
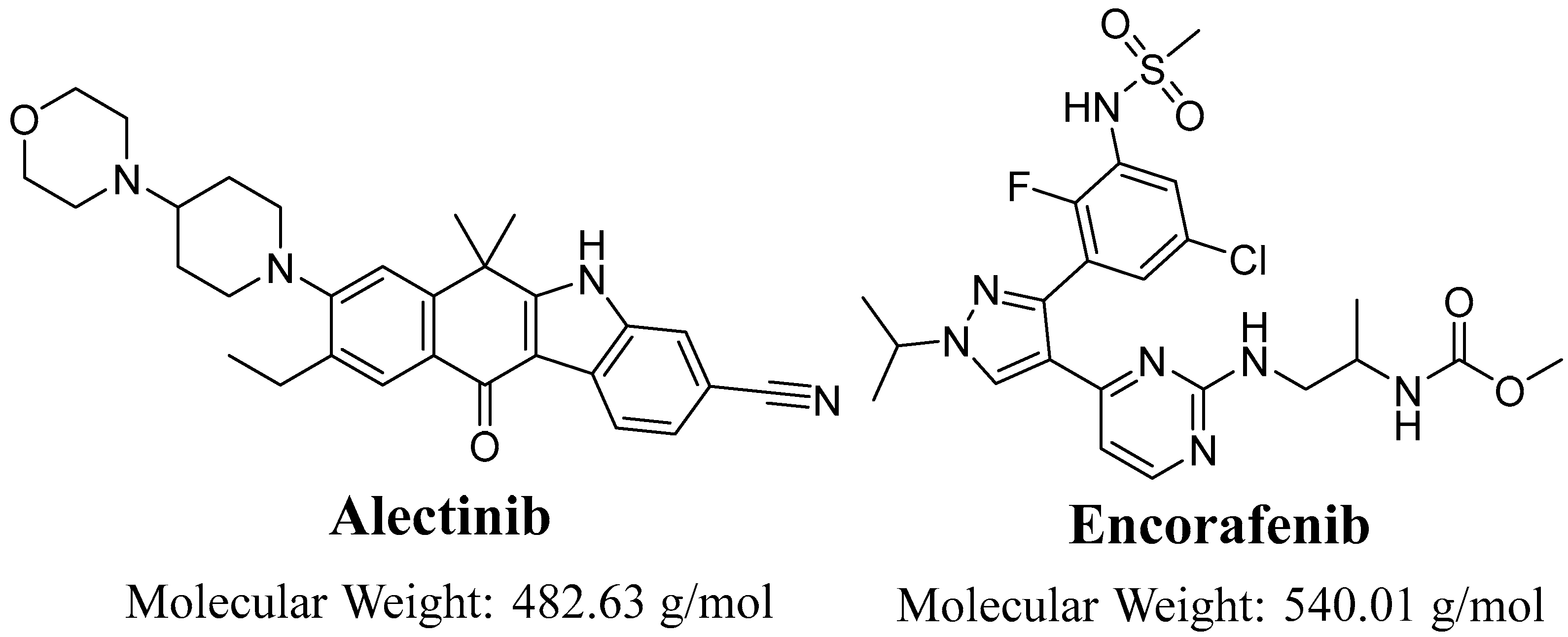
Evaluation of Alectinib Metabolic Stability in HLMs Using Fast LC-MS/MS Method: In Silico ADME Profile, P450 Metabolic Lability, and Toxic Alerts Screening

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. In Silico Assessment of ALC Metabolic Stability
2.4. In Silico Screening of the Toxicity of ALC Using DEREK Software
2.5. In Silico ADME Analysis
2.6. LC-MS/MS Instrumental Characteristics
2.7. ALC and EFB Working Dilutions
2.8. Construction of ALC Calibration Levels
2.9. Extracting the Target Analytes (ALC and EFB) from the Metabolic Matrix
2.10. Validation Features of the Proposed Analytical Method
2.10.1. Specificity
2.10.2. Linearity and Sensitivity
2.10.3. Accuracy and Precision
2.10.4. Matrix Effect and Extraction Recovery
2.10.5. Stability of ALC in the Stock and Working Preparations
2.11. In Vitro Determination of ALC Metabolic Stability
3. Results
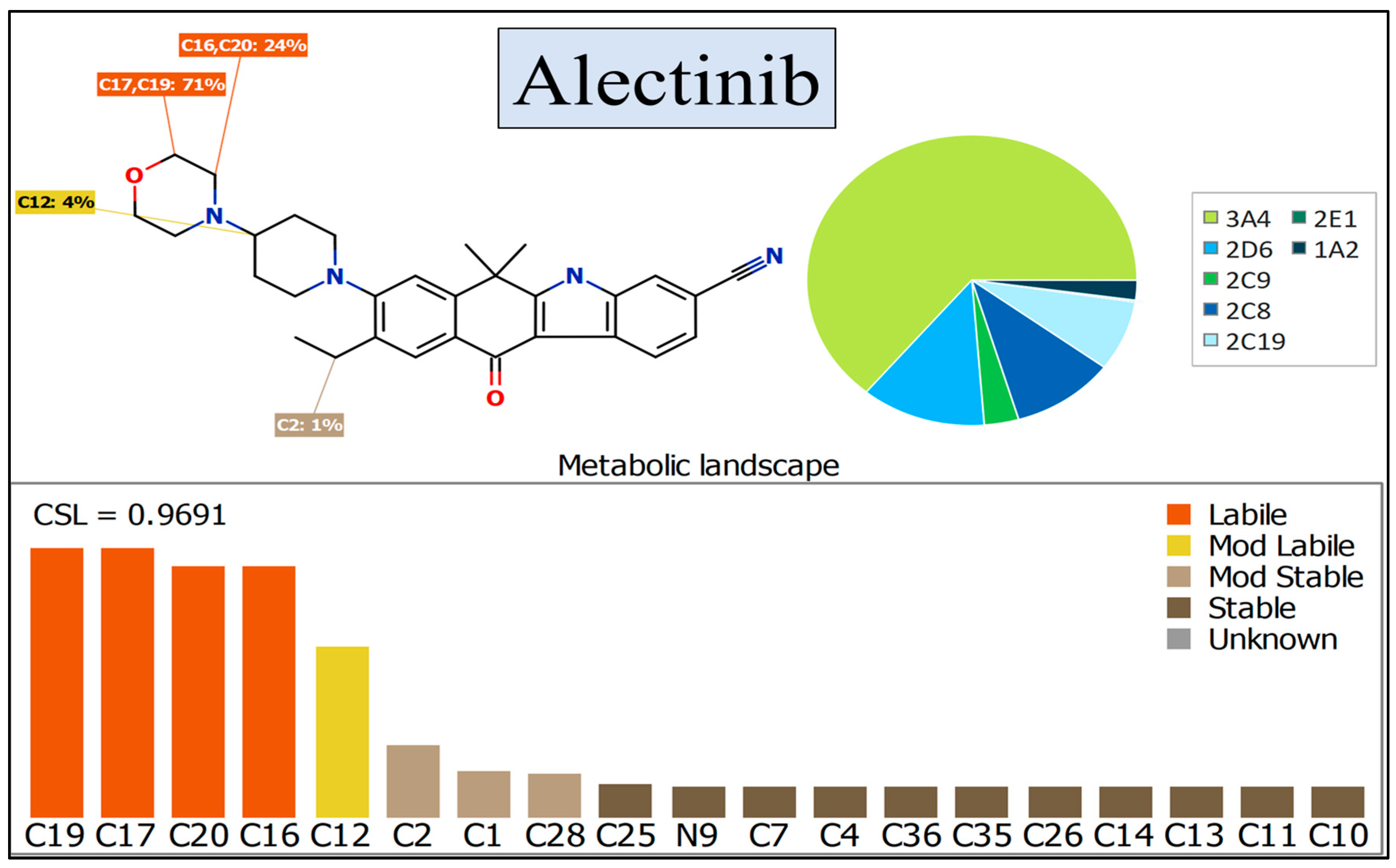
3.1. In Silico Assessment of ALC Metabolic Stability
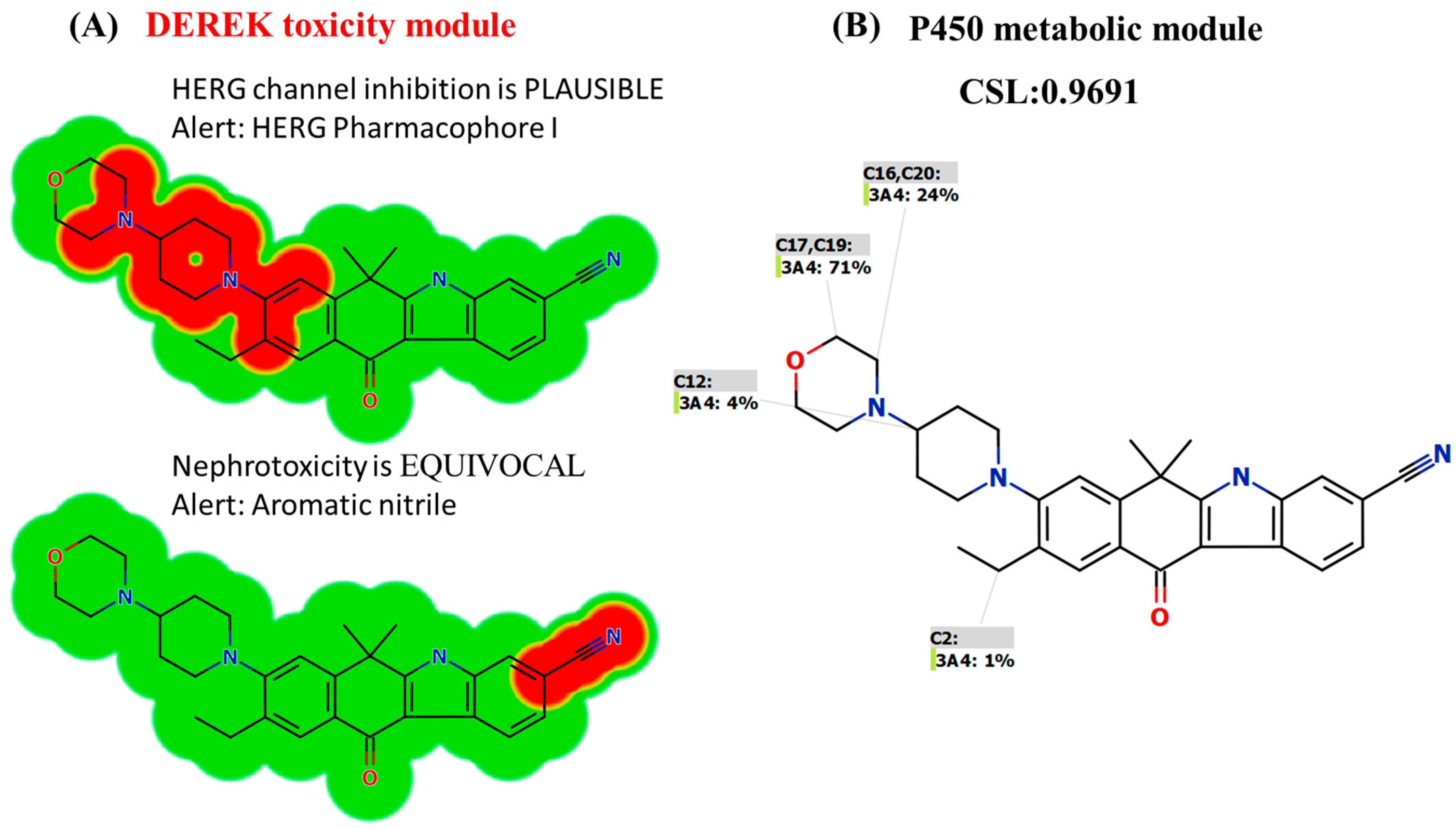
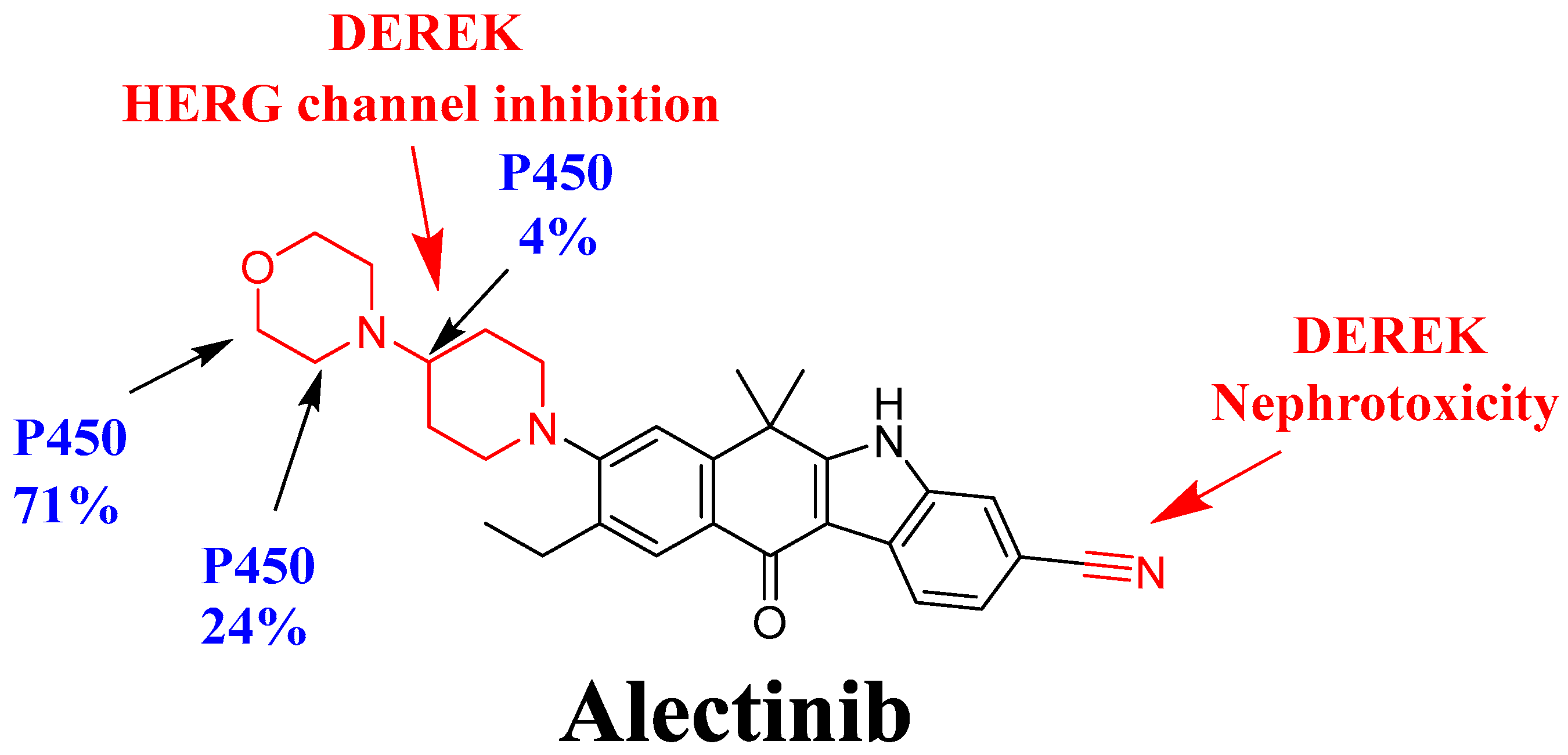
3.2. In Silico DEREK Module Prediction of ALC Toxic Alerts
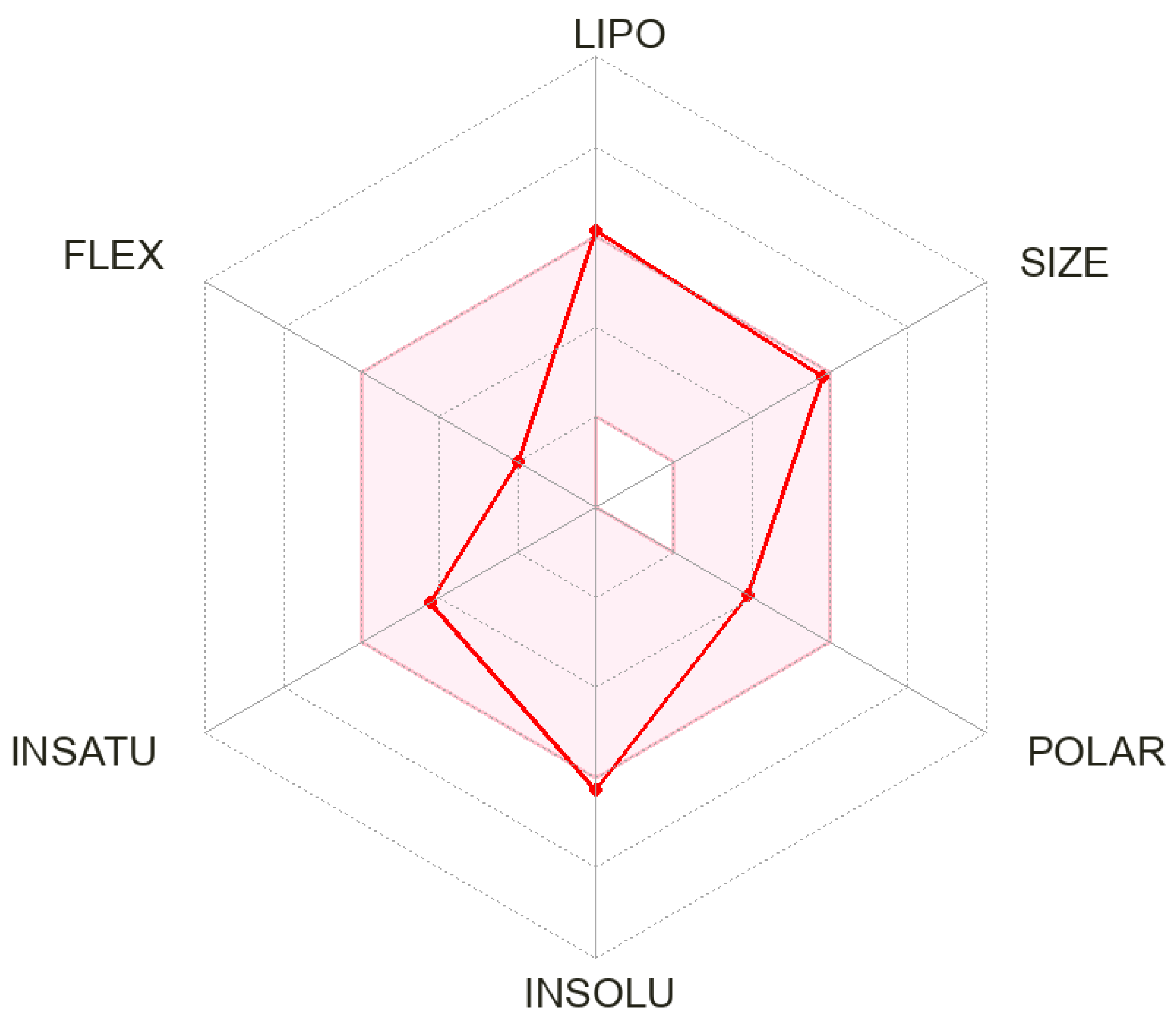
3.3. In Silico ADME Profile
3.4. LC-MS/MS Method
3.5. Validation Parameters of the Current LC-MS/MS Method
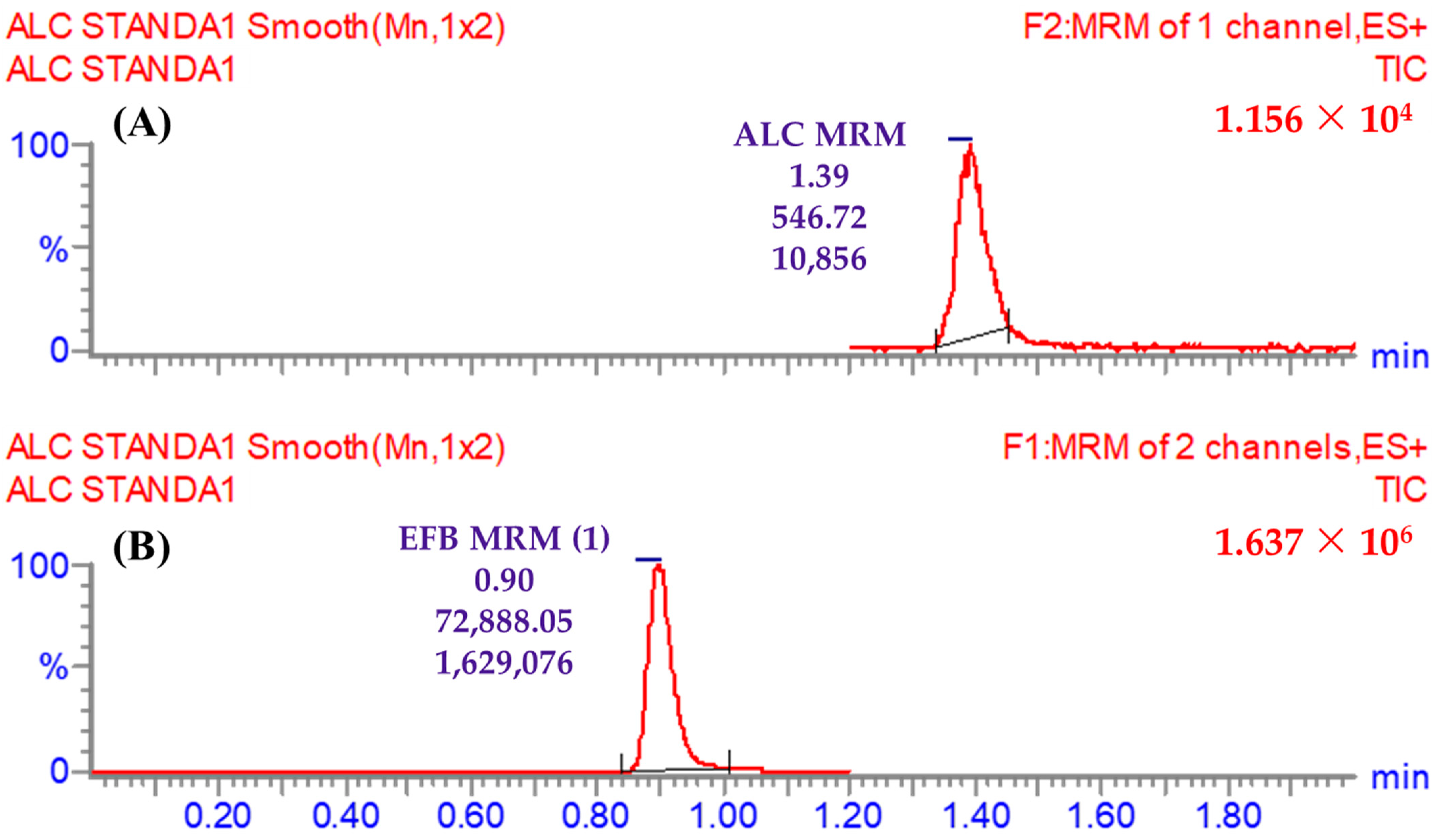
3.5.1. Specificity
3.5.2. Linearity and Sensitivity
3.5.3. Precision and Accuracy
3.5.4. Extraction and Recovery of ALC in the Proposed LC-MS/MS Method
3.5.5. ALC was Stable in the Stock and Working Preparations
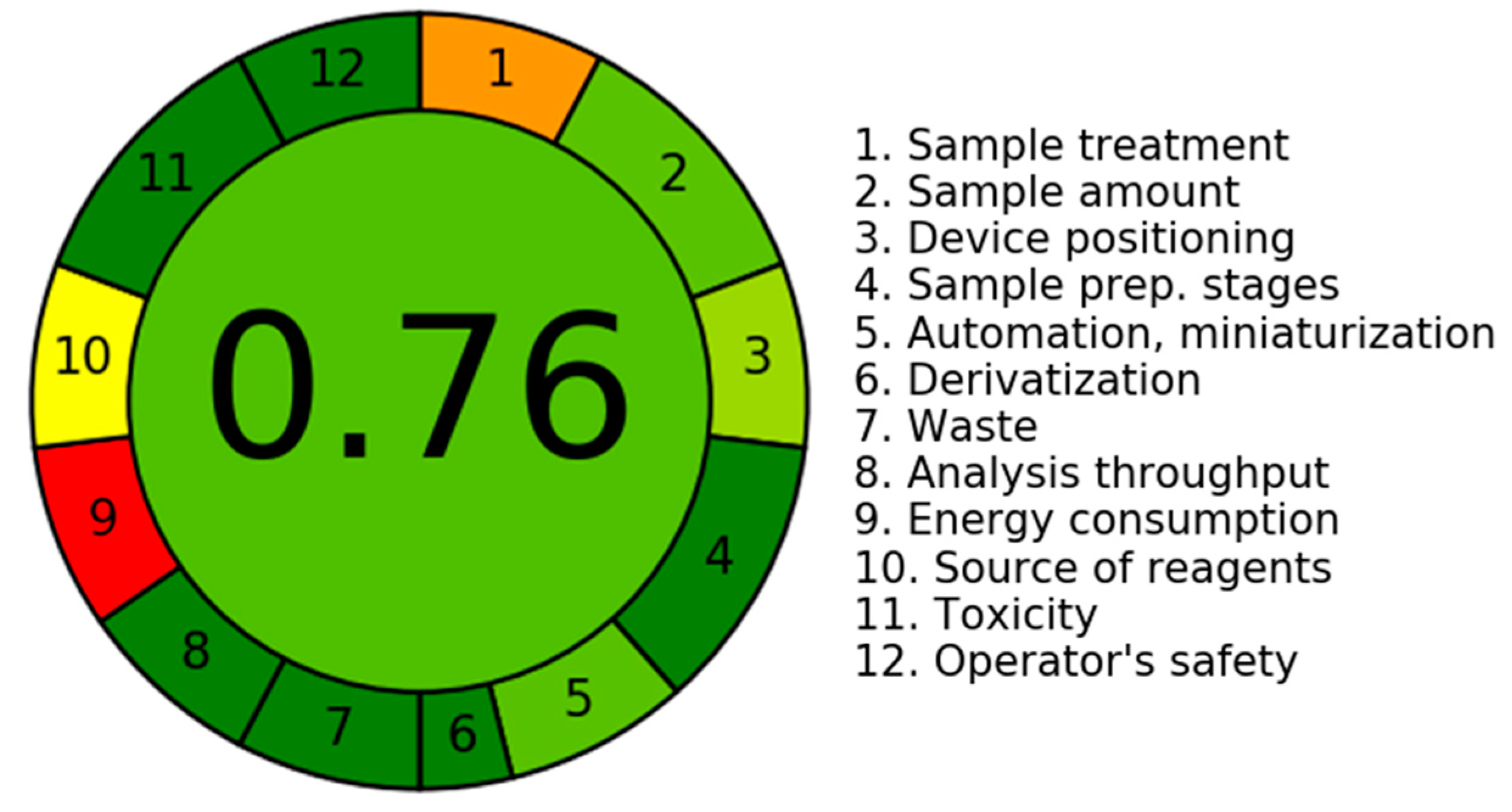
3.5.6. An Assessment of the Environmental Sustainability of the Current LC-MS/MS Technology Utilizing the AGREE Program
3.6. In Vitro Incubations of ALC with Metabolic HLM Matrix
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef] [PubMed]
