Structural Modeling Insights into Human VKORC1 Phenotypes

Abstract
:1. Introduction
2. The Crystal Structure of Synechococcus VKOR—A Homolog to hVKORC1
3. The Human VKORC1 Homology Model
4. Conserved Amino Acid Residues of Human VKORC1
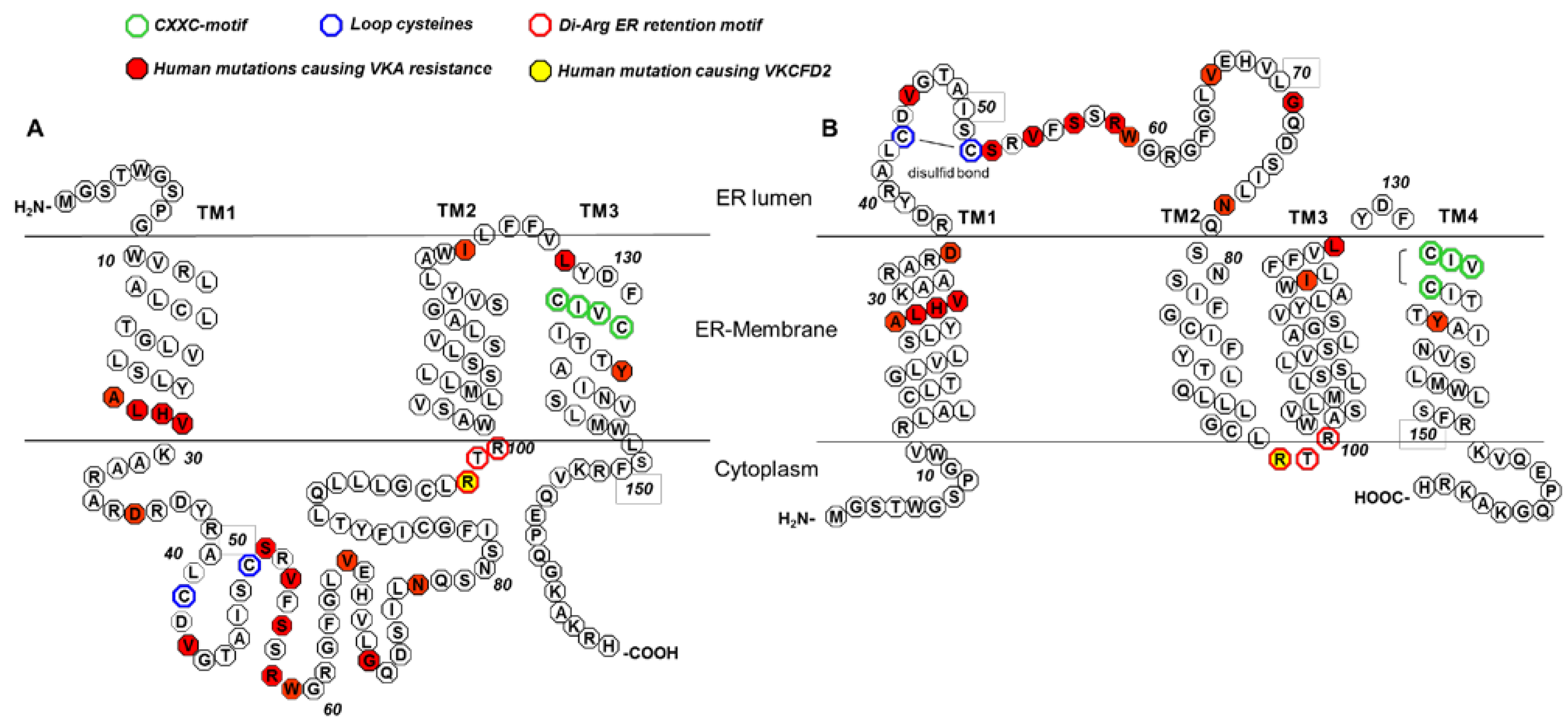

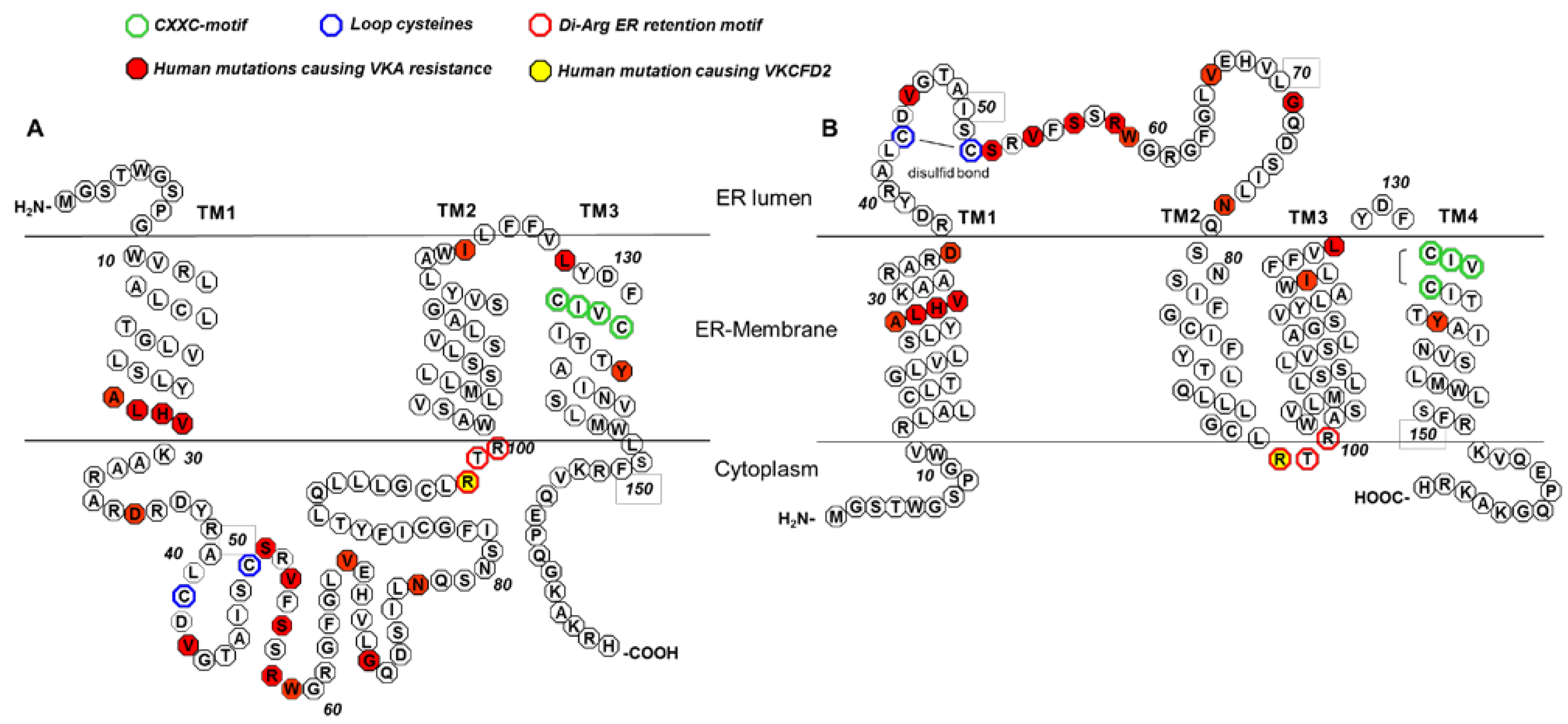{kind=link}
| Publication | Rost et al. [30] | Jin et al. [3] | Rishavy et al. [33] | Tie et al. [26] | Tie et al. [34] | Tie et al. [36] |
|---|---|---|---|---|---|---|
| Type of VKOR assay | DTT-driven assay | DTT-driven assay | DTT-driven assay | Cell-based assay | DTT-driven assay/cell-based assay | Cell-based assay |
| Reductant | DTT | DTT | DTT/Trx/TrxR | - | - | - |
| Cell line | HEK293 cells | Sf9 cells | Sf21 cells | HEK293 cells | HEK293 cells | C1 + L1 DKO HEK cells |
| Cysteine residue variants: | ||||||
| Cys43Ala | 20% | 25% | ~85%/0% | <5% | 25%/<5% | <5% |
| Cys43Ser | 20% | |||||
| Cys51Ala | 100% | ~50%/0% | 95% | 100%/100% | 105% | |
| Cys51Ser | <5% | |||||
| Cys43Ala + Cys51Ala | 112% | 60% | 85%/110% | 90% | ||
| Cys43_Cys51del | 85% | 85%/60% | ||||
| Cys132Ala/Ser | <5% | 0% | 0%/0% | |||
| Cys135Ala/Ser | <5% | 0% | 0%/0% |
5. The 3TM VKORC1 Topology Model
6. The 4TM VKORC1 Topology Model
| Arguments for 3TM hVKORC1 Structure | Arguments for 4TM hVKORC1 Structure |
|---|---|
| Location of the C-Terminus of VKORC1 in the cytoplasm and of the N-Terminus in the ER-lumen; FFP assay [26] | siRNA knock-down of PDI located in the ER lumen results in reduced VKOR activity [35] |
| Cys51Ala exhibits VKOR activity = Cys51 is not required for VKOR activity, DTT and cell-based assays [3,26,36] | Cys43Ala/Ser and Cys51Ala/Ser exhibit no VKOR activity = Cys43 and Cys51 are required for VKOR activity, DTT and Trx/TrxR assays [30,33] |
| hVKOR model, prediction program TOPCONS [37] | Cys43 forms a disulfide bond with four PDIs, immunoprecipitation [27] |
| 3.6 Å crystal structure of the bacterial homologue of VKOR from Synechococcus sp. in conjunction with multiple sequence alignments [19] | |
| hVKORC1 model based on crystal structure of synVKOR and putative warfarin binding interfaces that correspond to the reported WR mutations [23] |
7. Warfarin Binding and Mutations Causing Warfarin Resistance
| hVKORC1 Variant | Mean Patient Dosage in HDT Multiples [Drug] for n = Number of Reported Patients [11] | Warfarin IC50 by DTT-Driven VKOR Assay [8,12] | Warfarin IC50 by Cell Based Assay [23] | Warfarin Phenotypes by Cell Based Assay [36] |
|---|---|---|---|---|
| Wild-type | 1.0 [W, P] (n = 77) | |||
| Ala26Pro | >3.0 [W] (n = 1) | 11.2-fold increased Ki[12] | 49.6-fold increased IC50 | n.d. |
