nitrogen atmosphere. The moles of water added to the reaction mixture were calculated in order to generate the couple of hepta- and dihydrated phosphates in the case of Na2HPO4 (5 moles of water per mol of salt, aw ~ 0.57) and the couple of tri- and anhydrous acetate in the case of NaAc (1.5 moles of water per mol of salt, aw ~ 0.25) [50]. After overnight equilibration, the last substrate was added (freshly distilled and dry vinyl acetate) to begin the reaction. In the case of the dry system, no water was added (aw < 0.1).
Transesterification Catalyzed by Lipases in Organic Solvents under Fixed Water Activities
The protocol was adapted from [47]. The reaction conditions included substrates 1-propanol, 1-octanol, 2-octanol or benzylacohol (100 mM), vinyl acetate freshly distilled (1 or 5 equiv. in respect to the initial substrate concentration) and decane (ISTD, 500 mM final concentration). A range of 0.5–1 U of purified enzymes, 1 mg of immobilized Novozym 435 were tested as catalysts. Toluene or methyl-t-butyl ether were used as media (1 mL total volume in GC airtight vials). Reactions were carried out for 25 h, 30 °C and 1000 rpm (thermoblock Eppendorf, Hamburg, Germany). Negative controls were run for both substrates in absence of enzyme. All reactions were performed in duplicate and monitored over time by gas chromatography.
Analytics: GC and GC-MS
Gas chromatograph (GC) and gas chromatograph-mass spectrometry (GC-MS) methods were adapted from [47]. Samples were injected in a gas chromatograph (GC-2014, Shimadzu, Kyoto, Japan) equipped with a CP Sil 5 column (50 m × 0.53 mm × 1.0 um). Injector and detector temperatures were set to 340 and 360 °C, respectively. The initial column temperature was set to 35 °C for 5 min, followed by an increase of 15 °C/min up to 60 °C for 0.5 min and 15 °C/min up to 160 °C and hold for 2 min. Finally, a burnout was introduced, 30 °C/min up to 325 °C. The retention times for 1-propanol, vinyl acetate, toluene, decane, 1-octanol and 1-octylacetate were 1.69, 1.87, 6.64, 10.52, 11.19 and 12.79 min, respectively. To confirm the product’s structure, samples were also injected in a gas chromatograph-mass spectrometer (GC-MS QP2010s, Kyoto, Japan) equipped with a CP Sil 5 (25 m × 0.25 mm × 0.4 µm). The injector, interface and ion source temperatures were set to 315, 250 and 200 °C, respectively. The retention times for vinyl acetate, 1-octanol and 1-octylacetate were 1.78, 11.57 and 12.93 min, respectively.
Amidase Activity Assay: Hydrolysis of Benzyl Chloroacetamide
This protocol was adapted from the literature [36]. The biocatalysis conditions included a total volume 500 μL in a 2 mL Eppendorf tube containing 5 mM benzyl chloroacetamide (stock solution of 500 mM in THF), 100 μg/mL enzyme (as quantified by Bradford assay), in 25 mM potassium phosphate buffer pH 7.0 with 10 % v/v THF. The conversion was carried out for 24 h at 37 °C and 500 rpm. Afterwards, the derivatization of 200 μL of reaction mixture was carried out with 50 μL of NBDCl (20 mM in DMSO) for 1 h, at 37 °C and 500 rpm. UV detection at 475 nm was performed.
BSLA Structure
The structure of BSLA was taken from the Protein Data Bank (PDB). The structure downloaded (1R50) was treated by removing all molecules but the protein chain with the software PyMOL. The thus-generated structure was used for visual inspection with PyMOL as well as input for the BioGPS analysis.
