
Polymer Delivery Systems for Long-Acting Antiretroviral Drugs
 , and
, and
Abstract
:1. Introduction
2. LA DDSs
3. Drug Release Kinetics from LA DDSs
4. Common Polymers in LA DDSs
4.1. Polyethylene Glycol (PEG)
4.2. Poloxamers
4.3. Ethylene-Vinyl Acetate (EVA)
4.4. Polyvinyl Alcohol (PVOH)
4.5. Polyurethanes (PUs)
4.6. Polyesters
4.7. Chitosan and Hyaluronic Acid (HA)
4.8. Polymers and Polymer Blends
5. LA ARV Delivery
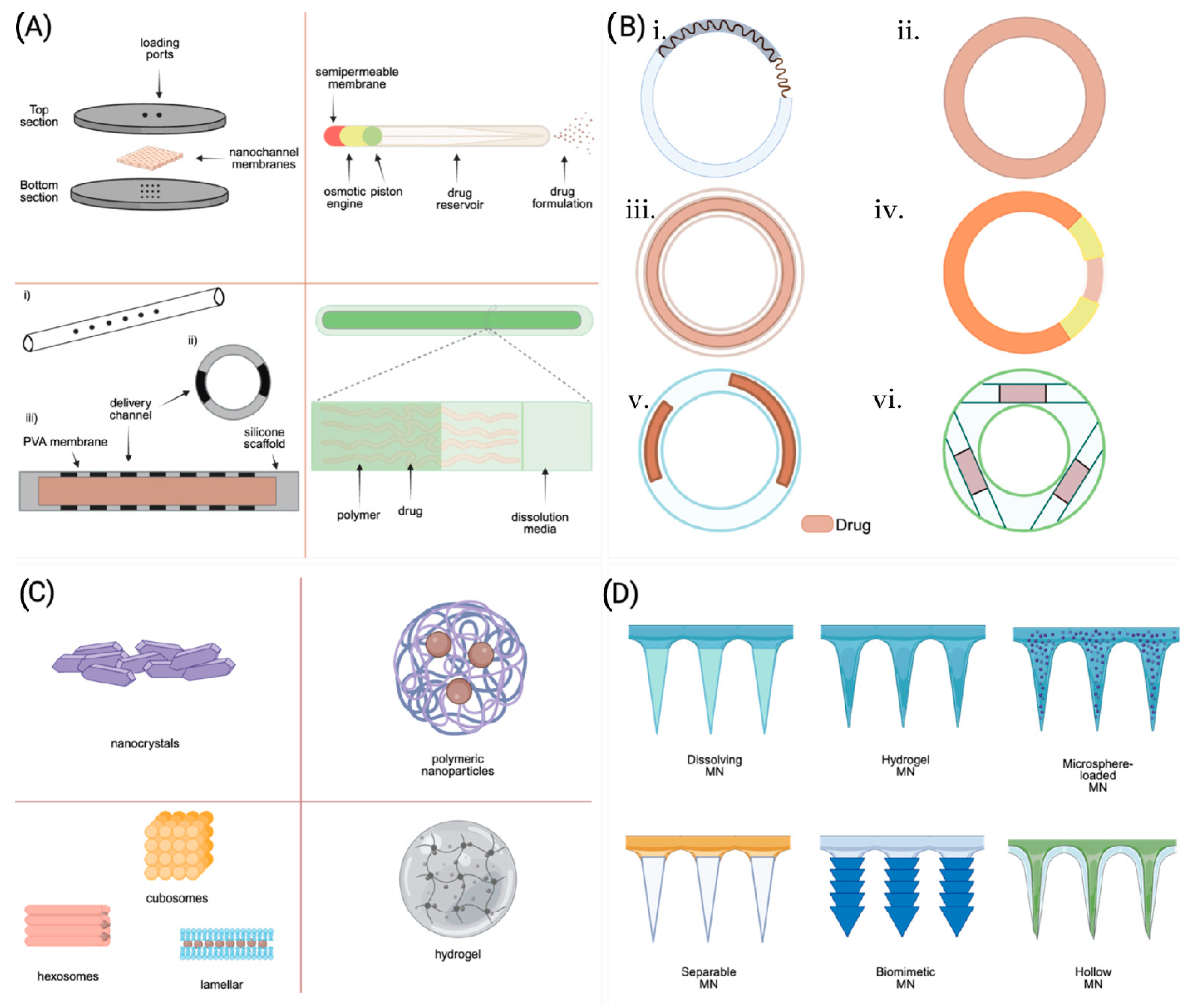
5.1. Implants

5.2. Vaginal Rings (VRs)
5.3. Microneedles (MNs)
5.4. Polymeric Micro- and Nanoparticles
5.4.1. PLGA-Based Micro- and Nanoparticles
5.4.2. Nanocrystal Formulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery System | ARV Drug | Polymer | Formulation Details | Predicted Dosage Regimen (Month) | Clinical Trial | Ref. |
|---|---|---|---|---|---|---|
| Implant | TAF | Silicone and PVA | TAF powder is encapsulated in a silicone tube, which has several drug delivery channels coated with PVA. Drug release rate can be controlled by controlling the number of channels or thickness of PVA membrane. | 6 | Phase I | [15] |
| TAF | Silicone | Transcutaneous refillable nanochannel delivery implant, where drug reservoir is made of titanium and drug is released through TaN-coated slit nanochannel. | 6 | Preclinical | [11] | |
| TAF | PUs | TAF formulation pellet is encased in PU-based drug reservoir and the drug is released through diffusion through the PU membrane. | 3 | [162] | ||
| CAB | CAB formulation is converted into pellets and then encased in PU membrane containing drug reservoir. PU membrane acts as RCM for controlling drug release through diffusion. | 3 | [168] | |||
| TAF | PCL | TAF formulation containing TAF and castor oil was loaded into PCL tubes and the drug was released through the diffusion and bulk erosion of PCL. Drug release rate can be controlled through controlling MW of PCL. | 3 | [160] | ||
| ISL | EVA | Crystalline ISL is uniformly dispersed in EVA and the implant is prepared through hot melt extrusion. | 12 | On hold | [165] | |
| ISFIs | CAB | PLGA | CAB and PLGA were solubilized in NMP:DMSO and injected subcutaneously. Upon injection, the solution went through phase inversion and formed solid implant. | 6 | Preclinical | [51] |
| DTG | Same as CAB PLGA ISFI | 6 | [180] | |||
| VRs | TDF | PUs | TDF formulation was loaded in hydrophilic PU tubes and the PU tubes were end-sealed using induction welding. | 1 | [203] | |
| TDF | PUs | TDF and NaCl formulation was loaded onto PU extruded tubes and end-sealed to prepare the VRs. | 1 | [17] | ||
| MNs | CAB | PVP and PVA | MN tips were generated by using a hydrogel composed of 20:20:60 PVA:PVP:CAB and the baseplate of MNs was made using PVP and PVA. The MN tips dissolved quickly to release the CAB and the CAB formed drug depot at the administration site. | 1 | Preclinical | [22] |
| BIC | MNs tips were made of PVP, PVA, and BIC. The baseplate was made of PVP and glycerol. | 1 | [230] | |||
| RPV | MN tips were made of PVP and RPV. The baseplate was made of PVA and glycerol. | 1 | [23] | |||
| Microparticle | DTG | PLGA | DTG was transformed into a hydrophobic prodrug and encapsulated into PLGA-based microparticles by organic and aqueous solvent emulsification–evaporation. | 3 | [251] | |
| Prodrug nanocrystal | TFV | Poloxamer, Polysorbate and PEG | TFV was converted into lipophilic ProTide and then formulated as aqueous nanocrystals. | ≥3 | [272] | |
| CAB | Lipohilic ester prodrug cabotegravir was synthesized and formulated as aqueous nanocrystals. | 12 | [9] | |||
| DTG | Fatty acid ester prodrug DTG was synthesized and nanoformulated as aqueous nanocrystals. | ≥6 | [269] |
6. Conclusions and Future Perspective of LA Drug Delivery Systems
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deanesly, R.; Parkes, A.S. Testosterone. Br. Med. J. 1936, 1, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Paolini, M.S.; Fenton, O.S.; Bhattacharya, C.; Andresen, J.L.; Langer, R. Polymers for extended-release administration. Biomed. Microdevices 2019, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Swindells, S.; Andrade-Villanueva, J.-F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masiá, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. N. Engl. J. Med. 2020, 382, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.R.; Henry, W.K.; Podzamczer, D.; Masiá, M.D.M.; Bettacchi, C.J.; Arasteh, K.; Jaeger, H.; Khuong-Josses, M.A.; Montes-Ramírez, M.L.; Stellbrink, H.J.; et al. Efficacy, Safety, and Durability of Long-Acting Cabotegravir and Rilpivirine in Adults with Human Immunodeficiency Virus Type 1 Infection: 5-Year Results from the LATTE-2 Study. Open Forum Infect. Dis. 2021, 8, ofab439. [Google Scholar] [CrossRef]
- Parikh, U.M.; Koss, C.A.; Mellors, J.W. Long-Acting Injectable Cabotegravir for HIV Prevention: What Do We Know and Need to Know about the Risks and Consequences of Cabotegravir Resistance? Curr. HIV/AIDS Rep. 2022, 19, 384–393. [Google Scholar] [CrossRef]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.-J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Orkin, C.; Oka, S.; Philibert, P.; Brinson, C.; Bassa, A.; Gusev, D.; Degen, O.; García, J.G.; Morell, E.B.; Tan, D.H.S.; et al. Long-acting cabotegravir plus rilpivirine for treatment in adults with HIV-1 infection: 96-week results of the randomised, open-label, phase 3 FLAIR study. Lancet HIV 2021, 8, e185–e196. [Google Scholar] [CrossRef]
- Orkin, C.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Overton, E.T.; Girard, P.-M.; Oka, S.; Walmsley, S.; Bettacchi, C.; Brinson, C. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. N. Engl. J. Med. 2020, 382, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.A.; Bade, A.N.; Sillman, B.; Shetty, B.L.D.; Wojtkiewicz, M.S.; Gautam, N.; Hilaire, J.R.; Sravanam, S.; Szlachetka, A.; Lamberty, B.G. A year-long extended release nanoformulated cabotegravir prodrug. Nat. Mater. 2020, 19, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Young, I.C.; Massud, I.; Cottrell, M.L.; Shrivastava, R.; Maturavongsadit, P.; Prasher, A.; Wong-Sam, A.; Dinh, C.; Edwards, T.; Mrotz, V.; et al. Ultra-long-acting in-situ forming implants with cabotegravir protect female macaques against rectal SHIV infection. Nat. Commun. 2023, 14, 708. [Google Scholar] [CrossRef]
- Chua, C.Y.X.; Jain, P.; Ballerini, A.; Bruno, G.; Hood, R.L.; Gupte, M.; Gao, S.; Di Trani, N.; Susnjar, A.; Shelton, K.; et al. Transcutaneously refillable nanofluidic implant achieves sustained level of tenofovir diphosphate for HIV pre-exposure prophylaxis. J. Control. Release 2018, 286, 315–325. [Google Scholar] [CrossRef]
- Johnson, L.M.; Krovi, S.A.; Li, L.; Girouard, N.; Demkovich, Z.R.; Myers, D.; Creelman, B.; van der Straten, A. Characterization of a Reservoir-Style Implant for Sustained Release of Tenofovir Alafenamide (TAF) for HIV Pre-Exposure Prophylaxis (PrEP). Pharmaceutics 2019, 11, 315. [Google Scholar] [CrossRef]
- Simpson, S.M.; Widanapathirana, L.; Su, J.T.; Sung, S.; Watrous, D.; Qiu, J.; Pearson, E.; Evanoff, A.; Karunakaran, D.; Chacon, J.E.; et al. Design of a Drug-Eluting Subcutaneous Implant of the Antiretroviral Tenofovir Alafenamide Fumarate. Pharm. Res. 2020, 37, 83. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, M.; Grobler, J.A. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Curr. Opin. HIV AIDS 2020, 15, 27–32. [Google Scholar] [CrossRef]
- Gunawardana, M.; Remedios-Chan, M.; Miller, C.S.; Fanter, R.; Yang, F.; Marzinke, M.A.; Hendrix, C.W.; Beliveau, M.; Moss, J.A.; Smith, T.J. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrob. Agents Chemother. 2015, 59, 3913–3919. [Google Scholar] [CrossRef]
- Baeten, J.M.; Palanee-Phillips, T.; Mgodi, N.M.; Mayo, A.J.; Szydlo, D.W.; Ramjee, G.; Gati Mirembe, B.; Mhlanga, F.; Hunidzarira, P.; Mansoor, L.E.; et al. Safety, uptake, and use of a dapivirine vaginal ring for HIV-1 prevention in African women (HOPE): An open-label, extension study. Lancet HIV 2021, 8, e87–e95. [Google Scholar] [CrossRef]
- Keller, M.J.; Wood, L.; Billingsley, J.M.; Ray, L.L.; Goymer, J.; Sinclair, S.; McGinn, A.P.; Marzinke, M.A.; Frank, B.; Srinivasan, S.; et al. Tenofovir disoproxil fumarate intravaginal ring for HIV pre-exposure prophylaxis in sexually active women: A phase 1, single-blind, randomised, controlled trial. Lancet HIV 2019, 6, e498–e508. [Google Scholar] [CrossRef]
