
Highway to Cell: Selection of the Best Cell-Penetrating Peptide to Internalize the CFTR-Stabilizing iCAL36 Peptide

 ,
,
Abstract
: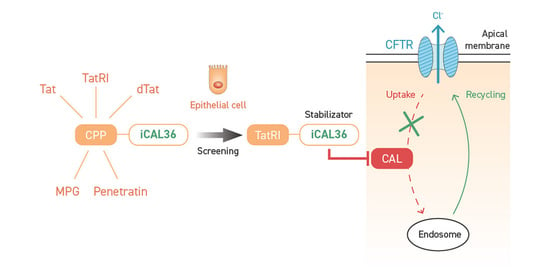
1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Cell Culture Conditions
2.3. Cell Cytotoxicity Measurements
2.4. Cell Transfection Conditions for Flow Cytometry Acquisition
2.5. Cell Transfection Conditions for Confocal Microscopy Imaging
2.6. Cell Transfection Conditions for CFTR Quantification
2.7. Statistical Analysis
3. Results and Discussion
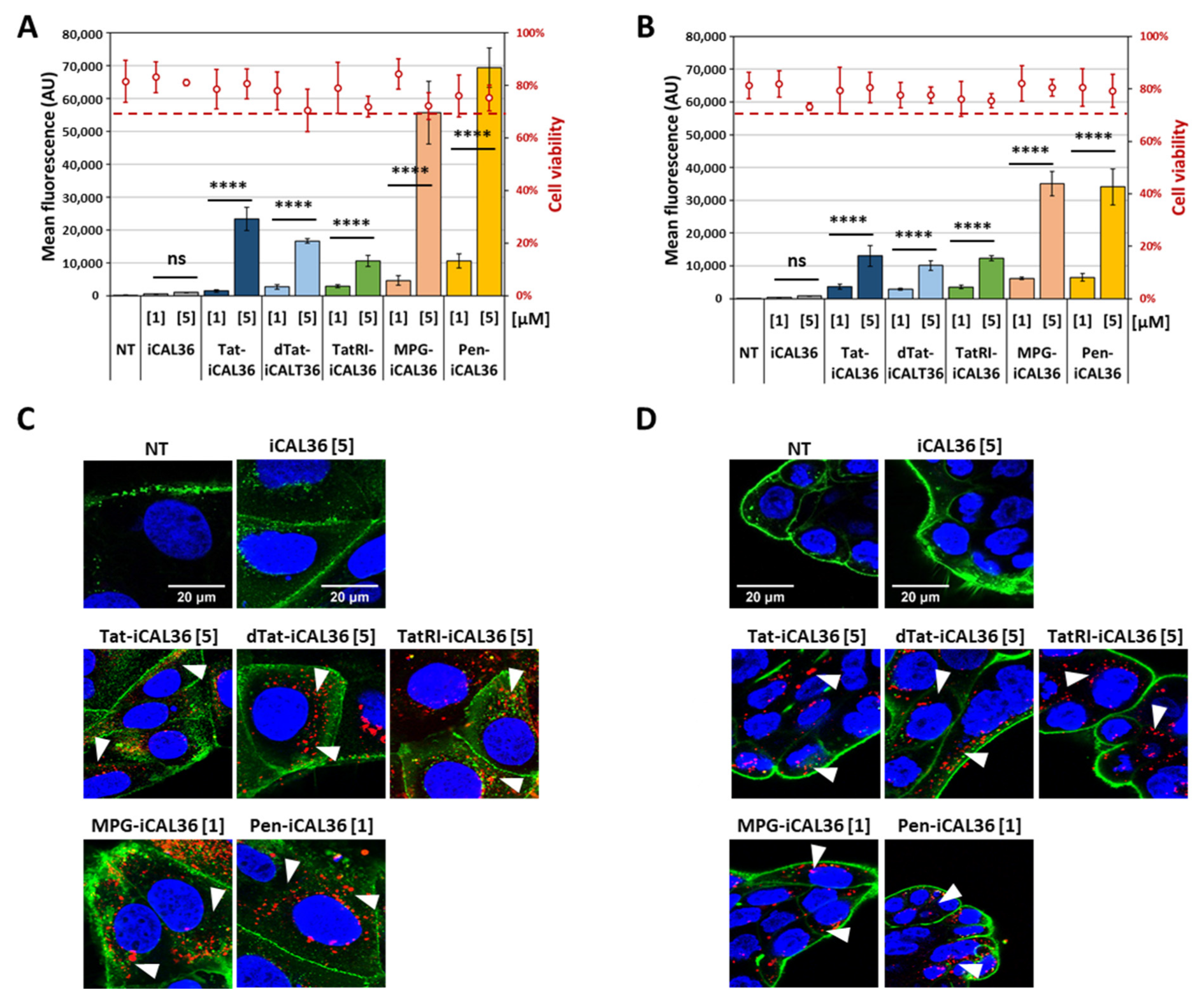
3.1. CPP Selection for iCAL36 Coupling and Evaluation of Their Effects on Cell Viability
3.2. Comparison of CPP-iCAL36 Internalization and Cellular Localization
3.3. Internalization of the CPP-iCAL36 Peptides in the Presence of Serum
3.4. Evaluation of TatRI-iCAL36 and Pen-iCAL36 Membrane Interaction
3.5. Dissecting the Internalization Mechanism of TatRI-iCAL36 and Pen-iCAL36
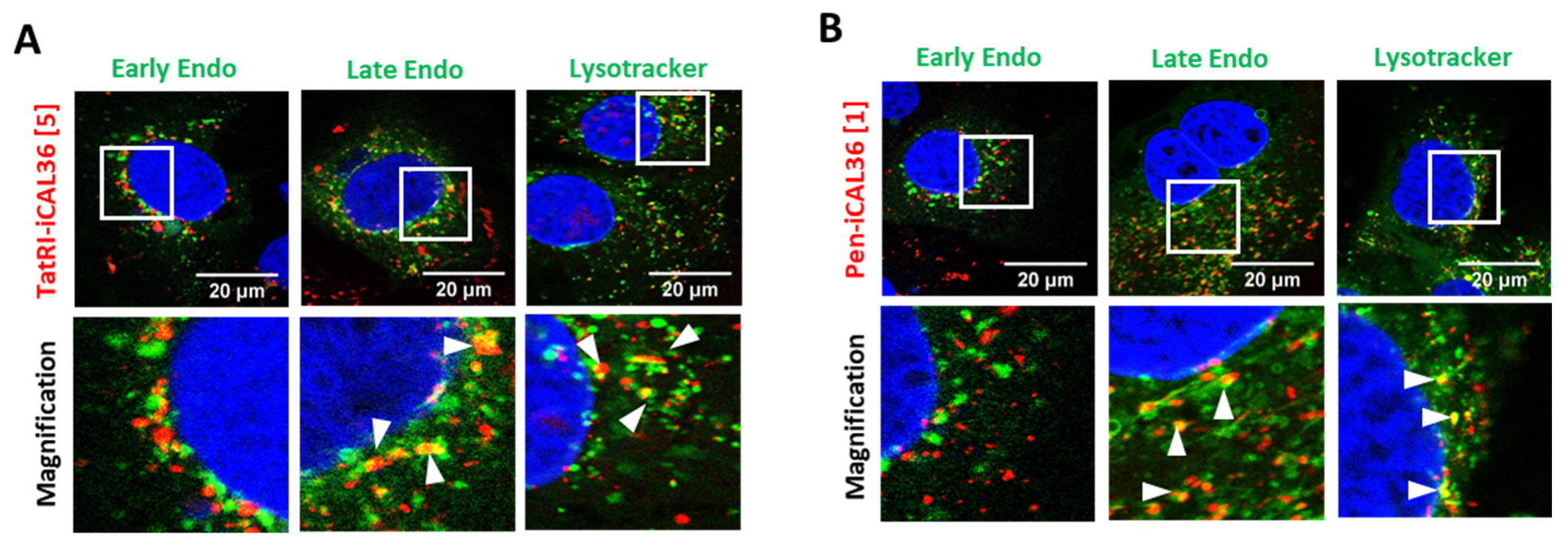
3.6. Intracellular Fate of TatRI-iCAL36 and Pen-iCAL36
3.7. Stabilizing Effect of TatRI-iCAL36 Increases CFTR Amount
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide Chemistry Toolbox—Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Terryah, S.T.; Fellner, R.C.; Ahmad, S.; Moore, P.J.; Reidel, B.; Sesma, J.I.; Kim, C.S.; Garland, A.L.; Scott, D.W.; Sabater, J.R.; et al. Evaluation of a SPLUNC1-Derived Peptide for the Treatment of Cystic Fibrosis Lung Disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L192–L205. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Chen, J.; Chmiel, J.F.; Dorkin, H.L.; Flume, P.A.; Griffin, R.; Nichols, D.; Donaldson, S.H. Inhaled Alpha1-Proteinase Inhibitor Therapy in Patients with Cystic Fibrosis. J. Cyst. Fibros. 2016, 15, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappiello, F.; Grazia, A.D.; Segev-Zarko, L.; Scali, S.; Ferrera, L.; Galietta, L.; Pini, A.; Shai, Y.; Di, Y.P.; Mangoni, M.L. Esculentin-1a-Derived Peptides Promote Clearance of Pseudomonas Aeruginosa Internalized in Bronchial Cells of Cystic Fibrosis Patients and Lung Cell Migration: Biochemical Properties and a Plausible Mode of Action. Antimicrob. Agents Chemother. 2016, 60, 7252–7262. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, J.; Roscia, G.; Lampronti, I.; Gambari, R.; Quercini, L.; Falciani, C.; Bracci, L.; Pini, A. Immunomodulatory and Anti-Inflammatory Activity in Vitro and in Vivo of a Novel Antimicrobial Candidate. J. Biol. Chem. 2016, 291, 25742–25748. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, N.; Aranzana-Climent, V.; Magréault, S.; Marchand, S.; Couet, W. Clinical Pharmacokinetics and Pharmacodynamics of Colistin. Clin. Pharmacokinet. 2017, 56, 1441–1460. [Google Scholar] [CrossRef]