4.3. BioGPS Computational Analysis
The BioGPS analysis and projection was taken from the previous published work. The BSLA BioGPS analysis was performed using the BioGPS software provided by Molecular Discovery Ltd. (Borehamwood, Hertfordshire, UK) by projecting the enzyme according to its active site properties in the previously performed analysis. The identification of the BSLA active site and the calculation of its properties has been performed as previously described. Specifically, FLAPsite was used for automatic active site identification. The active site was mapped using a GRID approach and the resulting computed properties were considered as electrondensity-like fields centered on each atom, which correspond to the so-called pseudo-molecular interaction fields (pseudo-MIFs). Four different properties were mapped: the active site shape (H probe), H-bond donor properties (O probe), H-bond acceptor capabilities (N1 probe), and hydrophobicity (DRY probe). The magnitude of the interaction of the N1 and O probes also includes, implicitly, information about the charge contribution, as these probes already have a partially positive and negative charge, respectively. The pseudo-MIF points were filtered, by means of a weighted energy-based and space-coverage function, and then used for the generation of quadruplets obtained from all possible combinations of the four pseudo-MIF points. Thus, the BSLA active site was described by a series of quadruplets. Finally, BSLA was projected according to its series of quadruples and scored by the previously performed BioGPS analysis.
5. Conclusions
The longstanding question “what differentiates lipases from esterases?” has led to a list of six parameters that are indicative, but not decisive. Here, we have probed BioGPS and aw as parameters to distinguish between lipases and esterases, utilizing the minimal serine hydrolase with an α/β fold, BSLA, as a test enzyme. While BioGPS has been used successfully to address similar questions earlier, it was not indicative in this case. The clear assignment of BSLA as either esterase or lipase was a challenging task. The high catalytic activity of BSLA at low aw clearly demonstrated this serine hydrolase to be a lipase. In future studies, activity at low aw should, therefore, be utilized to support the differentiation of lipases and esterases.
Supplementary Materials
The following file is available online at https://www.mdpi.com/2073-4344/10/3/308/s1, Figure S1: Bio GPS of 43 serine hydrolases, for BSLA the data of pdb 1R50 were utilized: (a) H-bond donor; (b) H-bond acceptor; (c) Hydrophobicity; Table S1: Enzymes utilized for the Bio GPS study with the relevant PDB codes.
Author Contributions
Conceptualization, U.H., L.G., P.B. and G.T.; methodology, P.B., V.F. and G.T.; validation, N.v.M., E.A., P.B. and V.F.; formal analysis, P.B., U.H. and L.G.; investigation, N.v.M., E.A., P.B. and V.F.; resources, U.H. and L.G.; data curation, N.v.M., E.A., P.B. and V.F.; writing—original draft preparation, U.H., P.B. and V.F.; writing—review and editing, U.H. and L.G.; visualization, U.H. and L.G.; supervision, P.B., U.H. and L.G.; project administration, U.H. and L.G.; funding acquisition, U.H. and L.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by BE-BASIC, grant number FES-0905 to P.B. and G.T. L.G. received funding from the University of Trieste, FRA 2018.
Acknowledgments
Excellent technical support by the technicians of the BOC group and by Linda Otten is gratefully acknowledged. Bauke Dijkstra and Wim Quax, University of Groningen, the Netherlands kindly provided the Escherichia coli (E. coli) HB2151 and E. coli HB2151 pCANTAB 5E bsla. L.G. is grateful to Molecular Discovery Ltd. for providing software access.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bornscheuer, U.T.; Kazlauskas, R. Hydrolases in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2005; pp. 61–184. [Google Scholar]
