Catalytic Pyrolysis of Aliphatic Carboxylic Acids into Symmetric Ketones over Ceria-Based Catalysts: Kinetics, Isotope Effect and Mechanism


Abstract
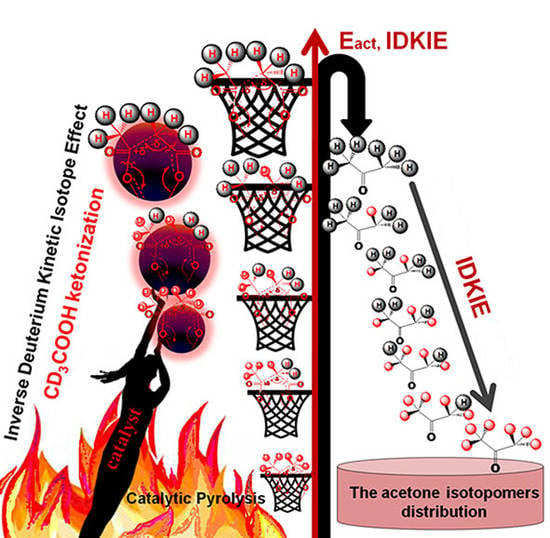
:
1. Introduction
2. Results and Discussion
2.1. Kinetic Study
2.2. Analysis of Kinetics Data
2.3. Analysis of Substituent Effects (Taft Plots) or Linear Free Energy Relationships (LFERs)
2.4. Study of DKIE During Catalytic Ketonization of CD3COOH (D3)
2.4.1. The Distribution of Acetone Isotopomers
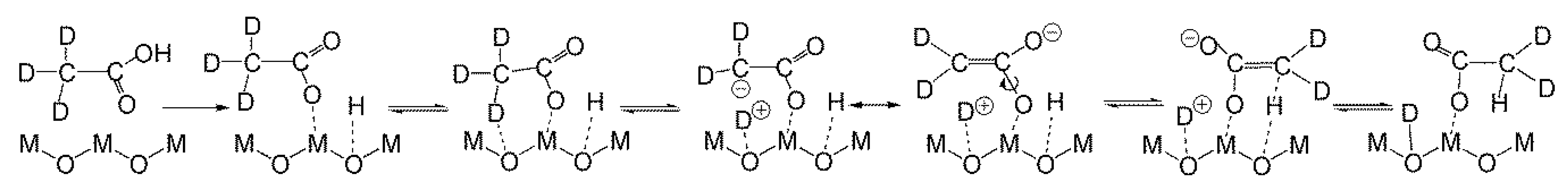
2.4.2. Study of the Magnitude and the Origin of DKIE
3. Materials and Methods
3.1. Materials
3.2. Temperature-Programmed Desorption Mass Spectrometry and Kinetic Parameters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bridgwater, A.V. Review of fast pyrolysis of biomass and product upgrading. Biomass Bioenergy 2012, 38, 68–94. [Google Scholar] [CrossRef]
- Kataki, R.; Chutia, R.S.; Mishra, M.; Bordoloi, N.; Saikia, R.; Bhaskar, T. Feedstock suitability for thermochemical processes. In Recent Advances in Thermo-Chemical Conversion of Biomass, 1st ed.; Pandey, A., Bhaskar, T., Stöcker, M., Sukumaran, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 31–74. [Google Scholar]
- Zhang, S.; Yang, X.; Zhang, H.; Chu, C.; Zheng, K.; Ju, M.; Liu, L. Liquefaction of biomass and upgrading of bio-oil: A review. Molecules 2019, 24, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Huber, G.W. The critical role of heterogeneous catalysis in lignocellulosic biomass conversion. Energy Environ. Sci. 2009, 2, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, K.O.; Olarte, M.V.; Wang, H. Upgrading Fast Pyrolysis Liquids. In Thermochemical Processing of Biomass: Conversion into Fuels, Chemicals and Power, 2nd ed.; Brown, R.C., Ed.; Wiley Online Library: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Sheldon, R.A. Green chemistry, catalysis and valorization of waste biomass. J. Mol. Catal. A Chem. 2016, 422, 3–12. [Google Scholar] [CrossRef]
- Climent, M.J.; Corma, A.; Iborra, S. Conversion of biomass platform molecules into fuel additives and liquid hydrocarbon fuels. Green Chem. 2014, 16, 516–547. [Google Scholar] [CrossRef] [Green Version]
- De Wit, M.; Faaij, A. European biomass resource potential and costs. Biomass Bioenergy 2010, 34, 188–202. [Google Scholar] [CrossRef]
- Nanda, S.; Mohanty, P.; Kozinski, J.A.; Dalai, A.K. Hydrothermal and Thermochemical Synthesis of Bio-Oil from Lignocellulosic Biomass: Composition, Engineering and Catalytic Upgrading. In Industrial Biotechnology, 1st ed.; Thangadurai, D., Sangeetha, J., Eds.; Apple Academic Press: New York, NY, USA, 2017; pp. 345–390. [Google Scholar]