- Barinaga, M. From Bench Top to Bedside. Science 1997, 278, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Akerley, W.; Bepler, G.; Blum, M.G.; Chang, A.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Ganti, A.K.P. Non–small cell lung cancer. J. Natl. Compr. Cancer Netw. 2010, 8, 740–801. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Cascone, T.; Gerber, D.E.; Heymach, J.V.; Minna, J.D. Targeted therapies for lung cancer: Clinical experience and novel agents. Cancer J. 2011, 17, 512. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kono, S.A.; Lu, X.; Okuyama, S.; Barón, A.E.; Oton, A.B.; Davies, A.M.; Varella-Garcia, M.; Franklin, W.; Doebele, R.C. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J. Thorac. Oncol. 2011, 6, 774–780. [Google Scholar] [CrossRef]
- Cooper, A.J.; Sequist, L.V.; Lin, J.J. Third-generation EGFR and ALK inhibitors: Mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 2022, 19, 499–514. [Google Scholar] [CrossRef]
- Larkins, E.; Blumenthal, G.M.; Chen, H.; He, K.; Agarwal, R.; Gieser, G.; Stephens, O.; Zahalka, E.; Ringgold, K.; Helms, W.; et al. FDA approval: Alectinib for the treatment of metastatic, ALK-positive non–small cell lung cancer following crizotinib. Clin. Cancer Res. 2016, 22, 5171–5176. [Google Scholar] [CrossRef]
- Herden, M.; Waller, C.F. Alectinib. In Small Molecules in Oncology; Springer: Cham, Switzerland, 2018; pp. 247–256. [Google Scholar]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Santarpia, M.; Altavilla, G.; Rosell, R. Alectinib: A selective, next-generation ALK inhibitor for treatment of ALK-rearranged non-small-cell lung cancer. Expert. Rev. Respir. Med. 2015, 9, 255–268. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, X.; Dong, Y.; Yang, J.; Zhang, J.; He, S.; Yang, F.; Wang, Z.; Dong, Y. A Green HPLC Method for Determination of Nine Sulfonamides in Milk and Beef, and Its Greenness Assessment with Analytical Eco-Scale and Greenness Profile. J. AOAC Int. 2020, 103, 1181–1189. [Google Scholar] [CrossRef]
- Tyzack, J.D.; Kirchmair, J. Computational methods and tools to predict cytochrome P450 metabolism for drug discovery. Chem. Biol. Drug Des. 2019, 93, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Marothu Vamsi, K.; Kantamaneni, P.; Gorrepati, M. In vitro Metabolic Stability of Drugs and Applications of LC-MS in Metabolite Profiling. In Drug Metabolism; Katherine, D., Ed.; IntechOpen: Rijeka, Croatia, 2021; p. Ch. 5. [Google Scholar]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 1994, 47, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [PubMed]
