
Atorvastatin Enhances the Efficacy of Immune Checkpoint Therapy and Suppresses the Cellular and Extracellular Vesicle PD-L1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Transmission Electron Microscopic Imaging
2.3. Cell Cytotoxicity Assay
2.4. Isolation of EVs
2.5. NTA
2.6. Western Blot Analysis
2.7. Flow Cytometry Analysis
2.8. CD8+ T Cell-Mediated Cancer Cell Killing Assay
2.9. Animal Experiments
2.10. Immune Phenotyping and Flow Cytometry Analysis
2.11. Statistical Analysis
3. Results
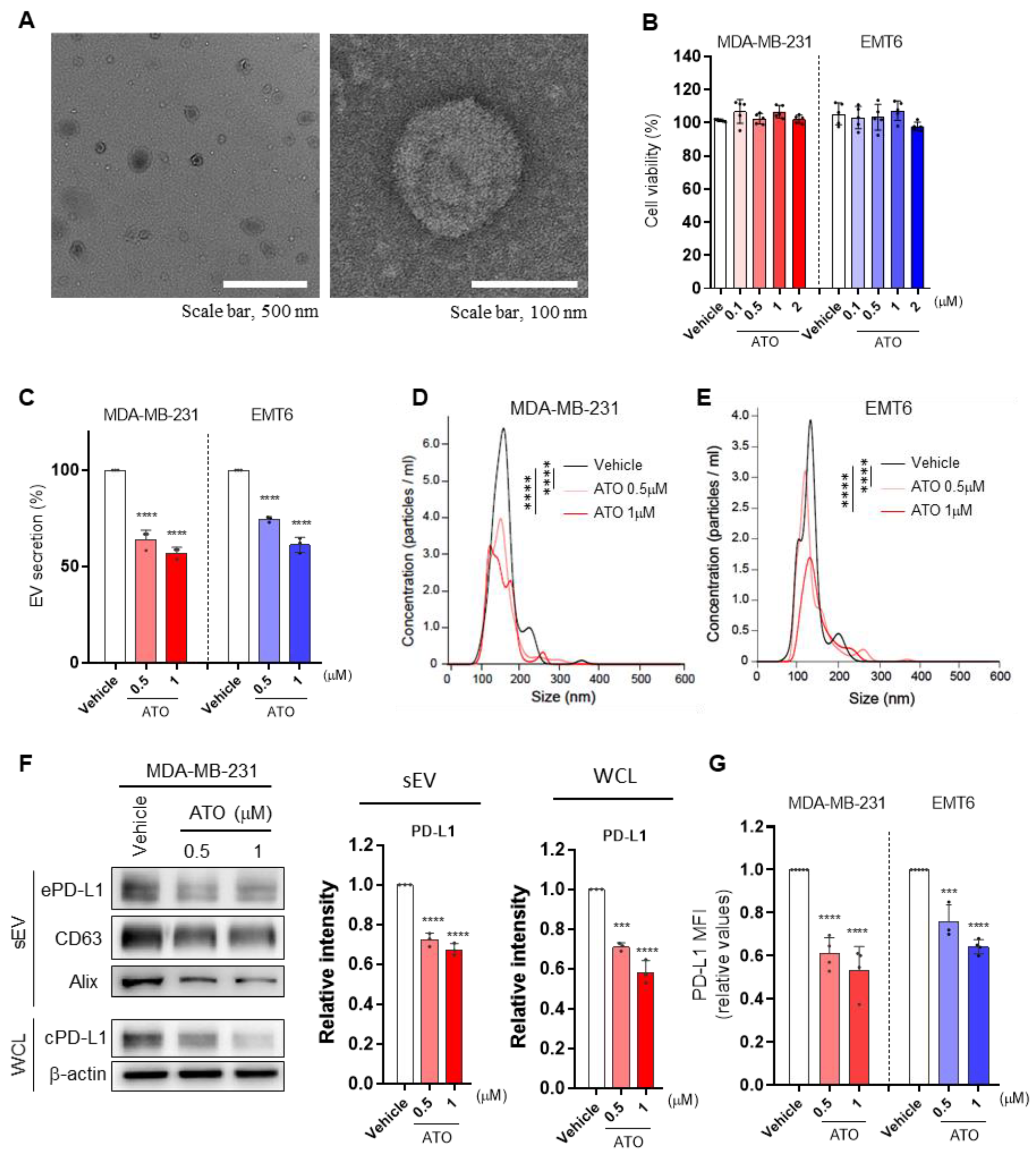
3.1. ATO Suppresses EV PD-L1 by Inhibiting EV Secretion and PD-L1 Expression
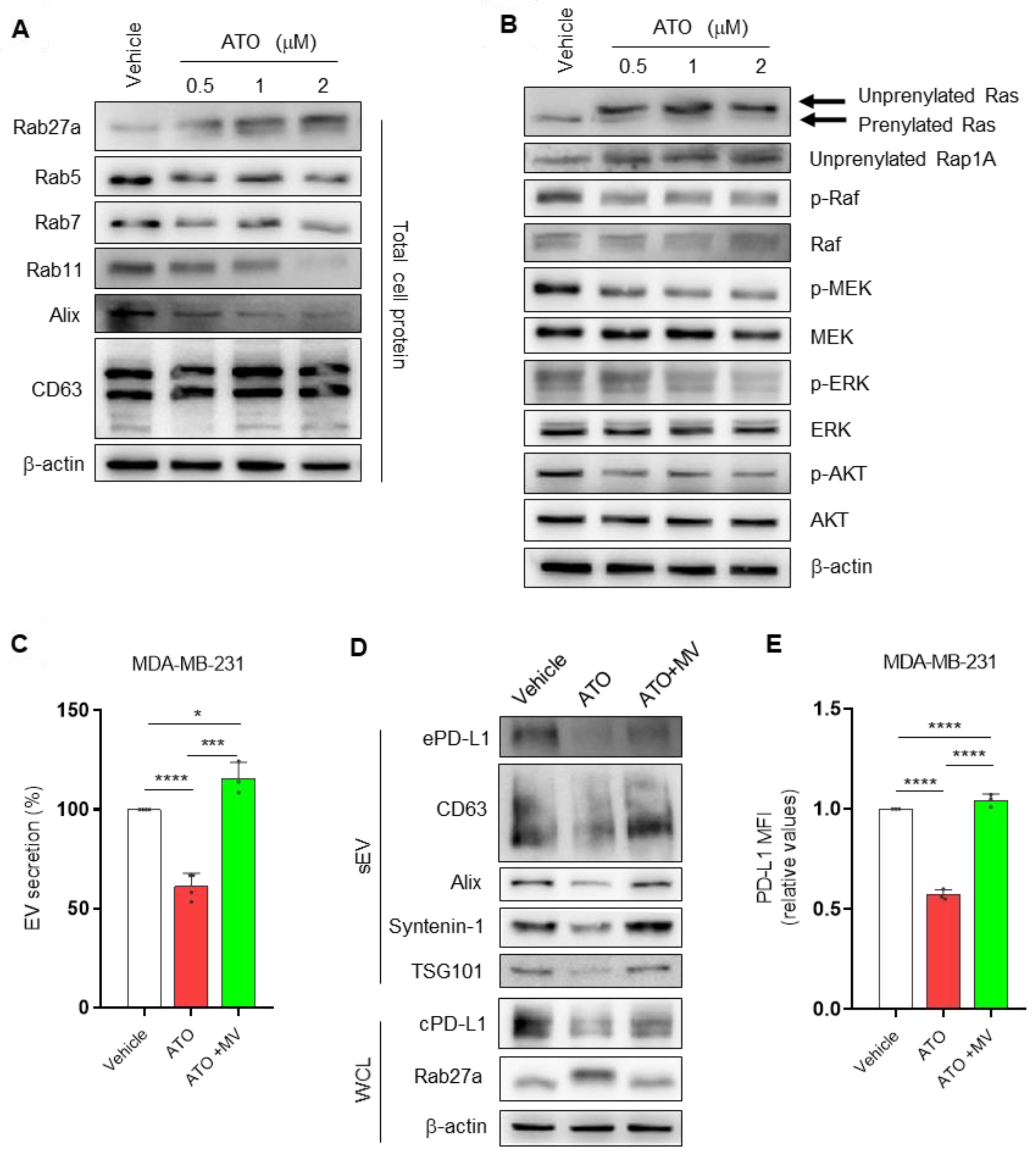
3.2. ATO Suppresses EV PD-L1 by Regulating EV Biogenesis and the Mevalonate Pathway
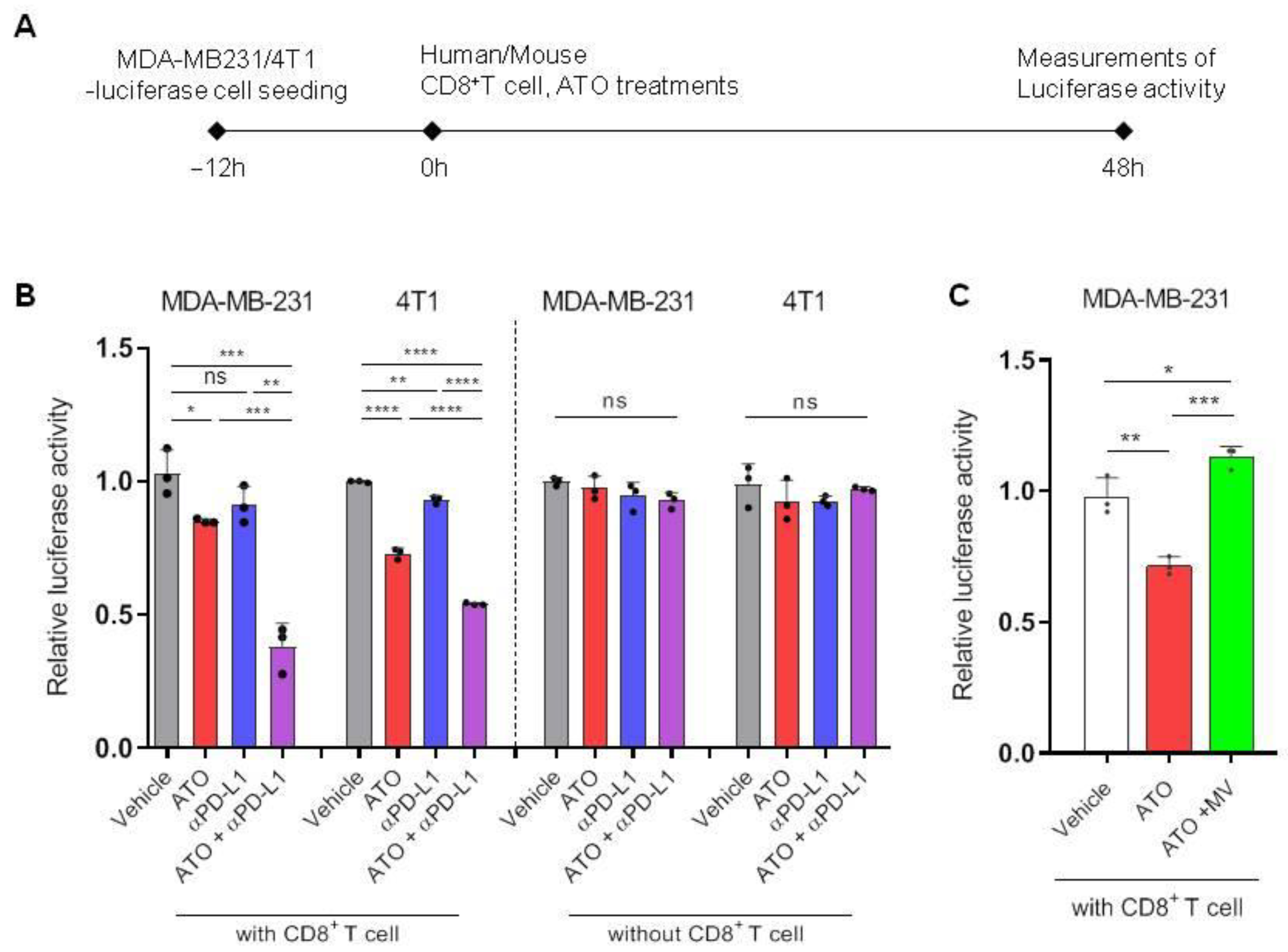
3.3. ATO Combined with Anti-PD-L1 Antibody Enhanced CD8+ T Cell-Mediated Cytotoxic Effects
3.4. ATO Treatment Improves the Therapeutic Effect of Anti-PD-L1 Antibody
3.5. Combination Therapy with ATO and Anti-PD-L1 Antibody Increases Immune Response
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.-Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.-J.; Ji, J. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: Mechanisms, predictive factors, and future perspectives. Biomark. Res. 2020, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, O.; Feldt, M.; Inasu, M.; Bendahl, P.-O.; Elebro, K.; Kimbung, S.; Borgquist, S. Statin use, HMGCR expression, and breast cancer survival–The Malmö Diet and Cancer Study. Sci. Rep. 2020, 10, 558. [Google Scholar] [CrossRef]
- Kimbung, S.; Lettiero, B.; Feldt, M.; Bosch, A.; Borgquist, S. High expression of cholesterol biosynthesis genes is associated with resistance to statin treatment and inferior survival in breast cancer. Oncotarget 2016, 7, 59640–59651. [Google Scholar] [CrossRef]
