


Engineered Hybrid Vesicles and Cellular Internalization in Mammary Cancer Cells
 , , and
, , and 
Abstract
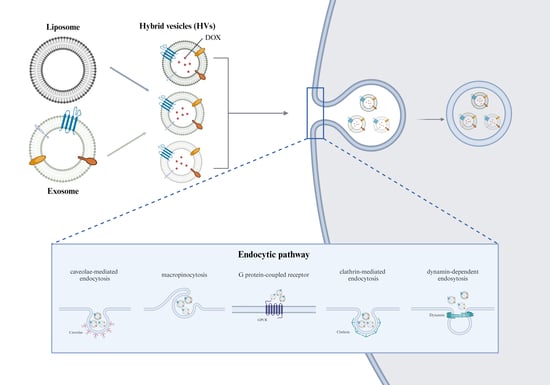
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. In Vitro Cell Culture
2.3. Isolation of the Extracellular Vesicle
2.4. Preparation of the Liposomes
2.5. Preparation of the HVs
2.6. Physicochemical Properties of the Lipid Vesicles
2.6.1. Size and Zeta Potential Values of the Lipid Vesicles
2.6.2. Cryo-TEM Analysis of the Lipid Vesicles
2.7. Quantification of DOX
2.8. Physical Stability of the Lipid Vesicles
2.9. In Vitro Cell Viability of the Lipid Vesicles
2.10. Measurement of the Cellular Uptake of the Lipid Vesicles
2.11. Cellular Uptake Mechanism of the Lipid Vesicles
2.12. Statistics
3. Results
3.1. Physicochemical Characterization of the HVs
3.2. Physical Stability of the Lipid Vesicles
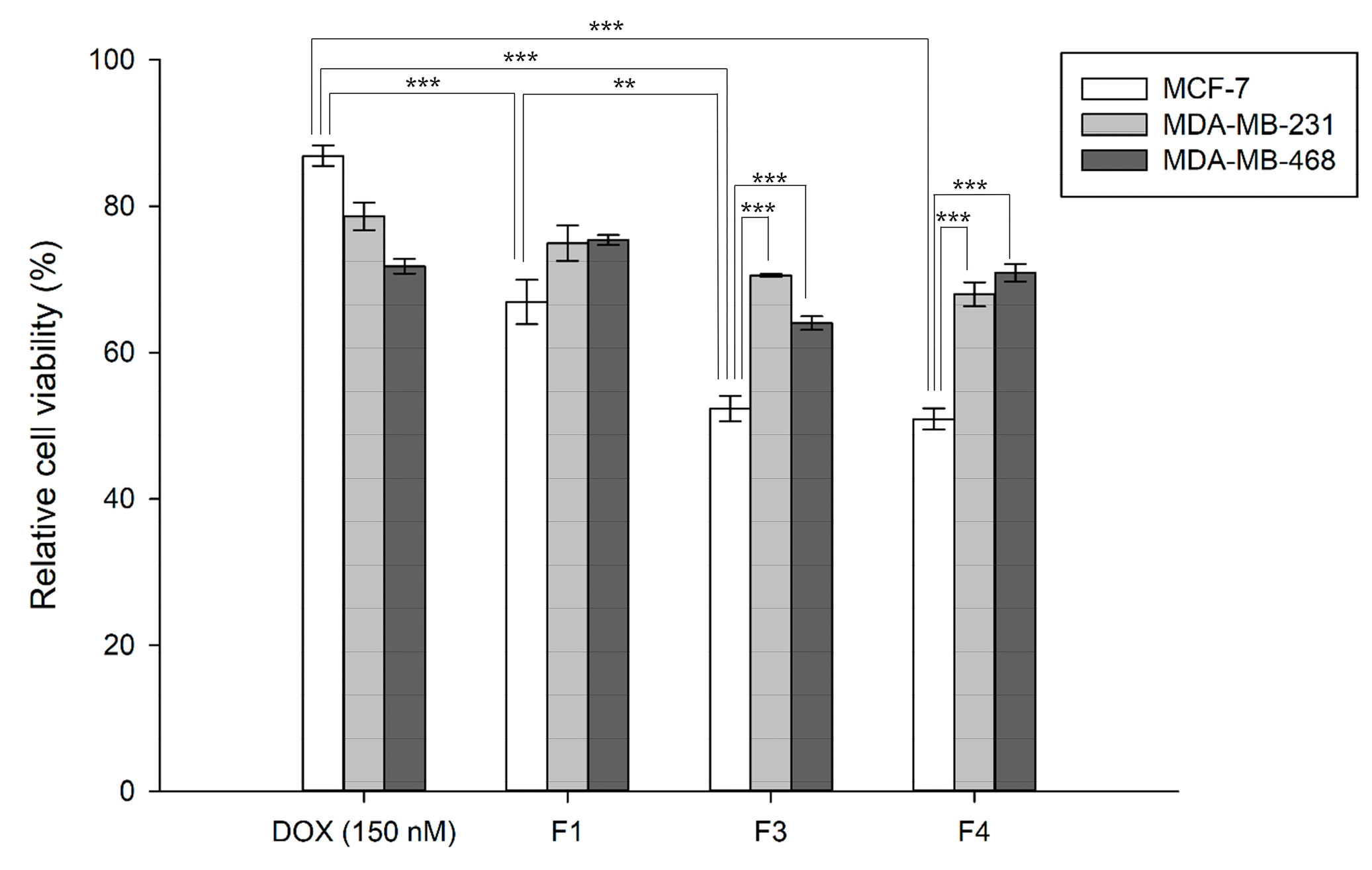
3.3. In Vitro Cytotoxicity of the DOX-Loaded Lipid Vesicles
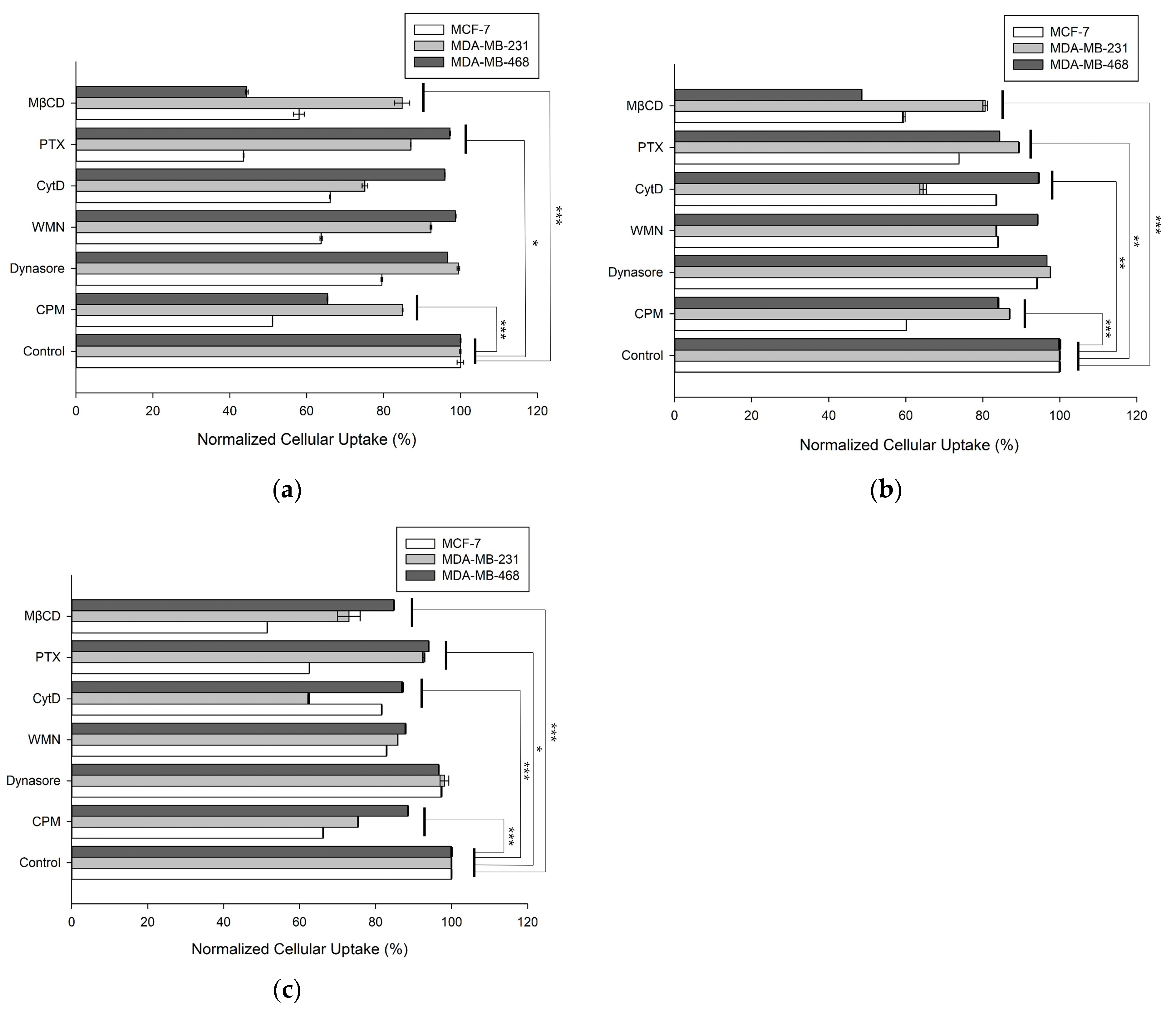
3.4. Cellular Uptake Mechanisms of the DOX-Loaded Vesicles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell–cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Dagur, R.S.; Liao, K.; Peeples, E.S.; Hu, G.; Periyasamy, P.; Buch, S. Strategies for the use of Extracellular Vesicles for the Delivery of Therapeutics. J. Neuroimmune Pharmacol. 2020, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Moreira, J.N.; Rodrigues, L.R. New advances in exosome-based targeted drug delivery systems. Crit. Rev. Oncol. Hematol. 2022, 172, 103628. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, X.; Han, G.; Liang, M.; Yang, Z.; Zhang, C.; Huang, S.; Tai, S.; Yu, S. The therapeutic potential and clinical significance of exosomes as carriers of drug delivery system. Pharmaceutics 2023, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513. [Google Scholar] [CrossRef]
- Ganesh, V.; Seol, D.; Gomez-Contreras, P.C.; Keen, H.L.; Shin, K.; Martin, J.A. Exosome-based cell homing and angiogenic differentiation for dental pulp regeneration. Int. J. Mol. Sci. 2023, 24, 466. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, R.; Yoshida, S.; Sawada, S.-i.; Sasaki, Y.; Akiyoshi, K. Fusogenic hybrid extracellular vesicles with PD-1 membrane proteins for the cytosolic delivery of cargos. Cancers 2022, 14, 2635. [Google Scholar] [CrossRef]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- Lennaárd, A.J.; Mamand, D.R.; Wiklander, R.J.; Andaloussi, S.E.L.; Wiklander, O.P.B. Optimised electroporation for loading of extracellular vesicles with doxorubicin. Pharmaceutics 2022, 14, 38. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Pilgaard, L.; Moos, T.; Duroux, M. A comprehensive overview of exosomes as drug delivery vehicles—Endogenous nanocarriers for targeted cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 75–87. [Google Scholar] [CrossRef]