- Ben Ali, Y.; Verger, R.; Abousalham, A. Lipases or Esterases: Does It Really Matter? Toward a New Bio-Physico-Chemical Classification. In Lipases and Phospholipases: Methods and Protocols; Book Series: Methods in Molecular Biology; Sandoval, G., Ed.; Springer Science+Business Media: New York, NY, USA, 2012; Volume 861, pp. 31–51. [Google Scholar]
- Paravidino, M.; Bohm, P.; Gröger, H.; Hanefeld, U. Hydrolysis and Formation of Carboxylic Acid Esters. In Enzyme Catalysis in Organic Synthesis; Wiley: Hoboken, NJ, USA, 2012; pp. 249–362. [Google Scholar]
- Arpigny, J.L.; Jaeger, K.E. Bacterial lipolytic enzymes: Classification and properties. Biochem. J. 1999, 343, 177–183. [Google Scholar] [CrossRef]
- Jaeger, K.-E.; Kovacic, F. Determination of Lipolytic Enzyme Activities. In Pseudomonas Methods and Protocols; Book Series: Methods in Molecular Biology; Filloux, A., Ramos, J.L., Eds.; Springer Science+Business Media: New York, NY, USA, 2014; Volume 1149, pp. 111–134. [Google Scholar]
- Holwerda, K.; Verkade, P.E.; De Willigen, A.H.A. Vergleichende Untersuchungen über die Verseifungsgeschwindigkeit einiger Einsäuriger Triglyceride unter Einfluss von Pankreasextrakt. I. Der Einfluss des Verteilungszustandes der Triglyceride auf die Verseifungsgeschwindigkeit. Rec. Trav. Chim. Pays Bas 1936, 55, 43–57. [Google Scholar] [CrossRef]
- Kirk, O.; Christensen, M.W. Lipases from Candida antarctica: Unique Biocatalysts from a Unique Origin. Org. Proc. Res. Develop. 2002, 6, 446–451. [Google Scholar] [CrossRef]
- Grochulski, P.; Li, Y.; Schrag, J.D.; Cygler, M. Two conformational states of Candida rugosa lipase. Protein Sci. 1994, 3, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Eggert, T.; van Pouderoyen, G.; Pencreac’h, G.; Douchet, I.; Verger, R.; Dijkstra, B.W.; Jaeger, K.-E. Biochemical properties and three-dimensional structures of two extracellular lipolytic enzymes from Bacillus subtilis. Colloids Surf. B Biointerfaces 2002, 26, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Adlercreutz, P. Comparison of lipases and glycoside hydrolases as catalysts in synthesis reactions. Appl. Microbiol. Biotechnol. 2017, 101, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Lopes, D.B.; Fraga, L.P.; Fleuri, L.F.; Macedo, G.A. Lipase and esterase—To what extent can this classification be applied accurately? Ciênc. Tecnol. Aliment. Camp. 2011, 31, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Berlemont, R.; Spee, O.; Delsaute, M.; Lara, Y.; Schuldes, J.; Simon, C.; Power, P.; Daniel, R.; Galleni, M. Novel organic solvent-tolerant esterase isolated by metagenomics: Insights into the lipase/esterase classification. Rev. Argent. Microbiol. 2013, 45, 3–12. [Google Scholar]
- Adlercreutz, P. Fundamentals of Biocatalysis in Neat Organic Solvents. In Organic Synthesis with Enzymes in Non-Aqueous Media; Carrea, G., Riva, S., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 3–24. [Google Scholar]
- Serdakowski, A.L.; Dordick, J.S. Activating Enzymes for Use in Organic Solvents. In Organic Synthesis with Enzymes in Non-Aqueous Media; Carrea, G., Riva, S., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 47–74. [Google Scholar]
- Svensson, I.; Wehtje, E.; Adlercreutz, P.; Mattiasson, B. Effects of water activity on reaction rates and equilibrium positions in enzymatic esterifications. Biotechnol. Bioeng. 1994, 44, 549–556. [Google Scholar] [CrossRef]
- Wehtje, E.; Svensson, I.; Adlercreutz, P.; Mattiasson, B. Continuous control of water activity during biocatalysis in organic media. Biotechnol. Tech. 1993, 7, 873–878. [Google Scholar] [CrossRef]