- Boyd, P.; Variano, B.; Spence, P.; McCoy, C.F.; Murphy, D.J.; Dallal Bashi, Y.H.; Malcolm, R.K. In vitro release testing methods for drug-releasing vaginal rings. J. Control. Release 2019, 313, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, E.T.; van der Straten, A.; Chitukuta, M.; Reddy, K.; Woeber, K.; Atujuna, M.; Bekker, L.G.; Etima, J.; Nakyanzi, T.; Mayo, A.J.; et al. Acceptability and use of a dapivirine vaginal ring in a phase III trial. Aids 2017, 31, 1159–1167. [Google Scholar] [CrossRef]
- Nel, A.; van Niekerk, N.; Kapiga, S.; Bekker, L.-G.; Gama, C.; Gill, K.; Kamali, A.; Kotze, P.; Louw, C.; Mabude, Z.; et al. Safety and Efficacy of a Dapivirine Vaginal Ring for HIV Prevention in Women. N. Engl. J. Med. 2016, 375, 2133–2143. [Google Scholar] [CrossRef]
- Vora, L.K.; Moffatt, K.; Tekko, I.A.; Paredes, A.J.; Volpe-Zanutto, F.; Mishra, D.; Peng, K.; Raj Singh Thakur, R.; Donnelly, R.F. Microneedle array systems for long-acting drug delivery. Eur. J. Pharm. Biopharm. 2021, 159, 44–76. [Google Scholar] [CrossRef]
- Tekko, I.A.; Vora, L.K.; Volpe-Zanutto, F.; Moffatt, K.; Jarrahian, C.; McCarthy, H.O.; Donnelly, R.F. Novel Bilayer Microarray Patch-Assisted Long-Acting Micro-Depot Cabotegravir Intradermal Delivery for HIV Pre-Exposure Prophylaxis. Adv. Funct. Mater. 2022, 32, 2106999. [Google Scholar] [CrossRef]
- Mc Crudden, M.T.C.; Larrañeta, E.; Clark, A.; Jarrahian, C.; Rein-Weston, A.; Lachau-Durand, S.; Niemeijer, N.; Williams, P.; Haeck, C.; McCarthy, H.O.; et al. Design, formulation and evaluation of novel dissolving microarray patches containing a long-acting rilpivirine nanosuspension. J. Control. Release 2018, 292, 119–129. [Google Scholar] [CrossRef]
- Deanesly, R.; Parkes, A.S. Biological properties of some new derivatives of testosterone. Biochem. J. 1937, 31, 1161. [Google Scholar] [CrossRef]
- Chien, Y.W. Novel drug delivery systems. Drugs Pharm. Sci. 1992, 50, 797. [Google Scholar]
- Dreyfuss, J.; Ross, J.J., Jr.; Shaw, J.M.; Miller, I.; Schreiber, E.C. Release and elimination of 14C-fluphenazine enanthate and decanoate esters administered in sesame oil to dogs. J. Pharm. Sci. 1976, 65, 502–507. [Google Scholar] [CrossRef]
- Wright, J.C.; Hoffman, A.S. Historical overview of long acting injections and implants. In Long Acting Injections and Implants; Springer: Berlin/Heidelberg, Germany, 2011; pp. 11–24. [Google Scholar]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Folkman, J.; Long, D.M., Jr.; Rosenbaum, R. Silicone rubber: A new diffusion property useful for general anesthesia. Science 1966, 154, 148–149. [Google Scholar] [CrossRef]
- Heilmann, K. Therapeutic Systems: Rate-Controlled Drug Delivery: Concept and Development; George Thieme Verlag: Erlangen, Germany, 1984. [Google Scholar]
- Frazza, E.; Schmitt, E. A new absorbable suture. J. Biomed. Mater. Res. 1971, 5, 43–58. [Google Scholar] [CrossRef]
- Kent, J.S.; Lewis, D.H.; Sanders, L.M.; Tice, T.R. Microencapsulation of Water Soluble Active Polypeptides. U.S. Patent 4,675,189A, 23 June 1987. [Google Scholar]
- Hoffman, A.S. The origins and evolution of “controlled” drug delivery systems. J. Control. Release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Nowacek, A.S.; Miller, R.L.; McMillan, J.; Kanmogne, G.; Kanmogne, M.; Mosley, R.L.; Ma, Z.; Graham, S.; Chaubal, M.; Werling, J.; et al. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine 2009, 4, 903–917. [Google Scholar] [CrossRef]
- Baert, L.; van‘t Klooster, G.; Dries, W.; François, M.; Wouters, A.; Basstanie, E.; Iterbeke, K.; Stappers, F.; Stevens, P.; Schueller, L. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur. J. Pharm. Biopharm. 2009, 72, 502–508. [Google Scholar] [CrossRef]
- Penrose, K.J.; Parikh, U.M.; Hamanishi, K.A.; Else, L.; Back, D.; Boffito, M.; Jackson, A.; Mellors, J.W. Selection of rilpivirine-resistant HIV-1 in a seroconverter from the SSAT 040 trial who received the 300-mg dose of long-acting rilpivirine (TMC278LA). J. Infect. Dis. 2016, 213, 1013–1017. [Google Scholar] [CrossRef]
- Venkatesan, P. Long-acting injectable ART for HIV: A (cautious) step forward. Lancet Microbe 2022, 3, e94. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Rahnfeld, L.; Luciani, P. Injectable lipid-based depot formulations: Where do we stand? Pharmaceutics 2020, 12, 567. [Google Scholar] [CrossRef]
- United States Pharmacopeia. <1151> Pharmaceutical dosage forms. In United States Pharmacopeia; USP-NF, Ed.; The United States Pharmacopeial Convention: Rockville, MD, USA, 2021. [Google Scholar]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef]
- Berchane, N.; Carson, K.; Rice-Ficht, A.; Andrews, M. Effect of mean diameter and polydispersity of PLG microspheres on drug release: Experiment and theory. Int. J. Pharm. 2007, 337, 118–126. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef]
- Webber, W.L.; Lago, F.; Thanos, C.; Mathiowitz, E. Characterization of soluble, salt-loaded, degradable PLGA films and their release of tetracycline. J. Biomed. Mater. Res. Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. 1998, 41, 18–29. [Google Scholar] [CrossRef]
- Marin, E.; Briceño, M.I.; Caballero-George, C. Critical evaluation of biodegradable polymers used in nanodrugs. Int. J. Nanomed. 2013, 8, 3071–3091. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef]
- Maroni, A.; Zema, L.; Cerea, M.; Foppoli, A.; Palugan, L.; Gazzaniga, A. Erodible drug delivery systems for time-controlled release into the gastrointestinal tract. J. Drug Deliv. Sci. Technol. 2016, 32, 229–235. [Google Scholar] [CrossRef]
- Keraliya, R.A.; Patel, C.; Patel, P.; Keraliya, V.; Soni, T.G.; Patel, R.C.; Patel, M. Osmotic drug delivery system as a part of modified release dosage form. Int. Sch. Res. Not. 2012, 2012, 528079. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Saltzman, W.M. Drug Delivery: Engineering Principles for Drug Therapy; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Kim, J.M.; Seo, K.S.; Jeong, Y.K.; Lee, H.B.; Kim, Y.S.; Khang, G. Co-effect of aqueous solubility of drugs and glycolide monomer on in vitro release rates from poly (D, L-lactide-co-glycolide) discs and polymer degradation. J. Biomater. Sci. Polym. Ed. 2005, 16, 991–1007. [Google Scholar] [CrossRef]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.-L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly (DL-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Ehrenstein, G.W. Polymeric Materials: Structure, Properties, Applications; Carl Hanser Verlag GmbH Co KG: Munich, Germany, 2012. [Google Scholar]
- Odian, G. Principles of Polymerization; John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Allcock, H.R.; Lampe, F.W.; Mark, J.E.; Allcock, H. Contemporary Polymer Chemistry; Prentice-Hall Englewood Cliffs: Upper Saddle River, NJ, USA, 1981. [Google Scholar]
- Muller, W.E.; Wang, X.; Schroder, H. Biomedical Inorganic Polymers; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Ginjupalli, K.; Shavi, G.V.; Averineni, R.K.; Bhat, M.; Udupa, N.; Upadhya, P.N. Poly (α-hydroxy acid) based polymers: A review on material and degradation aspects. Polym. Degrad. Stab. 2017, 144, 520–535. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Dash, A.; Cudworth II, G. Therapeutic applications of implantable drug delivery systems. J. Pharmacol. Toxicol. Methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Gan, M.; Zhou, Q.; Ge, J.; Zhao, J.; Wang, Y.; Yan, Q.; Wu, C.; Yu, H.; Xiao, Q.; Wang, W. Precise in-situ release of microRNA from an injectable hydrogel induces bone regeneration. Acta Biomater. 2021, 135, 289–303. [Google Scholar] [CrossRef]
- Rahme, K.; Dagher, N. Chemistry Routes for Copolymer Synthesis Containing PEG for Targeting, Imaging, and Drug Delivery Purposes. Pharmaceutics 2019, 11, 327. [Google Scholar] [CrossRef]
- Sanchez-Cano, C.; Carril, M. Recent Developments in the Design of Non-Biofouling Coatings for Nanoparticles and Surfaces. Int. J. Mol. Sci. 2020, 21, 1007. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Toh, K.; Li, J.; Osawa, S.; Tockary, T.A.; Liu, X.; Abbasi, S.; Hayashi, K.; Mochida, Y. Transient stealth coating of liver sinusoidal wall by anchoring two-armed PEG for retargeting nanomedicines. Sci. Adv. 2020, 6, eabb8133. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, H.; Geng, K.; Shen, J.; Feng, X.; Xu, P.; Duan, Y.; Li, Y.; Wu, R.; Gou, Z. Large fuzzy biodegradable polyester microspheres with dopamine deposition enhance cell adhesion and bone regeneration in vivo. Biomaterials 2021, 272, 120783. [Google Scholar] [CrossRef]
- Liu, S.; Li, K.; Hussain, I.; Oderinde, O.; Yao, F.; Zhang, J.; Fu, G. A Conductive Self-Healing Double Network Hydrogel with Toughness and Force Sensitivity. Chem. Eur. J. 2018, 24, 6632–6638. [Google Scholar] [CrossRef]