- Vouilleme, L.; Cushing, P.R.; Volkmer, R.; Madden, D.R.; Boisguerin, P. Engineering Peptide Inhibitors to Overcome PDZ Binding Promiscuity. Angew. Chem. 2010, 49, 9912–9916. [Google Scholar] [CrossRef]
- Cushing, P.R.; Vouilleme, L.; Pellegrini, M.; Boisguerin, P.; Madden, D.R. A Stabilizing Influence: CAL PDZ Inhibition Extends the Half-Life of ΔF508-CFTR. Angew. Chem. 2010, 49, 9907–9911. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.; Xu, X.; Amacher, J.F.; Madden, D.R.; Cormet-Boyaka, E.; Pei, D. Intracellular Delivery of Peptidyl Ligands by Reversible Cyclization: Discovery of a PDZ Domain Inhibitor That Rescues CFTR Activity. Angew. Chem. Int. Ed. 2015, 54, 5874–5878. [Google Scholar] [CrossRef]
- Dougherty, P.G.; Wellmerling, J.H.; Koley, A.; Lukowski, J.K.; Hummon, A.B.; Cormet-Boyaka, E.; Pei, D. Cyclic Peptidyl Inhibitors against CAL/CFTR Interaction for Treatment of Cystic Fibrosis. J. Med. Chem. 2020, 63, 15773–15784. [Google Scholar] [CrossRef] [PubMed]
- Amacher, J.F.; Cushing, P.R.; Weiner, J.A.; Madden, D.R. Crystallization and Preliminary Diffraction Analysis of the CAL PDZ Domain in Complex with a Selective Peptide Inhibitor. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Wang, H.; Guggino, W.B. Modulation of Mature Cystic Fibrosis Transmembrane Regulator Protein by the PDZ Domain Protein CAL. J. Biol. Chem. 2004, 279, 1892–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatecka-Urban, A.; Duhaime, M.; Coutermarsh, B.; Karlson, K.H.; Collawn, J.; Milewski, M.; Cutting, G.R.; Guggino, W.B.; Langford, G.; Stanton, B.A. PDZ Domain Interaction Controls the Endocytic Recycling of the Cystic Fibrosis Transmembrane Conductance Regulator. J. Biol. Chem. 2002, 277, 40099–40105. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Moyer, B.D.; Milewski, M.; Loffing, J.; Ikeda, M.; Mickle, J.E.; Cutting, G.R.; Li, M.; Stanton, B.A.; Guggino, W.B. A Golgi-Associated PDZ Domain Protein Modulates Cystic Fibrosis Transmembrane Regulator Plasma Membrane Expression. J. Biol. Chem. 2002, 277, 3520–3529. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Cushing, P.R.; Smithson, D.C.; Pellegrini, M.; Pletnev, A.A.; Al-Ayyoubi, S.; Grassetti, A.V.; Gerber, S.A.; Guy, R.K.; Madden, D.R. Cysteine Modifiers Suggest an Allosteric Inhibitory Site on the CAL PDZ Domain. Biosci. Rep. 2018, 38, BSR20180231. [Google Scholar] [CrossRef] [Green Version]
- Chellappan, D.K.; Prasher, P.; Saravanan, V.; Vern Yee, V.S.; Wen Chi, W.C.; Wong, J.W.; Wong, J.K.; Wong, J.T.; Wan, W.; Chellian, J.; et al. Protein and Peptide Delivery to Lungs by Using Advanced Targeted Drug Delivery. Chem. Biol. Interact. 2022, 351, 109706. [Google Scholar] [CrossRef]