- Nanda, S.; Mohanty, P.; Kozinski, J.A.; Dalai, A.K. Physico-Chemical Properties of Bio-Oils from Pyrolysis of Lignocellulosic Biomass with High and Slow Heating Rate. Energy Environ. Res. 2014, 21, 21–32. [Google Scholar] [CrossRef]
- Novakovskiy, D.J.; Jones, J.M. Uncatalysed and potassium-catalysed pyrolysis of the cell-wall constituents of biomass and their model compounds. J. Anal. Appl. Pyrol. 2008, 83, 12–25. [Google Scholar]
- Serrano-Ruiz, J.C.; Braden, D.J.; West, R.M.; Dumesic, J.A. Conversion of cellulose to hydrocarbon fuels by progressive removal of oxygen. Appl. Catal. B 2010, 100, 184–189. [Google Scholar] [CrossRef]
- Zhang, Y.; Cui, H.; Yi, W.; Song, F.; Zhao, P.; Wang, L.; Cui, J. Highly effective decarboxylation of the carboxylic acids in fast pyrolysis oil of rice husk towards ketones using CaCO3 as a recyclable agent. Biomass Bioenergy 2017, 102, 13–22. [Google Scholar] [CrossRef]
- Lange, J.P.; Price, R.; Ayoub, P.M.; Louis, J.; Petrus, L.; Clarke, L.; Gosselink, H. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels. Angew. Chem. Int. Ed. 2010, 49, 4479–4483. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Xi, Z.; Lu, F.; Huang, Z.; Yang, Y.; Sun, J.; Liao, Z.; Wang, J.; Yang, Y. Ce/MgAl mixed oxides derived from hydrotalcite LDH precursors as highly efficient catalysts for ketonization of carboxylic acid. Catal. Sci. Technol. 2019, 9, 6335–6344. [Google Scholar] [CrossRef]
- Ling, H.; Wang, Z.; Wang, L.; Stampfl, C.; Wang, D.; Chen, J.; Huang, J. Composition-structure-function correlation of Ca/Zn/AlOx catalysts for the ketonization of acetic acid. Catal. Today 2019. [Google Scholar] [CrossRef]
- Ignatchenko, A.V. Multiscale approach for the optimization of ketones production from carboxylic acids by the decarboxylative ketonization reaction. Catal. Today 2019, 338, 3–17. [Google Scholar] [CrossRef]
- Psarras, A.C.; Michailof, C.M.; Iliopoulou, E.F.; Kalogiannis, K.G.; Lappas, A.A.; Heracleous, E.; Triantafyllidis, K.S. Acetic acid conversion reactions on basic and acidic catalysts under biomass fast pyrolysis conditions. J. Mol. Catal. 2019, 465, 33–42. [Google Scholar] [CrossRef]
- Pham, T.N.; Sooknoi, T.; Crossley, S.P.; Resasco, D.E. Ketonization of carboxylic acids: Mechanisms, catalysts, and implications for biomass conversion. ACS Catal. 2013, 3, 2456–2473. [Google Scholar] [CrossRef]
- Renz, M. Ketonization of carboxylic acids by decarboxylation: Mechanism and scope. Eur. J. Org. Chem. 2005, 6, 979–988. [Google Scholar] [CrossRef]
- Kumar, R.; Enjamuri, N.; Shah, S.; Al-Fatesh, A.S.; Bravo-Suárez, J.J.; Chowdhury, B. Ketonization of oxygenated hydrocarbons on metal oxide based catalysts. Catal. Today 2018, 302, 16–49. [Google Scholar] [CrossRef]