- Baek, A.E.; Krawczynska, N.; Das Gupta, A.; Dvoretskiy, S.V.; You, S.; Park, J.; Deng, Y.-H.; Sorrells, J.E.; Smith, B.P.; Ma, L. The cholesterol metabolite 27HC increases secretion of extracellular vesicles which promote breast cancer progression. Endocrinology 2021, 162, bqab095. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Yu, P.; Chen, X.-S.; Wei, H.; Cao, S.-J.; Zhang, M.; Zhang, Y.; Tao, Y.-G.; Cao, D.-S.; Qiu, F. Identification of HMGCR as the anticancer target of physapubenolide against melanoma cells by in silico target prediction. Acta Pharmacol. Sin. 2022, 43, 1594–1604. [Google Scholar] [CrossRef]
- Jiang, P.; Mukthavaram, R.; Chao, Y.; Nomura, N.; Bharati, I.; Fogal, V.; Pastorino, S.; Teng, D.; Cong, X.; Pingle, S. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br. J. Cancer 2014, 111, 1562–1571. [Google Scholar] [CrossRef]
- Iannelli, F.; Lombardi, R.; Milone, M.R.; Pucci, B.; De Rienzo, S.; Budillon, A.; Bruzzese, F. Targeting mevalonate pathway in cancer treatment: Repurposing of statins. Recent Pat. Anti-Cancer Drug Discov. 2018, 13, 184–200. [Google Scholar] [CrossRef]
- Emberson, J.R.; Kearney, P.M.; Blackwell, L.; Newman, C.; Reith, C.; Bhala, N.; Holland, L.; Peto, R.; Keech, A.; Collins, R.; et al. Lack of effect of lowering LDL cholesterol on cancer: Meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS ONE 2012, 7, e29849. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Cui, X.-X.; Gao, Z.; Zhao, Y.; Lin, Y.; Shih, W.J.; Huang, M.-T.; Liu, Y.; Rabson, A.; Reddy, B. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer Prev. Res. 2010, 3, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh-Choudhury, N.; Mandal, C.C.; Ghosh-Choudhury, N.; Choudhury, G.G. Simvastatin induces derepression of PTEN expression via NFκB to inhibit breast cancer cell growth. Cell. Signal. 2010, 22, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantini, L.; Pecci, F.; Hurkmans, D.; Copparoni, C.; Aerts, S.; Belderbos, R.A.; Cornelissen, R.; Dumoulin, D.P.; Fiordoliva, I.; Rinaldi, S.; et al. Statin treatment improves response to anti-PD1 agents in patients with malignant pleural mesothelioma and non-small cell lung cancer. J. Clin. Oncol. 2020, 38, 3074. [Google Scholar] [CrossRef]