- Ahn, S.-H.; Ryu, S.-W.; Choi, H.; You, S.; Park, J.; Choi, C. Manufacturing therapeutic exosomes: From bench to industry. Mol. Cells 2022, 45, 284–290. [Google Scholar] [CrossRef]
- Sato, Y.T.; Umezaki, K.; Sawada, S.; Mukai, S.-a.; Sasaki, Y.; Harada, N.; Shiku, H.; Akiyoshi, K. Engineering hybrid exosomes by membrane fusion with liposomes. Sci. Rep. 2016, 6, 21933. [Google Scholar] [CrossRef]
- Liu, A.; Yang, G.; Liu, Y.; Liu, T. Research progress in membrane fusion-based hybrid exosomes for drug delivery systems. Front. Bioeng. Biotechnol. 2022, 10, 939441. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Silva, A.K.A.; Wilhelm, C.; Gazeau, F.; Tareste, D. Modification of extracellular vesicles by fusion with liposomes for the design of personalized biogenic drug delivery systems. ACS Nano 2018, 12, 6830–6842. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.A.; Vader, P. Extracellular vesicle-based hybrid systems for advanced drug delivery. Pharmaceutics 2022, 14, 267. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, X.; Tang, J.; Lv, Q.; Liu, J. Gene-engineered exosomes-thermosensitive liposomes hybrid nanovesicles by the blockade of CD47 signal for combined photothermal therapy and cancer immunotherapy. Biomaterials 2021, 275, 120964. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome–liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef]
- Lentz, B.R. PEG as a tool to gain insight into membrane fusion. Eur. Biophys. J. 2007, 36, 315–326. [Google Scholar] [CrossRef]
- Kannavou, M.; Marazioti, A.; Stathopoulos, G.T.; Antimisiaris, S.G. Engineered versus hybrid cellular vesicles as efficient drug delivery systems: A comparative study with brain targeted vesicles. Drug Deliv. Transl. Res. 2021, 11, 547–565. [Google Scholar] [CrossRef]
- Jhan, Y.-Y.; Prasca-Chamorro, D.; Zuniga, G.P.; Moore, D.M.; Kumar, S.A.; Gaharwar, A.K.; Bishop, C.J. Engineered extracellular vesicles with synthetic lipids via membrane fusion to establish efficient gene delivery. Int. J. Pharm. 2020, 573, 118802. [Google Scholar] [CrossRef]