- Dooley, K.M. Catalysis of acid/aldehyde/alcohol condensations to ketones. Catalysis 2004, 17, 293–319. [Google Scholar]
- Simakova, I.L.; Murzin, D.Y. Transformation of bio-derived acids into fuel-like alkanes via ketonic decarboxylation and hydrodeoxygenation: Design of multifunctional catalyst, kinetic and mechanistic aspects. J. Energy Chem. 2017, 25, 208–224. [Google Scholar] [CrossRef]
- Hites, R.A.; Biemann, K. Mechanism of ketonic decarboxylation. Pyrolysis of calcium decanoate. J. Am. Chem. Soc. 1972, 94, 5772–5777. [Google Scholar] [CrossRef]
- Rand, L.; Wagner, W.; Warner, P.O.; Kovac, L.R. Reactions catalyzed by potassium fluoride. II. The conversion of adipic acid to cyclopentanone. J. Org. Chem. 1962, 27, 1034–1035. [Google Scholar] [CrossRef]
- Gumidyala, A.; Sooknoi, T.; Crossley, S. Selective ketonization of acetic acid over HZSM-5: The importance of acyl species and the influence of water. J. Catal. 2016, 340, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Hendren, T.S.; Dooley, K.M. Kinetics of catalyzed acid/acid and acid/aldehyde condensation reactions to non-symmetric ketones. Catal. Today 2003, 85, 333–351. [Google Scholar] [CrossRef]
- Pestman, R.; Koster, R.M.; van Duijne, A.; Pieterse, J.A.Z.; Ponec, V. Reactions of Carboxylic Acids on Oxides. 2. Bimolecular Reaction of Aliphatic Acids to Ketones. J. Catal. 1997, 168, 265–272. [Google Scholar] [CrossRef]
- González, F.; Munuera, G.; Prieto, J.A. Mechanism of Ketonization of Acetic Acid on Anatase Ti02 Surfaces. J. Chem. Soc. Faraday Trans. 1978, 74, 1517–1529. [Google Scholar] [CrossRef]
- Pulido, A.; Oliver-Tomas, B.; Renz, M.; Boronat, M.; Corma, A. Ketonic decarboxylation reaction mechanism: A combined experimental and DFT study. ChemSusChem 2013, 6, 141–151. [Google Scholar] [CrossRef]
- Pham, T.N.; Shi, D.; Resasco, D.E. Kinetics and Mechanism of Ketonization of Acetic Acid on Ru/TiO2 Catalyst. Top. Catal. 2014, 57, 706–714. [Google Scholar] [CrossRef]
- Ignatchenko, A.V.; McSally, J.P.; Bishop, M.D.; Zweigle, J. Ab initio study of the mechanism of carboxylic acids cross-ketonization on monoclinic zirconia via condensation to beta-keto acids followed by decarboxylation. J. Mol. Catal. 2017, 441, 35–62. [Google Scholar] [CrossRef] [Green Version]
- Ignatchenko, A.V.; Kozliak, E.I. Distinguishing enolic and carbonyl components in the mechanism of carboxylic acid ketonization on monoclinic zirconia. ACS Catal. 2012, 2, 1555–1562. [Google Scholar] [CrossRef]
- Ignatchenko, A.V.; Cohen, A.J. Reversibility of the catalytic ketonization of carboxylic acids and of beta-keto acids decarboxylation. Catal. Commun. 2018, 111, 104–107. [Google Scholar] [CrossRef]