- Takada, K.; Shimokawa, M.; Takamori, S.; Shimamatsu, S.; Hirai, F.; Tagawa, T.; Okamoto, T.; Hamatake, M.; Tsuchiya-Kawano, Y.; Otsubo, K.; et al. A propensity score-matched analysis of the impact of statin therapy on the outcomes of patients with non-small-cell lung cancer receiving anti-PD-1 monotherapy: A multicenter retrospective study. BMC Cancer 2022, 22, 503. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.-J.; Lee, M.; Oh, Y.; Fang, X.-Q.; Lee, S.; Lim, C.-H.; Park, J.; Lim, J.-H. Statins Decrease Programmed Death-Ligand 1 (PD-L1) by inhibiting AKT and β-Catenin signaling. Cells 2021, 10, 2488. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, A.; Singh, S.; Ahmad, M.; Khanna, K.; Ahmad, T.; Agrawal, A.; Ghosh, B. Simvastatin mediates inhibition of exosome synthesis, localization and secretion via multicomponent interventions. Sci. Rep. 2019, 9, 16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyder, T.; Marti, J.L.G.; Nasrazadani, A.; Brufsky, A.M. Statins and endocrine resistance in breast cancer. Cancer Drug Resist. 2021, 4, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Im, E.-J.; Lee, C.-H.; Moon, P.-G.; Rangaswamy, G.G.; Lee, B.; Lee, J.M.; Lee, J.-C.; Jee, J.-G.; Bae, J.-S.; Kwon, T.-K. Sulfisoxazole inhibits the secretion of small extracellular vesicles by targeting the endothelin receptor A. Nat. Commun. 2019, 10, 1387. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Lee, C.-H.; Bae, J.-H.; Choe, E.-J.; Park, J.-M.; Park, S.-S.; Cho, H.J.; Song, B.-J.; Baek, M.-C. Macitentan improves antitumor immune responses by inhibiting the secretion of tumor-derived extracellular vesicle PD-L1. Theranostics 2022, 12, 1971–1987. [Google Scholar] [CrossRef]
- Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar]