- Attwa, M.W.; Abdelhameed, A.S.; Alsaif, N.A.; Kadi, A.A.; AlRabiah, H. A validated LC-MS/MS analytical method for the quantification of pemigatinib: Metabolic stability evaluation in human liver microsomes. RSC Adv. 2022, 12, 20387–20394. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef]
- Darwish, I.A.; Alzoman, N.Z.; Almomen, A.; Almehizia, A.A.; Attwa, M.W.; Darwish, H.W.; Sayed, A.Y. Development and validation of an UPLC-ESI-MS/MS method for quantification of duvelisib in plasma: Application to pharmacokinetic study in rats. RSC Adv. 2023, 13, 7929–7938. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules 2020, 25, 5004. [Google Scholar] [CrossRef]
- Alrabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation EGFR-TKI naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019, 9, 4862–4869. [Google Scholar] [CrossRef]
- Attwa, M.W.; Mostafa, G.A.E.; AlRabiah, H.; Kadi, A.A. An LC–MS/MS Analytical Method for Quantifying Tepotinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation. Separations 2023, 10, 330. [Google Scholar]
- Marchant, C.A.; Briggs, K.A.; Long, A. In Silico Tools for Sharing Data and Knowledge on Toxicity and Metabolism: Derek for Windows, Meteor, and Vitic. Toxicol. Mech. Methods 2008, 18, 177–187. [Google Scholar] [CrossRef]
- AlRabiah, H.; Kadi, A.A.; Attwa, M.W.; Mostafa, G.A.E. Development and validation of an HPLC-MS/MS method for the determination of filgotinib, a selective Janus kinase 1 inhibitor: Application to a metabolic stability study. J. Chromatogr. B 2020, 1154, 122195. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, S.P.; de Bruijn, P.; Koolen, S.L.W.; Dingemans, A.-M.C.; Mathijssen, R.H.J.; Veerman, G.D.M. Quantitation of osimertinib, alectinib and lorlatinib in human cerebrospinal fluid by UPLC-MS/MS. J. Pharm. Biomed. Anal. 2023, 225, 115233. [Google Scholar] [CrossRef]