- Apostolides, D.E.; Patrickios, C.S.; Sakai, T.; Guerre, M.; Lopez, G.R.; Améduri, B.; Ladmiral, V.; Simon, M.; Gradzielski, M.; Clemens, D. Near-model amphiphilic polymer conetworks based on four-arm stars of poly (vinylidene fluoride) and poly (ethylene glycol): Synthesis and characterization. Macromolecules 2018, 51, 2476–2488. [Google Scholar] [CrossRef]
- Xue, S.; Li, X.; Li, S.; Chen, N.; Zhan, Q.; Long, L.; Zhao, J.; Hou, X.; Yuan, X. Bone fracture microenvironment responsive hydrogel for timing sequential release of cargoes. Colloids Surf. A Physicochem. Eng. Asp. 2021, 629, 127413. [Google Scholar] [CrossRef]
- Le Fer, G.; Dilla, R.A.; Wang, Z.; King, J.; Chuang, S.S.; Becker, M.L. Clustering and Hierarchical Organization of 3D Printed Poly (propylene fumarate)-block-PEG-block-poly (propylene fumarate) ABA Triblock Copolymer Hydrogels. Macromolecules 2021, 54, 3458–3468. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Future perspectives and recent advances in stimuli-responsive materials. Prog. Polym. Sci. 2010, 35, 278–301. [Google Scholar] [CrossRef]
- Alexandridis, P.; Zhou, D.; Khan, A. Lyotropic liquid crystallinity in amphiphilic block copolymers: Temperature effects on phase behavior and structure for poly (ethylene oxide)-b-poly (propylene oxide)-b-poly (ethylene oxide) copolymers of different composition. Langmuir 1996, 12, 2690–2700. [Google Scholar] [CrossRef]
- Pozzo, D.C.; Hollabaugh, K.R.; Walker, L.M. Rheology and phase behavior of copolymer-templated nanocomposite materials. J. Rheol. 2005, 49, 759–782. [Google Scholar] [CrossRef]
- Bodratti, A.M.; Alexandridis, P. Formulation of poloxamers for drug delivery. J. Funct. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, X.; Lin, X.; Yao, C.; Shen, L.; Feng, Y. Poloxamer-based in situ hydrogels for controlled delivery of hydrophilic macromolecules after intramuscular injection in rats. Drug Deliv. 2015, 22, 375–382. [Google Scholar] [CrossRef]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef]
- Van Hemelryck, S.; Dewulf, J.; Niekus, H.; van Heerden, M.; Ingelse, B.; Holm, R.; Mannaert, E.; Langguth, P. In vitro evaluation of poloxamer in situ forming gels for bedaquiline fumarate salt and pharmacokinetics following intramuscular injection in rats. Int. J. Pharm. X 2019, 1, 100016. [Google Scholar] [CrossRef]
- Demarteau, J.; Kermagoret, A.; Jérôme, C.; Detrembleur, C.; Debuigne, A. Controlled Synthesis of Ethylene-Vinyl Acetate Based Copolymers by Organometallic Mediated Radical Polymerization. In Controlled Radical Polymerization: Materials; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2015; Volume 1188, pp. 47–61. [Google Scholar]
- Schneider, C.; Langer, R.; Loveday, D.; Hair, D. Applications of ethylene vinyl acetate copolymers (EVA) in drug delivery systems. J. Control. Release 2017, 262, 284–295. [Google Scholar] [CrossRef]
- Arsac, A.; Carrot, C.; Guillet, J. Determination of primary relaxation temperatures and melting points of ethylene vinyl acetate copolymers. J. Therm. Anal. Calorim. 2000, 61, 681–685. [Google Scholar] [CrossRef]
- Wang, X.; Venkatraman, S.S.; Boey, F.Y.; Loo, J.S.; Tan, L.P. Controlled release of sirolimus from a multilayered PLGA stent matrix. Biomaterials 2006, 27, 5588–5595. [Google Scholar] [CrossRef]
- Zhang, Q.; Lin, W.; Yang, G.; Chen, Q. Studies on the phase structure of ethylene-vinyl acetate copolymers by solid-state 1H and 13C NMR. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2199–2207. [Google Scholar] [CrossRef]
- Wang, L.; Fang, P.; Ye, C.; Feng, J. Solid-state NMR characterizations on phase structures and molecular dynamics of poly(ethylene-co-vinyl acetate). J. Polym. Sci. Part B Polym. Phys. 2006, 44, 2864–2879. [Google Scholar] [CrossRef]
- Almeida, A.; Possemiers, S.; Boone, M.N.; De Beer, T.; Quinten, T.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. Eur. J. Pharm. Biopharm. 2011, 77, 297–305. [Google Scholar] [CrossRef] [PubMed]
- McKeen, L.W. The Effect of Sterilization on Plastics and Elastomers; William Andrew: Norwich, NY, USA, 2018. [Google Scholar]
- Gaaz, T.S.; Sulong, A.B.; Akhtar, M.N.; Kadhum, A.A.; Mohamad, A.B.; Al-Amiery, A.A. Properties and Applications of Polyvinyl Alcohol, Halloysite Nanotubes and Their Nanocomposites. Molecules 2015, 20, 22833–22847. [Google Scholar] [CrossRef]
- Song, S.I.; Kim, B.C. Characteristic rheological features of PVA solutions in water-containing solvents with different hydration states. Polymer 2004, 45, 2381–2386. [Google Scholar] [CrossRef]
- Marin, E.; Rojas, J.; Ciro, Y. A review of polyvinyl alcohol derivatives: Promising materials for pharmaceutical and biomedical applications. Afr. J. Pharm. Pharmacol. 2014, 8, 674–684. [Google Scholar]
- Rowe, R.C.; Sheskey, P.; Quinn, M. Handbook of Pharmaceutical Excipients; Libros Digitales-Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Dediu, V.; Busila, M.; Tucureanu, V.; Bucur, F.I.; Iliescu, F.S.; Brincoveanu, O.; Iliescu, C. Synthesis of ZnO/Au nanocomposite for antibacterial applications. Nanomaterials 2022, 12, 3832. [Google Scholar] [CrossRef]
- Fatema, U.K.; Rahman, M.M.; Islam, M.R.; Mollah, M.Y.A.; Susan, M.A.B.H. Silver/poly (vinyl alcohol) nanocomposite film prepared using water in oil microemulsion for antibacterial applications. J. Colloid Interface Sci. 2018, 514, 648–655. [Google Scholar] [CrossRef]
- Meira, J.; Madeira, C.; Falcão-Reis, F.; Figueira, L. Sustained control from recurring non-infectious uveitic macular edema with 0.19 mg fluocinolone acetonide intravitreal implant—A case report. Ophthalmol. Ther. 2019, 8, 635–641. [Google Scholar] [CrossRef]
- Wang, L.L.; Highley, C.B.; Yeh, Y.C.; Galarraga, J.H.; Uman, S.; Burdick, J.A. Three-dimensional extrusion bioprinting of single-and double-network hydrogels containing dynamic covalent crosslinks. J. Biomed. Mater. Res. Part A 2018, 106, 865–875. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Liu, J.; Long, Y.; Fang, L.; Wang, Q.; Liu, T. Highly compressible and superior low temperature tolerant supercapacitors based on dual chemically crosslinked PVA hydrogel electrolytes. J. Mater. Chem. A 2020, 8, 6219–6228. [Google Scholar] [CrossRef]
- Chao, Y.; Chen, Q.; Liu, Z. Smart injectable hydrogels for cancer immunotherapy. Adv. Funct. Mater. 2020, 30, 1902785. [Google Scholar] [CrossRef]
- Lanman, T.; Martin, N.; Vinters, H. The pathology of encephalic arteriovenous malformations treated by prior embolotherapy. Neuroradiology 1988, 30, 1–10. [Google Scholar] [CrossRef]
- Hulman, G.; Kirkham, J. Ivalon sponge presenting as an extrarectal mass. J. Pathol. 1990, 160, A171. [Google Scholar]
- Fassi, A.; Naidoo, N. Irritation associated with tear-replacement ophthalmic drops-a pharmaceutical and subjective investigation. S. Afr. Med. J. 1989, 75, 233–235. [Google Scholar]
- Singh, B.; Varshney, L.; Francis, S. Designing tragacanth gum based sterile hydrogel by radiation method for use in drug delivery and wound dressing applications. Int. J. Biol. Macromol. 2016, 88, 586–602. [Google Scholar] [CrossRef]
- Nasef, S.M.; Khozemy, E.E.; Kamoun, E.A.; El-Gendi, H. Gamma radiation-induced crosslinked composite membranes based on polyvinyl alcohol/chitosan/AgNO3/vitamin E for biomedical applications. Int. J. Biol. Macromol. 2019, 137, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.J.; Wei, Z.J.; Yu, X.Z. Study on Polyvinyl Alcohol Hydrogel Materials for Improving the Performance of Artificial Articular Cartilage. Adv. Mater. Res. 2013, 703, 29–32. [Google Scholar] [CrossRef]
- DeMerlis, C.; Schoneker, D. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Rodwell, D.; Kelly, C.; DeMerlis, C.; Schoneker, D.; Borzelleca, J. Effects of polyvinyl alcohol administered in the diet to rats on fertility, early embryonic development, growth and development. Food Chem. Toxicol. 2003, 41, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; DeMerlis, C.; Schoneker, D.; Borzelleca, J. Subchronic toxicity study in rats and genotoxicity tests with polyvinyl alcohol. Food Chem. Toxicol. 2003, 41, 719–727. [Google Scholar] [CrossRef]
- Sun, W.; Tian, J.; Chen, L.; He, S.; Wang, J. Improvement of biodegradability of PVA-containing wastewater by ionizing radiation pretreatment. Environ. Sci. Pollut. Res. 2012, 19, 3178–3184. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.-L.; Wang, C.-T.; Huang, K.-Y. Investigation of process parameters for the removal of polyvinyl alcohol from aqueous solution by iron electrocoagulation. Desalination 2010, 251, 12–19. [Google Scholar] [CrossRef]
- Jabbari, E.; Khakpour, M. Morphology of and release behavior from porous polyurethane microspheres. Biomaterials 2000, 21, 2073–2079. [Google Scholar] [CrossRef]