- Xu, J.; Khan, A.R.; Fu, M.; Wang, R.; Ji, J.; Zhai, G. Cell-Penetrating Peptide: A Means of Breaking through the Physiological Barriers of Different Tissues and Organs. J. Control. Release 2019, 309, 106–124. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-Independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [Green Version]
- Langel, Ü. (Ed.) Cell-Penetrating Peptides; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2015; Volume 1324, ISBN 978-1-4939-2805-7. [Google Scholar]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milletti, F. Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization Mechanisms of Cell-Penetrating Peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Seisel, Q.; Pelletier, F.; Deshayes, S.; Boisguerin, P. How to Evaluate the Cellular Uptake of CPPs with Fluorescence Techniques: Dissecting Methodological Pitfalls Associated to Tryptophan-Rich Peptides. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1533–1545. [Google Scholar] [CrossRef]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of Cellular Uptake Using 22 CPPs in 4 Different Cell Lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Jafari, M.; Karunaratne, D.N.; Sweeting, C.M.; Chen, P. Modification of a Designed Amphipathic Cell-Penetrating Peptide and Its Effect on Solubility, Secondary Structure, and Uptake Efficiency. Biochemistry 2013, 52, 3428–3435. [Google Scholar] [CrossRef]
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I.F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-Based Nanoparticles to Rapidly and Efficiently “Wrap ‘n Roll” SiRNA into Cells. Bioconjug. Chem. 2019, 30, 592–603. [Google Scholar] [CrossRef]
- Meloni, B.P.; Craig, A.J.; Milech, N.; Hopkins, R.M.; Watt, P.M.; Knuckey, N.W. The Neuroprotective Efficacy of Cell-Penetrating Peptides TAT, Penetratin, Arg-9, and Pep-1 in Glutamic Acid, Kainic Acid, and in Vitro Ischemia Injury Models Using Primary Cortical Neuronal Cultures. Cell. Mol. Neurobiol. 2014, 34, 173–181. [Google Scholar] [CrossRef]
- Tünnemann, G.; Martin, R.M.; Haupt, S.; Patsch, C.; Edenhofer, F.; Cardoso, M.C. Cargo-Dependent Mode of Uptake and Bioavailability of TAT-Containing Proteins and Peptides in Living Cells. FASEB J. 2006, 20, 1775–1784. [Google Scholar] [CrossRef] [Green Version]
