
Development of Oral Tablets of Nebivolol with Improved Dissolution Properties, Based on Its Combinations with Cyclodextrins

 , , ,
, , ,
Abstract
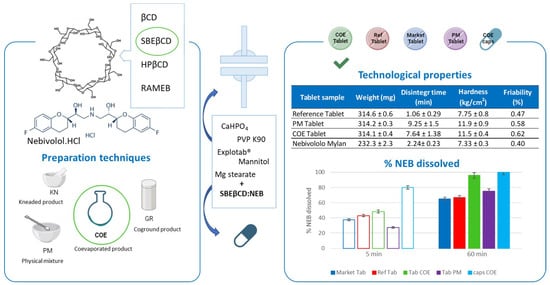
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Phase-Solubility Studies
2.3. Preparation of Drug–CD Solid Systems
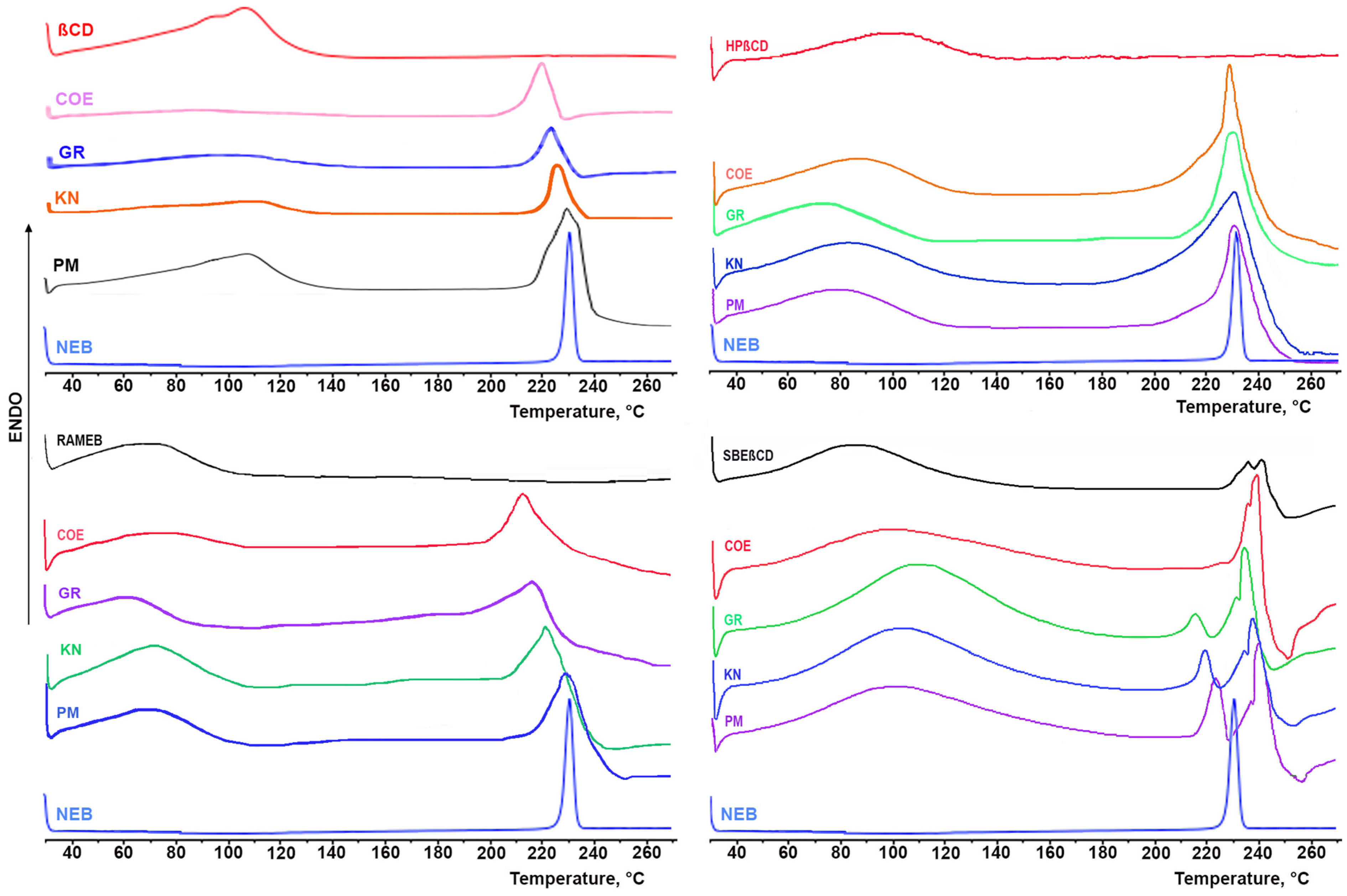
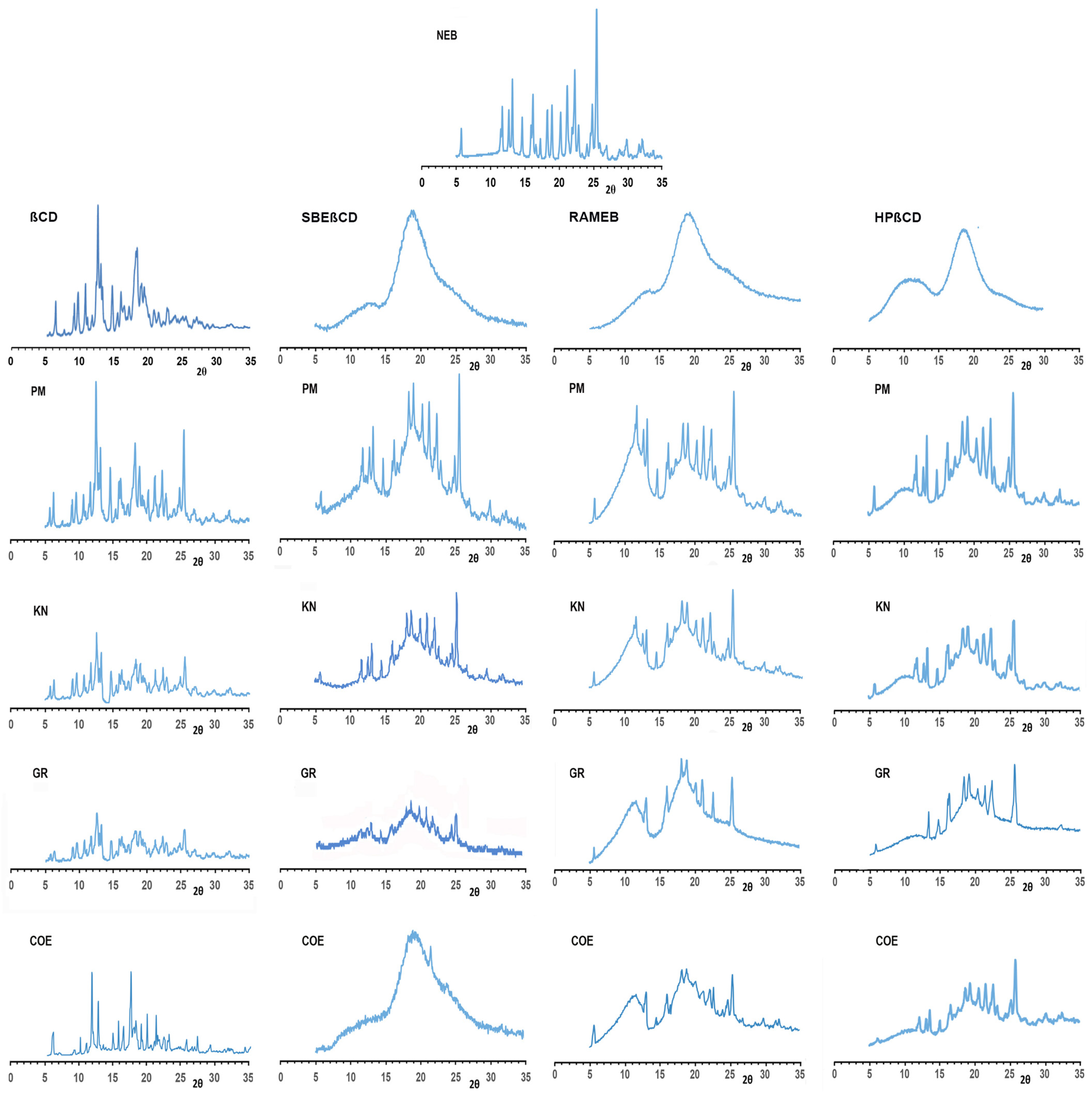
2.4. Solid-State Characterization of Drug–CD Binary Systems
2.5. Dissolution Studies
2.6. Preparation and Characterization of Tablets
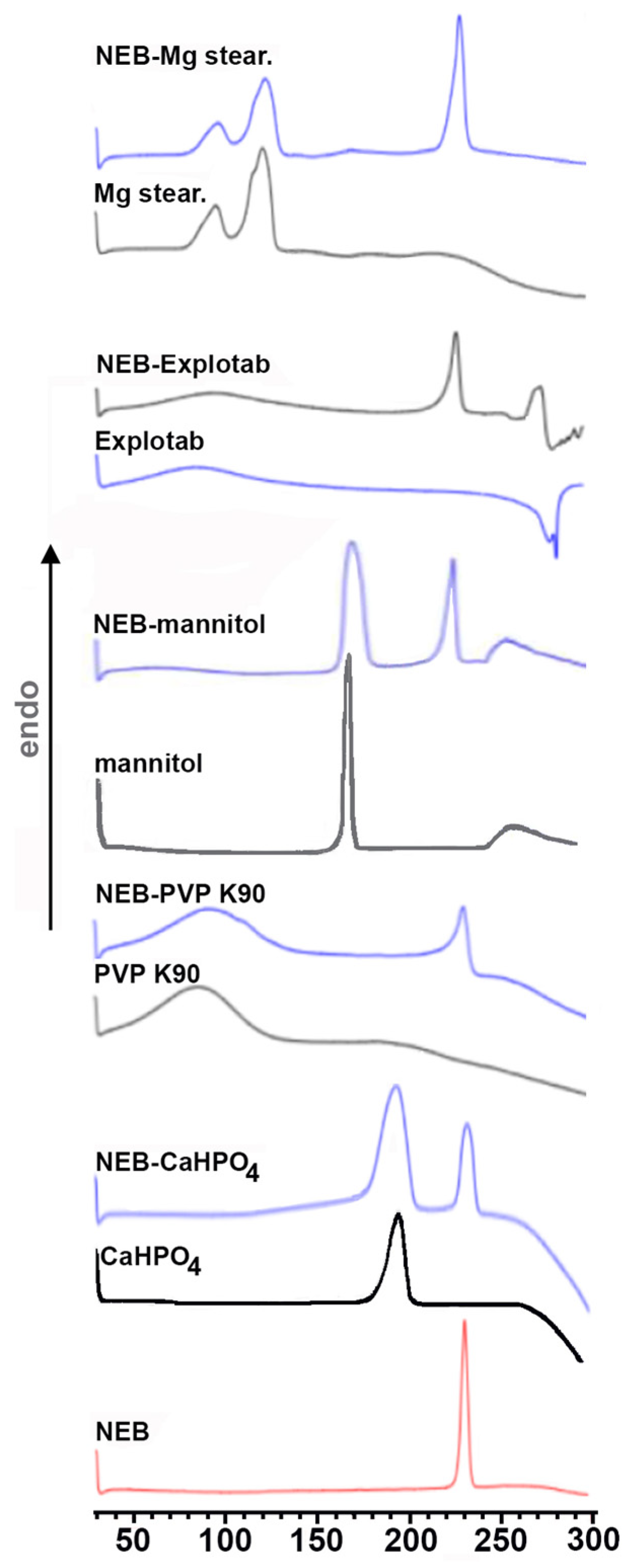
2.7. Compatibility Studies
3. Results
3.1. Phase-Solubility Studies
3.2. Preparation and Characterization of Drug–CD Solid Systems
3.2.1. Solid-State Studies
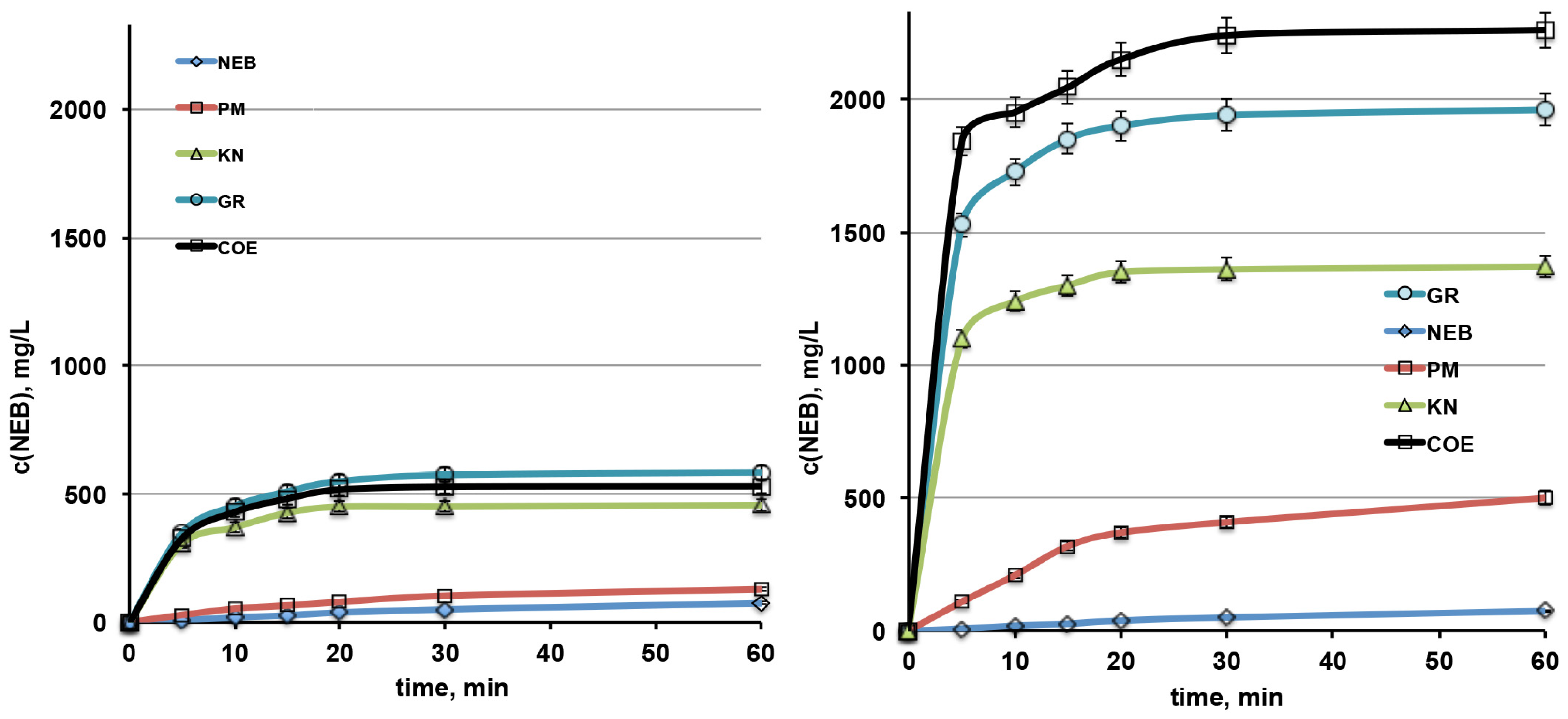
3.2.2. Dissolution Rate Studies
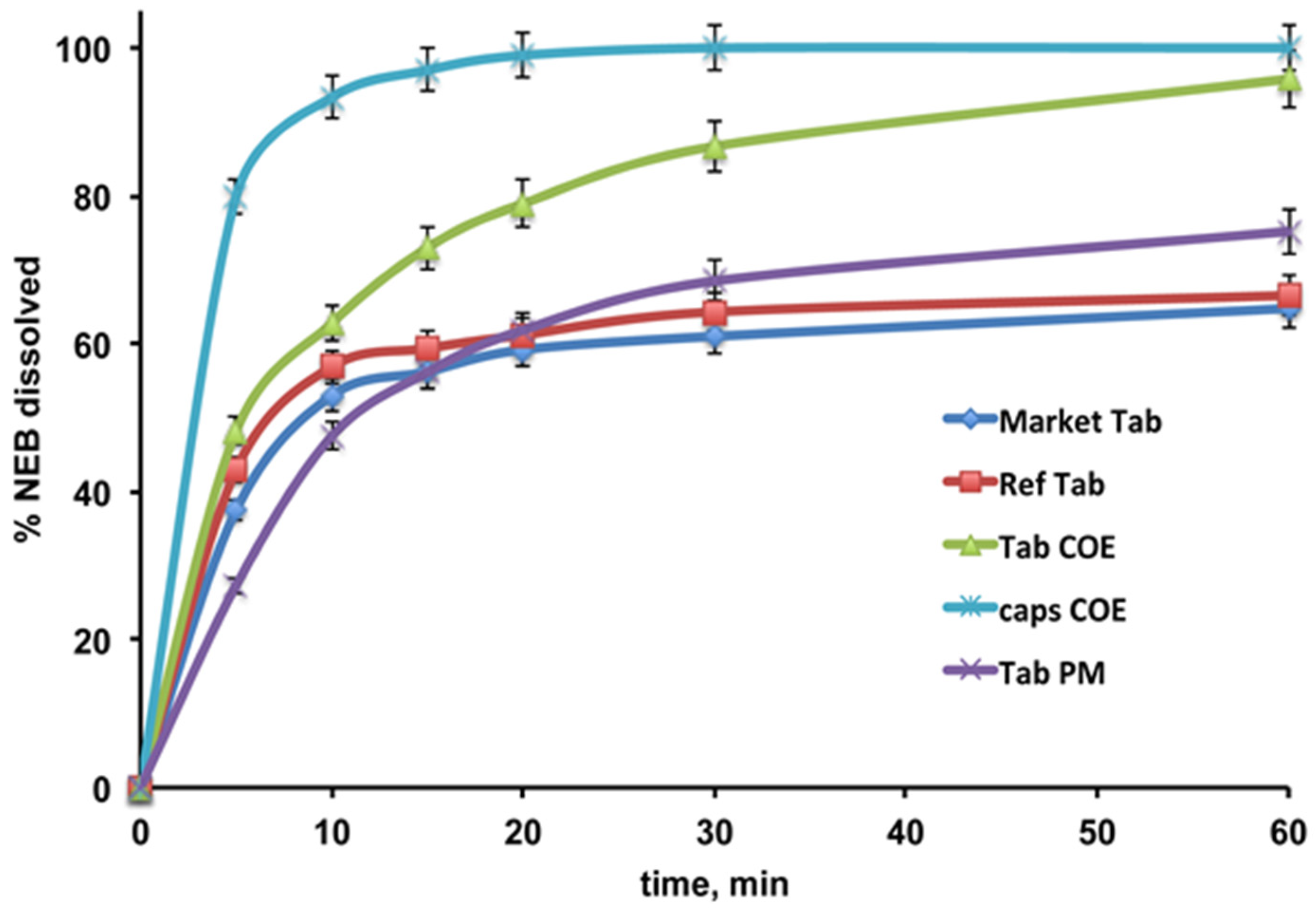
3.3. Preparation and Characterization of NEB Tablets
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NEB | Nebivolol HCl |
| CD | Cyclodextrin |