- Kääriä, K.; Hirvonen, A.; Norppa, H.; Piirilä, P.; Vainio, H.; Rosenberg, C. Exposure to 4,4′-methylenediphenyl diisocyanate (MDI) during moulding of rigid polyurethane foam: Determination of airborne MDI and urinary 4,4′-methylenedianiline (MDA). Analyst 2001, 126, 476–479. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Wei, K.; Zhao, N.; Zhang, S.; Chen, J. Drug distribution within poly (ɛ-caprolactone) microspheres and in vitro release. J. Mater. Process. Technol. 2009, 209, 348–354. [Google Scholar] [CrossRef]
- Thompson, D.G.; Osborn, J.C.; Kober, E.M.; Schoonover, J.R. Effects of hydrolysis-induced molecular weight changes on the phase separation of a polyester polyurethane. Polym. Degrad. Stab. 2006, 91, 3360–3370. [Google Scholar] [CrossRef]
- Kaur, M.; Gupta, K.M.; Poursaid, A.E.; Karra, P.; Mahalingam, A.; Aliyar, H.A.; Kiser, P.F. Engineering a degradable polyurethane intravaginal ring for sustained delivery of dapivirine. Drug Deliv. Transl. Res. 2011, 1, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.M.; Dadsetan, M.; Wiggins, M.; Anderson, J.M.; Hiltner, A. Poly (carbonate urethane) and poly (ether urethane) biodegradation: In vivo studies. J. Biomed. Mater. Res. Part A Off. J. Soc. Biomater. Jpn. Soc. Biomater. Aust. Soc. Biomater. Korean Soc. Biomater. 2004, 69, 407–416. [Google Scholar] [CrossRef]
- Rychlý, J.; Lattuati-Derieux, A.; Lavédrine, B.; Matisová-Rychlá, L.; Malíková, M.; Csomorová, K.; Janigová, I. Assessing the progress of degradation in polyurethanes by chemiluminescence and thermal analysis. II. Flexible polyether-and polyester-type polyurethane foams. Polym. Degrad. Stab. 2011, 96, 462–469. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, L.Y.; Wang, C.-H. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 1274–1325. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Gupta, K.M.; Fabian, J.; Albright, T.H.; Kiser, P.F. Segmented polyurethane intravaginal rings for the sustained combined delivery of antiretroviral agents dapivirine and tenofovir. Eur. J. Pharm. Sci. 2010, 39, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.F.; Hickey, T.P. Degradable polyurethane networks based on d, l-lactide, glycolide, ε-caprolactone, and trimethylene carbonate homopolyester and copolyester triols. Polymer 1994, 35, 830–838. [Google Scholar] [CrossRef]
- Guelcher, S.; Srinivasan, A.; Hafeman, A.; Gallagher, K.; Doctor, J.; Khetan, S.; McBride, S.; Hollinger, J. Synthesis, in vitro biocompatibility and biodegradation, and mechanical properties of two-component polyurethane scaffolds: Effects of water and polyol composition. Tissue Eng. 2007, 13, 2321–2333. [Google Scholar] [CrossRef]
- Labow, R.S.; Erfle, D.J.; Santerre, J.P. Elastase-induced hydrolysis of synthetic solid substrates: Poly (ester-urea-urethane) and poly (ether-urea-urethane). Biomaterials 1996, 17, 2381–2388. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical Applications of Biodegradable Polymers. J. Polym. Sci. B Polym. Phys. 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Feng, P.; Jia, J.; Liu, M.; Peng, S.; Zhao, Z.; Shuai, C. Degradation mechanisms and acceleration strategies of poly (lactic acid) scaffold for bone regeneration. Mater. Des. 2021, 210, 110066. [Google Scholar] [CrossRef]
- Kuppermann, B.D.; Patel, S.S.; Boyer, D.S.; Augustin, A.J.; Freeman, W.R.; Kerr, K.J.; Guo, Q.; Schneider, S.; López, F.J. Phase 2 study of the safety and efficacy of brimonidine drug delivery system (brimo dds) generation 1 in patients with geographic atrophy secondary to age-related macular degeneration. Retina 2021, 41, 144–155. [Google Scholar] [CrossRef]
- Pişkin, E. Biodegradable polymers as biomaterials. J. Biomater. Sci. Polym. Ed. 1995, 6, 775–795. [Google Scholar] [CrossRef]
- Pisecky, L.; Luger, M.; Klasan, A.; Gotterbarm, T.; Klotz, M.C.; Hochgatterer, R. Bioabsorbable implants in forefoot surgery: A review of materials, possibilities and disadvantages. EFORT Open Rev. 2021, 6, 1132–1139. [Google Scholar] [CrossRef]
- Wan, B.; Bao, Q.; Burgess, D. Long-acting PLGA microspheres: Advances in excipient and product analysis toward improved product understanding. Adv. Drug Deliv. Rev. 2023, 198, 114857. [Google Scholar] [CrossRef]
- Blasi, P.; D’Souza, S.S.; Selmin, F.; DeLuca, P.P. Plasticizing effect of water on poly (lactide-co-glycolide). J. Control. Release 2005, 108, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Rustgi, R. Biodegradable polymers. Prog. Polym. Sci. 1998, 23, 1273–1335. [Google Scholar] [CrossRef]
- Cayuela, J.; Bounor-Legaré, V.; Cassagnau, P.; Michel, A. Ring-opening polymerization of ε-caprolactone initiated with titanium n-propoxide or titanium phenoxide. Macromolecules 2006, 39, 1338–1346. [Google Scholar] [CrossRef]
- Natta, F.J.v.; Hill, J.W.; Carothers, W.H. Studies of polymerization and ring formation. XXIII. 1 ε-Caprolactone and its polymers. J. Am. Chem. Soc. 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Sinha, V.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-ϵ-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Lu, X.; Dalai, S.; Zhang, J. Thermophysical properties of polycaprolactone/chitosan blend membranes. Thermochim. Acta 2009, 487, 33–38. [Google Scholar] [CrossRef]
- Perez, M.H.; Zinutti, C.; Lamprecht, A.; Ubrich, N.; Astier, A.; Hoffman, M.; Bodmeier, R.; Maincent, P. The preparation and evaluation of poly (ϵ-caprolactone) microparticles containing both a lipophilic and a hydrophilic drug. J. Control. Release 2000, 65, 429–438. [Google Scholar] [CrossRef]
- Gorna, K.; Gogolewski, S. In vitro degradation of novel medical biodegradable aliphatic polyurethanes based on ϵ-caprolactone and Pluronics® with various hydrophilicities. Polym. Degrad. Stab. 2002, 75, 113–122. [Google Scholar] [CrossRef]
- Ren, D.; Yi, H.; Wang, W.; Ma, X. The enzymatic degradation and swelling properties of chitosan matrices with different degrees of N-acetylation. Carbohydr. Res. 2005, 340, 2403–2410. [Google Scholar] [CrossRef]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. React. Funct. Polym. 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef]
- Fonte, P.; Araújo, F.; Silva, C.; Pereira, C.; Reis, S.; Santos, H.A.; Sarmento, B. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnol. Adv. 2015, 33, 1342–1354. [Google Scholar] [CrossRef]
- Li, P.; Fujimoto, K.; Bourguingnon, L.; Yukl, S.; Deeks, S.; Wong, J.K. Exogenous and endogenous hyaluronic acid reduces HIV infection of CD4(+) T cells. Immunol. Cell Biol. 2014, 92, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, V.; Meng, J.; Ezoulin, M.J.; Youm, I.; Dim, D.C.; Molteni, A.; Hung, W.T.; Christenson, L.K.; Youan, B.C. Stimuli-sensitive thiolated hyaluronic acid based nanofibers: Synthesis, preclinical safety and in vitro anti-HIV activity. Nanomedicine 2016, 11, 2935–2958. [Google Scholar] [CrossRef] [PubMed]
- Jouyban, A.; Fakhree, M.A.A.; Shayanfar, A. Review of pharmaceutical applications of N-methyl-2-pyrrolidone. J. Pharm. Pharm. Sci. 2010, 13, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Indivior UK Limited. Highlights of Prescribing Information—SublocadeTM; Indivior UK Limited: Hull, UK, 2017. [Google Scholar]
- Rivera-Briso, A.L.; Serrano-Aroca, Á. Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate): Enhancement Strategies for Advanced Applications. Polymers 2018, 10, 732. [Google Scholar] [CrossRef]
- Avella, M.; Martuscelli, E.; Raimo, M. Review Properties of blends and composites based on poly (3-hydroxy) butyrate (PHB) and poly (3-hydroxybutyrate-hydroxyvalerate)(PHBV) copolymers. J. Mater. Sci. 2000, 35, 523–545. [Google Scholar] [CrossRef]
- Mofokeng, J.; Luyt, A. Morphology and thermal degradation studies of melt-mixed poly (hydroxybutyrate-co-valerate)(PHBV)/poly (ε-caprolactone)(PCL) biodegradable polymer blend nanocomposites with TiO2 as filler. J. Mater. Sci. 2015, 50, 3812–3824. [Google Scholar] [CrossRef]
- Hedayati, A.; Yazdi, S.G.; Dehghankelishadi, P.; Javan, N.B.; Akbari, H.; Dorkoosh, F.A. Preparation, Optimization and Physicochemical Characterization of Aripiprazole Loaded Nano-porous in situ Forming Implant. Pharm. Nanotechnol. 2017, 5, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Acemoglu, M.; Nimmerfall, F.; Bantle, S.; Stoll, G.H. Poly(ethylene carbonate)s, part I: Syntheses and structural effects on biodegradation. J. Control. Release 1997, 49, 263–276. [Google Scholar] [CrossRef]