- Oliver-Tomas, B.; Renz, M.; Corma, A. Ketone formation from carboxylic acids by Ketonic decarboxylation: The exceptional case of the tertiary carboxylic acids. Chem. Eur. J. 2017, 23, 12900–12908. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Iglesia, E. Experimental and theoretical assessment of the mechanism and site requirements for ketonization of carboxylic acids on oxides. J. Catal. 2017, 345, 183–206. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Zhao, J.; Yu, Q. Effect of Zirconia Polymorph on Vapor-Phase Ketonization of Propionic Acid. Catalysts 2019, 9, 768. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, O.; Sato, S.; Takahashi, R.; Sodesawa, T. Ketonization of carboxylic acids over CeO2-based composite oxides. J. Mol. Catal. A Chem. 2005, 227, 231–239. [Google Scholar] [CrossRef]
- Almutairi, S.T.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Ketonisation of acetic acid on metal oxides: Catalyst activity, stability and mechanistic insights. Appl. Catal. A 2018, 565, 135–145. [Google Scholar] [CrossRef]
- Hasan, M.A.; Zaki, M.I.; Pasupulety, L. Oxide-catalyzed conversion of acetic acid into acetone: An FTIR spectroscopic investigation. Appl. Catal. A 2003, 243, 81–92. [Google Scholar] [CrossRef]
- Kulyk, K.; Palianytsia, B.; Alexander, J.D.; Azizova, L.; Borysenko, M.; Kartel, M.; Larsson, M.; Kulik, T. Kinetics of valeric acid ketonization and ketenization in catalytic pyrolysis on nanosized SiO2, γ-Al2O3, CeO2/SiO2, Al2O3/SiO2 and TiO2/SiO2. ChemPhysChem 2017, 18, 1943–1955. [Google Scholar] [CrossRef]
- Anslyn, E.V.; Dougherty, D.A. Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, USA, 2006. [Google Scholar] [CrossRef] [Green Version]
- Kulik, T.V. Use of TPD-MS and linear free energy relationships for assessing the reactivity of aliphatic carboxylic acids on a silica surface. J. Phys. Chem. C 2012, 116, 570–580. [Google Scholar] [CrossRef]
- Dupre, G.D.; Schlosberg, R.H.; Pancirov, R.J.; Ashe, T.R.; Baset, Z. Pyrolysis studies of organic oxygenates VI. thermal Chemistry of long chain aliphatic acids. Liq. Fuels Technol. 1985, 3, 323–344. [Google Scholar] [CrossRef]
- Kim, K.S.; Barteau, M.A. Pathways for carboxylic acid decomposition on titania. Langmuir 1988, 4, 945–953. [Google Scholar] [CrossRef]
- García-Sánchez, M.; Sales-Cruz, M.; Lopez-Arenas, T.; Viveros-García, T.; Pérez-Cisneros, E.S. An Intensified Reactive Separation Process for Bio-Jet Diesel Production. Processes 2019, 7, 655. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Huff, M.C.; Barteau, M.A. Synthesis of ketenes from carboxylic acids on functionalized silica monoliths at short contact time. Appl. Catal. A 2000, 200, 79–88. [Google Scholar] [CrossRef]