- Ferrario, V.; Pellis, A.; Cespugli, M.; Guebitz, G.; Gardossi, L. Nature Inspired Solutions for Polymers: Will Cutinase Enzymes Make Polyesters and Polyamides Greener? Catalysts 2016, 6, 205. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Ferrario, V.; Zartl, B.; Brandauer, M.; Gamerith, C.; Acero, E.H.; Ebert, C.; Gardossi, L.; Guebitz, G.M. Enlarging the tools for efficient enzymatic polycondensation: Structural and catalytic features of cutinase 1 from Thermobifida cellulosilytica. Catal. Sci. Technol. 2016, 6, 3430–3442. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Ferrario, V.; Cespugli, M.; Corici, L.; Guarneri, A.; Zartl, B.; Acero, E.H.; Ebert, C.; Guebitz, G.; Gardossi, L. Fully renewable polyesters via polycondensation catalyzed by Thermobifida cellulosilytica cutinase 1: An integrated approach. Green Chem. 2017, 19, 490–502. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Peilow, A.D.; Macrae, A.R. Relationship between water activity and catalytic activity of lipases in organic media. Effects of supports, loading and enzyme preparation. Eur. J. Biochem. 1994, 222, 461–466. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Peilow, A.D.; Macrae, A.R. Lipases from different sources vary widely in dependence of catalytic activity on water activity. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1992, 1122, 143–146. [Google Scholar] [CrossRef]
- Gupta, R.; Gupta, N.; Rathi, P. Bacterial lipases: An overview of production, purification and biochemical properties. Appl. Microbiol. Biotechnol. 2004, 64, 763–781. [Google Scholar] [CrossRef]
- Eggert, T.; van Pouderoyen, G.; Dijkstra, B.W.; Jaeger, K.-E. Lipolytic enzymes LipA and LipB from Bacillus subtilis differ in regulation of gene expression, biochemical properties, and three-dimensional structure. FEBS Lett. 2001, 502, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Boersma, Y.L.; Pijning, T.; Bosma, M.S.; van der Sloot, A.; Godinho, L.F.; Dröge, M.J.; Winter, R.T.; van Pouderoyen, G.; Dijkstra, B.W.; Quax, W.J. Loop Grafting of Bacillus subtilis Lipase A: Inversion of Enantioselectivity. Chem. Biol. 2008, 15, 782–789. [Google Scholar] [CrossRef] [Green Version]
- van Pouderoyen, G.; Eggert, T.; Jaeger, K.-E.; Dijkstra, B.W. The crystal structure of Bacillus subtilis lipase: A minimal α/β hydrolase fold enzyme. J. Mol. Biol. 2001, 309, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Augustyniak, W.; Brzezinska, A.A.; Pijning, T.; Wienk, H.; Boelens, R.; Dijkstra, B.W.; Reetz, M.T. Biophysical characterization of mutants of Bacillus subtilis lipase evolved for thermostability: Factors contributing to increased activity retention. Protein Sci. 2012, 21, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Kamal, Z.; Ahmad, S.; Molugu, T.R.; Vijayalakshmi, A.; Deshmukh, M.V.; Sankaranarayanan, R.; Rao, N.M. In Vitro Evolved Non-Aggregating and Thermostable Lipase: Structural and Thermodynamic Investigation. J. Mol. Biol. 2011, 413, 726–741. [Google Scholar] [CrossRef]
- Ahmad, S.; Kamal, Z.; Sankaranarayanan, R.; Rao, N.M. Thermostable Bacillus subtilis Lipases: In Vitro Evolution and Structural Insight. J. Mol. Biol. 2008, 381, 324–340. [Google Scholar] [CrossRef]
- Rajakumara, E.; Acharya, P.; Ahmad, S.; Sankaranaryanan, R.; Rao, N.M. Structural basis for the remarkable stability of Bacillus subtilis lipase (Lip A) at low pH. Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 302–311. [Google Scholar] [CrossRef]
- Frauenkron-Machedjou, V.J.; Fulton, A.; Zhao, J.; Weber, L.; Jaeger, K.E.; Schwaneberg, U.; Zhu, L. Exploring the full natural diversity of single amino acid exchange reveals that 40–60% of BSLA positions improve organic solvents resistance. Bioresour. Bioprocess. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Markel, U.; Zhu, L.; Frauenkron-Machedjou, V.J.; Zhao, J.; Bocola, M.; Davari, M.D.; Jaeger, K.-E.; Schwaneberg, U. Are Directed Evolution Approaches Efficient in Exploring Nature’s Potential to Stabilize a Lipase in Organic Cosolvents? Catalysts 2017, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Laane, C.; Boeren, S.; Vos, K.; Veeger, C. Rules for optimization of biocatalysis in organic solvents. Biotechnol. Bioeng. 1987, 30, 81–87. [Google Scholar] [CrossRef]