- Sasanuma, Y.; Takahashi, Y. Structure–Property Relationships of Poly(ethylene carbonate) and Poly(propylene carbonate). ACS Omega 2017, 2, 4808–4819. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.H.; Nimmerfall, F.; Acemoglu, M.; Bodmer, D.; Bantle, S.; Müller, I.; Mahl, A.; Kolopp, M.; Tullberg, K. Poly(ethylene carbonate)s, part II: Degradation mechanisms and parenteral delivery of bioactive agents. J. Control. Release 2001, 76, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kemmer, A.; Keim, K.; Curdy, C.; Petersen, H.; Kissel, T. Poly(ethylene carbonate) as a surface-eroding biomaterial for in situ forming parenteral drug delivery systems: A feasibility study. Eur. J. Pharm. Biopharm. 2010, 76, 222–229. [Google Scholar] [CrossRef]
- Panja, S.; Maji, S.; Maiti, T.K.; Chattopadhyay, S. A Smart Magnetically Active Nanovehicle for on-Demand Targeted Drug Delivery: Where van der Waals Force Balances the Magnetic Interaction. ACS Appl. Mater. Interfaces 2015, 7, 24229–24241. [Google Scholar] [CrossRef]
- Panja, S.; Dey, G.; Bharti, R.; Mandal, P.; Mandal, M.; Chattopadhyay, S. Metal Ion Ornamented Ultrafast Light-Sensitive Nanogel for Potential In Vivo Cancer Therapy. Chem. Mater. 2016, 28, 8598–8610. [Google Scholar] [CrossRef]
- Panja, S.; Dey, G.; Bharti, R.; Kumari, K.; Maiti, T.K.; Mandal, M.; Chattopadhyay, S. Tailor-Made Temperature-Sensitive Micelle for Targeted and On-Demand Release of Anticancer Drugs. ACS Appl. Mater. Interfaces 2016, 8, 12063–12074. [Google Scholar] [CrossRef]
- Lan, T.; Guo, Q. Phenylboronic acid-decorated polymeric nanomaterials for advanced bio-application. Nanotechnol. Rev. 2019, 8, 548–561. [Google Scholar] [CrossRef]
- Feng, R.; Zhu, L.; Teng, F.; Wang, M.; Chen, S.; Song, Z.; Li, H. Phenylboronic acid-modified polymaleic anhydride-F127 micelles for pH-activated targeting delivery of doxorubicin. Colloids Surf. B Biointerfaces 2022, 216, 112559. [Google Scholar] [CrossRef]
- Mahalingam, A.; Jay, J.I.; Langheinrich, K.; Shukair, S.; McRaven, M.D.; Rohan, L.C.; Herold, B.C.; Hope, T.J.; Kiser, P.F. Inhibition of the transport of HIV in vitro using a pH-responsive synthetic mucin-like polymer system. Biomaterials 2011, 32, 8343–8355. [Google Scholar] [CrossRef]
- Jay, J.I.; Shukair, S.; Langheinrich, K.; Hanson, M.C.; Cianci, G.C.; Johnson, T.J.; Clark, M.R.; Hope, T.J.; Kiser, P.F. Modulation of Viscoelasticity and HIV Transport as a Function of pH in a Reversibly Crosslinked Hydrogel. Adv. Funct. Mater. 2009, 19, 2969–2977. [Google Scholar] [CrossRef]
- Folkman, J.; Long, D.M. The use of silicone rubber as a carrier for prolonged drug therapy. J. Surg. Res. 1964, 4, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, E.; Johengen, D.; Luecke, E.; Rothrock, G.; McGowan, I.; van der Straten, A.; Desai, T. A tunable, biodegradable, thin-film polymer device as a long-acting implant delivering tenofovir alafenamide fumarate for HIV pre-exposure prophylaxis. Pharm. Res. 2016, 33, 1649–1656. [Google Scholar] [CrossRef]
- Li, L.; Johnson, L.M.; Krovi, S.A.; Demkovich, Z.R.; van der Straten, A. Performance and stability of tenofovir alafenamide formulations within subcutaneous biodegradable implants for HIV pre-exposure prophylaxis (PrEP). Pharmaceutics 2020, 12, 1057. [Google Scholar] [CrossRef]
- Su, J.T.; Simpson, S.M.; Sung, S.; Tfaily, E.B.; Veazey, R.; Marzinke, M.; Qiu, J.; Watrous, D.; Widanapathirana, L.; Pearson, E. A subcutaneous implant of tenofovir alafenamide fumarate causes local inflammation and tissue necrosis in rabbits and macaques. Antimicrob. Agents Chemother. 2020, 64, 10-1128. [Google Scholar] [CrossRef]
- Joiner, J.B.; Prasher, A.; Young, I.C.; Kim, J.; Shrivastava, R.; Maturavongsadit, P.; Benhabbour, S.R. Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics. Pharmaceutics 2022, 14, 1188. [Google Scholar] [CrossRef]
- Romano, J.W.; Baum, M.M.; Demkovich, Z.R.; Diana, F.; Dobard, C.; Feldman, P.L.; Garcia-Lerma, J.G.; Grattoni, A.; Gunawardana, M.; Ho, D.K.; et al. Tenofovir Alafenamide for HIV Prevention: Review of the Proceedings from the Gates Foundation Long-Acting TAF Product Development Meeting. AIDS Res. Hum. Retroviruses 2021, 37, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.E.; Teller, R.S.; Forster, S.P.; Li, L.; Mackey, M.A.; Skomski, D.; Yang, Z.; Fillgrove, K.L.; Doto, G.J.; Wood, S.L.; et al. Extended-Duration MK-8591-Eluting Implant as a Candidate for HIV Treatment and Prevention. Antimicrob. Agents Chemother. 2018, 62, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, R.K.; Fetherston, S.M.; McCoy, C.F.; Boyd, P.; Major, I. Vaginal rings for delivery of HIV microbicides. Int. J. Womens Health 2012, 4, 595–605. [Google Scholar] [CrossRef]
- Ullah Nayan, M.; Sillman, B.; Hasan, M.; Deodhar, S.; Das, S.; Sultana, A.; Thai Hoang Le, N.; Soriano, V.; Edagwa, B.; Gendelman, H.E. Advances in long-acting slow effective release antiretroviral therapies for treatment and prevention of HIV infection. Adv. Drug Deliv. Rev. 2023, 200, 115009. [Google Scholar] [CrossRef]
- Karunakaran, D.; Simpson, S.M.; Su, J.T.; Bryndza-Tfaily, E.; Hope, T.J.; Veazey, R.; Dobek, G.; Qiu, J.; Watrous, D.; Sung, S.; et al. Design and Testing of a Cabotegravir Implant for HIV Prevention. J. Control. Release 2021, 330, 658–668. [Google Scholar] [CrossRef]
- Nevoralová, M.; Koutný, M.; Ujčić, A.; Starý, Z.; Šerá, J.; Vlková, H.; Šlouf, M.; Fortelný, I.; Kruliš, Z. Structure Characterization and Biodegradation Rate of Poly(ε-caprolactone)/Starch Blends. Front. Mater. 2020, 7, 141. [Google Scholar] [CrossRef]
- Markowitz, M.; Sarafianos, S.G. EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591): A novel HIV-1 reverse transcriptase translocation inhibitor. Curr. Opin. HIV AIDS 2018, 13, 294. [Google Scholar] [CrossRef]
- Wang, X.; Burgess, D.J. Drug release from in situ forming implants and advances in release testing. Adv. Drug Deliv. Rev. 2021, 178, 113912. [Google Scholar] [CrossRef]
- Phaechamud, T.; Senarat, S.; Puyathorn, N.; Praphanwittaya, P. Solvent exchange and drug release characteristics of doxycycline hyclate-loaded bleached shellac in situ-forming gel and-microparticle. Int. J. Biol. Macromol. 2019, 135, 1261–1272. [Google Scholar] [CrossRef]
- Amini-Fazl, M.S. Biodegradation study of PLGA as an injectable in situ depot-forming implant for controlled release of paclitaxel. Polym. Bull. 2021, 79, 2763–2776. [Google Scholar] [CrossRef]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Kanwar, N.; Sinha, V.R. In situ forming depot as sustained-release drug delivery systems. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 93–136. [Google Scholar] [CrossRef]
- Yu, L.; Ding, J. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37, 1473–1481. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Ayoub, M.M.; El-Bassossy, H.M.; El-Nahas, H.M.; Gomaa, E. Investigation of Alogliptin-Loaded In Situ Gel Implants by 23 Factorial Design with Glycemic Assessment in Rats. Pharmaceutics 2022, 14, 1867. [Google Scholar] [CrossRef] [PubMed]
- Wex, J.; Sidhu, M.; Odeyemi, I.; Abou-Setta, A.M.; Retsa, P.; Tombal, B. Leuprolide acetate 1-, 3-and 6-monthly depot formulations in androgen deprivation therapy for prostate cancer in nine European countries: Evidence review and economic evaluation. Clin. Outcomes Res. 2013, 5, 257–269. [Google Scholar] [CrossRef]
- Kim, M.; Johnson, C.E.; Schmalstig, A.A.; Annis, A.; Wessel, S.E.; Van Horn, B.; Schauer, A.; Exner, A.A.; Stout, J.E.; Wahl, A. A long-acting formulation of rifabutin is effective for prevention and treatment of Mycobacterium tuberculosis. Nat. Commun. 2022, 13, 4455. [Google Scholar] [CrossRef]
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nat. Commun. 2019, 10, 4324. [Google Scholar] [CrossRef]
- Cox, M.C.; Scripture, C.D.; Figg, W.D. Leuprolide acetate given by a subcutaneous extended-release injection: Less of a pain? Expert Rev. Anticancer Ther. 2005, 5, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Rupenthal, I.D. Injectable implants for the sustained release of protein and peptide drugs. Drug Discov Today 2013, 18, 337–349. [Google Scholar] [CrossRef]
- Packhaeuser, C.; Schnieders, J.; Oster, C.; Kissel, T. In situ forming parenteral drug delivery systems: An overview. Eur. J. Pharm. Biopharm. 2004, 58, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.B.; Ridgeway, K.; Montgomery, E.; Torjesen, K.; Clark, R.; Peterson, J.; Baggaley, R.; van der Straten, A. Vaginal ring acceptability and related preferences among women in low-and middle-income countries: A systematic review and narrative synthesis. PLoS ONE 2019, 14, e0224898. [Google Scholar] [CrossRef] [PubMed]
- MACHT, D.I. On the absorption of drugs and poisons through the vagina. J. Pharmacol. Exp. Ther. 1918, 10, 509–522. [Google Scholar]