| SBEβCD | Sulfobutylether-βCD |
| HPβCD | Hydroxypropyl-βCD |
| RAMEB | Randomly substituted methyl-βCD |
| PM | Physical mixture |
| GR | Coground products |
| COE | Coevaporated products |
| KN | Kneaded products |
| CE | Complexation efficiency |
| DE | Dissolution efficiency |
| DSC | Differential scanning calorimetry |
| PXRD | Powder X-ray diffractometry |
References
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J. Hypertens. 2018, 36, 1953–2041. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Gallo, G.; Tocci, G. Is early and fast blood pressure control important in hypertension management? Int. J. Cardiol. 2018, 254, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.W.M. Nebivolol: A third-generation β-blocker for hypertension. Clin. Ther. 2009, 31, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Fongemie, J.; Felix-Getzik, E. A Review of Nebivolol Pharmacology and Clinical Evidence. Drugs 2015, 75, 1349–1371. [Google Scholar] [CrossRef]
- Moen, M.D.; Wagstaff, A.J. Nebivolol: A Review of its Use in the Management of Hypertension and Chronic Heart Failure. Drugs 2006, 66, 1389–1409. [Google Scholar] [CrossRef] [PubMed]
- Olawi, N.; Krüger, M.; Grimm, D.; Infanger, M.; Wehland, M. Nebivolol in the treatment of arterial hypertension. Basic Clin. Pharmacol. Toxicol. 2019, 125, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Gulcan, E.; Ilhan, D.; Toker, S. Nebivolol Might Be Beneficial in the Prevention and Treatment of Diabetic Neuropathy. Am. J. Ther. 2016, 23, e240. [Google Scholar] [CrossRef]
- Gielen, W.; Cleophas, T.J.; Agrawal, R. Nebivolol: A review of its clinical and pharmacological characteristics. Int. J. Clin. Pharmacol. Ther. 2006, 44, 344–357. [Google Scholar] [CrossRef]
- Shah, H.; Shah, V.; Bhutani, S.; Parikh, D.; Mehta, T. Dissolution improvement of nebivolol hydrochloride using solid dispersion adsorbate technique. Asian J. Pharm. 2015, 9, 49–55. [Google Scholar] [CrossRef]
