
Quality Control of Emerging Contaminants in Marine Aquaculture Systems by Spot Sampling-Optimized Solid Phase Extraction and Passive Sampling

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Sampling
2.3. Solid-Phase Extraction (SPE)
2.3.1. Pesticides
2.3.2. Pharmaceuticals
2.4. Passive Sampling Procedure
2.5. LC–MS Analysis
3. Results and Discussions
3.1. Optimization and Validation of SPE Method for Pesticides
3.2. Optimization and Validation of SPE Method for the Determination of Pharmaceuticals
3.3. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernando, M.D.; Martínez-Bueno, M.J.; Fernández-Alba, A.R. Seawater Quality Control of Microcontaminants in Fish Farm Cage Systems: Application of Passive Sampling Devices. Bol. Inst. Esp. Oceanogr. 2005, 21, 37–46. [Google Scholar]
- European Commission. Directive 2008/56/EC, Official Journal of the European Union, L 164/19, 25.6.2008. 2008. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2008:164:0019:0040:EN:PDF (accessed on 31 January 2021).
- Batt, A.L.; Furlong, E.T.; Mash, H.E.; Glassmeyer, S.T.; Kolpin, D.W. The Importance of Quality Control in Validating Concentrations of Contaminants of Emerging Concern in Source and Treated Drinking Water Samples. Sci. Total Environ. 2017, 579, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldas, S.S.; Bolzan, C.M.; Guilherme, J.R.; Silveira, M.A.K.; Escarrone, A.L.V.; Primel, E.G. Determination of Pharmaceuticals, Personal Care Products, and Pesticides in Surface and Treated Waters: Method Development and Survey. Environ. Sci. Pollut. Res. 2013, 20, 5855–5863. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.; Lapworth, D.; Crane, E.; Hart, A. Review of Risk from Potential Emerging Contaminants in UK Groundwater. Sci. Total Environ. 2012, 416, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Vergeynst, L.; Haeck, A.; De Wispelaere, P.; Van Langenhove, H.; Demeestere, K. Multi-Residue Analysis of Pharmaceuticals in Wastewater by Liquid Chromatography–Magnetic Sector Mass Spectrometry: Method Quality Assessment and Application in a Belgian Case Study. Chemosphere 2015, 119, S2–S8. [Google Scholar] [CrossRef]