- Malcolm, R.K.; Boyd, P.J.; McCoy, C.F.; Murphy, D.J. Microbicide vaginal rings: Technological challenges and clinical development. Adv. Drug Deliv. Rev. 2016, 103, 33–56. [Google Scholar] [CrossRef] [PubMed]
- McConville, C.; Andrews, G.P.; Laverty, T.P.; Woolfson, A.D.; Malcolm, R.K. Rheological evaluation of the isothermal cure characteristics of medical grade silicone elastomers. J. Appl. Polym. Sci. 2010, 116, 2320–2327. [Google Scholar] [CrossRef]
- Monteiro, I.; Guazzelli, C.F.; Bahamondes, L. Advances in contraceptive vaginal rings: What does the future hold? Expert Opin. Pharmacother. 2018, 19, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Speroff, L.; Group, U.S.V.I. Efficacy and tolerability of a novel estradiol vaginal ring for relief of menopausal symptoms. Obstet. Gynecol. 2003, 102, 823–834. [Google Scholar]
- Woolfson, A.; Malcolm, R.; Gallagher, R. Design of a silicone reservoir intravaginal ring for the delivery of oxybutynin. J. Control. Release 2003, 91, 465–476. [Google Scholar] [CrossRef]
- Leonhard, M.; Moser, D.; Reumueller, A.; Mancusi, G.; Bigenzahn, W.; Schneider-Stickler, B. Comparison of biofilm formation on new phonax and provox 2 voice prostheses—A pilot study. Head Neck 2010, 32, 886–895. [Google Scholar] [CrossRef]
- MacCallum, N.; Howell, C.; Kim, P.; Sun, D.; Friedlander, R.; Ranisau, J.; Ahanotu, O.; Lin, J.J.; Vena, A.; Hatton, B. Liquid-infused silicone as a biofouling-free medical material. ACS Biomater. Sci. Eng. 2015, 1, 43–51. [Google Scholar] [CrossRef]
- Fundeanu, I.; Klee, D.; Schouten, A.J.; Busscher, H.J.; Van der Mei, H.C. Solvent-free functionalization of silicone rubber and efficacy of PAAm brushes grafted from an amino-PPX layer against bacterial adhesion. Acta Biomater. 2010, 6, 4271–4276. [Google Scholar] [CrossRef]
- Rodrigues, L.; Van der Mei, H.; Teixeira, J.A.; Oliveira, R. Biosurfactant from Lactococcus lactis 53 inhibits microbial adhesion on silicone rubber. Appl. Microbiol. Biotechnol. 2004, 66, 306–311. [Google Scholar] [CrossRef]
- Everaert, E.; Belt-Gritter, B.V.D.; Van der Mei, H.; Busscher, H.; Verkerke, G.; Dijk, F.; Mahieu, H.; Reitsma, A. In vitro and in vivo microbial adhesion and growth on argon plasma-treated silicone rubber voice prostheses. J. Mater. Sci. Mater. Med. 1998, 9, 147–157. [Google Scholar] [CrossRef]
- Everaert, E.P.; Mahieu, H.F.; van de Belt-Gritter, B.; Peeters, A.J.G.; Verkerke, G.J.; van der Mei, H.C.; Busscher, H.J. Biofilm formation in vivo on perfluoro-alkylsiloxane–modified voice prostheses. Arch. Otolaryngol. Head Neck Surg. 1999, 125, 1329–1332. [Google Scholar] [CrossRef]
- Traore, Y.L.; Chen, Y.; Bernier, A.-M.; Ho, E.A. Impact of hydroxychloroquine-loaded polyurethane intravaginal rings on lactobacilli. Antimicrob. Agents Chemother. 2015, 59, 7680–7686. [Google Scholar] [CrossRef]
- Roddy, R.E.; Zekeng, L.; Ryan, K.A.; Tamoufe, U.; Weir, S.S.; Wong, E.L. A controlled trial of nonoxynol 9 film to reduce male-to-female transmission of sexually transmitted diseases. N. Engl. J. Med. 1998, 339, 504–510. [Google Scholar] [CrossRef]
- Malcolm, R.K.; Woolfson, A.D.; Toner, C.F.; Morrow, R.J.; McCullagh, S.D. Long-term, controlled release of the HIV microbicide TMC120 from silicone elastomer vaginal rings. J. Antimicrob. Chemother. 2005, 56, 954–956. [Google Scholar] [CrossRef]
- Nel, A.; Smythe, S.; Young, K.; Malcolm, K.; McCoy, C.; Rosenberg, Z.; Romano, J. Safety and Pharmacokinetics of Dapivirine Delivery from Matrix and Reservoir Intravaginal Rings to HIV-Negative Women. JAIDS J. Acquir. Immune Defic. Syndr. 2009, 51, 416–423. [Google Scholar] [CrossRef]
- Nel, A.; Bekker, L.-G.; Bukusi, E.; Hellström, E.; Kotze, P.; Louw, C.; Martinson, F.; Masenga, G.; Montgomery, E.; Ndaba, N. Safety, acceptability and adherence of dapivirine vaginal ring in a microbicide clinical trial conducted in multiple countries in Sub-Saharan Africa. PLoS ONE 2016, 11, e0147743. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Palanee-Phillips, T.; Brown, E.R.; Schwartz, K.; Soto-Torres, L.E.; Govender, V.; Mgodi, N.M.; Matovu Kiweewa, F.; Nair, G.; Mhlanga, F. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. N. Engl. J. Med. 2016, 375, 2121–2132. [Google Scholar] [CrossRef]
- Johnson, T.J.; Clark, M.R.; Albright, T.H.; Nebeker, J.S.; Tuitupou, A.L.; Clark, J.T.; Fabian, J.; McCabe, R.T.; Chandra, N.; Doncel, G.F.; et al. A 90-day tenofovir reservoir intravaginal ring for mucosal HIV prophylaxis. Antimicrob. Agents Chemother. 2012, 56, 6272–6283. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, P.M.; Rastogi, R.; Segarra, T.J.; Teller, R.S.; Torres, N.M.; Huber, A.M.; Kiser, P.F.; Herold, B.C. Intravaginal ring delivery of tenofovir disoproxil fumarate for prevention of HIV and herpes simplex virus infection. J. Antimicrob. Chemother. 2012, 67, 1730–1738. [Google Scholar] [CrossRef]
- Smith, J.M.; Rastogi, R.; Teller, R.S.; Srinivasan, P.; Mesquita, P.M.; Nagaraja, U.; McNicholl, J.M.; Hendry, R.M.; Dinh, C.T.; Martin, A.; et al. Intravaginal ring eluting tenofovir disoproxil fumarate completely protects macaques from multiple vaginal simian-HIV challenges. Proc. Natl. Acad. Sci. USA 2013, 110, 16145–16150. [Google Scholar] [CrossRef]
- Su, J.T.; Teller, R.S.; Srinivasan, P.; Zhang, J.; Martin, A.; Sung, S.; Smith, J.M.; Kiser, P.F. A Dose Ranging Pharmacokinetic Evaluation of IQP-0528 Released from Intravaginal Rings in Non-Human Primates. Pharm. Res. 2017, 34, 2163–2171. [Google Scholar] [CrossRef]
- Quinn, H.L.; Kearney, M.-C.; Courtenay, A.J.; McCrudden, M.T.; Donnelly, R.F. The role of microneedles for drug and vaccine delivery. Expert Opin. Drug Deliv. 2014, 11, 1769–1780. [Google Scholar] [CrossRef]
- Permana, A.D.; McCrudden, M.T.; Donnelly, R.F. Enhanced intradermal delivery of nanosuspensions of antifilariasis drugs using dissolving microneedles: A proof of concept study. Pharmaceutics 2019, 11, 346. [Google Scholar] [CrossRef]
- Harvey, A.J.; Kaestner, S.A.; Sutter, D.E.; Harvey, N.G.; Mikszta, J.A.; Pettis, R.J. Microneedle-based intradermal delivery enables rapid lymphatic uptake and distribution of protein drugs. Pharm. Res. 2011, 28, 107–116. [Google Scholar] [CrossRef]
- Moga, K.A.; Bickford, L.R.; Geil, R.D.; Dunn, S.S.; Pandya, A.A.; Wang, Y.; Fain, J.H.; Archuleta, C.F.; O’Neill, A.T.; DeSimone, J.M. Rapidly–dissolvable microneedle patches via a highly scalable and reproducible soft lithography approach. Adv. Mater. 2013, 25, 5060–5066. [Google Scholar] [CrossRef]
- Römgens, A.; Bader, D.; Bouwstra, J.; Baaijens, F.; Oomens, C. Monitoring the penetration process of single microneedles with varying tip diameters. J. Mech. Behav. Biomed. Mater. 2014, 40, 397–405. [Google Scholar] [CrossRef]
- Gill, H.; Prausnitz, M. Effect of microneedle design on pain in human subjects. Clin. J. Pain 2008, 24, 585–594. [Google Scholar] [CrossRef]
- Moffatt, K.; Quinn, C.; McCague, P.J.; Donnelly, R.F. Exploration into the opinions of patients with HIV, healthcare professionals and the lay public of the use of microneedles in clinical practice: Highlighting the translational potential for their role in HIV infection. Drug Deliv. Transl. Res. 2021, 11, 1199–1217. [Google Scholar] [CrossRef]
- Chen, M.; Quan, G.; Sun, Y.; Yang, D.; Pan, X.; Wu, C. Nanoparticles-encapsulated polymeric microneedles for transdermal drug delivery. J. Control. Release 2020, 325, 163–175. [Google Scholar] [CrossRef]
- Xie, L.; Zeng, H.; Sun, J.; Qian, W. Engineering microneedles for therapy and diagnosis: A survey. Micromachines 2020, 11, 271. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, J.-H.; Prausnitz, M.R. Dissolving microneedles for transdermal drug delivery. Biomaterials 2008, 29, 2113–2124. [Google Scholar] [CrossRef]
- Ito, Y.; Hirono, M.; Fukushima, K.; Sugioka, N.; Takada, K. Two-layered dissolving microneedles formulated with intermediate-acting insulin. Int. J. Pharm. 2012, 436, 387–393. [Google Scholar] [CrossRef]
- Chu, L.Y.; Choi, S.-O.; Prausnitz, M.R. Fabrication of dissolving polymer microneedles for controlled drug encapsulation and delivery: Bubble and pedestal microneedle designs. J. Pharm. Sci. 2010, 99, 4228–4238. [Google Scholar] [CrossRef]
- Chen, W.; Wang, C.; Yan, L.; Huang, L.; Zhu, X.; Chen, B.; Sant, H.J.; Niu, X.; Zhu, G.; Yu, K. Improved polyvinylpyrrolidone microneedle arrays with non-stoichiometric cyclodextrin. J. Mater. Chem. B 2014, 2, 1699–1705. [Google Scholar] [CrossRef]
- Vora, L.K.; Vavia, P.R.; Larrañeta, E.; Bell, S.E.; Donnelly, R.F. Novel nanosuspension-based dissolving microneedle arrays for transdermal delivery of a hydrophobic drug. J. Interdiscip. Nanomed. 2018, 3, 89–101. [Google Scholar] [CrossRef]