- Thone, M.N.; Kwon, Y.J. Extracellular blebs: Artificially-induced extracellular vesicles for facile production and clinical translation. Methods 2020, 177, 135–145. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.-K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Verdera, H.C.; Gitz-Francois, J.J.; Schiffelers, R.M.; Vader, P. Cellular uptake of extracellular vesicles is mediated by clathrin-independent endocytosis and macropinocytosis. J. Control Release 2017, 266, 100–108. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Rosenblum, D.; Weinstein, S.; Bairey, O.; Raanani, P.; Peer, D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015, 364, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Albero, M.; Navascués, N.; Mendoza, G.; Sebastián, V.; Arruebo, M.; Martín-Duque, P.; Santamaría, J. Exosome origin determines cell targeting and the transfer of therapeutic nanoparticles towards target cells. J. Nanobiotechnol. 2019, 17, 16. [Google Scholar] [CrossRef]
- Lu, M.; Huang, Y. Bioinspired exosome-like therapeutics and delivery nanoplatforms. Biomaterials 2020, 242, 119925. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.W.; Sceneay, J.; Lima, L.G.; Wong, C.S.F.; Becker, M.; Krumeich, S.; Lobb, R.J.; Castillo, V.; Wong, K.N.; Ellis, S.; et al. The biodistribution and immune suppressive effects of breast cancer–derived exosomes. Cancer Res. 2016, 76, 6816–6827. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, S.; Zhu, D.; Li, J.; Cheng, K.; Liu, G. Comparison of extruded cell nanovesicles and exosomes in their molecular cargos and regenerative potentials. Nano Res. 2023, 16, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.S.; de Oliveira Silva, J.; Fernandes, R.S.; Miranda, S.E.M.; Leite, E.A.; de Farias, M.A.; Portugal, R.V.; Cassali, G.D.; Townsend, D.M.; Oliveira, M.C.; et al. PEGylated versus non-PEGylated pH-sensitive liposomes: New insights from a comparative antitumor activity study. Pharmaceutics 2022, 14, 272. [Google Scholar] [CrossRef]
- Lin, W.-C.; Blanchette, C.D.; Ratto, T.V.; Longo, M.L. Lipid asymmetry in DLPC/DSPC-supported lipid bilayers: A combined AFM and fluorescence microscopy study. Biophys. J. 2006, 90, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Pitchaimani, A.; Nguyen, T.D.T.; Aryal, S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials 2018, 160, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.R.; Souza, F.R.; Cassali, G.D.; de Paula Sabino, A.; de Barros, A.L.B.; Oliveira, M.C. Investigation of the Antitumor Activity and Toxicity of Tumor-Derived Exosomes Fused with Long-Circulating and pH-Sensitive Liposomes Containing Doxorubicin. Pharmaceutics 2022, 14, 2256. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lee, C.; Singh, S. Versatile Encapsulation and Synthesis of Potent Therapeutic Liposomes by Thermal Equilibration. bioRxiv 2021. [Google Scholar] [CrossRef]