- Attwa, M.W.; Darwish, H.W.; Al-Shakliah, N.S.; Kadi, A.A. A Validated LC–MS/MS Assay for the Simultaneous Quantification of the FDA-Approved Anticancer Mixture (Encorafenib and Binimetinib): Metabolic Stability Estimation. Molecules 2021, 26, 2717. [Google Scholar] [CrossRef]
- Busby, W.F., Jr.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999, 27, 246–249. [Google Scholar] [PubMed]
- Störmer, E.; Roots, I.; Brockmöller, J. Benzydamine N-oxidation as an index reaction reflecting FMO activity in human liver microsomes and impact of FMO3 polymorphisms on enzyme activity. Br. J. Clin. Pharmacol. 2000, 50, 553–561. [Google Scholar] [CrossRef]
- Fouin-Fortunet, H.; Tinel, M.; Descatoire, V.; Letteron, P.; Larrey, D.; Geneve, J.; Pessayre, D. Inactivation of cytochrome P-450 by the drug methoxsalen. J. Pharmacol. Exp. Ther. 1986, 236, 237–247. [Google Scholar]
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation: Guidance for Industry; Food and Drug Administration: Washington, DC, USA, 2018. [Google Scholar]
- Smith, G. European Medicines Agency guideline on bioanalytical method validation: What more is there to say? Bioanalysis 2012, 4, 865–868. [Google Scholar] [CrossRef]
- McNaney, C.A.; Drexler, D.M.; Hnatyshyn, S.Y.; Zvyaga, T.A.; Knipe, J.O.; Belcastro, J.V.; Sanders, M. An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay. Drug Dev. Technol. 2008, 6, 121–129. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Kirchmair, J. Software for metabolism prediction. In Drug Metabolism Prediction; Wiley-VCH: Weinheim, Germany, 2014; pp. 27–52. [Google Scholar]
- Hunt, P.A.; Segall, M.D.; Tyzack, J.D. WhichP450: A multi-class categorical model to predict the major metabolising CYP450 isoform for a compound. J. Comput. Aided Mol. Des. 2018, 32, 537–546. [Google Scholar] [CrossRef] [PubMed]
- G Shin, Y.; Le, H.; Khojasteh, C.; ECA Hop, C. Comparison of metabolic soft spot predictions of CYP3A4, CYP2C9 and CYP2D6 substrates using MetaSite and StarDrop. Comb. Chem. High. Throughput Screen. 2011, 14, 811–823. [Google Scholar] [CrossRef]