- El-Andaloussi, S.; Järver, P.; Johansson, H.J.; Langel, U. Cargo-Dependent Cytotoxicity and Delivery Efficacy of Cell-Penetrating Peptides: A Comparative Study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Duchardt, F.; Ruttekolk, I.R.; Verdurmen, W.P.R.; Lortat-Jacob, H.; Bürck, J.; Hufnagel, H.; Fischer, R.; van den Heuvel, M.; Löwik, D.W.P.M.; Vuister, G.W.; et al. A Cell-Penetrating Peptide Derived from Human Lactoferrin with Conformation-Dependent Uptake Efficiency. J. Biol. Chem. 2009, 284, 36099–36108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechinger, B.; Aisenbrey, C. The Polymorphic Nature of Membrane-Active Peptides from Biophysical and Structural Investigations. Curr. Protein Pept. Sci. 2012, 13, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.; Gräslund, A. The Position of the Cell Penetrating Peptide Penetratin in SDS Micelles Determined by NMR. FEBS Lett. 2001, 497, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Magzoub, M.; Eriksson, L.E.G.; Gräslund, A. Conformational States of the Cell-Penetrating Peptide Penetratin When Interacting with Phospholipid Vesicles: Effects of Surface Charge and Peptide Concentration. Biochim. Biophys. Acta 2002, 1563, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Balayssac, S.; Burlina, F.; Convert, O.; Bolbach, G.; Chassaing, G.; Lequin, O. Comparison of Penetratin and Other Homeodomain-Derived Cell-Penetrating Peptides: Interaction in a Membrane-Mimicking Environment and Cellular Uptake Efficiency. Biochemistry 2006, 45, 1408–1420. [Google Scholar] [CrossRef]
- Konate, K.; Seisel, Q.; Vivès, E.; Boisguérin, P.; Deshayes, S. Fluorescent Leakage Assay to Investigate Membrane Destabilization by Cell-Penetrating Peptide. J. Vis. Exp. 2020, 166, e62028. [Google Scholar] [CrossRef]
- Hoyer, J.; Neundorf, I. Peptide Vectors for the Nonviral Delivery of Nucleic Acids. Acc. Chem. Res. 2012, 45, 1048–1056. [Google Scholar] [CrossRef]
- Langel, U. (Ed.) Cell-Penetrating Peptides; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Brock, R. The Uptake of Arginine-Rich Cell-Penetrating Peptides: Putting the Puzzle Together. Bioconjug. Chem. 2014, 25, 863–868. [Google Scholar] [CrossRef]
- Jones, A.T.; Sayers, E.J. Cell Entry of Cell Penetrating Peptides: Tales of Tails Wagging Dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef]
- Vercauteren, D.; Rejman, J.; Martens, T.F.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. On the Cellular Processing of Non-Viral Nanomedicines for Nucleic Acid Delivery: Mechanisms and Methods. J. Control. Release 2012, 161, 566–581. [Google Scholar] [CrossRef]