- Trissel, L.A.; Xu, Q.A.; Gilbert, D.L. Compatibility and stability of paclitaxel combined with doxorubicin hydrochloride in infusion solutions. Ann. Pharmacother. 1998, 32, 1013–1016. [Google Scholar] [CrossRef]
- Surma, M.A.; Szczepaniak, A.; Króliczewski, J. Comparative studies on detergent-assisted apocytochrome b6 reconstitution into liposomal bilayers monitored by Zetasizer instruments. PLoS ONE 2014, 9, e111341. [Google Scholar] [CrossRef]
- Vázquez-Ríos, A.J.; Molina-Crespo, Á.; Bouzo, B.L.; López-López, R.; Moreno-Bueno, G.; de la Fuente, M. Exosome-mimetic nanoplatforms for targeted cancer drug delivery. J. Nanobiotechnol. 2019, 17, 85. [Google Scholar] [CrossRef]
- Naik, H.; Sonju, J.J.; Singh, S.; Chatzistamou, I.; Shrestha, L.; Gauthier, T.; Joi, S. Lipidated peptidomimetic ligand-functionalized HER2 targeted liposome as nano-carrier designed for doxorubicin delivery in cancer therapy. Pharmaceuticals 2021, 14, 221. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Chen, H.; Wang, X.; Chen, T. Regorafenib induces Bim-mediated intrinsic apoptosis by blocking AKT-mediated FOXO3a nuclear export. Cell Death Discov. 2023, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.-U.; Allen, T.; Shen, D.; et al. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474–3487. [Google Scholar] [CrossRef] [PubMed]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key principles and methods for studying the endocytosis of biological and nanoparticle therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Kim, S.T.; Yan, B.; Miranda, O.R.; Alfonso, F.S.; Shlosman, D.; Rotello, V.M. Surface functionality of nanoparticles determines cellular uptake mechanisms in mammalian cells. Small 2013, 9, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.W.; Monteiro-Riviere, N.A. Mechanisms of quantum dot nanoparticle cellular uptake. Toxicol. Sci. 2009, 110, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef]
- Xu, X.; Xu, L.; Wen, C.; Xia, J.; Zhang, Y.; Liang, Y. Programming assembly of biomimetic exosomes: An emerging theranostic nanomedicine platform. Mater. Today Bio 2023, 22, 100760. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Hu, B.; Zhao, D.; Liu, W.; Liu, Q.; Huang, Y.; Ruan, S. Recent progresses of exosome-liposome fusions in drug delivery. Chin. Chem. Lett. 2024, 35, 108647. [Google Scholar] [CrossRef]