- Vanryckeghem, F.; Huysman, S.; Van Langenhove, H.; Vanhaecke, L.; Demeestere, K. Multi-Residue Quantification and Screening of Emerging Organic Micropollutants in the Belgian Part of the North Sea by Use of Speedisk Extraction and Q-Orbitrap HRMS. Mar. Pollut. Bull. 2019, 142, 350–360. [Google Scholar] [CrossRef]
- Belmonte Vega, A.; Garrido Frenich, A.; Martínez Vidal, J.L. Monitoring of Pesticides in Agricultural Water and Soil Samples from Andalusia by Liquid Chromatography Coupled to Mass Spectrometry. Anal. Chim. Acta 2005, 538, 117–127. [Google Scholar] [CrossRef]
- Larson, S.J.; Capel, P.D.; Goolsby, D.A.; Zaugg, S.D.; Sandstrom, M.W. Relations between Pesticide Use and Riverine Flux in the Mississippi River Basin. Chemosphere 1995, 31, 3305–3321. [Google Scholar] [CrossRef]
- Schulz, R. Rainfall-Induced Sediment and Pesticide Input from Orchards into the Lourens River, Western Cape, South Africa: Importance of a Single Event. Water Res. 2001, 35, 1869–1876. [Google Scholar] [CrossRef]
- Kearney, P.; Wauchope, R. Disposal Options Based on Properties of Pesticides in Soil and Water. In Pesticide Remediation in Soils and Water, 1st ed.; Kearney, P., Roberts, T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Flores, C.; Morgante, V.; González, M.; Navia, R.; Seeger, M. Adsorption Studies of the Herbicide Simazine in Agricultural Soils of the Aconcagua Valley, Central Chile. Chemosphere 2009, 74, 1544–1549. [Google Scholar] [CrossRef]
- Kalogridi, E.-C.; Christophoridis, C.; Bizani, E.; Drimaropoulou, G.; Fytianos, K. Part I: Temporal and Spatial Distribution of Multiclass Pesticide Residues in Lake Waters of Northern Greece: Application of an Optimized SPE-UPLC-MS/MS Pretreatment and Analytical Method. Environ. Sci. Pollut. Res. 2014, 21, 7239–7251. [Google Scholar] [CrossRef]
- Casado, J.; Santillo, D.; Johnston, P. Multi-Residue Analysis of Pesticides in Surface Water by Liquid Chromatography Quadrupole-Orbitrap High Resolution Tandem Mass Spectrometry. Anal. Chim. Acta 2018, 1024, 1–17. [Google Scholar] [CrossRef]
- Köck-Schulmeyer, M.; Postigo, C.; Farré, M.; Barceló, D.; López de Alda, M. Medium to Highly Polar Pesticides in Seawater: Analysis and Fate in Coastal Areas of Catalonia (NE Spain). Chemosphere 2019, 215, 515–523. [Google Scholar] [CrossRef]