- Meesters, R.; Voswinkel, S. Bioanalytical method development and validation: From the USFDA 2001 to the USFDA 2018 guidance for industry. J. Appl. Bioanal. 2018, 4, 67–73. [Google Scholar] [CrossRef]
- Leahy, D.E. Integrating invitro ADMET data through generic physiologically based pharmacokinetic models. Expert. Opin. Drug Metab. Toxicol. 2006, 2, 619–628. [Google Scholar] [CrossRef]
- Kadi, A.A.; Amer, S.M.; Darwish, H.W.; Attwa, M.W. LC-MS/MS reveals the formation of aldehydes and iminium reactive intermediates in foretinib metabolism: Phase I metabolic profiling. RSC Adv. 2017, 7, 36279–36287. [Google Scholar] [CrossRef]
- Mašič, L.P. Role of cyclic tertiary amine bioactivation to reactive iminium species: Structure toxicity relationship. Curr. Drug Metab. 2011, 12, 35–50. [Google Scholar] [CrossRef]
- Kodama, T.; Hasegawa, M.; Takanashi, K.; Sakurai, Y.; Kondoh, O.; Sakamoto, H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother. Pharmacol. 2014, 74, 1023–1028. [Google Scholar] [CrossRef]
- Stergiopoulos, C.; Tsopelas, F.; Valko, K. Prediction of hERG inhibition of drug discovery compounds using biomimetic HPLC measurements. Admet Dmpk 2021, 9, 191–207. [Google Scholar] [CrossRef]
- Prado-Mel, E.; Ciudad-Gutiérrez, P.; Sánchez-Martín, A.; Martín, L.A.-K. Alectinib–induced acute renal failure. J. Oncol. Pharm. Pract. 2023. Epub ahead of print. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LC (H10UPH) | MS/MS (QBB1203) | ||
|---|---|---|---|
| Binary mobile phase | 45% line A: ammonium acetate in H2O (pH: 6.0) | ESI | Positive ESI source |
| 55% line B: ACN | Cone gas: 100 L/H flow rate | ||
| 0.5 mL/min flow rate | The voltage of RF lens: 0.1 (V) | ||
| Injection volume: 5.0 μL | The voltage of extractor: 3.0 (V) | ||
| Eclipse plus-C8 column | 50.0 mm long | Capillary voltage: 4 KV | |
| 2.1 mm i.d. | Nitrogen (drying gas; 350 °C) at 100 L/h | ||
| 3.5 μm | Mode of detection | MRM | |
| 23.0 ± 1.0 °C | Collision cell | Argon gas (0.14 mL/min) | |
| Time | Retention Time | MRM Mass Transitions | ||
|---|---|---|---|---|
| Time segments | 1.2 to 2.0 min | ALC (1.39 min) | One mass transition (m/z) | 483 → 396 (CE a: 18 and CV b: 50) |
