
Integrating Clopidogrel’s First-Pass Effect in a Joint Semi-Physiological Population Pharmacokinetic Model of the Drug and Its Inactive Carboxylic Acid Metabolite
 , ,
, , 
Abstract
:1. Introduction
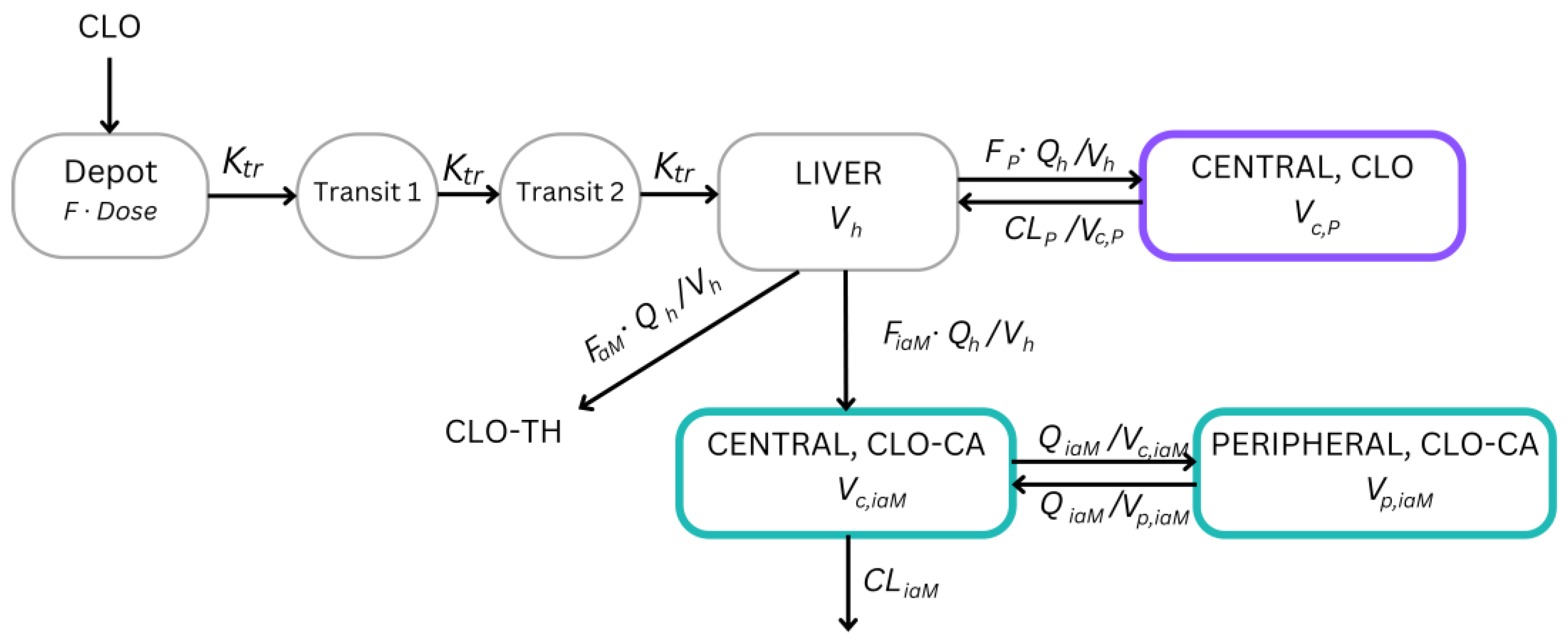
2. Materials and Methods
2.1. Subjects and Data
2.2. Data Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Medicine Agency (EMA). Plavix 75 mg Film-Coated Tablets. EPAR Product Information EMEA/H/C/000174. Available online: https://www.ema.europa.eu/en/documents/product-information/plavix-epar-product-information_en.pdf (accessed on 3 April 2024).