| Ala26Thr | >2.0 [P] (n = 1) | sensitive as wt [12] | 3.0-fold increased IC50 | n.d. |
| Leu27Val | >3.0 [F], 1.0 [W] (n = 1) * | sensitive as wt [12] | 2.5-fold increased IC50 | n.d. |
| His28Gln | 3.5 [P] (n = 1) | more sensitive than wt [ 12] | 2.9-fold increased IC50 | n.d. |
| Val29Leu | 2.0 [W] (n = 1) | absence of expression [12]/low VKOR activity and more sensitive than wt [8] | 5.5-fold increased IC50 | n.d. |
| Ala34Pro | 3.8 [W] (n = 1) | n.d. | n.d. | n.d. |
| Asp36Gly | 3.0 [W] (n = 1) | more sensitive than wt [ 12] | 3.2-fold increased IC50 | n.d. |
| Asp36Tyr | 1.5–3.5 [W] (n = 10) | sensitive as wt [12] | 3.8-fold increased IC50 | n.d. |
| Val45Ala | >2.0 [W] (n = 1) | low VKOR activity [ 8], more sensitive than wt [8,12] | 6.2-fold increased IC50 | n.d. |
| Ser52Leu | >3.0 [P] (n = 1) | low VKOR activity, Ki determination not possible [ 12] | 7.4-fold increased IC50 | moderate resistance |
| Ser52Trp | 3.5 [P] (n = 1) | low VKOR activity, Ki determination not possible [ 12] | 5.7-fold increased IC50 | sensitive as wt |
| Val54Leu | 1.5–5.5 [W] (n = 2) | 4.6-fold increased Ki[12] | 4.5-fold increased IC50 | n.d. |
| Ser56Phe | >5.0 [P] (n = 1) | more sensitive than wt [12] | 6.8-fold increased IC50 | n.d. |
| Arg58Gly | 5.0 [W] (n = 1) | low VKOR activity [8], more sensitive than wt [8 and 12] | 3.4-fold increased IC50 | n.d. |
| Trp59Arg | 7.0 [P] (n = 1) | low VKOR activity, Ki determination not possible [12] | 17.5-fold increased IC50 | high resistance |
| Trp59Cys | >3.5 [P] (n = 1) | more sensitive than wt [12] | 7.6-fold increased IC50 | n.d. |
| Trp59Leu | >5.0 [P] (n = 1) | low VKOR activity, Ki determination not possible [12] | 75.2-fold increased IC50 | high VKOR activity, high resistance |
| Val66Gly | 2.5 [P] (n = 1) | low VKOR activity, Ki determination not possible [12] | 2.8-fold increased IC50 | sensitive as wt |
| Val66Met | 3.0–6.0 [W] (n = 7) | low VKOR activity, Ki determination not possible [12] | 5.4-fold increased IC50 | sensitive as wt |
| Gly71Ala | >2.0 [P] (n = 1) | low VKOR activity, Ki determination not possible [12] | 5.1-fold increased IC50 | sensitive as wt |
| Asn77Ser | >3.0 [P] (n = 1) | low VKOR activity, Ki determination not possible [12] | 5.3-fold increased IC50 | moderate resistance |
| Asn77Tyr | 3.5 [W] (n = 1) | low VKOR activity, Ki determination not possible [12] | 3.9-fold increased IC50 | sensitive as wt |
| Ile123Asn | >7.0 [P] (n = 1) | 2.4-fold increased Ki[12] | 8.5-fold increased IC50 | n.d. |
| Leu128Arg | >4.0–7.0 [W] (n = 5) | low VKOR activity [8,12], Ki determination not possible [12]/more sensitive than wt [8] | 49.7-fold increased IC50 | high VKOR activity, high resistance |