- Gros, M.; Petrović, M.; Barceló, D. Wastewater treatment plants as a pathway for aquatic contamination by pharmaceuticals in the Ebro river basis (Northeast Spain). Environ. Toxicol. Chem. 2007, 26, 1553. [Google Scholar] [CrossRef]
- López-Pacheco, I.Y.; Silva-Núñez, A.; Salinas-Salazar, C.; Arévalo-Gallegos, A.; Lizarazo-Holguin, L.A.; Barceló, D.; Iqbal, H.M.N.; Parra-Saldívar, R. Anthropogenic Contaminants of High Concern: Existence in Water Resources and Their Adverse Effects. Sci. Total Environ. 2019, 690, 1068–1088. [Google Scholar] [CrossRef]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Ferro-García, M.Á.; Prados-Joya, G.; Ocampo-Pérez, R. Pharmaceuticals as Emerging Contaminants and Their Removal from Water. A Review. Chemosphere 2013, 93, 1268–1287. [Google Scholar] [CrossRef]
- Liu, H.-H.; Wong, C.S.; Zeng, E.Y. Recognizing the Limitations of Performance Reference Compound (PRC)-Calibration Technique in Passive Water Sampling. Environ. Sci. Technol. 2013, 47, 10104–10105. [Google Scholar] [CrossRef]
- Boxall, A.B.A.; Rudd, M.A.; Brooks, B.W.; Caldwell, D.J.; Choi, K.; Hickmann, S.; Innes, E.; Ostapyk, K.; Staveley, J.P.; Verslycke, T.; et al. Pharmaceuticals and Personal Care Products in the Environment: What Are the Big Questions? Environ. Health Perspect. 2012, 120, 1221–1229. [Google Scholar] [CrossRef]
- Hass, U.; Duennbier, U.; Massmann, G. Occurrence and Distribution of Psychoactive Compounds and Their Metabolites in the Urban Water Cycle of Berlin (Germany). Water Res. 2012, 46, 6013–6022. [Google Scholar] [CrossRef]
- Fono, L.J.; Kolodziej, E.P.; Sedlak, D.L. Attenuation of Wastewater-Derived Contaminants in an Effluent-Dominated River. Environ. Sci. Technol. 2006, 40, 7257–7262. [Google Scholar] [CrossRef]
- Li, Y.; Yao, C.; Zha, D.; Yang, W.; Lu, G. Selection of Performance Reference Compound (PRC) for Passive Sampling of Pharmaceutical Residues in an Effluent Dominated River. Chemosphere 2018, 211, 884–892. [Google Scholar] [CrossRef]
- Zhang, R.; Du, J.; Dong, X.; Huang, Y.; Xie, H.; Chen, J.; Li, X.; Kadokami, K. Occurrence and Ecological Risks of 156 Pharmaceuticals and 296 Pesticides in Seawater from Mariculture Areas of Northeast China. Sci. Total Environ. 2021, 792, 148375. [Google Scholar] [CrossRef]