| 0.0 to 1.2 min | EFB (IS; 0.9 min) | First mass transition (m/z) | 540 → 359 (CE: 32 and CV: 56) | |
| Second mass transition (m/z) | 540 → 116 (CE: 36 and CV: 56) | |||
| Physicochemical Properties | Water Solubility | ||
|---|---|---|---|
| Formula | C30H34N4O2 | LogS (ESOL) | −6.25 |
| Molecular weight | 482.62 g/mol | Solubility | 2.71 × 10−4 mg/mL; 5.62 × 10−7 mol/L |
| Num. arom. heavy atoms | 15 | Class | Poorly soluble |
| Num. heavy atoms | 36 | LogS (Ali) | −6.52 |
| Num. rotatable bonds | 3 | Solubility | 1.46 × 10−4 mg/mL; 3.03 × 10−7 mol/L |
| Fraction Csp3 | 0.47 | Class | Poorly soluble |
| Num. H-bond donors | 1 | Solubility | 1.91 × 10−6 mg/mL; 3.96 × 10−9 mol/L |
| Num. H-bond acceptors | 4 | Class | Poorly soluble |
| TPSA | 72.36 Å2 | Medicinal Chemistry | |
| Molar refractivity | 149.63 | PAINS | 0 alert |
| Lipophilicity | Leadlikeness | No; 2 violations: MW > 350, XLOGP3 > 3.5 | |
| LogPo/w (XLOGP3) | 5.25 | Brenk | 0 alert |
| LogPo/w (MLOGP) | 2.39 | Synthetic accessibility | 3.92 |
| LogPo/w (iLOGP) | 4.08 | Pharmacokinetics | |
| LogPo/w (SILICOS-IT) | 5.93 | GI absorption | High |
| LogPo/w (WLOGP) | 4.01 | P-gp substrate | Yes |
| Consensus LogPo/w | 4.33 | BBB permeant | Yes |
| Druglikeness | CYP2C9 inhibitor | Yes | |
| Ghose | No; 2 violations: MW > 480, MR > 130 | CYP1A2 inhibitor | No |
| Muegge | No; 1 violation: XLOGP3 > 5 | CYP2C19 inhibitor | Yes |
| Egan | Yes | CYP2D6 inhibitor | No |
| Veber | Yes | CYP3A4 inhibitor | No |
| Lipinski | Yes; 0 violation | LogKp (skin permeation) | −5.52 cm/s |
| Bioavailability score | 0.55 | ||
| ALC (ng/mL) | Mean | SD | RSD (%) | Accuracy (%) | Recovery |
|---|---|---|---|---|---|
| 1.0 | 1.02 | 0.04 | 3.45 | 1.67 | 101.67 |
| 15.0 | 14.76 | 0.09 | 0.58 | −1.60 | 98.40 |
| 50.0 | 52.57 | 0.87 | 1.66 | 5.13 | 105.13 |
| 200.0 | 194.46 | 1.48 | 0.76 | −2.77 | 97.23 |
| 500.0 | 491.42 | 5.14 | 1.05 | −1.72 | 98.28 |
| 1500.0 | 1511.66 | 16.56 | 1.10 | 0.78 | 100.78 |
| 3000.0 | 3011.51 | 14.12 | 0.47 | 0.38 | 100.38 |
| % Recovery | 100.27 ± 2.65 |