| Tyr139His | >3.0 [W] (n = 1) | 3.6-fold increased Ki[12] | 4.6-fold increased IC50 | n.d. |
8. VKCFD2
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wajih, N.; Sane, D.C.; Hutson, S.M.; Wallin, R. Engineering of a recombinant vitamin K-dependent gamma-carboxylation system with enhanced gamma-carboxyglutamic acid forming capacity: Evidence for a functional CXXC redox center in the system. J. Biol. Chem. 2005, 280, 10540–10547. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Bevans, C.G.; Müller, C.R.; Watzka, M. Vitamin K epoxide reductase complex subunit 1 (VKORC1): The key protein of the vitamin K cycle. Antioxid. Redox Signal. 2006, 8, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.-Y.; Tie, J.-K.; Stafford, D.W. The conversion of vitamin K epoxide to vitamin K quinone and vitamin K quinone to vitamin K hydroquinone uses the same active site cysteines. Biochemistry 2007, 46, 7279–7283. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Marinova, M.; Müller-Reible, C.; Watzka, M. The vitamin K cycle. Vitam. Horm. 2008, 78, 35–62. [Google Scholar] [PubMed]
- Gundberg, C.M.; Lian, J.B.; Booth, S.L. Vitamin K-dependent carboxylation of osteocalcin: Friend or foe? Adv. Nutr. 2012, 3, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Cancela, M.L.; Laizé, V.; Conceição, N. Matrix Gla protein and osteocalcin: From gene duplication to neofunctionalization. Arch. Biochem. Biophys. 2014, 561, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ferland, G. Vitamin K and the nervous system: An overview of its actions. Adv. Nutr. 2012, 3, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hörtnagel, K.; Pelz, H.J.; Lappegard, K.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chang, C.Y.; Jin, D.Y.; Lin, P.J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nature 2004, 427, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Fregin, A.; Rost, S.; Wolz, W.; Krebsova, A.; Muller, C.R.; Oldenburg, J. Homozygosity mapping of a second gene locus for hereditary combined deficiency of vitamin K-dependent clotting factors to the centromeric region of chromosome 16. Blood 2002, 100, 3229–3232. [Google Scholar] [CrossRef] [PubMed]