- Park, J.-H.; Allen, M.G.; Prausnitz, M.R. Polymer microneedles for controlled-release drug delivery. Pharm. Res. 2006, 23, 1008–1019. [Google Scholar] [CrossRef]
- Abdelghany, S.; Tekko, I.A.; Vora, L.; Larrañeta, E.; Permana, A.D.; Donnelly, R.F. Nanosuspension-based dissolving microneedle arrays for intradermal delivery of curcumin. Pharmaceutics 2019, 11, 308. [Google Scholar] [CrossRef]
- Li, W.; Terry, R.N.; Tang, J.; Feng, M.R.; Schwendeman, S.P.; Prausnitz, M.R. Rapidly separable microneedle patch for the sustained release of a contraceptive. Nat. Biomed. Eng. 2019, 3, 220–229. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, S.; Yang, G.; Zhou, Z.; Gao, Y. Exploring Trehalose on the release of levonorgestrel from implantable PLGA microneedles. Polymers 2020, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Yang, G.; Zhao, X.; Zhang, S.; Gao, Y. Intradermal implantable PLGA microneedles for etonogestrel sustained release. J. Pharm. Sci. 2020, 109, 1958–1966. [Google Scholar] [CrossRef]
- Than, A.; Liang, K.; Xu, S.; Sun, L.; Duan, H.; Xi, F.; Xu, C.; Chen, P. Transdermal delivery of anti-obesity compounds to subcutaneous adipose tissue with polymeric microneedle patches. Small Methods 2017, 1, 1700269. [Google Scholar] [CrossRef]
- Chen, M.-C.; Wang, K.-W.; Chen, D.-H.; Ling, M.-H.; Liu, C.-Y. Remotely triggered release of small molecules from LaB6@ SiO2-loaded polycaprolactone microneedles. Acta Biomater. 2015, 13, 344–353. [Google Scholar] [CrossRef]
- Mc Crudden, M.T.C.; Larrañeta, E.; Clark, A.; Jarrahian, C.; Rein-Weston, A.; Creelman, B.; Moyo, Y.; Lachau-Durand, S.; Niemeijer, N.; Williams, P.; et al. Design, Formulation, and Evaluation of Novel Dissolving Microarray Patches Containing Rilpivirine for Intravaginal Delivery. Adv. Healthc. Mater. 2019, 8, 1801510. [Google Scholar] [CrossRef]
- Abbate, M.T.A.; Ramöller, I.K.; Sabri, A.H.; Paredes, A.J.; Hutton, A.J.; McKenna, P.E.; Peng, K.; Hollett, J.A.; McCarthy, H.O.; Donnelly, R.F. Formulation of antiretroviral nanocrystals and development into a microneedle delivery system for potential treatment of HIV-associated neurocognitive disorder (HAND). Int. J. Pharm. 2023, 640, 123005. [Google Scholar] [CrossRef]
- Zhang, C.; Vora, L.K.; Tekko, I.A.; Volpe-Zanutto, F.; Peng, K.; Paredes, A.J.; McCarthy, H.O.; Donnelly, R.F. Development of dissolving microneedles for intradermal delivery of the long-acting antiretroviral drug bictegravir. Int. J. Pharm. 2023, 642, 123108. [Google Scholar] [CrossRef]
- Donnelly, R.F.; McCrudden, M.T.; Zaid Alkilani, A.; Larrañeta, E.; McAlister, E.; Courtenay, A.J.; Kearney, M.-C.; Singh, T.R.R.; McCarthy, H.O.; Kett, V.L. Hydrogel-forming microneedles prepared from “super swelling” polymers combined with lyophilised wafers for transdermal drug delivery. PLoS ONE 2014, 9, e111547. [Google Scholar] [CrossRef]
- Wang, M.; Hu, L.; Xu, C. Recent advances in the design of polymeric microneedles for transdermal drug delivery and biosensing. Lab Chip 2017, 17, 1373–1387. [Google Scholar] [CrossRef]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for biomedical applications: Their characteristics and the mechanisms behind them. Gels 2017, 3, 6. [Google Scholar] [CrossRef]
- Lin, C.-C.; Metters, A.T. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Garland, M.J.; Migalska, K.; Majithiya, R.; McCrudden, C.M.; Kole, P.L.; Mahmood, T.M.T.; McCarthy, H.O.; Woolfson, A.D. Hydrogel-forming microneedle arrays for enhanced transdermal drug delivery. Adv. Funct. Mater. 2012, 22, 4879–4890. [Google Scholar] [CrossRef]
- Blasi, P. Poly (lactic acid)/poly (lactic-co-glycolic acid)-based microparticles: An overview. J. Pharm. Investig. 2019, 49, 337–346. [Google Scholar] [CrossRef]
- Abdelkader, H.; Fathalla, Z.; Seyfoddin, A.; Farahani, M.; Thrimawithana, T.; Allahham, A.; Alani, A.W.G.; Al-Kinani, A.A.; Alany, R.G. Polymeric long-acting drug delivery systems (LADDS) for treatment of chronic diseases: Inserts, patches, wafers, and implants. Adv. Drug Deliv. Rev. 2021, 177, 113957. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K. Functional coatings and microencapsulation: A general perspective. In Functional Coatings: By Polymer Microencapsulation; Wiley-VCH: Weinheim, Germany, 2006; pp. 1–28. [Google Scholar] [CrossRef]
- Yeo, Y.; Baek, N.; Park, K. Microencapsulation methods for delivery of protein drugs. Biotechnol. Bioprocess Eng. 2001, 6, 213–230. [Google Scholar] [CrossRef]
- Tomaro-Duchesneau, C.; Saha, S.; Malhotra, M.; Kahouli, I.; Prakash, S. Microencapsulation for the therapeutic delivery of drugs, live mammalian and bacterial cells, and other biopharmaceutics: Current status and future directions. J. Pharm. 2013, 2013, 103527. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, N.V.N.; Prasanna, P.M.; Sakarkar, S.N.; Prabha, K.S.; Ramaiah, P.S.; Srawan, G. Microencapsulation techniques, factors influencing encapsulation efficiency. J. Microencapsul. 2010, 27, 187–197. [Google Scholar] [CrossRef]
- Bansode, S.; Banarjee, S.; Gaikwad, D.; Jadhav, S.; Thorat, R. Microencapsulation: A review. Int. J. Pharm. Sci. Rev. Res. 2010, 1, 38–43. [Google Scholar]
- Dubey, R. Microencapsulation technology and applications. Def. Sci. J. 2009, 59, 82. [Google Scholar]
- Sharifi, F.; Otte, A.; Yoon, G.; Park, K. Continuous in-line homogenization process for scale-up production of naltrexone-loaded PLGA microparticles. J. Control. Release 2020, 325, 347–358. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Vandermeulen, G.; Préat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair Regen. 2016, 24, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, F.; Chen, W.; van Nostrum, C.F.; Storm, G.; Kiessling, F.; Lammers, T.; Hennink, W.E.; Kok, R.J. Strategies for encapsulation of small hydrophilic and amphiphilic drugs in PLGA microspheres: State-of-the-art and challenges. Int. J. Pharm. 2016, 499, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kwon, H.J.; Ji, H.; Cho, S.H.; Cho, E.-H.; Han, H.D.; Shin, B.C. Marbofloxacin-encapsulated microparticles provide sustained drug release for treatment of veterinary diseases. Mater. Sci. Eng. C 2016, 60, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Shahjin, F.; Patel, M.; Machhi, J.; Cohen, J.D.; Nayan, M.U.; Yeapuri, P.; Zhang, C.; Waight, E.; Hasan, M.; Abdelmoaty, M.M.; et al. Multipolymer microsphere delivery of SARS-CoV-2 antigens. Acta Biomater. 2023, 158, 493–509. [Google Scholar] [CrossRef] [PubMed]
- van de Weert, M.; Hennink, W.E.; Jiskoot, W. Protein instability in poly (lactic-co-glycolic acid) microparticles. Pharm. Res. 2000, 17, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, L.I.; Gracia, I.; de Lucas, A.; Rodríguez, J.F. Novel model for the description of the controlled release of 5-fluorouracil from PLGA and PLA foamed scaffolds impregnated in supercritical CO2. Ind. Eng. Chem. Res. 2014, 53, 15374–15382. [Google Scholar] [CrossRef]
- Khuroo, T.; Dharani, S.; Mohamed, E.M.; Immadi, S.; Wu, Z.; Khan, M.A.; Lu, D.; Nehete, P.; Rahman, Z. Ultra-long acting prodrug of dolutegravir and delivery system—Physicochemical, pharmacokinetic and formulation characterizations. Int. J. Pharm. 2021, 607, 120889. [Google Scholar] [CrossRef]
- Collett, B.M. Scanning electron microscopy: A review and report of research in wood science. Wood Fiber Sci. 1970, 2, 113–133. [Google Scholar]
- Doty, A.C.; Weinstein, D.G.; Hirota, K.; Olsen, K.F.; Ackermann, R.; Wang, Y.; Choi, S.; Schwendeman, S.P. Mechanisms of in vivo release of triamcinolone acetonide from PLGA microspheres. J. Control. Release 2017, 256, 19–25. [Google Scholar] [CrossRef]
- Shah, R.B.; Schwendeman, S.P. A biomimetic approach to active self-microencapsulation of proteins in PLGA. J. Control. Release 2014, 196, 60–70. [Google Scholar] [CrossRef]
- Srikar, G.; Rani, A.P. Study on influence of polymer and surfactant on in vitro performance of biodegradable aqueous-core nanocapsules of tenofovirdisoproxil fumarate by response surface methodology. Braz. J. Pharm. Sci. 2019, 55, e18736. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Landin, M.; Lathuile, A.; Veldhuis, G.J.; Rahimian, S.; Hennink, W.E.; Kok, R.J.; van Nostrum, C.F. Computer modeling assisted design of monodisperse PLGA microspheres with controlled porosity affords zero order release of an encapsulated macromolecule for 3 months. Pharm. Res. 2014, 31, 2844–2856. [Google Scholar] [CrossRef]