| ALC (ng/mL) | Intra-Day (Twelve Groups on One Day) | Inter-Day (Six Groups in Three Days) | ||||||
|---|---|---|---|---|---|---|---|---|
| QCs | 1 | 3 | 900 | 2400 | 1 | 3 | 900 | 2400 |
| Mean | 1.02 | 2.92 | 900.33 | 2388.72 | 2.89 | 892.77 | 2389.93 | 1.04 |
| SD | 0.04 | 0.03 | 11.01 | 15.82 | 0.05 | 5.19 | 3.70 | 0.01 |
| Precision (%RSD) | 3.45 | 1.05 | 1.22 | 0.66 | 1.64 | 0.58 | 0.15 | 1.11 |
| % Accuracy | 1.67 | −2.56 | 0.04 | −0.47 | −3.78 | −0.80 | −0.42 | 4.33 |
| Recovery (%) | 101.67 | 97.44 | 100.04 | 99.53 | 96.22 | 99.20 | 99.58 | 104.33 |
| Stability Features | 3.0 | 2400.0 | 3.0 | 2400.0 | 3.0 | 2400.0 | 3.0 | 2400.0 |
|---|---|---|---|---|---|---|---|---|
| Mean | SD | RSD (%) | Accuracy (%) | |||||
| Freeze–Thaw Stability (three cycles at −80 °C) | 3.07 | 2421.62 | 0.04 | 5.17 | 1.42 | 0.21 | 2.33 | 0.90 |
| Autosampler Stability (24 h at 15 °C) | 3.05 | 2411.13 | 0.12 | 15.06 | 3.91 | 0.62 | 1.78 | 0.46 |
| Long-Term Stability (−80 °C for 28 d) | 3.01 | 2418.62 | 0.03 | 10.85 | 1.01 | 0.45 | 0.44 | 0.78 |
| Short-Term Stability (4 h at room temperature) | 2.93 | 2403.33 | 0.04 | 5.66 | 1.23 | 0.24 | −2.33 | 0.14 |
| Criteria | Score | Weight |
|---|---|---|
| 1. It is recommended to utilize direct analytical procedures in order to minimize the necessity for sample treatment. | 0.3 | 2 |
| 2. The aims of this investigation are to attain a limited sample size and reduce the quantity of samples. | 0.75 | 3 |
| 3. Ideally, it is recommended to perform measurements inside their original contextual environment. | 0.66 | |
| 4. The incorporation of analytical measures with operational tactics has been observed to decrease in reagent depletion and produce energy preservation. | 1.0 | 2 |
| 5. It is advisable to consider the adoption of automated and streamlined processes. | 0.75 | 3 |
| 6. It is recommended to abstain from utilizing derivatization processes. | 1.0 | 2 |
| 7. The minimization of the production of a significant volume of analytical surplus and the adoption of efficient solutions for its proper disposal are of paramount significance. | 1.0 | 1 |