- Lai, W.W.-P.; Lin, Y.-C.; Wang, Y.-H.; Guo, Y.L.; Lin, A.Y.-C. Occurrence of Emerging Contaminants in Aquaculture Waters: Cross-Contamination between Aquaculture Systems and Surrounding Waters. Water. Air. Soil Pollut. 2018, 229, 249. [Google Scholar] [CrossRef]
- Xie, H.; Hao, H.; Xu, N.; Liang, X.; Gao, D.; Xu, Y.; Gao, Y.; Tao, H.; Wong, M. Pharmaceuticals and Personal Care Products in Water, Sediments, Aquatic Organisms, and Fish Feeds in the Pearl River Delta: Occurrence, Distribution, Potential Sources, and Health Risk Assessment. Sci. Total Environ. 2019, 659, 230–239. [Google Scholar] [CrossRef]
- Ismail, N.A.H.; Wee, S.Y.; Kamarulzaman, N.H.; Aris, A.Z. Quantification of Multi-Classes of Endocrine-Disrupting Compounds in Estuarine Water. Environ. Pollut. 2019, 249, 1019–1028. [Google Scholar] [CrossRef]
- Togola, A.; Budzinski, H. Development of Polar Organic Integrative Samplers for Analysis of Pharmaceuticals in Aquatic Systems. Anal. Chem. 2007, 79, 6734–6741. [Google Scholar] [CrossRef]
- Martinezbueno, M.; Hernando, M.; Aguera, A.; Fernandezalba, A. Application of Passive Sampling Devices for Screening of Micro-Pollutants in Marine Aquaculture Using LC–MS/MS. Talanta 2009, 77, 1518–1527. [Google Scholar] [CrossRef]
- Valenzuela, E.F.; Menezes, H.C.; Cardeal, Z.L. Passive and Grab Sampling Methods to Assess Pesticide Residues in Water. A Review. Environ. Chem. Lett. 2020, 18, 1019–1048. [Google Scholar] [CrossRef]
- Miège, C.; Mazzella, N.; Allan, I.; Dulio, V.; Smedes, F.; Tixier, C.; Vermeirssen, E.; Brant, J.; O’Toole, S.; Budzinski, H.; et al. Position paper on passive sampling techniques for the monitoring of contaminants in the aquatic environment—Achievements to date and perspectives. Trends Environ. Anal. Chem. 2015, 8, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Nannou, C.I.; Kosma, C.I.; Albanis, T.A. Albanis. Occurrence of pharmaceuticals in surface waters: Analytical method development and environmental risk assessment. Int. J. Environ. Anal. Chem. 2015, 95, 1242–1262. [Google Scholar] [CrossRef]