- Ferrario, V.; Siragusa, L.; Ebert, C.; Baroni, M.; Foscato, M.; Cruciani, G.; Gardossi, L. BioGPS Descriptors for Rational Engineering of Enzyme Promiscuity and Structure Based Bioinformatic Analysis. PLoS ONE 2014, 9, 109354. [Google Scholar] [CrossRef]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef]
- Veum, L.; Kanerva, L.T.; Halling, P.J.; Maschmeyer, T.; Hanefeld, U. Optimisation of the Enantioselective Synthesis of Cyanohydrin Esters. Adv. Synth. Catal. 2005, 347, 1015–1021. [Google Scholar] [CrossRef]
- Henke, E.; Bornscheuer, U.T. Fluorophoric Assay for the High-Throughput Determination of Amidase Activity. Anal. Chem. 2003, 75, 255–260. [Google Scholar] [CrossRef]
- Mathews, I.; Soltis, M.; Saldajeno, M.; Ganshaw, G.; Sala, R.; Weyler, W.; Cervin, M.A.; Whited, G.; Bott, R. Structure of a Novel Enzyme That Catalyzes Acyl Transfer to Alcohols in Aqueous Conditions. Biochemistry 2007, 46, 8969–8979. [Google Scholar] [CrossRef] [PubMed]
- Mestrom, L.; Claessen, J.G.R.; Hanefeld, U. Enzyme-Catalyzed Synthesis of Esters in Water. ChemCatChem 2019, 11, 2004–2010. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Hendil-Forssell, P.; Martinelle, M.; Berglund, P. One-pot biocatalytic amine transaminase/acyl transferase cascade for aqueous formation of amides from aldehydes or ketones. Catal. Sci. Technol. 2016, 6, 2897–2900. [Google Scholar] [CrossRef] [Green Version]
- Contente, M.L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Flow-based enzymatic synthesis of melatonin and other high value tryptamine derivatives: A five-minute intensified process. Green Chem. 2019, 21, 3263–3266. [Google Scholar] [CrossRef] [Green Version]
- Hanefeld, U. Reagents for (ir)reversible enzymatic acylations. Org. Biomol. Chem. 2003, 1, 2405–2415. [Google Scholar] [CrossRef]
- Paravidino, M.; Hanefeld, U. Enzymatic acylation: Assessing the greenness of different acyl donors. Green Chem. 2011, 13, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, E.L.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef] [Green Version]
- Basso, A.; Braiuca, P.; Cantone, S.; Ebert, C.; Linda, P.; Spizzo, P.; Caimi, P.; Hanefeld, U.; Degrassi, G.; Gardossi, L. In Silico Analysis of Enzyme Surface and Glycosylation Effect as a Tool for Efficient Covalent Immobilisation of CalB and PGA on Sepabeads®. Adv. Synth. Catal. 2007, 349, 877–886. [Google Scholar] [CrossRef]
- Ma, L.; Persson, M.; Adlercreutz, P. Water activity dependence of lipase catalysis in organic media explains successful transesterification reactions. Enzym. Microb. Technol. 2002, 31, 1024–1029. [Google Scholar] [CrossRef]
- Paravidino, M.; Sorgedrager, M.J.; Orru, R.V.A.; Hanefeld, U. Activity and Enantioselectivity of the Hydroxynitrile Lyase MeHNL in Dry Organic Solvents. Chem. Eur. J. 2010, 16, 7596–7604. [Google Scholar] [CrossRef] [Green Version]
- Secundo, F.; Carrea, G.; Soregaroli, C.; Varinelli, D.; Morrone, R. Activity of different Candida antarctica lipase B formulations in organic solvents. Biotechnol. Bioeng. 2001, 73, 157–163. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Macrae, A.R. Reaction rate with suspended lipase catalyst shows similar dependence on water activity in different organic solvents. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1992, 1118, 218–222. [Google Scholar] [CrossRef]
- Fontes, N.; Partridge, J.; Halling, P.J.; Barreiros, S. Zeolite molecular sieves have dramatic acid-base effects on enzymes in nonaqueous media. Biotechnol. Bioeng. 2002, 77, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Halling, P.J. Salt hydrates for water activity control with biocatalysts in organic media. Biotechnol. Tech. 1992, 6, 271–276. [Google Scholar] [CrossRef]
- Greenspan, L. Humidity fixed points of binary saturated aqueous solutions. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem. 1977, 81, 89–96. [Google Scholar] [CrossRef]