- Kazui, M.; Nishiya, Y.; Ishizuka, T.; Hagihara, K.; Farid, N.A.; Okazaki, O.; Ikeda, T.; Kurihara, A. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab. Dispos. 2010, 38, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Loer, H.L.H.; Turk, D.; Gomez-Mantilla, J.D.; Selzer, D.; Lehr, T. Physiologically Based Pharmacokinetic (PBPK) Modeling of Clopidogrel and Its Four Relevant Metabolites for CYP2B6, CYP2C8, CYP2C19, and CYP3A4 Drug-Drug-Gene Interaction Predictions. Pharmaceutics 2022, 14, 915. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Jin, B.H.; Park, M.S.; Kim, C.O.; Chae, D. Population pharmacokinetic-pharmacodynamic modeling of clopidogrel for dose regimen optimization based on CYP2C19 phenotypes: A proof of concept study. CPT Pharmacomet. Syst. Pharmacol. 2023, 13, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Karazniewicz-Lada, M.; Danielak, D.; Burchardt, P.; Kruszyna, L.; Komosa, A.; Lesiak, M.; Glowka, F. Clinical pharmacokinetics of clopidogrel and its metabolites in patients with cardiovascular diseases. Clin. Pharmacokinet. 2014, 53, 155–164. [Google Scholar] [CrossRef] [PubMed]
- European Medicine Agency (EMA). Clinical Pharmacology and Pharmacokinetics: Questions and Answers. Bioequivalence Studies for Generic Products Containing Clopidogrel. Available online: https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/scientific-guidelines/clinical-pharmacology-pharmacokinetics/clinical-pharmacology-pharmacokinetics-questions-answers (accessed on 3 April 2024).
- Mani, H.; Toennes, S.W.; Linnemann, B.; Urbanek, D.A.; Schwonberg, J.; Kauert, G.F.; Lindhoff-Last, E. Determination of clopidogrel main metabolite in plasma: A useful tool for monitoring therapy? Ther. Drug Monit. 2008, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Pejcic, Z.; Vucicevic, K.; Garcia-Arieta, A.; Miljkovic, B. Adjusted indirect comparisons to assess bioequivalence between generic clopidogrel products in Serbia. Br. J. Clin. Pharmacol. 2019, 85, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Heads of Medicines Agencies (HMA). Public Assessment Report Scientific Discussion Agregex 75 mg Film-Coated Tablets, DK/H/1624/001/DC. Available online: http://www.hma.eu/fileadmin/dateien/pipar/dk1624/parmod5_dk1624agregex.pdf (accessed on 30 March 2024).
- Heads of Medicines Agencies (HMA). Public Assessment Report Scientific Discussion Clopidogrel Sandoz 75 mg Film-Coated Tablets, AT/H/1449/001. Available online: https://mri.cts-mrp.eu/portal/details?productnumber=AT/H/1449/001 (accessed on 29 March 2024).
- Bahrami, G.; Mohammadi, B.; Sisakhtnezhad, S. High-performance liquid chromatographic determination of inactive carboxylic acid metabolite of clopidogrel in human serum: Application to a bioequivalence study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 864, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Kang, W.; Lee, H.W.; Park, D.J.; Ahn, J.H.; Kim, M.J.; Kim, E.Y.; Kim, S.W.; Nam, H.S.; Na, H.J.; et al. Bioequivalence and tolerability of two clopidogrel salt preparations, besylate and bisulfate: A randomized, open-label, crossover study in healthy Korean male subjects. Clin. Ther. 2009, 31, 793–803. [Google Scholar] [CrossRef] [PubMed]
- El Ahmady, O.; Ibrahim, M.; Hussein, A.M.; Bustami, R.T. Bioequivalence of two oral formulations of clopidogrel tablets in healthy male volunteers. Int. J. Clin. Pharmacol. Ther. 2009, 47, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Filipe, A.; Almeida, S.; Franco Spinola, A.C.; Neves, R.; Tanguay, M.; Jimenez, C.; Shink, E. Single-dose randomized, open-label, 2-way crossover bioequivalence study of clopidogrel 75 mg tablet in healthy volunteers under fasting conditions. Int. J. Clin. Pharmacol. Ther. 2009, 47, 187–194. [Google Scholar] [CrossRef]
- European Medicine Agency (EMA). CHMP Assessment Report for Clopidogrel Teva Film-Coated Tablets 75 mg, EPAR Scientific Discussion, EMEA/H/C/001053. Available online: https://www.ema.europa.eu/en/documents/assessment-report/clopidogrel-teva-hydrogen-sulphate-epar-public-assessment-report_en.pdf (accessed on 9 April 2024).
- European Medicine Agency (EMA). CHMP Assessment Report for Zyllt Film-Coated Tablets 75 mg, EPAR—Scientific Discussion, EMEA/H/C/1058. Available online: https://www.ema.europa.eu/en/documents/assessment-report/zyllt-epar-public-assessment-report_en.pdf (accessed on 9 April 2024).