- Libby, M.C.; Watson, P.C.; Barteau, M.A. Synthesis of Ketenes with oxide catalysts. Ind. Eng. Chem. Res. 1994, 33, 2904–2912. [Google Scholar] [CrossRef]
- Azizova, L.R.; Kulik, T.V.; Palyanytsya, B.B.; Lipkovska, N.A. Thermal and hydrolytic stability of grafted ester groups of carboxylic acids on the silica surface. J. Therm. Anal. Calorim. 2015, 122, 517–523. [Google Scholar] [CrossRef]
- Brei, V.V.; Brichka, A.V. A Study of the Brönsted Site Acidity of Crystalline and Amorphous Aluminosilicates: 2. Thermal Decomposition of Grafted Acetyl Groups. Adsorp. Sci. Technol. 1996, 14, 359–362. [Google Scholar] [CrossRef]
- Kulik, T.V.; Barvinchenko, V.N.; Palyanitsa, B.B.; Smirnova, O.V.; Pogorelyi, V.K.; Chuiko, A.A. A desorption mass spectrometry study of the interaction of cinnamic acid with a silica surface. Russ. J. Phys. Chem. 2007, 8, 83–90. [Google Scholar] [CrossRef]
- Kulik, T.V.; Lipkovska, N.A.; Barvinchenko, V.N.; Palyanytsya, B.B.; Kazakova, O.A.; Dovbiy, O.A.; Pogorelyi, V.K. Interactions between bioactive ferulic acid and fumed silica by UV–vis spectroscopy, FT–IR, TPD MS investigation and quantum chemical methods. J. Colloid Interface Sci. 2009, 339, 60–68. [Google Scholar] [CrossRef]
- Kulik, T.V.; Lipkovska, N.O.; Barvinchenko, V.M.; Palyanytsya, B.B.; Kazakova, O.A.; Dudik, O.O.; Menyhárd, A.; László, K. Thermal transformation of bioactive caffeic acid on fumed silica seen by UV–Vis spectroscopy, thermogravimetric analysis, temperature programmed desorption mass spectrometry and quantum chemical methods. J. Colloid Interface Sci. 2016, 470, 132–141. [Google Scholar] [CrossRef]
- Nastasiienko, N.; Palianytsia, B.; Kartel, M.; Larsson, M.; Kulik, T. Thermal transformation of caffeic acid on the nanosized cerium dioxide studied by temperature programmed desorption mass-spectrometry, thermogravimetric analysis and FTIR-spectroscopy. Colloids Interfaces 2019, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Vlasenko, N.V.; Kyriienko, P.I.; Yanushevska, O.I.; Valihura, K.V.; Soloviev, S.O.; Strizhak, P.E. The Effect of Ceria Content on the Acid–Base and Catalytic Characteristics of ZrO2–CeO2 Oxide Compositions in the Process of Ethanol to n-Butanol Condensation. Catal. Lett. 2019, 1–9. [Google Scholar] [CrossRef]
- Vivier, L.; Duprez, D. Ceria-Based Solid Catalysts for Organic Chemistry. ChemSusChem 2010, 3, 654–678. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Mann, A.K.; Li, M.; Overbury, S.H. Spectroscopic investigation of surface-dependent acid–base property of ceria nanoshapes. J. Phys. Chem. 2015, C 119, 7340–7350. [Google Scholar] [CrossRef]