- Johnson, J.R.; Affsprung, H.E.; Christian, S.D. The molecular complexity of water in organic solvents. Part II. J. Chem. Soc. A 1966, 77–78. [Google Scholar] [CrossRef]
- Gupta, R.; Rathi, P.; Gupta, N.; Bradoo, S. Lipase assays for conventional and molecular screening: An overview. Biotechnol. Appl. Biochem. 2003, 37, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Díaz, E.; Rodríguez, J.A.; de los Ángeles Camacho-Ruiz, M.; Mateos-Díaz, J.C. High-Throughput Screening Method for Lipases/Esterases. In Lipases and Phospholipases: Methods and Protocols; Book Series: Methods in Molecular Biology; Sandoval, G., Ed.; Springer Science + Business Media: New York, NY, USA, 2012; Volume 861, pp. 89–100. [Google Scholar]
- Híreš, M.; Rapavá, N.; Šimkovič, M.; Varečka, Ľ.; Berkeš, D.; Kryštofová, S. Development and Optimization of a High-Throughput Screening Assay for Rapid Evaluation of Lipstatin Production by Streptomyces Strains. Curr. Microbiol. 2018, 75, 580–587. [Google Scholar] [CrossRef]
- Nalder, T.D.; Ashton, T.D.; Pfeffer, F.M.; Marshall, S.N.; Barrow, C.J. 4-Hydroxy-N-propyl-1,8-naphthalimide esters: New fluorescence-based assay for analysing lipase and esterase activity. Biochimie 2016, 128–129, 127–132. [Google Scholar] [CrossRef]
- Corici, L.; Ferrario, V.; Pellis, A.; Ebert, C.; Lotteria, S.; Cantone, S.; Voinovich, D.; Gardossi, L. Large scale applications of immobilized enzymes call for sustainable and inexpensive solutions: Rice husk as renewable alternative to fossil-based organic resins. RSC Adv. 2016, 6, 63256–63270. [Google Scholar] [CrossRef]
- Secundo, F.; Carrea, G. Lipase activity and conformation in neat organic solvents. J. Mol. Catal. B Enzym. 2002, 19–20, 93–102. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Stauch, B.; Fisher, S.J.; Cianci, M. Open and closed states of Candida antarctica lipase B: Protonation and the mechanism of interfacial activation. J. Lipid Res. 2015, 56, 2348–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, S.D.; Nordblad, M.; Woodley, J.M.; Peters, G.H. Effect of Water Clustering on the Activity of Candida antarctica Lipase B in Organic Medium. Catalysts 2017, 7, 227. [Google Scholar] [CrossRef] [Green Version]
- Ebert, C.; Gardossi, L.; Linda, P. Control of enzyme hydration in penicillin amidase catalysed synthesis of amide bond. Tetrahedron Lett. 1996, 37, 9377–9380. [Google Scholar] [CrossRef]
- Ru, M.T.; Dordick, J.S.; Reimer, J.A.; Clark, D.S. Optimizing the salt-induced activation of enzymes in organic solvents: Effects of lyophilization time and water content. Biotechnol. Bioeng. 1999, 63, 233–241. [Google Scholar] [CrossRef]
- Partridge, J.; Dennison, P.R.; Moore, B.D.; Halling, P.J. Activity and mobility of subtilisin in low water organic media: Hydration is more important than solvent dielectric. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 1998, 1386, 79–89. [Google Scholar] [CrossRef]
- Valivety, R.H.; Halling, P.J.; Macrae, A.R. Rhizomucor miehei lipase remains highly active at water activity below 0.0001. FEBS Lett. 1992, 301, 258–260. [Google Scholar] [CrossRef] [Green Version]
- Syrén, P.-O.; Hult, K. Amidases Have a Hydrogen Bond that Facilitates Nitrogen Inversion, but Esterases Have Not. ChemCatChem 2011, 3, 853–860. [Google Scholar] [CrossRef]
- Larsen, M.W.; Zielinska, D.F.; Martinelle, M.; Hidalgo, A.; Jensen, L.J.; Bornscheuer, U.T.; Hult, K. Suppression of Water as a Nucleophile in Candida antarctica Lipase B Catalysis. ChemBioChem 2010, 11, 796–801. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
Figure 1.
Bacillus subtilis lipase A (BSLA) is the smallest serine hydrolase with an α/β hydrolase fold. With only 181 amino acids, it has a molecular weight of 19 kDa. The depicted BSLA structure is pdb 1R50 and the catalytic triad His156, Ser77 and Asp133 and the oxyanion hole Ile12 and Met78 are highlighted. The figure was created with PyMOL.