- Papadakis, E.N.; Tsaboula, A.; Vryzas, Z.; Kotopoulou, A.; Kintzikoglou, K.; Papadopoulou-Mourkidou, E. Pesticides in the rivers and streams of two river basins in northern Greece. Sci. Total Environ. 2018, 624, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Lambropoulou, D.; Hela, D.; Koltsakidou, A.; Konstantinou, I. Overview of the Pesticide Residues in Greek Rivers: Occurrence and Environmental Risk Assessment. In The Rivers of Greece. The Handbook of Environmental Chemistry; Skoulikidis, N., Dimitriou, E., Karaouzas, I., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 59. [Google Scholar] [CrossRef]
- Nannou, C. Modern Analytical Methods for the Determination of Pesticide and Pharmaceutical Residues in Natural Waters and Sediments. Ph.D. Thesis, Department of Chemistry, University of Ioannina, Ioannina, Greece, 2018. [Google Scholar]
- European Commission. SANTE/12682/2019, Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticide Residues and Analysis in Food and Feed. 2019. Available online: https://www.eurl-pesticides.eu/userfiles/file/EurlALL/AqcGuidance_SANTE_2019_12682.pdf (accessed on 30 January 2021).
- Alvarez, D.A.; Petty, J.D.; Huckins, J.N.; Jones-Lepp, T.L.; Getting, D.T.; Goddard, J.P.; Manahan, S.E. development of a passive, in situ, integrative sampler for hydrophilic organic contaminants in aquatic environments. Environ. Toxicol. Chem. 2004, 23, 1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomatou, A.A.; Zacharias, I.; Hela, D.; Konstantinou, I. Passive sampling of selected pesticides in aquatic environment using polar oragnic chemical integrative samplers. Environ. Sci. Pollut. Res. 2011, 18, 1222–1233. [Google Scholar] [CrossRef] [PubMed]
- Vrana, B.; Urík, J.; Fedorova, G.; Švecová, H.; Grabicová, K.; Golovko, O.; Randák, T.; Grabic, R. In Situ Calibration of Polar Organic Chemical Integrative Sampler (POCIS) for Monitoring of Pharmaceuticals in Surface Waters. Environ. Pollut. 2021, 269, 116121. [Google Scholar] [CrossRef]
- Sadutto, D.; Pico, Y. Sample Preparation to Determine Pharmaceutical and Personal Care Products in an All-Water Matrix: Solid Phase Extraction. Molecules 2020, 25, 5204. [Google Scholar] [CrossRef]