- Raj, A.L.; Kumar, Y.S. Preparation and Evaluation of Solid Dispersion of Nebivolol Using Solvent Evaporation Method. Int. J. Pharm. Sci. Drug Res. 2018, 10, 322–328. [Google Scholar] [CrossRef]
- Kiran, T.; Sailu, C.; Aukunuru, J. Formulation, optimization and evaluation of oral nanosuspension tablets of nebivolol hydrochloride for enhancement of dissolution rate. Pharma Lett. 2015, 7, 71–84. [Google Scholar]
- Ryakala, H.; Dineshmohan, S.; Ramesh, A.; Gupta, V.R.M. Formulation and In Vitro Evaluation of Bilayer Tablets of Nebivolol Hydrochloride and Nateglinide for the Treatment of Diabetes and Hypertension. J. Drug. Deliv. 2015, 2015, 827859. [Google Scholar] [CrossRef] [PubMed]
- Sipos, E.; Szabó, Z.I.; Rédai, E.; Szabó, P.; Sebe, I.; Zelkó, R. Preparation and characterization of nanofibrous sheets for enhanced oral dissolution of nebivolol hydrochloride. J. Pharm. Biomed. Anal. 2016, 12, 224–228. [Google Scholar] [CrossRef]
- Sabri, L.A.; Hussein, A.A. Formulation and In-Vitro Characterization of Solidified Nebivolol Self-nanoemulsion using Liquisolid Technique. Syst. Rev. Pharm. 2020, 11, 261–268. [Google Scholar]