- Ette, E.I.; Williams, P.J. Population pharmacokinetics I: Background, concepts, and models. Ann. Pharmacother. 2004, 38, 1702–1706. [Google Scholar] [CrossRef]
- Roganović, M.; Homšek, A.; Jovanović, M.; Topić Vučenović, V.; Ćulafić, M.; Miljković, B.; Vučićević, K. Concept and utility of population pharmacokinetic and pharmacokinetic/pharmacodynamic models in drug development and clinical practice. Arch. Pharm. 2021, 71, 336–353. [Google Scholar] [CrossRef]
- Yousef, A.M.; Melhem, M.; Xue, B.; Arafat, T.; Reynolds, D.K.; Van Wart, S.A. Population pharmacokinetic analysis of clopidogrel in healthy Jordanian subjects with emphasis optimal sampling strategy. Biopharm. Drug Dispos. 2013, 34, 215–226. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.; Kang, W.; Seong, S.J.; Lim, M.S.; Lee, H.W.; Yim, D.S.; Sohn, D.R.; Han, S.; Yoon, Y.R. Population pharmacokinetic/pharmacodynamic modeling of clopidogrel in Korean healthy volunteers and stroke patients. J. Clin. Pharmacol. 2012, 52, 985–995. [Google Scholar] [CrossRef]
- Ernest, C.S., 2nd; Small, D.S.; Rohatagi, S.; Salazar, D.E.; Wallentin, L.; Winters, K.J.; Wrishko, R.E. Population pharmacokinetics and pharmacodynamics of prasugrel and clopidogrel in aspirin-treated patients with stable coronary artery disease. J. Pharmacokinet. Pharmacodyn. 2008, 35, 593–618. [Google Scholar] [CrossRef]
- Danielak, D.; Karazniewicz-Lada, M.; Komosa, A.; Burchardt, P.; Lesiak, M.; Kruszyna, L.; Graczyk-Szuster, A.; Glowka, F. Influence of genetic co-factors on the population pharmacokinetic model for clopidogrel and its active thiol metabolite. Eur. J. Clin. Pharmacol. 2017, 73, 1623–1632. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, H.; Liu, Y.; Lai, X.; Gong, Y.; Liu, X.; Li, Y.G.; He, Y.; Zhang, E.Y.; Yan, X. Semi-mechanistic population pharmacokinetics analysis reveals distinct CYP2C19 dependency in the bioactivation of vicagrel and clopidogrel to active metabolite M15-2. Eur. J. Pharm. Sci. 2022, 177, 106264. [Google Scholar] [CrossRef]
- Jiang, X.L.; Samant, S.; Lewis, J.P.; Horenstein, R.B.; Shuldiner, A.R.; Yerges-Armstrong, L.M.; Peletier, L.A.; Lesko, L.J.; Schmidt, S. Development of a physiology-directed population pharmacokinetic and pharmacodynamic model for characterizing the impact of genetic and demographic factors on clopidogrel response in healthy adults. Eur. J. Pharm. Sci. 2016, 82, 64–78. [Google Scholar] [CrossRef]
- Djebli, N.; Fabre, D.; Boulenc, X.; Fabre, G.; Sultan, E.; Hurbin, F. Physiologically based pharmacokinetic modeling for sequential metabolism: Effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab. Dispos. 2015, 43, 510–522. [Google Scholar] [CrossRef]
- Duong, J.K.; Nand, R.A.; Patel, A.; Della Pasqua, O.; Gross, A.S. A physiologically based pharmacokinetic model of clopidogrel in populations of European and Japanese ancestry: An evaluation of CYP2C19 activity. Pharmacol. Res. Perspect. 2022, 10, e00946. [Google Scholar] [CrossRef]
- Xu, R.J.; Kong, W.M.; An, X.F.; Zou, J.J.; Liu, L.; Liu, X.D. Physiologically-Based Pharmacokinetic-Pharmacodynamics Model Characterizing CYP2C19 Polymorphisms to Predict Clopidogrel Pharmacokinetics and Its Anti-Platelet Aggregation Effect Following Oral Administration to Coronary Artery Disease Patients with or without Diabetes. Front. Pharmacol. 2020, 11, 593982. [Google Scholar]
- World Medical Association. Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects. Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 13 April 2024).
- European Medicines Agency (EMA). Guideline for Good Clinical Practice E6(R2). EMA/CPMP/ICH/135/95. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-good-clinical-practice-e6r2-step-5_en.pdf (accessed on 7 April 2024).
- European Medicine Agency (EMA). Guideline on the Investigation of Bioequivalence. CPMP/EWP/QWP/1401/98 Rev. 1/Corr**. Available online: https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf (accessed on 10 April 2024).
- Beal, S.; Sheiner, L.; Boeckmann, A.; Bauer, R. NONMEM 7.4 User’s Guides; Icon Development Solutions: Ellicott City, MD, USA, 1989–2018. [Google Scholar]
- Owen, J.; Fiedler-Kelly, J. Introduction to Population Pharmacokinetic/Pharmacodynamic Analysis with Nonlinear Mixed Effects Models; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: http://www.R-project.org (accessed on 1 October 2023).