- Detter, J.C.; Zhang, Q.; Mules, E.H.; Novak, E.K.; Mishra, V.S.; Li, W.; McMurtrie, E.B.; Tchernev, V.T.; Wallace, M.R.; Seabra, M.C. Rab geranylgeranyl transferase α mutation in the gunmetal mouse reduces Rab prenylation and platelet synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 4144–4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, P.; Wei, W.; Long, C.; Li, J. Atorvastatin augments temozolomide’s efficacy in glioblastoma via prenylation-dependent inhibition of Ras signaling. Biochem. Biophys. Res. Commun. 2017, 489, 293–298. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 distribution and perspective for cancer immunotherapy—Blockade, knockdown, or inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Shen, H.; Huang, H.; Yang, Z.; Zhou, Y.; Zhao, G. Cholesterol-lowering drug pitavastatin targets lung cancer and angiogenesis via suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling. Anti-Cancer Drugs 2020, 31, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef]
- Mo, H.; Jeter, R.; Bachmann, A.; Yount, S.T.; Shen, C.-L.; Yeganehjoo, H. The potential of isoprenoids in adjuvant cancer therapy to reduce adverse effects of statins. Front. Pharmacol. 2019, 9, 1515. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Seol, Y.M.; Choi, Y.J.; Shin, H.-J.; Chung, J.S.; Shin, N.; Kim, A.; Kim, J.Y.; Kim, K.Y.; Bae, Y. HMG CoA reductase expression as a prognostic factor in Korean patients with breast cancer: A retrospective study. Medicine 2019, 98, e14968. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Thurnher, M. Mevalonate metabolism in cancer. Cancer Lett. 2015, 356, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Kantor, E.D.; Lipworth, L.; Fowke, J.H.; Giovannucci, E.L.; Mucci, L.A.; Signorello, L.B. Statin use and risk of prostate cancer: Results from the Southern Community Cohort Study. Prostate 2015, 75, 1384–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedland, S.; Hamilton, R.; Gerber, L.; Banez, L.; Moreira, D.; Andriole, G.; Rittmaster, R. Statin