- Jimah, J.R.; Schlesinger, P.H.; Tolia, N.H. Liposome disruption assay to examine lytic properties of biomolecules. Bio Protoc. 2017, 7, e2433. [Google Scholar] [CrossRef] [PubMed]
- van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021, 288, 6095–6111. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Chandra, A.; Kaur, A.; Sabnis, N.; Lacko, A.; Gryczynski, Z.; Fudala, R.; Gryczynski, I. Fluorescence properties of doxorubicin in PBS buffer and PVA films. J. Photochem. Photobiol. B 2017, 170, 65–69. [Google Scholar] [CrossRef]
- Gandek, T.B.; van der Koog, L.; Nagelkerke, A. A comparison of cellular uptake mechanisms, delivery efficacy, and intracellular fate between liposomes and extracellular vesicles. Adv. Healthc. Mater. 2023, 12, 2300319. [Google Scholar] [CrossRef]
- Pérez-Arnaiz, C.; Busto, N.; Leal, J.M.; Garcia, B. New insights into the mechanisms of the DNA/doxorubicin interaction. J. Phys. Chem. B 2014, 118, 1288–1295. [Google Scholar] [CrossRef]
- Aminipour, Z.; Khorshid, M.; Keshvari, H.; Bonakdar, S.; Wagner, P.; van der Bruggen, B. Passive permeability assay of doxorubicin through model cell membranes under cancerous and normal membrane potential conditions. Eur. J. Pharm. Biopharm. 2020, 146, 133–142. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Composition 1 | Size (nm) | Polydispersity Index (PDI) | Zeta Potential Value (ζ, mV) |
|---|---|---|---|---|
| F1 | Liposome | 134.90 ± 0.49 | 0.10 ± 0.01 | −5.37 ± 0.24 |
| F2 | Lipo:Exo (10:1) | 129.17 ± 0.45 | 0.08 ± 0.00 | −6.16 ± 0.22 |
| F3 | Lipo:Exo (5:1) | 119.03 ± 0.34 | 0.09 ± 0.01 | −6.05 ± 0.35 |
| F4 | Lipo:Exo (2:1) | 113.60 ± 0.52 | 0.11 ± 0.01 | −9.75 ± 0.58 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.Y.; Guk, D.; Jeong, Y.; Kim, E.; Kim, H.; Kim, S.T. Engineered Hybrid Vesicles and Cellular Internalization in Mammary Cancer Cells. Pharmaceutics 2024, 16, 440. https://doi.org/10.3390/pharmaceutics16040440
Kim SY, Guk D, Jeong Y, Kim E, Kim H, Kim ST. Engineered Hybrid Vesicles and Cellular Internalization in Mammary Cancer Cells. Pharmaceutics. 2024; 16(4):440. https://doi.org/10.3390/pharmaceutics16040440
Chicago/Turabian StyleKim, So Yun, Dagyeong Guk, Youngdo Jeong, Eunji Kim, Hansol Kim, and Sung Tae Kim. 2024. "Engineered Hybrid Vesicles and Cellular Internalization in Mammary Cancer Cells" Pharmaceutics 16, no. 4: 440. https://doi.org/10.3390/pharmaceutics16040440
APA StyleKim, S. Y., Guk, D., Jeong, Y., Kim, E., Kim, H., & Kim, S. T. (2024). Engineered Hybrid Vesicles and Cellular Internalization in Mammary Cancer Cells. Pharmaceutics, 16(4), 440. https://doi.org/10.3390/pharmaceutics16040440