- Wu, Z.; Li, M.; Mullins, D.R.; Overbury, S.H. Probing the surface sites of CeO2 nanocrystals with well-defined surface planes via methanol adsorption and desorption. ACS Catal. 2012, 2, 2224–2234. [Google Scholar] [CrossRef]
- Martin, D.; Duprez, D. Evaluation of the acid-base surface properties of several oxides and supported metal catalysts by means of model reactions. J. Mol. Catal. A Chem. 1997, 118, 113–128. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Calaza, F.C.; Chen, T.L.; Mullins, D.R.; Xu, Y.; Overbury, S.H. Reactivity and reaction intermediates for acetic acid adsorbed on CeO2 (111). Catal. Today 2015, 253, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Stein, E. NIST Chemistry WebBook. IR and Mass Spectra, Database Number 69; Mallard, W.G., Linstrom, P.J., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2000. [Google Scholar]
- Gliński, M.; Kijeński, J.; Jakubowski, A. Ketones from monocarboxylic acids: Catalytic ketonization over oxide systems. Appl. Catal. A Gen. 1995, 128, 209–217. [Google Scholar] [CrossRef]
- Deng, L.; Fu, Y.; Guo, Q.X. Upgraded acidic components of bio-oil through catalytic ketonic condensation. Energy Fuels 2008, 23, 564–568. [Google Scholar] [CrossRef]
- Woodruff, D.P.; Delchar, T.A. Modern Techniques of Surface Science; Cambridge University Press: London, UK, 1986. [Google Scholar] [CrossRef]
- Redhead, P.A. Thermal Desorption of Gases. Vacuum 1962, 12, 203–211. [Google Scholar] [CrossRef]
- Kislyuk, M.U.; Rozanov, V.V. Temperature-programmed desorption and temperature-programmed reaction - methods of studying of kinetics and mechanisms of catalytic processes. Kinet. Catal. 1995, 36, 89–98. [Google Scholar]
- Carter, G. Thermal resolution of desorption energy spectra. Vacuum 1962, 12, 245–254. [Google Scholar] [CrossRef]
- Leardini, F.; Fernández, J.F.; Bodega, J.; Sanchez, C. Isotope effects in the kinetics of simultaneous H and D thermal desorption from Pd. J. Phys. Chem. Solid 2008, 69, 116–127. [Google Scholar] [CrossRef]
- Houk, K.N.; Gustafson, S.M.; Black, K.A. Theoretical secondary kinetic isotope effects and the interpretation of transition state geometries. 1. The Cope rearrangement. J. Am. Chem. Soc. 1992, 114, 8565–8572. [Google Scholar] [CrossRef]
- Hoque, M.E.U.; Guha, A.K.; Kim, C.K.; Lee, B.S.; Lee, H.W. Concurrent primary and secondary deuterium kinetic isotope effects in anilinolysis of O-aryl methyl phosphonochloridothioates. Org. Biomol. Chem. 2009, 7, 2919–2925. [Google Scholar] [CrossRef]