- Nikam, V.J.; Patil, S.B. Pharmaceutical cocrystals of nebivolol hydrochloride with enhanced solubility. J. Cryst. Growth 2020, 534, 125488. [Google Scholar] [CrossRef]
- Kaur, G.; Saifi, A.; Kumar, K.; Teotia, D. Development and Evaluation of Micro Emulsion Formulations of Nebivolol for Solubility Enhancement. J. Drug Deliv. Ther. 2021, 11, 84–89. [Google Scholar] [CrossRef]
- Sura, R.S.; Subrahmanyam, C.; Rachamalla, S.S. Design and evaluation of liquisolid compacts of nebivolol hydrochloride. Int. J. Appl. Pharm. 2022, 7, 293–307. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef]
- Maheriya, P.M. Cyclodextrin: A promising candidate in enhancing oral bioavailability of poorly water soluble drugs. MOJ Bioequiv. Bioavailab. 2017, 3, 60–63. [Google Scholar] [CrossRef]
- Del Valle, E.M.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Jambhekar, S.S.; Breen, P. Cyclodextrins in pharmaceutical formulations II: Solubilization, binding constant, and complexation efficiency. Drug Discov. Today 2016, 21, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef]
- Bhopate, S.B.; Dhole, S.N. Preparation and characterization of β-cyclodextrin- nebivolol inclusion complex. Int. J. Pharm. Sci. Res. 2015, 6, 2205–2213. [Google Scholar]
- Zhou, Y.; Fu, X.D.; Bi, S.T. Preparation and characterization of nebivolol hydrochloride HP-β-CD inclusion complexes. Chin. J. New Drugs 2018, 27, 954–959. [Google Scholar]
- Duchêne, D.; Bochot, A. Thirty years of cyclodextrins. Int. J. Pharm. 2016, 514, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Rajewski, R.A. Sulfobutylether-β-cyclodextrin. Int. J. Pharm. 2020, 583, 119396. [Google Scholar] [CrossRef]
- ICH Harmonization for Better Health—EMA/CPMP/ICH/82072/2006. ICH Q2(R2) Guideline on Validation of Analytical Procedures. 24 March 2022. Available online: https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline (accessed on 17 April 2024).