use and risk of prostate cancer and high-grade prostate cancer: Results from the REDUCE study. Prostate Cancer Prostatic Dis. 2013, 16, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Maisano, D.; Mimmi, S.; Russo, R.; Fioravanti, A.; Fiume, G.; Vecchio, E.; Nisticò, N.; Quinto, I.; Iaccino, E. Uncovering the exosomes diversity: A window of opportunity for tumor progression monitoring. Pharmaceuticals 2020, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-S.; Jeong, E.; Boyiadzis, M.; Whiteside, T.L. Increased small extracellular vesicle secretion after chemotherapy via upregulation of cholesterol metabolism in acute myeloid leukaemia. J. Extracell. Vesicles 2020, 9, 1800979. [Google Scholar] [CrossRef]
- Thurnher, M.; Nussbaumer, O.; Gruenbacher, G. Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clin. Cancer Res. 2012, 18, 3524–3531. [Google Scholar] [CrossRef] [Green Version]
- Bobrie, A.; Krumeich, S.; Reyal, F.; Recchi, C.; Moita, L.F.; Seabra, M.C.; Ostrowski, M.; Théry, C. Rab27a supports exosome-dependent and-independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012, 72, 4920–4930. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Fang, R.; Fang, J.; He, S.; Liu, T. Functional implications of Rab27 GTPases in cancer. Cell Commun. Signal. 2018, 16, 44. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.Q.; Ali, B.R.; Ramalho, J.S.; Godfrey, R.F.; Barral, D.C.; Hume, A.N.; Seabra, M.C. Membrane targeting of Rab GTPases is influenced by the prenylation motif. Mol. Biol. Cell 2003, 14, 1882–1899. [Google Scholar] [CrossRef] [Green Version]
- Iotefa, T.; Guillaume, S.; Liza, F.; Véronique, M.; Philippe, R. Atorvastatin-Induced Inhibition of Human Melanoma. Immunother. Open Access 2016, 2, 1000111. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.-Y.; Zhuang, L.-H.; Wang, D.-D.; Zhou, T.-Y.; Chang, L.-L.; Gai, R.-H.; Zhu, D.-F.; Yang, B.; Zhu, H.; He, Q.