- Borysenko, M.V.; Kulyk, K.S.; Ignatovych, M.V.; Poddenezhny, E.N.; Boiko, A.A.; Dobrodey, A.O. Sol–gel synthesis of silica glasses, doped with nanoparticles of cerium oxide. In Nanomaterials and Supramolecular Structures; Shpak, A.P., Gorbyk, P.P., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 227–233. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acid | ΣEs | Σσ* | Tmax, °C | m/z | n | E≠, kJ·mol−1 | ν0, s−1, (n = 1); mol−1s−1, (n = 2) | dS≠, cal·K−1 mol−1 | D± %, | R2 a | E≠ b, kJ·mol−1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acetic C2 | 3.72 | 1.2 | 247 | 58 | 1 | 119 | 5.3 × 109 | −15 | 2.5 | 0.982 | 115.1 |
| 2 | 212 | 3.5 × 1019 | 30 | 11 | 0.957 | - | |||||
| Propionic C3 | 2.48 | 0.8 | 266 | 86 | 1 | 122 | 4.4 × 109 | −16 | 4 | 0.970 | 119.4 |
| 2 | 271 | 6.7 × 1024 | 54 | 43 | 0.879 | - | |||||
| Butyric C4 | 2.41 | 0.7. | 262 | 114 | 1 | 113 | 2.4 × 109 | −17 | 3.4 | 0.972 | 118.5 |
| 2 | 252 | 9.7 × 1022 | 45 | 21 | 0.922 | - | |||||
| Isobutyric C4 | 1.24 | 0.4 | 284 | 114 | 1 | 128 | 7.4 × 109 | −15 | 0.4 | 0.995 | 123.6 |
| 2 | 241 | 8.7 × 1020 | 36 | 6 | 0.962 | - | |||||
| Valeric C5 | 2.12 | 0.685 | 268 | 142 | 1 | 123 | 4.8 × 109 | −15 | 2.5 | 0.964 | 119.9 |
| 2 | 254 | 5.2 × 1022 | 44 | 10 | 0.930 | - | |||||
| Pivalic C5 | 0 | 0 | 290 | 57 c | 1 | 130 | 5.2 × 108 | −20 | 4.8 | 0.962 | - |
| 2 | 251 | 4.2 × 1019 | 30 | 40 | 0.853 | - | |||||
| Hexanoic C6 | 2.09 | 0.67 | 269 | 85 | 1 | 120 | 1.3 × 109 | −18 | 4.7 | 0.958 | 120.1 |
| 2 | 205 | 1.0 × 1017 | 18 | 13 | 0.933 | - | |||||
| Heptanoic C7 | 2.08 | 0.55 | 272 | 142 | 1 | 122 | 1.6 × 109 | −18 | 6 | 0.947 | 120.8 |
| 2 | 282 | 1.7 × 1025 | 56 | 20 | 0.920 | - | |||||
| Octanoic C8 | - | 0.49 | 282 | 85 | 1 | 122 | 1.4 × 109 | −18 | 9.0 | 0.940 | 123.1 |
| 2 | 250 | 5.3 × 1021 | 40 | 32 | 0.900 | - | |||||
| Nonanoic C9 | - | 0.43 | 290 | 85 | 1 | 119 | 1.5 × 109 | −18 | 4.9 | 0.966 | 124.9 |
| 2 | 209 | 2.0 × 1017 | 18 | 14 | 0.944 | - | |||||
| Decanoic C10 | - | 0.45 | 292 | 72 | 1 | 124 | 5.2 × 109 | −15 | 3.5 | 0.960 | 125.4 |
| 2 | 295 | 5.2 × 1026 | 62 | 21 | 0.903 | - |
| Peak a | Process/Reaction | Product, m\z | Tmax (°C) TPD-Curve | Tmax (°C) P/T Curve | Peak Area (a.u.), Σ (Peak Areas) | % |
|---|---|---|---|---|---|---|
| Desorption of C2H5COOH in molecular form | ||||||
| 1 | Desorption physically adsorbed acid | C2H5COOH | 62 | 65 | 0.06329 | 13 |
| m/z 74 | 63 | |||||
| 3 | Desorption of H-bonded acid | C2H5COOH m/z 29 | 131 | 133 | 0.09624 | 20 |
| Σ (1+3) = 0.15953 | 33 | |||||
| Catalytic conversion of C2H5COOH | ||||||
| 4 | Decarboxylation with ethane formation | C2H6 m/z 29 CO2 m/z 44 | 196 196 | 195 | 0.00511 | ~1 |
| 5 | Ketonic decarboxylation | (C2H5)2CO m/z 86 CO2 m/z44 | 265 267 | 264 | 0.31606 | 66 |
| Σ (4+5) = 0.32117 | 67 | |||||
| Σ (1+3+4+5) = 0.48070 | 100 | |||||
| H2O desorption | ||||||
| 2 | Desorption physically adsorbed water | H2O | 87 | 81 | 0.06071 | 26 |