- Wu, J.; Qian, X.; Yang, Z.; Zhang, L. Study on the matrix effect in the determination of selected pharmaceutical residues in seawater by solid-phase extraction and ultra-high-performance liquid chromatography–electrospray ionization low-energy collision-induced dissociation tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 1471–1475. [Google Scholar]
- Muñoz, I.; Martínez Bueno, M.J.; Agüera, A.; Fernández-Alba, A.R. Environmental and Human Health Risk Assessment of Organic Micro-Pollutants Occurring in a Spanish Marine Fish Farm. Environ. Pollut. 2010, 158, 1809–1816. [Google Scholar] [CrossRef]
- Ojemaye, C.Y.; Petrik, L. Pharmaceuticals in the marine environment: A review. Environ. Rev. 2019, 27, 151–165. [Google Scholar] [CrossRef]
- Carballeira, C.; De Orte, M.R.; Viana, I.G.; DelValls, T.A.; Carballeira, A. Assessing the toxicity of chemical compounds associated with land-based marine fish farms: The sea urchin embryo bioassay with Paracentrotus lividus and Arbacia lixula. Arch. Environ. Contam. Toxicol. 2012, 63, 249–261. [Google Scholar] [CrossRef]
- EU. Cybutryne EQS Dossier 2011 Prepared by the Sub-Group on Review of the Priority Substances List (under Working Group E of the Common Implementation Strategy for the Water Framework Directive). 2011. Available online: https://circabc.europa.eu/sd/d/1eb5aa3b-bf6c-48ca-8ce0-00488a0c2905/Cybutryne%20EQS%20%20dossier%202011.pdf (accessed on 30 January 2011).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Min | Max | Average | SD |
|---|---|---|---|---|
| Temperature (°C) | 14.9 | 17.5 | 15.9 | 0.82 |
| TDS (mg L−1) | 77,000 | 91,000 | 85,542 | 4496.3 |
| Conductivity (mS cm−1) | 45.6 | 50.5 | 47.8 | 1.97 |
| Salinity (‰) | 35.3 | 41.2 | 38.9 | 1.98 |
| pH | 6.5 | 7.5 | 6.8 | 0.4 |
| Compound | 25 ng L−1 | 100 ng L−1 | 250 ng L−1 | ||||
|---|---|---|---|---|---|---|---|
| (%) R | RSDr (%) | (%) R | RSDr (%) | RSDIP (%) | (%) R | RSDr (%) | |
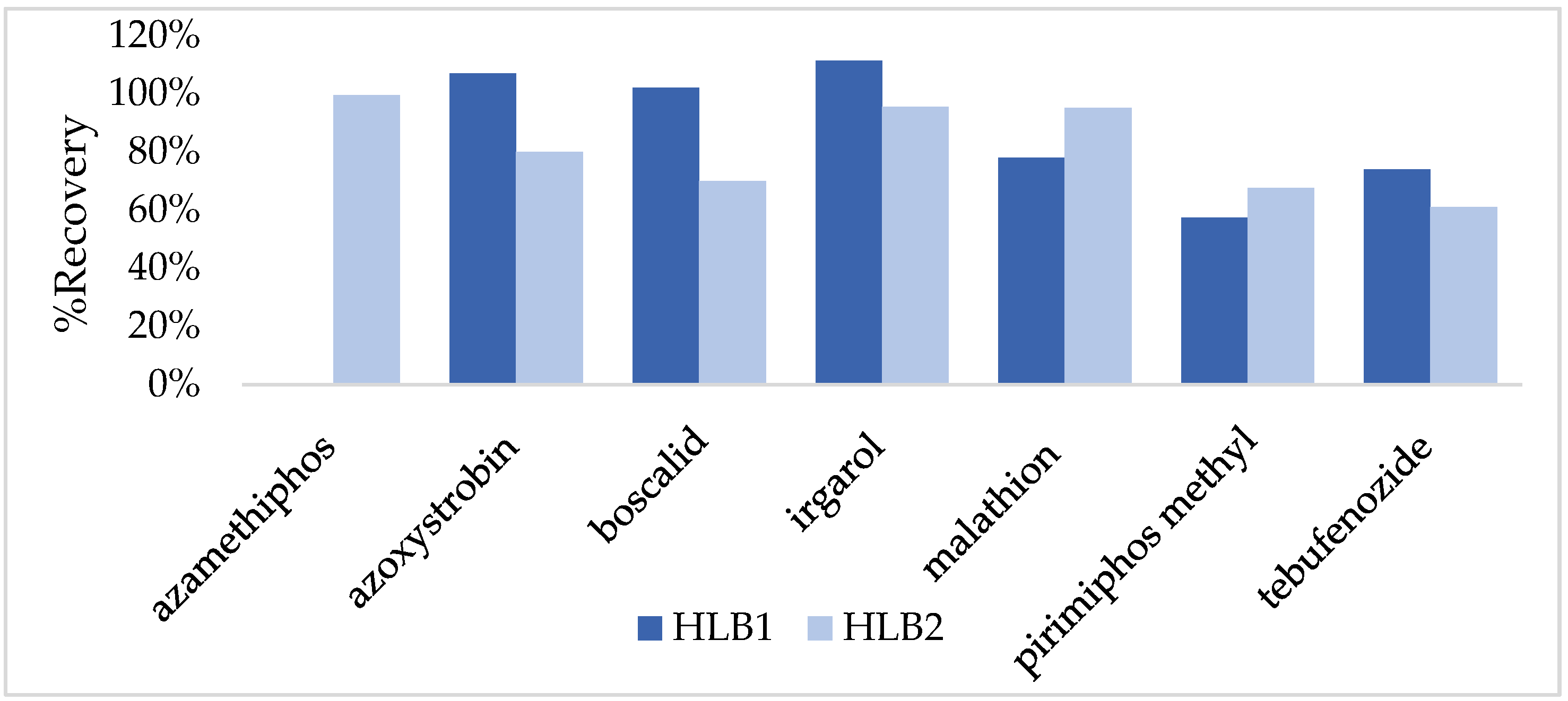
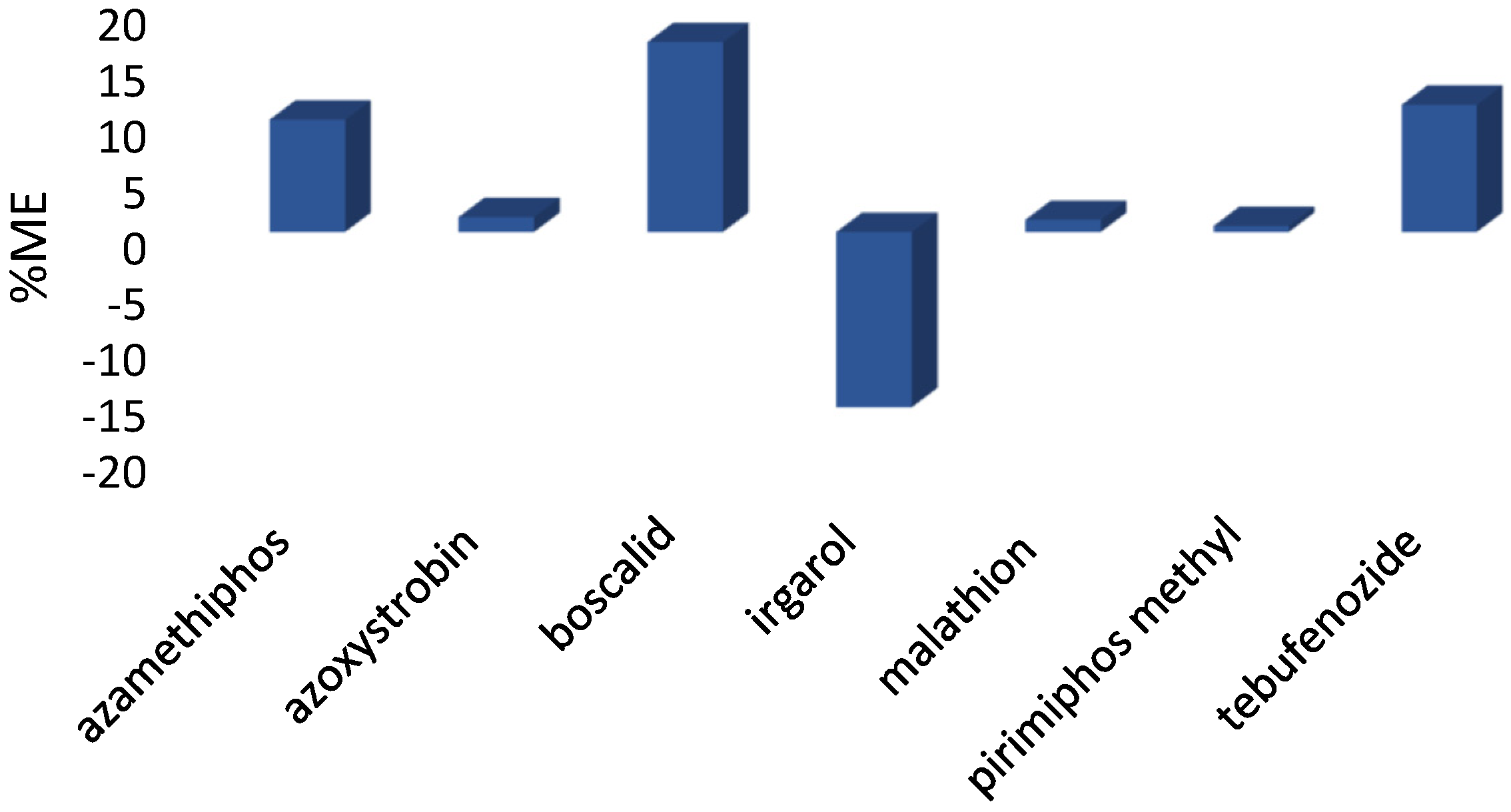
| Azamethipos | 80.5 | 10.4 | 79.9 | 4.0 | 5.2 | 70.6 | 7.9 |
| Azoxystrobin | 72.3 | 13.8 | 83.6 | 8.4 | 11.4 | 59.7 | 0.7 |
| Boscalid | 68.3 | 7.8 | 70.2 | 5.2 | 7.0 | 69.6 | 19.5 |
| Irgarol | 68.2 | 8.0 | 69.1 | 10.6 | 2.5 | 74.7 | 5.4 |
| Malathion | 81.4 | 2.9 | 95.0 | 5.4 | 7.7 | 78.3 | 17.6 |
| Pirimiphos-methyl | 98.9 | 4.9 | 82.0 | 2.9 | 9.4 | 77.2 | 8.8 |