| 8. The inclination is towards employing multi-analyte or multi-parameter methodologies rather than relying solely on single-analyte approaches. | 1.0 | 2 |
| 9. The prioritization of endeavors aimed at minimizing energy use is of paramount significance. | 0.0 | 2 |
| 10. It is recommended to give priority to the utilization of reagents obtained from environmentally friendly sources. | 0.5 | 2 |
| 11. The prioritizing of the removal or replacement of hazardous substances is of utmost importance. | 1.0 | 3 |
| 12. There exists a significant imperative to augment the safety practices for operators. | 1.0 | 3 |
| Time (Min) | Mean a | X b | LN X | Linearity Characteristics |
|---|---|---|---|---|
| 0.00 | 473.85 | 100.00 | 4.61 | Regression line equation: y = −0.0311x + 4.639 |
| 2.50 | 453.85 | 95.78 | 4.56 | |
| 5.00 | 423.86 | 89.45 | 4.49 | R2 = 0.9951 |
| 7.50 | 392.11 | 82.75 | 4.42 | |
| 15.00 | 311.51 | 65.74 | 4.19 | Slope: −0.0311 |
| 20.00 | 270.81 | 57.15 | 4.05 | |
| 30.00 | 186.93 | 39.45 | 3.68 | t1/2: 22.28 min and |
| 40.00 | 136.23 | 28.75 | 3.36 | Clint: 36.37 mL/min/kg |
| 50.00 | 117.28 | 24.75 | 3.21 | |
| 60.00 | 107.94 | 22.78 | 3.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attwa, M.W.; AlRabiah, H.; Mostafa, G.A.E.; Kadi, A.A. Evaluation of Alectinib Metabolic Stability in HLMs Using Fast LC-MS/MS Method: In Silico ADME Profile, P450 Metabolic Lability, and Toxic Alerts Screening. Pharmaceutics 2023, 15, 2449. https://doi.org/10.3390/pharmaceutics15102449
Attwa MW, AlRabiah H, Mostafa GAE, Kadi AA. Evaluation of Alectinib Metabolic Stability in HLMs Using Fast LC-MS/MS Method: In Silico ADME Profile, P450 Metabolic Lability, and Toxic Alerts Screening. Pharmaceutics. 2023; 15(10):2449. https://doi.org/10.3390/pharmaceutics15102449
Chicago/Turabian StyleAttwa, Mohamed W., Haitham AlRabiah, Gamal A. E. Mostafa, and Adnan A. Kadi. 2023. "Evaluation of Alectinib Metabolic Stability in HLMs Using Fast LC-MS/MS Method: In Silico ADME Profile, P450 Metabolic Lability, and Toxic Alerts Screening" Pharmaceutics 15, no. 10: 2449. https://doi.org/10.3390/pharmaceutics15102449