Figure 1.
Bacillus subtilis lipase A (BSLA) is the smallest serine hydrolase with an α/β hydrolase fold. With only 181 amino acids, it has a molecular weight of 19 kDa. The depicted BSLA structure is pdb 1R50 and the catalytic triad His156, Ser77 and Asp133 and the oxyanion hole Ile12 and Met78 are highlighted. The figure was created with PyMOL.

Figure 2.
BioGPS of 43 serine hydrolases, for BSLA the data of pdb 1R50 were utilized (global score). Each analyzed enzyme structure is placed within a multidimensional space. Relative distances between each enzyme and all the other enzymes are determined by a statistical principal component analysis. The pdb codes of the processed enzyme structures are indicated in different colors according to their class: lipases in blue, amidases in red, proteases in cyan and esterases in green; the BSLA structure is in black.
Figure 2.
BioGPS of 43 serine hydrolases, for BSLA the data of pdb 1R50 were utilized (global score). Each analyzed enzyme structure is placed within a multidimensional space. Relative distances between each enzyme and all the other enzymes are determined by a statistical principal component analysis. The pdb codes of the processed enzyme structures are indicated in different colors according to their class: lipases in blue, amidases in red, proteases in cyan and esterases in green; the BSLA structure is in black.

Scheme 1.
Amidase activity assay [36]. The assay can be quantified spectrophotometrically. BSLA showed no activity in this assay, ruling out amidase activity.
Scheme 1.
Amidase activity assay [36]. The assay can be quantified spectrophotometrically. BSLA showed no activity in this assay, ruling out amidase activity.

Scheme 2.
Test reaction for the activity of BSLA at low aw. The reaction was performed in toluene at 30 °C, with a ratio of 1-octanol to vinyl acetate of 1:5 and aw < 0.1, 0.23 and 0.75.
Scheme 2.
Test reaction for the activity of BSLA at low aw. The reaction was performed in toluene at 30 °C, with a ratio of 1-octanol to vinyl acetate of 1:5 and aw < 0.1, 0.23 and 0.75.

Figure 3.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels of purified BSLA (19 kDa) and Candida antarctica lipase B (CALB) (33 kDa).
Figure 3.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels of purified BSLA (19 kDa) and Candida antarctica lipase B (CALB) (33 kDa).

Figure 4.
Activity of BSLA, CALB and Novozym 435 in toluene with different aw. U = µmol butyric acid × min−1 in tributyrin activity assay, 0.5–1.2 U of catalyst, 1-octanol (100 mM), vinyl acetate (5 eq.), ISTD: Decane (500 mM), 1 mL reaction volume, 24 h, 30 °C and 1000 rpm. Blanks were performed in the absence of enzyme and showed no conversion. Final conversions are given as inset; the color corresponds to the aw.
Figure 4.
Activity of BSLA, CALB and Novozym 435 in toluene with different aw. U = µmol butyric acid × min−1 in tributyrin activity assay, 0.5–1.2 U of catalyst, 1-octanol (100 mM), vinyl acetate (5 eq.), ISTD: Decane (500 mM), 1 mL reaction volume, 24 h, 30 °C and 1000 rpm. Blanks were performed in the absence of enzyme and showed no conversion. Final conversions are given as inset; the color corresponds to the aw.

Figure 5.
Activity of BSLA, CALB and Novozym 435 in toluene with different aw. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 500 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.
Figure 5.
Activity of BSLA, CALB and Novozym 435 in toluene with different aw. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 500 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.

Figure 6.
Activity of BSLA, toluene at aw < 0.1. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 100 mM or 500 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.
Figure 6.
Activity of BSLA, toluene at aw < 0.1. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 100 mM or 500 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.

Figure 7.
Activity of BSLA co-lyophilized with the appropriate salt, toluene at aw < 0.1 or 0.57. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 100 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme, i.e., in the presence of salt, and showed no conversion. After 8 h (480 min) an additional equivalent of vinyl acetate was added.
Figure 7.