-J. Nuclear translocation and activation of YAP by hypoxia contributes to the chemoresistance of SN38 in hepatocellular carcinoma cells. Oncotarget 2016, 7, 6933–6947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubatka, P.; Bojková, B.; Kassayová, M.; Orendáš, P.; Kajo, K.; Výbohová, D.; Kružliak, P.; Adamicová, K.; Péč, M.; Stollárová, N. Combination of Pitavastatin and melatonin shows partial antineoplastic effects in a rat breast carcinoma model. Acta Histochem. 2014, 116, 1454–1461. [Google Scholar] [CrossRef]
- Kitagawa, K.; Moriya, K.; Kaji, K.; Saikawa, S.; Sato, S.; Nishimura, N.; Namisaki, T.; Akahane, T.; Mitoro, A.; Yoshiji, H. Atorvastatin augments gemcitabine-mediated anti-cancer effects by inhibiting Yes-associated protein in human cholangiocarcinoma cells. Int. J. Mol. Sci. 2020, 21, 7588. [Google Scholar] [CrossRef]
- Okoye, I.; Namdar, A.; Xu, L.; Crux, N.; Elahi, S. Atorvastatin downregulates co-inhibitory receptor expression by targeting Ras-activated mTOR signalling. Oncotarget 2017, 8, 98215–98232. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, E.-J.; Lee, C.-H.; Bae, J.-H.; Park, J.-M.; Park, S.-S.; Baek, M.-C. Atorvastatin Enhances the Efficacy of Immune Checkpoint Therapy and Suppresses the Cellular and Extracellular Vesicle PD-L1. Pharmaceutics 2022, 14, 1660. https://doi.org/10.3390/pharmaceutics14081660
Choe E-J, Lee C-H, Bae J-H, Park J-M, Park S-S, Baek M-C. Atorvastatin Enhances the Efficacy of Immune Checkpoint Therapy and Suppresses the Cellular and Extracellular Vesicle PD-L1. Pharmaceutics. 2022; 14(8):1660. https://doi.org/10.3390/pharmaceutics14081660
Chicago/Turabian StyleChoe, Eun-Ji, Chan-Hyeong Lee, Ju-Hyun Bae, Ju-Mi Park, Seong-Sik Park, and Moon-Chang Baek. 2022. "Atorvastatin Enhances the Efficacy of Immune Checkpoint Therapy and Suppresses the Cellular and Extracellular Vesicle PD-L1" Pharmaceutics 14, no. 8: 1660. https://doi.org/10.3390/pharmaceutics14081660