- Higuchi, T.; Connors, K.A. Phase Solubility Techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. Evaluation of cyclodextrin solubilization of drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Nogami, H.; Nagai, T.; Yotsuyanagi, T. Dissolution Phenomena of Organic Medicinals involving Simultaneous Phase Changes. Chem. Pharm. Bull. 1969, 17, 499–509. [Google Scholar] [CrossRef]
- Maestrelli, F.; Mura, P.; Cirri, M.; Mennini, N.; Ghelardini, C.; Di Cesare Mannelli, L. Development and characterization of fast dissolving tablets of oxaprozin based on hybrid systems of the drug with cyclodextrins and nanoclays. Int. J. Pharm. 2017, 531, 640–649. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; Aguzzi, C.; Viseras, C. Hybrid systems based on “drug–in cyclodextrin–in nanoclays” for improving oxaprozin dissolution properties. Int. J. Pharm. 2016, 509, 8–15. [Google Scholar] [CrossRef]
- Maestrelli, F.; Cirri, M.; García-Villén, F.; Borrego-Sánchez, A.; Viseras Iborra, C.; Mura, P. Tablets of “Hydrochlorothiazide in Cyclodextrin in Nanoclay”: A New Nanohybrid System with Enhanced Dissolution Properties. Pharmaceutics. 2020, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Al-Marzouqi, A.H.; Jobe, B.; Dowaidar, A.; Maestrelli, F.; Mura, P. Evaluation of supercritical fluid technology as preparative technique of benzocaine–cyclodextrin complexes—Comparison with conventional methods. J. Pharm. Biomed. Anal. 2007, 43, 5665–5674. [Google Scholar] [CrossRef] [PubMed]
- Maestrelli, F.; Cecchi, M.; Cirri, M.; Capasso, G.; Mennini, N.; Mura, P. Comparative study of oxaprozin complexation with natural and chemically-modified cyclodextrins in solution and in the solid state. J. Incl. Phenom. Macrocycl. Chem. 2009, 63, 17–25. [Google Scholar] [CrossRef]
- Mennini, N.; Bragagni, M.; Maestrelli, F.; Mura, P. Physico-chemical characterization in solution and in the solid state of clonazepam complexes with native and chemically-modified cyclodextrins. J. Pharm. Biomed. Anal. 2014, 89, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Chadha, R.; Bhandari, S. Drug–excipient compatibility screening—Role of thermoanalytical and spectroscopic techniques. J. Pharm. Biomed. Anal. 2014, 87, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Tiţa, B.; Fuliaş, A.; Bandur, G.; Marian, E.; Tiţa, D. Compatibility study between ketoprofen and pharmaceutical excipients used in solid dosage forms. J. Pharm. Biomed. Anal. 2011, 56, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, L.M.; Fiorot, A.; Xavier, T.; Yoshida, M.I.; De Oliveira, M.A. Drug-excipient compatibility assessment of solid formulations containing meloxicam. Eur. J. Pharm. Sci. 2018, 112, 146–151. [Google Scholar] [CrossRef]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.M.; Lobo, J.M.S. Cyclodextrins as excipients in tablet formulations. Drug Discov. Today 2018, 23, 1274–1284. [Google Scholar] [CrossRef]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.M.; Lobo, J.M.S. Hydroxypropyl-β-Cyclodextrin and β-Cyclodextrin as Tablet Fillers for Direct Compression. AAPS PharmSciTech. 2018, 19, 2710–2718. [Google Scholar] [CrossRef]