| m/z 17 | ||||||
| 6 | Dehydration, dehydroxylation | H2O | 391 | 391 | 0.17421 | 74 |
| m/z 17 | ||||||
| Σ (2+6) = 0.23492 | 100 | |||||
| m/z Isotopomer of Acetone | Tmax | ∫ I(m/z), a.u. | Distr., % | R2 a |
|---|---|---|---|---|
| CD3COOH | ||||
| 58, C2D0H6CO | 269.9 | 10.21 | 26.20 | 0.9906 |
| 59, C2D1H5CO | 263.1 | 13.58 | 34.85 | 0.9929 |
| 60, C2D2H4CO | 250.4 | 9.17 | 23.53 | 0.9817 |
| 61, C2D3H3CO | 229.5 | 4.32 | 11.09 | 0.9774 |
| 62, C2D4H2CO | 208.1 | 1.44 | 3.70 | 0.9641 |
| 64, C2D6H0CO | - | <0.25 | <0.64 | - |
| ∑ ∫ I(m/z) | ~38.97 | ~100 | - | |
| CH313COOH | ||||
| (CH3)213CO | 255.7 | ~37.30 | ~100 | 0.9955 |
| CH3COOH | ||||
| (CH3)2CO | 256.4 | ~36.79 | ~100 | 0.9912 |
| m/z, Isotopomer of Acetone | Tmax, °C | k(H)/k(D) | n | E≠, kJ mol−1 | ν0, s−1 | dS≠, cal K−1 mol−1 | D±, % | R2 a | E≠ b, kJ mol−1 |
|---|---|---|---|---|---|---|---|---|---|
| CD3COOH | |||||||||
| 58, C2D0H6CO | 269.9 | - | 1 | 125 | 5.1 × 109 | −15 | 11 | 0.908 | 120.3 |
| 2 | 225 | 8.3 × 1019 | 31 | 24 | 0.854 | - | |||
| 59, C2D1H5CO | 263.1 | 1.3951 | 1 | 116 | 1.1 × 109 | −17 | 3 | 0.959 | 118.7 |
| 2 | 168 | 3.1 × 1019 | 6 | 8 | 0.901 | - | |||
| 60, C2D2H4CO | 250.4 | 0.7368 | 1 | 118 | 3.8 × 109 | −16 | 4 | 0.973 | 116.2 |
| 2 | 240 | 2.3 × 1022 | 45 | 30 | 0.922 | - | |||
| 61, C2D3H3CO | 229.5 | 0.2402 | 1 | - | - | - | - | - | 111.2 |
| 62, C2D4H2CO | 208.1 | 0.0689 | 1 | - | - | - | - | - | 106.4 |
| CH313COOH | |||||||||
| 59, CH313COCH3 | 255.7 | - | 1 | 117 | 1.8 × 109 | −17 | 7 | 0.960 | 117.0 |
| 2 | 204 | 2.5 × 1018 | 25 | 11 | 0.906 | - | |||
| CH3COOH | |||||||||
| 58, CH3COCH3 | 256.4 | - | 1 | 122 | 5.4 × 109 | −15 | 6 | 0.963 | 117.2 |
| 2 | 220 | 1.1 × 1020 | 32 | 13 | 0.900 | - | |||
| Catalysts 1 | Sa, m2g−1 | Amount of CeO2,% | dav,nm |
|---|---|---|---|
| lCeSi | 230 | 12.3 | 5 |
| hCeSi | 212 | 18.3 | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulik, T.; Palianytsia, B.; Larsson, M. Catalytic Pyrolysis of Aliphatic Carboxylic Acids into Symmetric Ketones over Ceria-Based Catalysts: Kinetics, Isotope Effect and Mechanism. Catalysts 2020, 10, 179. https://doi.org/10.3390/catal10020179
Kulik T, Palianytsia B, Larsson M. Catalytic Pyrolysis of Aliphatic Carboxylic Acids into Symmetric Ketones over Ceria-Based Catalysts: Kinetics, Isotope Effect and Mechanism. Catalysts. 2020; 10(2):179. https://doi.org/10.3390/catal10020179
Chicago/Turabian StyleKulik, Tetiana, Borys Palianytsia, and Mats Larsson. 2020. "Catalytic Pyrolysis of Aliphatic Carboxylic Acids into Symmetric Ketones over Ceria-Based Catalysts: Kinetics, Isotope Effect and Mechanism" Catalysts 10, no. 2: 179. https://doi.org/10.3390/catal10020179