| Tebufenozide | 58.5 | 1.2 | 61.9 | 7.1 | 8.3 | 58.6 | 14.7 |
| Compound | LOD (ng L−1) | LOQ (ng L−1) | Linear Range | R2 |
|---|---|---|---|---|
| Azamethipos | 7.5 | 25 | LOQ-500 | 0.9994 |
| Azoxystrobin | 0.3 | 1 | LOQ-500 | 0.9992 |
| Boscalid | 0.5 | 1.5 | LOQ-500 | 0.9996 |
| Irgarol | 0.2 | 0.5 | LOQ-500 | 0.9998 |
| Malathion | 0.5 | 1.5 | LOQ-500 | 0.9992 |
| Pirimiphos-methyl | 0.5 | 2 | LOQ-750 | 0.9996 |
| Tebufenozide | 3 | 10 | LOQ-500 | 0.9993 |
| Compound | 25 ng L−1 | 100 ng L−1 | 250 ng L−1 | ||||
|---|---|---|---|---|---|---|---|
| (%) R | RSDr (%) | (%) R | RSDr (%) | RSDIP (%) | (%) R | RSDr (%) | |
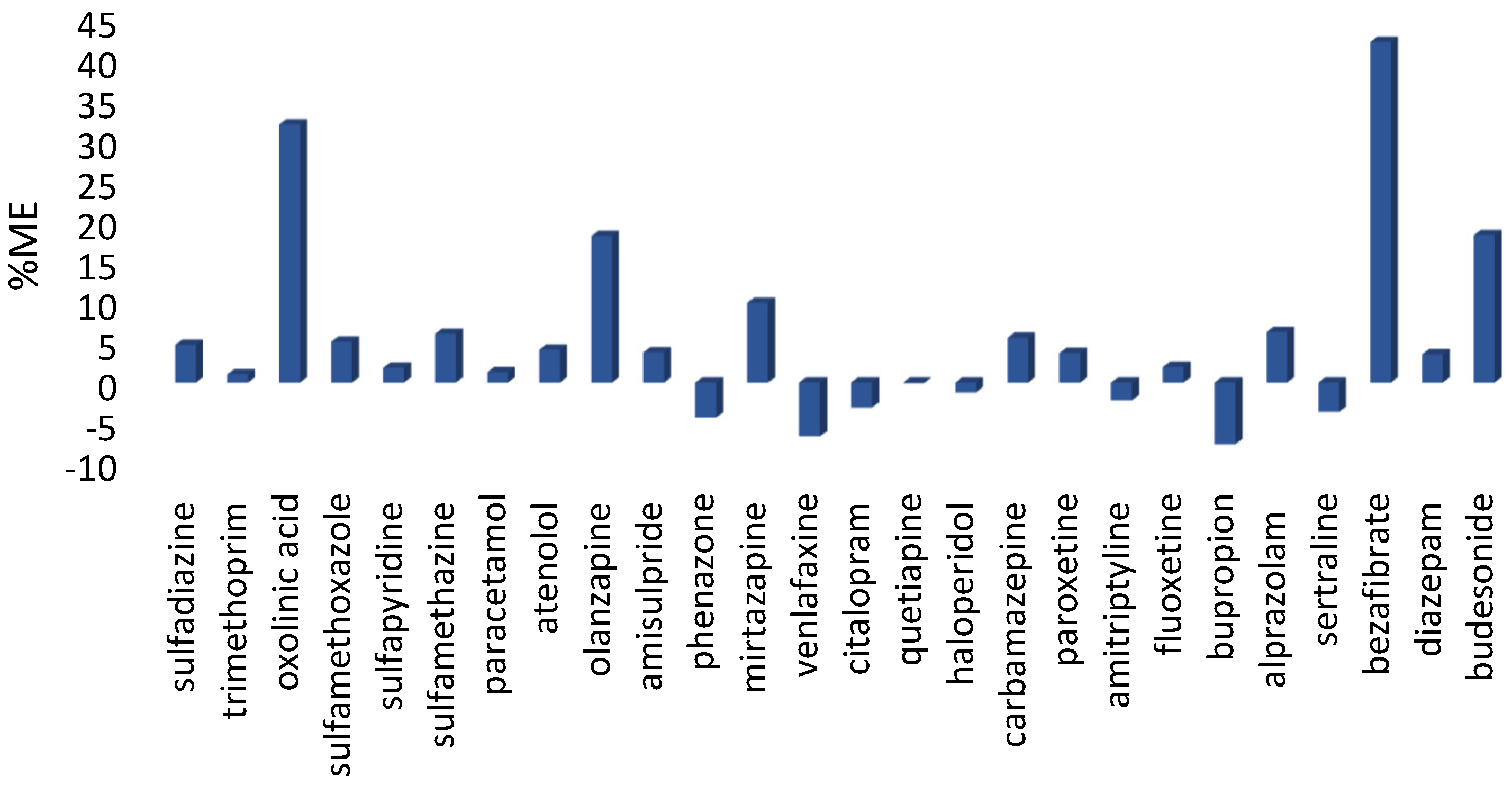
| Oxolinic acid | 105.6 | 12.2 | 80.8 | 15.8 | 7.2 | 118.3 | 2.1 |
| Sulfadiazine | − | − | 99.6 | 7.9 | 15.5 | 111.2 | 13.3 |
| Sulfamethazine | − | − | 91.9 | 8.1 | 10.3 | 73.2 | 3.5 |
| Sulfamethoxazole | − | − | 90.7 | 9.2 | 6.7 | 113.5 | 7.1 |
| Sulfapyridine | 88.8 | 8.7 | 98.1 | 11.3 | 18.2 | 118.8 | 3.1 |
| Trimethoprim | 61.6 | 9.8 | 78.4 | 9.0 | 9.5 | 95.4 | 0.1 |
| Paracetamol | − | − | 76.5 | 3.5 | 7.3 | 82.8 | 0.8 |
| Alprazolam | 105.1 | 10.7 | 94.2 | 7.5 | 6.2 | 81.6 | 5.4 |
| Amisulpride | 83.7 | 4.0 | 89.8 | 4.7 | 3.6 | 81.4 | 5.4 |
| Amitriptyline | 98.1 | 2.1 | 94.4 | 5.2 | 2.5 | 99.5 | 1.3 |
| Bupropion | 52.3 | 14.1 | 71.5 | 3.5 | 9.7 | 47.0 | 5.9 |
| Carbamazepine | 107.2 | 2.0 | 87.0 | 5.2 | 4.2 | 92.1 | 2.2 |
| Citalopram | 94.4 | 5.2 | 79.3 | 6.9 | 6.6 | 96.4 | 6.8 |
| Diazepam | 128.0 | 9.4 | 93.6 | 5.6 | 12.3 | 94.6 | 0.2 |
| Fluoxetine | 103.2 | 3.6 | 89.0 | 3.9 | 3.9 | 95.1 | 4.1 |
| Haloperidol | 96.0 | 4.2 | 87.4 | 4.6 | 5.6 | 100.5 | 5.4 |
| Mirtazapine | 113.3 | 7.5 | 82.2 | 6.5 | 10.1 | 119.0 | 3.6 |
| Olanzapine | 112.3 | 9.8 | 86.1 | 4.6 | 4.0 | 128.7 | 0.4 |
| Paroxetine | 104.0 | 4.3 | 79.6 | 9.6 | 7.3 | 118.8 | 19.2 |
| Quetiapine | 109.9 | 7.4 | 92.5 | 7.9 | 6.6 | 111.7 | 12.0 |
| Sertraline | 97.3 | 11.1 | 65.6 | 10.3 | 11.7 | 103.7 | 7.9 |
| Venlafaxine | 60.0 | 1.3 | 76.8 | 5.8 | 11.7 | 59.9 | 1.3 |
| Bezafibrate | 89.6 | 12.4 | 79.6 | 15.7 | 13.5 | 122.5 | 6.3 |
| Atenolol | 58.7 | 5.5 | 85.4 | 9.2 | 8.8 | 52.1 | 2.9 |
| Budesonide | 118.0 | 1.7 | 89.5 | 7.6 | 5.3 | 116.0 | 3.4 |