- Cleal, K.; He, L.; Watson, P.D.; Jones, A.T. Endocytosis, Intracellular Traffic and Fate of Cell Penetrating Peptide Based Conjugates and Nanoparticles. Curr. Pharm. Des. 2013, 19, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I. Pharmacological Inhibition of Endocytic Pathways: Is It Specific Enough to Be Useful? Methods Mol. Biol. 2008, 440, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, D.; Vandenbroucke, R.E.; Jones, A.T.; Rejman, J.; Demeester, J.; De Smedt, S.C.; Sanders, N.N.; Braeckmans, K. The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls. Mol. Ther. 2010, 18, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Rosazza, C.; Deschout, H.; Buntz, A.; Braeckmans, K.; Rols, M.-P.; Zumbusch, A. Endocytosis and Endosomal Trafficking of DNA After Gene Electrotransfer In Vitro. Mol. Ther. Nucleic Acids 2016, 5, e286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, Z.; Zhang, Y.; Naren, A.P. CFTR-NHERF2-LPA2 Complex in the Airway and Gut Epithelia. Int. J. Mol. Sci. 2017, 18, 1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Mandrup Bertozzi, S.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef]
- Amacher, J.F.; Zhao, R.; Spaller, M.R.; Madden, D.R. Chemically Modified Peptide Scaffolds Target the CFTR-Associated Ligand PDZ Domain. PLoS ONE 2014, 9, e103650. [Google Scholar] [CrossRef]
- Wang, Z.X. An Exact Mathematical Expression for Describing Competitive Binding of Two Different Ligands to a Protein Molecule. FEBS Lett. 1995, 360, 111–114. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Code | Sequence | Residues | pI |
|---|---|---|---|---|
| iCAL36 | 36 | ANSRWPTSII | 10 | 9.79 |
| Tat-iCAL36 1 | T36 | GRKKRRQRRRPPQ-ANSRWPTSII | 23 | 12.78 |
| dTat-iCAL36 | dT36 | grkkrrqrrrppq-ANSRWPTSII | 23 | 12.78 |
| TatRI-iCAL36 | TRI36 | qpprrrqrrkkrg-ANSRWPTSII | 23 | 12.78 |
| Pen-iCAL36 | P36 | RQILIWFQNRRMKWKK-ANSRWPTSII | 26 | 12.48 |
| MPG-iCAL36 1 | M36 | GALFLGWLGAAGSTMGAWSQPKKKRKV-ANSRWPTSII | 37 | 12.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seisel, Q.; Lakumpa, I.; Josse, E.; Vivès, E.; Varilh, J.; Taulan-Cadars, M.; Boisguérin, P. Highway to Cell: Selection of the Best Cell-Penetrating Peptide to Internalize the CFTR-Stabilizing iCAL36 Peptide. Pharmaceutics 2022, 14, 808. https://doi.org/10.3390/pharmaceutics14040808
Seisel Q, Lakumpa I, Josse E, Vivès E, Varilh J, Taulan-Cadars M, Boisguérin P. Highway to Cell: Selection of the Best Cell-Penetrating Peptide to Internalize the CFTR-Stabilizing iCAL36 Peptide. Pharmaceutics. 2022; 14(4):808. https://doi.org/10.3390/pharmaceutics14040808
Chicago/Turabian StyleSeisel, Quentin, Israpong Lakumpa, Emilie Josse, Eric Vivès, Jessica Varilh, Magali Taulan-Cadars, and Prisca Boisguérin. 2022. "Highway to Cell: Selection of the Best Cell-Penetrating Peptide to Internalize the CFTR-Stabilizing iCAL36 Peptide" Pharmaceutics 14, no. 4: 808. https://doi.org/10.3390/pharmaceutics14040808