- Watzka, M.; Geisen, C.; Bevans, C.G.; Sittinger, K.; Spohn, G.; Rost, S.; Seifried, E.; Müller, C.R.; Oldenburg, J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: Insights into improved patient diagnosis and treatment. J. Thromb. Haemost. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hodroge, A.; Matagrin, B.; Moreau, C.; Fourel, I.; Hammed, A.; Benoit, E.; Lattard, V. VKORC1 mutations detected in patients resistant to vitamin K antagonists are not all associated with a resistant VKOR activity. J. Thromb. Haemost. 2012, 10, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Pelz, H.-J.; Rost, S.; Hünerberg, M.; Fregin, A.; Heiberg, A.C.; Baert, K.; MacNicoll, A.D.; Prescott, C.V.; Walker, A.S.; Oldenburg, J.; et al. The genetic basis of resistance to anticoagulants in rodents. Genetics 2005, 170, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Pelz, H.-J.; Menzel, S.; MacNicoll, A.D.; León, V.; Song, K.J.; Jäkel, T.; Oldenburg, J.; Müller, C.R. Novel mutations in the VKORC1 gene of wild rats and mice—A response to 50 years of selection pressure by warfarin? BMC Genet. 2009, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, K.J.; Biswas, A.; Rost, S.; Watzka, M.; Oldenburg, J. The Arg98Trp mutation in human VKORC1 causing VKCFD2 disrupts a di-Arginine-based ER retention motif. Blood 2014, 124, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; von Brederlow, B.; Fregin, A.; Rost, S.; Wolz, W.; Eberl, W.; Eber, S.; Lenz, E.; Schwaab, R.; Brackmann, H.H.; et al. Congenital deficiency of vitamin K dependent coagulation factors in two families presents as a genetic defect of the vitamin K-epoxide-reductase-complex. Thromb. Haemost. 2000, 84, 937–941. [Google Scholar] [PubMed]
- Marchetti, G.; Caruso, P.; Lunghi, B.; Pinotti, M.; Lapecorella, M.; Napolitano, M.; Canella, A.; Mariani, G.; Bernardi, F. Vitamin K-induced modification of coagulation phenotype in VKORC1 homozygous deficiency. J. Thromb. Haemost. 2008, 6, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Pauli, R.M.; Lian, J.B.; Mosher, D.F.; Suttie, J.W. Association of congenital deficiency of multiple vitamin K-dependent coagulation factors and the phenotype of the warfarin embryopathy: Clues to the mechanism of teratogenicity of coumarin derivatives. Am. J. Hum. Genet. 1987, 41, 566–583. [Google Scholar] [PubMed]
- Li, W.; Schulman, S.; Dutton, R.J.; Boyd, D.; Beckwith, J.; Rapoport, T.A. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 2010, 463, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Goodstadt, L.; Ponting, C.P. Vitamin K epoxide reductase: Homology, active site and catalytic mechanism. Trends Biochem. Sci. 2004, 29, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cheng, W.; Fowle Grider, R.; Shen, G.; Li, W. Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nat. Commun. 2014, 5, 3110. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, W.D. Structural and functional insights into human vitamin K epoxide reductase and vitamin K epoxide reductase-like1. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, K.J.; Biswas, A.; Wendeln, A.C.; Westhofen, P.; Müller, C.R.; Watzka, M.; Oldenburg, J. Human VKORC1 mutations cause variable degrees of 4-hydroxycoumarin resistance and affect putative warfarin binding interfaces. Blood 2013, 122, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, S.; Davis, C.H.; Stafford, D.W.; Kulman, J.D.; Pedersen, L.G. A hetero-dimer model for concerted action of vitamin K carboxylase and vitamin K reductase in vitamin K cycle. J. Theor. Biol. 2011, 279, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Nicchitta, C.; von Heijne, G.; Stafford, D.W. Membrane topology mapping of vitamin K epoxide reductase by in vitro translation/cotranslocation. J. Biol. Chem. 2005, 280, 16410–16416. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Stafford, D.W. Human vitamin K epoxide reductase and its bacterial homologue have different membrane topologies and reaction mechanisms. J. Biol. Chem. 2012, 287, 33945–33955. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.M. Genes encoding vitamin-K epoxide reductase are present in Drosophila and trypanosomatid protists. Genetics 2004, 168, 1077–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldenburg, J.; Müller, C.R.; Rost, S.; Watzka, M.; Bevans, C.G. Comparative genetics of warfarin resistance. Hamostaseologie 2014, 34, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Hünerberg, M.; Bevans, C.G.; Müller, C.R.; Oldenburg, J. Site-directed mutagenesis of coumarin-type anticoagulant-sensitive VKORC1: Evidence that highly conserved amino acids define structural requirements for enzymatic activity and inhibition by warfarin. Thromb. Haemost. 2005, 94, 780–786. [Google Scholar] [PubMed]
- Du, J.J.; Zhan, C.Y.; Lu, Y.; Cui, H.R.; Wang, X.Y. The conservative cysteines in transmembrane domain of AtVKOR/LTO1 are critical for photosynthetic growth and photosystem II activity in Arabidopsis. Front. Plant Sci. 2015, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Cui, H.R.; Yu, Z.B.; Du, J.J.; Xu, J.N.; Wang, X.Y. Key amino acids of arabidopsis VKOR in the activity of phylloquinone reduction and disulfide bond formation. Pro. Pept. Lett. 2015, 22, 81–86. [Google Scholar] [CrossRef]