| Compound | LOD (ng L−1) | LOQ (ng L−1) | Linear Range | R2 |
|---|---|---|---|---|
| Oxolinic acid | 1.5 | 5 | LOQ-500 | 0.9992 |
| Sulfadiazine | 10 | 30 | LOQ-750 | 0.9998 |
| Sulfamethazine | 10 | 30 | LOQ-750 | 0.9990 |
| Sulfamethoxazole | 10 | 30 | LOQ-500 | 0.9986 |
| Sulfapyridine | 7.5 | 25 | LOQ-750 | 0.9997 |
| Trimethoprim | 0.5 | 2 | LOQ-500 | 0.9996 |
| Paracetamol | 5 | 12.5 | LOQ-750 | 0.9999 |
| Alprazolam | 1.5 | 5 | LOQ-250 | 0.9991 |
| Amisulpride | 1.5 | 5 | LOQ-250 | 0.9997 |
| Amitriptyline | 0.5 | 2 | LOQ-250 | 0.9998 |
| Bupropion | 3 | 10 | LOQ-250 | 0.9998 |
| Carbamazepine | 0.5 | 2 | LOQ-250 | 0.9997 |
| Citalopram | 1.5 | 5 | LOQ-250 | 0.9997 |
| Diazepam | 0.5 | 2 | LOQ-100 | 0.9980 |
| Fluoxetine | 1.5 | 5 | LOQ-250 | 0.9994 |
| Haloperidol | 5 | 12.5 | LOQ-250 | 1.000 |
| Mirtazapine | 7.5 | 25 | LOQ-250 | 0.9991 |
| Olanzapine | 0.5 | 2 | LOQ-250 | 0.9993 |
| Paroxetine | 7.5 | 25 | LOQ-750 | 1.000 |
| Quetiapine | 0.5 | 2 | LOQ-250 | 0.9984 |
| Sertraline | 1.5 | 5 | LOQ-250 | 0.9993 |
| Venlafaxine | 1.5 | 5 | LOQ-250 | 0.9998 |
| Bezafibrate | 7.5 | 25 | LOQ-750 | 0.9994 |
| Atenolol | 1.5 | 5 | LOQ-500 | 0.9984 |
| Budesonide | 3 | 10 | LOQ-500 | 0.9992 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinaiou, P.; Manoli, P.; Boti, V.; Hela, D.; Makou, E.; Albanis, T.; Konstantinou, I. Quality Control of Emerging Contaminants in Marine Aquaculture Systems by Spot Sampling-Optimized Solid Phase Extraction and Passive Sampling. Sustainability 2022, 14, 3452. https://doi.org/10.3390/su14063452
Martinaiou P, Manoli P, Boti V, Hela D, Makou E, Albanis T, Konstantinou I. Quality Control of Emerging Contaminants in Marine Aquaculture Systems by Spot Sampling-Optimized Solid Phase Extraction and Passive Sampling. Sustainability. 2022; 14(6):3452. https://doi.org/10.3390/su14063452
Chicago/Turabian StyleMartinaiou, Panagiota, Panagiota Manoli, Vasiliki Boti, Dimitra Hela, Elissavet Makou, Triantafyllos Albanis, and Ioannis Konstantinou. 2022. "Quality Control of Emerging Contaminants in Marine Aquaculture Systems by Spot Sampling-Optimized Solid Phase Extraction and Passive Sampling" Sustainability 14, no. 6: 3452. https://doi.org/10.3390/su14063452