- R Studio Team. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2019; Available online: http://www.rstudio.com (accessed on 1 October 2023).
- Savic, R.M.; Jonker, D.M.; Kerbusch, T.; Karlsson, M.O. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007, 34, 711–726. [Google Scholar] [CrossRef]
- Gibiansky, L.; Giraudon, M.; Rayner, C.R.; Brennan, B.J.; Subramoney, V.; Robson, R.; Kamal, M.A. Population pharmacokinetic analysis of oseltamivir and oseltamivir carboxylate following intravenous and oral administration to patients with and without renal impairment. J. Pharmacokinet. Pharmacodyn. 2015, 42, 225–236. [Google Scholar] [CrossRef]
- Yu, H.; Steeghs, N.; Kloth, J.S.; de Wit, D.; van Hasselt, J.G.; van Erp, N.P.; Beijnen, J.H.; Schellens, J.H.; Mathijssen, R.H.; Huitema, A.D. Integrated semi-physiological pharmacokinetic model for both sunitinib and its active metabolite SU12662. Br. J. Clin. Pharmacol. 2015, 79, 809–819. [Google Scholar] [CrossRef]
- Hooker, A.C.; Staatz, C.E.; Karlsson, M.O. Conditional weighted residuals (CWRES): A model diagnostic for the FOCE method. Pharm. Res. 2007, 24, 2187–2197. [Google Scholar] [CrossRef]
- Karlsson, M.O.; Savic, R.M. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 2007, 82, 17–20. [Google Scholar] [CrossRef]
- Gobburu, J.V.; Lawrence, J. Application of resampling techniques to estimate exact significance levels for covariate selection during nonlinear mixed effects model building: Some inferences. Pharm. Res. 2002, 19, 92–98. [Google Scholar] [CrossRef]
- Parke, J.; Holford, N.H.; Charles, B.G. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput. Methods Programs Biomed. 1999, 59, 19–29. [Google Scholar] [CrossRef]
- Bergstrand, M.; Hooker, A.C.; Wallin, J.E.; Karlsson, M.O. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011, 13, 143–151. [Google Scholar] [CrossRef]
- Taubert, D.; Kastrati, A.; Harlfinger, S.; Gorchakova, O.; Lazar, A.; von Beckerath, N.; Schomig, A.; Schomig, E. Pharmacokinetics of clopidogrel after administration of a high loading dose. Thromb. Haemost. 2004, 92, 311–316. [Google Scholar]
- Rousseau, A.; Leger, F.; Le Meur, Y.; Saint-Marcoux, F.; Paintaud, G.; Buchler, M.; Marquet, P. Population pharmacokinetic modeling of oral cyclosporin using NONMEM: Comparison of absorption pharmacokinetic models and design of a Bayesian estimator. Ther. Drug Monit. 2004, 26, 23–30. [Google Scholar] [CrossRef]
- Jiang, X.L.; Samant, S.; Lesko, L.J.; Schmidt, S. Clinical pharmacokinetics and pharmacodynamics of clopidogrel. Clin. Pharmacokinet. 2015, 54, 147–166. [Google Scholar] [CrossRef]
- Song, B.L.; Wan, M.; Tang, D.; Sun, C.; Zhu, Y.B.; Linda, N.; Fan, H.W.; Zou, J.J. Effects of CYP2C19 Genetic Polymorphisms on the Pharmacokinetic and Pharmacodynamic Properties of Clopidogrel and Its Active Metabolite in Healthy Chinese Subjects. Clin. Ther. 2018, 40, 1170–1178. [Google Scholar] [CrossRef]
- Zhu, H.J.; Wang, X.; Gawronski, B.E.; Brinda, B.J.; Angiolillo, D.J.; Markowitz, J.S. Carboxylesterase 1 as a determinant of clopidogrel metabolism and activation. J. Pharmacol. Exp. Ther. 2013, 344, 665–672. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Characteristics (Units) | Number (%)/Mean ± Standard Deviation (Range) | |