- Samadi, N.; Abbadessa, A.; Di Stefano, A.; Van Nostrum, C.; Vermonden, T.; Rahimian, S.; Teunissen, E.; Van Steenbergen, M.; Amidi, M.; Hennink, W. The effect of lauryl capping group on protein release and degradation of poly (d, l-lactic-co-glycolic acid) particles. J. Control. Release 2013, 172, 436–443. [Google Scholar] [CrossRef]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. J. Control. Release 2019, 294, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Kang, G.; Zhao, Y.; Yuan, Z.; Fan, W.; Li, Q.; Destache, C.J. Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission. AIDS 2017, 31, 469. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Sunagawa, S.W.; Destache, C.J. A concept evaluation study of a new combination bictegravir plus tenofovir alafenamide nanoformulation with prolonged sustained-drug-release potency for HIV-1 preexposure prophylaxis. Antimicrob. Agents Chemother. 2021, 65, 940. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Fan, W.; Li, Q.; Destache, C.J. Long-acting parenteral combination antiretroviral loaded nano-drug delivery system to treat chronic HIV-1 infection: A humanized mouse model study. Antivir. Res. 2018, 156, 85–91. [Google Scholar] [CrossRef]
- Prathipati, P.K.; Mandal, S.; Pon, G.; Vivekanandan, R.; Destache, C.J. Pharmacokinetic and Tissue Distribution Profile of Long Acting Tenofovir Alafenamide and Elvitegravir Loaded Nanoparticles in Humanized Mice Model. Pharm. Res. 2017, 34, 2749–2755. [Google Scholar] [CrossRef]
- DeYoung, M.B.; MacConell, L.; Sarin, V.; Trautmann, M.; Herbert, P. Encapsulation of exenatide in poly-(D, L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol. Ther. 2011, 13, 1145–1154. [Google Scholar] [CrossRef]
- Jindal, A.B.; Bhide, A.R.; Salave, S.; Rana, D.; Benival, D. Long-acting parenteral drug delivery systems for the treatment of chronic diseases. Adv. Drug Deliv. Rev. 2023, 198, 114862. [Google Scholar] [CrossRef]
- Trezza, C.; Ford, S.L.; Spreen, W.; Pan, R.; Piscitelli, S. Formulation and pharmacology of long-acting cabotegravir. Curr. Opin. HIV AIDS 2015, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Overton, E.T.; Richmond, G.; Rizzardini, G.; Jaeger, H.; Orrell, C.; Nagimova, F.; Bredeek, F.; García Deltoro, M.; Swindells, S.; Andrade-Villanueva, J.F.; et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: A randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet 2021, 396, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Delany-Moretlwe, S.; Hughes, J.P.; Bock, P.; Ouma, S.G.; Hunidzarira, P.; Kalonji, D.; Kayange, N.; Makhema, J.; Mandima, P.; Mathew, C. Cabotegravir for the prevention of HIV-1 in women: Results from HPTN 084, a phase 3, randomised clinical trial. Lancet 2022, 399, 1779–1789. [Google Scholar] [CrossRef] [PubMed]
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I. Cabotegravir for HIV prevention in cisgender men and transgender women. N. Engl. J. Med. 2021, 385, 595–608. [Google Scholar] [CrossRef]
- Deodhar, S.; Sillman, B.; Bade, A.N.; Avedissian, S.N.; Podany, A.T.; McMillan, J.M.; Gautam, N.; Hanson, B.; Dyavar Shetty, B.L.; Szlachetka, A. Transformation of dolutegravir into an ultra-long-acting parenteral prodrug formulation. Nat. Commun. 2022, 13, 3226. [Google Scholar] [CrossRef]
- Sillman, B.; Bade, A.N.; Dash, P.K.; Bhargavan, B.; Kocher, T.; Mathews, S.; Su, H.; Kanmogne, G.D.; Poluektova, L.Y.; Gorantla, S. Creation of a long-acting nanoformulated dolutegravir. Nat. Commun. 2018, 9, 443. [Google Scholar] [CrossRef]
- Zhou, T.; Su, H.; Dash, P.; Lin, Z.; Shetty, B.L.D.; Kocher, T.; Szlachetka, A.; Lamberty, B.; Fox, H.S.; Poluektova, L. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials 2018, 151, 53–65. [Google Scholar] [CrossRef]
- Cobb, D.A.; Smith, N.; Deodhar, S.; Bade, A.N.; Gautam, N.; Shetty, B.L.D.; McMillan, J.; Alnouti, Y.; Cohen, S.M.; Gendelman, H.E. Transformation of tenofovir into stable ProTide nanocrystals with long-acting pharmacokinetic profiles. Nat. Commun. 2021, 12, 5458. [Google Scholar] [CrossRef]
- Guo, D.; Zhou, T.; Araínga, M.; Palandri, D.; Gautam, N.; Bronich, T.; Alnouti, Y.; McMillan, J.; Edagwa, B.; Gendelman, H.E. Creation of a long-acting nanoformulated 2′, 3′-dideoxy-3′-thiacytidine. J. Acquir. Immune Defic. Syndr. (1999) 2017, 74, e75. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Bade, A.N.; Lin, Z.; Soni, D.; Wojtkiewicz, M.; Dyavar Shetty, B.L.; Gautam, N.; McMillan, J.M.; Alnouti, Y.; Edagwa, B.J. Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation. Int. J. Nanomed. 2019, 14, 6231–6247. [Google Scholar] [CrossRef]
- Ogunnaike, M.; Das, S.; Raut, S.S.; Sultana, A.; Nayan, M.U.; Ganesan, M.; Edagwa, B.J.; Osna, N.A.; Poluektova, L.Y. Chronic Hepatitis B Infection: New Approaches towards Cure. Biomolecules 2023, 13, 1208. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Wang, W.; Ganesan, M.; Fonseca-Lanza, F.; Cobb, D.A.; Bybee, G.; Sun, Y.; Guo, L.; Hanson, B.; Cohen, S.M. An ultralong-acting tenofovir ProTide nanoformulation achieves monthslong HBV suppression. Sci. Adv. 2022, 8, eade9582. [Google Scholar] [CrossRef] [PubMed]
- Spreen, W.; Ford, S.L.; Chen, S.; Wilfret, D.; Margolis, D.; Gould, E.; Piscitelli, S. GSK1265744 Pharmacokinetics in Plasma and Tissue after Single-Dose Long-Acting Injectable Administration in Healthy Subjects. JAIDS J. Acquir. Immune Defic. Syndr. 2014, 67, 481–486. [Google Scholar] [CrossRef]
- Ford, S.; Chiu, J.; Lovern, M.; Spreen, W.; Kim, J. Population PK approach to predict cabotegravir (CAB, GSK1265744) long-acting injectable doses for phase 2b. In Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington DC, USA, 30 October–2 November 2004; pp. 5–9. [Google Scholar]
- Soriano, V.; Barreiro, P.; Benitez, L.; Peña, J.M.; de Mendoza, C. New antivirals for the treatment of chronic hepatitis B. Expert Opin. Investig. Drugs 2017, 26, 843–851. [Google Scholar] [CrossRef]
- Scarsi, K.K. Chasing the cabotegravir tail: Implications for prevention. Lancet HIV 2020, 7, e451–e453. [Google Scholar] [CrossRef]






| Drugs | (CAB + RPV LA) | CAB LA |
| Brand name | Cabenuva. | Apretude or Vocabria. |
| Formulation characteristics | CAB nanocrystals are produced in an aqueous solution of polysorbate 20 (Tween 20), PEG3350, and mannitol with particle size of 200 nm. RPV nanocrystals are produced in an aqueous solution of Poloxamer 338 with an average particle size of 200 nm. | Same as Cabenuva. |
| Indication | Treatment of HIV-1 infection. | Prevention of HIV-1 infection |
| Population | Virologically suppressed PLWH. | Adults and adolescents weighing at least 35 kg (77 lbs) who are at risk of sexually acquiring HIV. |
| Dosage regimen | Two initial injections of CAB 600 mg/3 mL and RPV 900 mg/3 mL given 1 month apart for two consecutive months and then given every two months (CAB 600 mg/3 mL and 900 mg/3 mL) thereafter. Injection is given in separate gluteal muscles. | Two initial injections (600 mg; 3 mL) given one month apart for two consecutive months, followed by maintenance doses (600 mg; 3 mL) given every 2 months thereafter. An OLI of CAB tablets may, or may not, be given for one month prior to starting CAB to assess tolerability. |
| Approval | First approved by Health Canada in March 2020, followed by the EMA in October 2020 and the US FDA in January 2021. | First approved by the US FDA in December 2021 and the EMA in October 2022. |
| Efficacy in the clinical trials | Several phase IIb and phase III clinical trials (LATTE-2, FLAIR, ATLAS, ATLAS-2M) proved non-inferiority for maintaining viral suppression compared to standard daily oral therapy [3,6,8,266]. | Proved superior to approved oral PrEP agent TDF/FTC in 2 phase III clinical trials (HPTN-083 and 084) [267,268]. |
| Side effects | Mild ISRs were the most reported AEs during clinical trials, but none of them caused treatment withdrawal. | Same as Cabenuva. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nayan, M.U.; Panja, S.; Sultana, A.; Zaman, L.A.; Vora, L.K.; Sillman, B.; Gendelman, H.E.; Edagwa, B. Polymer Delivery Systems for Long-Acting Antiretroviral Drugs. Pharmaceutics 2024, 16, 183. https://doi.org/10.3390/pharmaceutics16020183
Nayan MU, Panja S, Sultana A, Zaman LA, Vora LK, Sillman B, Gendelman HE, Edagwa B. Polymer Delivery Systems for Long-Acting Antiretroviral Drugs. Pharmaceutics. 2024; 16(2):183. https://doi.org/10.3390/pharmaceutics16020183
Chicago/Turabian StyleNayan, Mohammad Ullah, Sudipta Panja, Ashrafi Sultana, Lubaba A. Zaman, Lalitkumar K. Vora, Brady Sillman, Howard E. Gendelman, and Benson Edagwa. 2024. "Polymer Delivery Systems for Long-Acting Antiretroviral Drugs" Pharmaceutics 16, no. 2: 183. https://doi.org/10.3390/pharmaceutics16020183