Activity of BSLA co-lyophilized with the appropriate salt, toluene at aw < 0.1 or 0.57. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 100 mM vinyl acetate, 500 mM decane (ISTD), in dry toluene (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme, i.e., in the presence of salt, and showed no conversion. After 8 h (480 min) an additional equivalent of vinyl acetate was added.

Figure 8.
Activity of BSLA, in MTBE and toluene aw < 0.1. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 500 mM vinyl acetate, 500 mM decane (ISTD), in dry solvent (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.
Figure 8.
Activity of BSLA, in MTBE and toluene aw < 0.1. Reaction conditions: 0.5–1.2 U of catalyst, 100 mM 1-octanol, 500 mM vinyl acetate, 500 mM decane (ISTD), in dry solvent (1 mL reaction) at 30 °C and 1000 rpm. U: µmol butyric acid × min−1. Blanks were performed in the absence of enzyme and showed no conversion.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
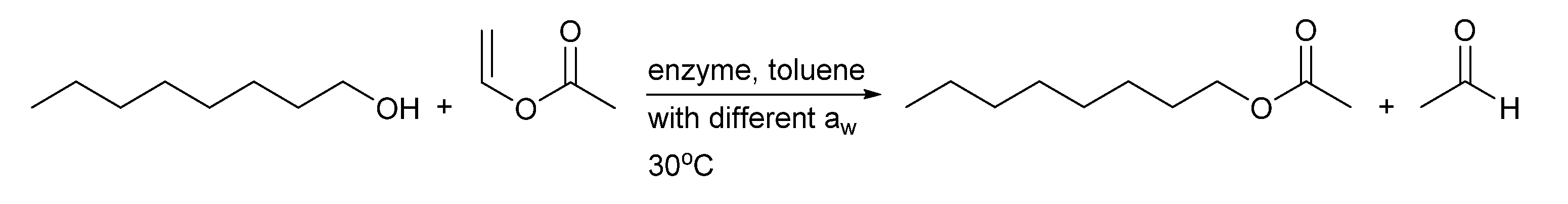{kind=link}
Table 1.
Equilibration to different aw via vapor phase over a saturated solution of salt [47,51] and via the salt pair method [50]. All reaction components, except the acyl donor, were mixed and equilibrated overnight at 30 °C. Finally, dried and freshly distilled vinyl acetate was added in order to start the reaction. The water content was determined by Karl Fischer Titration after 48 h.
Table 1.
Equilibration to different aw via vapor phase over a saturated solution of salt [47,51] and via the salt pair method [50]. All reaction components, except the acyl donor, were mixed and equilibrated overnight at 30 °C. Finally, dried and freshly distilled vinyl acetate was added in order to start the reaction. The water content was determined by Karl Fischer Titration after 48 h.
| aw | Agent (Vapor Phase or Salt Pair) | Moles of H2O/mol of Salt | Water Content (ppm) |
|---|---|---|---|
| <0.1 | Mol. sieves | 0 | ~20 |
| 0.25 | NaAc anhydr. (salt pair) | 1.5 | ~180 |
| 0.57 | Na2HPO4 anhydr. (salt pair) | 5.0 | ~360 |
| 0.23 | KAc (vapor phase) | NA a) | ~120 |
| 0.75 | NaCl (vapor phase) | NA a) | ~400 |
a) Not applicable (NA).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bracco, P.; van Midden, N.; Arango, E.; Torrelo, G.; Ferrario, V.; Gardossi, L.; Hanefeld, U. Bacillus subtilis Lipase A—Lipase or Esterase? Catalysts 2020, 10, 308. https://doi.org/10.3390/catal10030308
AMA Style
Bracco P, van Midden N, Arango E, Torrelo G, Ferrario V, Gardossi L, Hanefeld U. Bacillus subtilis Lipase A—Lipase or Esterase? Catalysts. 2020; 10(3):308. https://doi.org/10.3390/catal10030308
Chicago/Turabian StyleBracco, Paula, Nelleke van Midden, Epifanía Arango, Guzman Torrelo, Valerio Ferrario, Lucia Gardossi, and Ulf Hanefeld. 2020. "Bacillus subtilis Lipase A—Lipase or Esterase?" Catalysts 10, no. 3: 308. https://doi.org/10.3390/catal10030308
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.