- Rishavy, M.A.; Usubalieva, A.; Hallgren, K.W.; Berkner, K.L. Novel insight into the mechanism of the vitamin K oxidoreductase (VKOR): Electron relay through Cys43 and Cys51 reduces VKOR to allow vitamin K reduction and facilitation of vitamin K-dependent protein carboxylation. J. Biol. Chem. 2011, 286, 7267–7278. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Stafford, D.W. Mycobacterium tuberculosis vitamin K epoxide reductase homologue supports vitamin K-dependent carboxylation in mammalian cells. Antioxid. Redox. Signal. 2012, 16, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wajih, N.; Hutson, S.M.; Wallin, R. Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum. A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J. Biol. Chem. 2007, 282, 2626–2635. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.K.; Jin, D.Y.; Tie, K.; Stafford, D.W. Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J. Thromb. Haemost. 2013, 11, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tie, J.K.; Stafford, D.W.; Pedersen, L.G. Membrane topology for human vitamin K epoxide reductase. J. Thromb. Haemost. 2014, 12, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Fasco, M.J.; Principe, L.M.; Walsh, W.A.; Friedman, P.A. Warfarin inhibition of vitamin K 2,3-epoxide reductase in rat liver microsomes. Biochemistry 1983, 22, 5655–5660. [Google Scholar] [CrossRef] [PubMed]
- Fregin, A.; Czogalla, K.J.; Gansler, J.; Rost, S.; Taverna, M.; Watzka, M.; Bevans, C.G.; Müller, C.R.; Oldenburg, J. A new cell culture-based assay quantifies vitamin K 2,3-epoxide reductase complex subunit 1 function and reveals warfarin resistance phenotypes not shown by the dithiothreitol-driven VKOR assay. J. Thromb. Haemost. 2013, 11, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Müller, E.; Keller, A.; Fregin, A.; Müller, C.R.; Rost, S. Confirmation of warfarin resistance of naturally occurring VKORC1 variants by coexpression with coagulation factor IX and in silico protein modelling. BMC Genet. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, W.D. VKORC1 ER mislocalization causes rare disease. Blood 2014, 124, 1215–1216. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Shikano, S.; Li, M. Membrane receptor trafficking: Evidence of proximal and distal zones conferred by two independent endoplasmic reticulum localization signals. Proc. Natl. Acad. Sci. USA 2003, 100, 5783–5788. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czogalla, K.J.; Watzka, M.; Oldenburg, J. Structural Modeling Insights into Human VKORC1 Phenotypes. Nutrients 2015, 7, 6837-6851. https://doi.org/10.3390/nu7085313
Czogalla KJ, Watzka M, Oldenburg J. Structural Modeling Insights into Human VKORC1 Phenotypes. Nutrients. 2015; 7(8):6837-6851. https://doi.org/10.3390/nu7085313
Chicago/Turabian StyleCzogalla, Katrin J., Matthias Watzka, and Johannes Oldenburg. 2015. "Structural Modeling Insights into Human VKORC1 Phenotypes" Nutrients 7, no. 8: 6837-6851. https://doi.org/10.3390/nu7085313