|---|---|---|
| Gender | Male | 29 (58.00) |
| Female | 21 (42.00) | |
| Age (year) | 31.94 ± 8.51 (19–54) | |
| Body-weight (kg) | 74.1 ± 13.56 (47–100) | |
| Height (cm) | 177.26 ± 9.06 (155–194) | |
| Body mass index (kg/m2) | 23.40 ± 2.66 (19.10–29.30) | |
| Bilirubin (µmol/L) | 9.1 ± 4.43 (3–25) | |
| Serum creatinine (µmol/L) | 81.5 ± 17.70 (53–114) | |
| Alanine transaminase (ALT) (U/L) | 26.2 ± 9.85 (11–52) | |
| Aspartate transaminase (AST) (U/L) | 23.7 ± 4.80 (16–35) | |
| Parameters (Units) | Dataset | Sampling Importance Resampling (SIR) | ||
|---|---|---|---|---|
| Estimate | 95% CI | Median | 2.5–97.5 Percentile | |
| CLP (L/h/70 kg) | 89.5 FIX | - | - | - |
| Vc,P (L/70 kg) | 218 | 188–248 | 217 | 189–243 |
| MTT_st1 (h) | 0.470 | 0.425–0.515 | 0.471 | 0.429–0.516 |
| Fgen_st1 | 1.08 | 0.993–1.17 | 1.08 | 0.995–1.16 |
| FR1_st1 | 119 | 84.3–154 | 118 | 88.2–155 |
| CLiaM (L/h/70 kg) | 8.70 | 7.38–10.0 | 8.60 | 7.59–9.73 |
| Vc,iaM (L/70 kg) | 23.7 | 19.7–27.7 | 23.4 | 20.0–26.9 |
| QiaM (L/h/70 kg) | 10.8 | 8.02–13.6 | 10.8 | 8.94–13.0 |
| Vp,iaM (L/70 kg) | 61.3 | 50.3–72.3 | 60.9 | 52.4–70.2 |
| Qh (L/h) | 50 FIX | - | - | - |
| Vh (L/70 kg) | 1.5 FIX | - | - | - |
| MTT_st2 (h) | 0.410 | 0.381–0.439 | 0.411 | 0.385–0.438 |
| Fgen_st2 | 0.960 | 0.818–1.10 | 0.952 | 0.840–1.07 |
| FR1_st2 | 76.8 | 64.8–88.8 | 76.0 | 66.3–85.9 |
| Inter-Individual (IIV)/Inter-Occasional Variability (IOV) | Estimate CV (%) | RSE (%) | Median CV (%) | 2.5–97.5 Percentile |
| IIV (Vc,P) | 45.82 | 10.4 | 45.69 | 37.38–53.99 |
| IIV (Vc,iaM) | 25.06 | 13.2 | 25.30 | 18.54–30.38 |
| IIV (F_st1) | 42.66 | 13.8 | 42.40 | 29.99–51.26 |
| IIV (F_st2) | 25.88 | 24.1 | 26.31 | 14.16–35.57 |
| IOV (F_st1) | 8.83 | 32.9 | 9.29 | 3.03–13.33 |
| IOV (F_st2) | 23.24 | 13.6 | 23.76 | 18.21–28.27 |
| IOV (MTT_st1) | 25.44 | 16.8 | 25.58 | 16.22–32.97 |
| IOV (MTT_st2) | 27.48 | 9.9 | 27.46 | 21.86–32.39 |
| IIV (FR1_st1) | 72.80 | 10.5 | 73.59 | 57.52–89.97 |
| IIV (FR1_st2) | 27.86 | 13.0 | 28.36 | 20.25–34.18 |
| Residual Error | Estimate (%) | RSE (%) | Median CV (%) | 2.5–97.5 Percentile |
| Wp (st1) | 41.95 | 3.4 | 41.98 | 39.37–44.50 |
| Wp (st2) | 29.39 | 5.7 | 29.51 | 27.57–31.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pejčić, Z.; Topić Vučenović, V.; Miljković, B.; Vučićević, K.M. Integrating Clopidogrel’s First-Pass Effect in a Joint Semi-Physiological Population Pharmacokinetic Model of the Drug and Its Inactive Carboxylic Acid Metabolite. Pharmaceutics 2024, 16, 685. https://doi.org/10.3390/pharmaceutics16050685
Pejčić Z, Topić Vučenović V, Miljković B, Vučićević KM. Integrating Clopidogrel’s First-Pass Effect in a Joint Semi-Physiological Population Pharmacokinetic Model of the Drug and Its Inactive Carboxylic Acid Metabolite. Pharmaceutics. 2024; 16(5):685. https://doi.org/10.3390/pharmaceutics16050685
Chicago/Turabian StylePejčić, Zorica, Valentina Topić Vučenović, Branislava Miljković, and Katarina M. Vučićević. 2024. "Integrating Clopidogrel’s First-Pass Effect in a Joint Semi-Physiological Population Pharmacokinetic Model of the Drug and Its Inactive Carboxylic Acid Metabolite" Pharmaceutics 16, no. 5: 685. https://doi.org/10.3390/pharmaceutics16050685