- Late, S.G.; Banga, A.K. Response Surface Methodology to Optimize Novel Fast Disintegrating Tablets Using β Cyclodextrin as Diluent. AAPS PharmSciTech 2010, 11, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, Ł.; Kasperek, R.; Poleszak, E. Application of β-cyclodextrin in the formulation of ODT tablets containing ibuprofen. Polim Med. 2014, 44, 231–235. [Google Scholar] [PubMed]
- Vora, L.K.; Gholap, A.D.; Jetha, K.; Thakur, R.R.S.; Solanki, H.K.; Chavda, V.P. Artificial Intelligence in Pharmaceutical Technology and Drug Delivery Design. Pharmaceutics 2023, 15, 1916. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD Type | K1:1 (mM−1) | CE | Solubilizing Efficiency * |
|---|---|---|---|
| βCD | 644 | 0.110 | 8.2 |
| HPβCD | 654 | 0.112 | 15.3 |
| RAMEB | 1011 | 0.173 | 22.4 |
| SBEβCD | 2306 | 0.394 | 42.6 |
| Sample | PD 10 | PD 30 | DE 60 |
|---|---|---|---|
| NEB | 0.8 | 2.1 | 1.8 |
| NEB-βCD PM | 2.2 | 4.3 | 3.7 |
| NEB-βCD KN | 15.4 | 18.8 | 17.2 |
| NEB-βCD GR | 18.9 | 23.9 | 21.5 |
| NEB-βCD COE | 17.9 | 22.0 | 19.9 |
| NEB-SΒΕβCD PM | 8.7 | 17.0 | 15.0 |
| NEB-SΒΕβCD KN | 51.6 | 56.7 | 52.8 |
| NEB-SΒΕβCD GR | 71.9 | 80.8 | 74.8 |
| NEB-SΒΕβCD COE | 81.3 | 93.3 | 86.1 |
| Tablet Sample | Weight (mg) | Disintegration Time (min) | Hardness (kg/cm2) | Friability (%) |
|---|---|---|---|---|
| Reference Tablet | 314.6 ± 0.6 | 1.06 ± 0.29 | 7.75 ± 0.8 | 0.47 |
| PM Tablet | 314.2 ± 0.3 | 9.25 ± 1.50 | 11.9 ± 0.9 | 0.58 |
| COE Tablet | 314.1 ± 0.4 | 7.64 ± 1.38 | 11.5 ± 0.4 | 0.62 |
| Nebivololo Mylan | 232.3 ± 2.3 | 2.24 ± 0.23 | 7.33 ± 0.3 | 0.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestrelli, F.; Cirri, M.; Mennini, N.; Fiani, S.; Stoppacciaro, B.; Mura, P. Development of Oral Tablets of Nebivolol with Improved Dissolution Properties, Based on Its Combinations with Cyclodextrins. Pharmaceutics 2024, 16, 633. https://doi.org/10.3390/pharmaceutics16050633
Maestrelli F, Cirri M, Mennini N, Fiani S, Stoppacciaro B, Mura P. Development of Oral Tablets of Nebivolol with Improved Dissolution Properties, Based on Its Combinations with Cyclodextrins. Pharmaceutics. 2024; 16(5):633. https://doi.org/10.3390/pharmaceutics16050633
Chicago/Turabian StyleMaestrelli, Francesca, Marzia Cirri, Natascia Mennini, Silvia Fiani, Beatrice Stoppacciaro, and Paola Mura. 2024. "Development of Oral Tablets of Nebivolol with Improved Dissolution Properties, Based on Its Combinations with Cyclodextrins" Pharmaceutics 16, no. 5: 633. https://doi.org/10.3390/pharmaceutics16050633