Microwave-Induced In Situ Amorphization: A New Strategy for Tackling the Stability Issue of Amorphous Solid Dispersions


Abstract
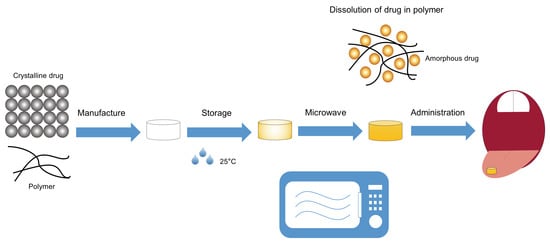
:
1. Introduction
2. Current Issues in the Development of ASDs
2.1. Stability Issues of ASDs
2.1.1. Thermodynamic Factors on the Physical Stability of ASDs
2.1.2. Kinetic Factors on the Physical Stability of ASDs
2.1.3. Environmental Factors Affecting the Physical Stability of ASDs
2.2. Downstream Processing Issues of ASDs
3. In situ Amorphization
3.1. The Pharmaceutical Significance of in situ Amorphization
3.2. General Development of in situ Amorphization
4. Microwave-Induced in situ Amorphization
4.1. Microwave Heating
4.2. Microwave Heating for Bulk Preparation of Amorphous Glass Solutions
- (i)
- Preparing the physically mixed API-carrier powders. It is worth noting that low Tg carriers (polymers, surfactants or the combination of both) have frequently been used (Table 2) as they can possibly be molten or softened at relatively low temperatures;
- (ii)
- Treating the physically mixed loose powders with continuous or intermittent microwaves to induce amorphization; and
- (iii)
- Cooling, pulverizing and sieving to get the final bulk ASD powders.
4.3. Microwave-Induced in Situ Amorphization within the Final Dosage Form
4.3.1. The Role of Microwave Energy Input and Storage Humidity
4.3.2. The Role of the Molecular Weight of Polymeric Carriers
4.3.3. Dissolution of the Drug into the Polymeric Carrier
4.3.4. Performance of Microwave-Induced ASDs Activated in situ
5. Challenges and Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Leuner, C.; Dressman, J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Abuzara, S.M.; Hyun, S.M.; Kim, J.H.; Park, H.J.; Kim, M.S.; Park, J.S.; Hwang, S.J. Enhancing the solubility and bioavailability of poorly water-soluble drugs using supercritical antisolvent (SAS) process. Int. J. Pharm. 2017, 538, 1. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Chen, Y.; Zhang, G.G.Z.; Yu, L.; Mantri, R.V. Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2016; p. 46. [Google Scholar]
- Broman, E.; Khoo, C.; Taylor, L.S. A comparison of alternative polymer excipients and processing methods for making solid dispersions of a poorly water soluble drug. Int. J. Pharm. 2001, 222, 139–151. [Google Scholar] [CrossRef]
- Yu, D.; Li, J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, E.; Démuth, B.; Galata, D.L.; Vass, P.; Hirsch, E.; Csontos, I.; Marosi, G.; Nagy, Z.K. Continuous Formulation Approaches of Amorphous Solid Dispersions: Significance of Powder Flow Properties and Feeding Performance. Pharmaceutics 2019, 11, 654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankar, S.D.; Rach, P.R. Solubility enhancement of poor water soluble drugs by solid dispersion: A review. J. Drug Deliv. Ther. 2018, 8, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Hu, Y.; Liu, L.; Su, L.; Li, N.; Yu, J.; Tang, B.; Yang, Z. Physical stability of amorphous solid dispersions: A physicochemical perspective with thermodynamic, kinetic and environmental aspects. Pharm. Res. 2018, 35, 125. [Google Scholar] [CrossRef] [PubMed]
- Jermain, S.V.; Brough, C.; Williams III, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery-An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef]
- Haser, A.; Zhang, F. New strategies for improving the development and performance of amorphous solid dispersions. AAPS Pharmscitech 2018, 19, 978–990. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [Green Version]
- Hancock, B.C.; Parks, M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Laitinen, R.; Priemel, P.A.; Surwase, S.; Graeser, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Theoretical considerations in developing amorphous solid dispersions. In Amorphous Solid Dispersions; Springer: New York, NY, USA, 2014; pp. 35–90. [Google Scholar] [CrossRef]
- Huang, S.; Williams, R.O. Effects of the preparation process on the properties of amorphous solid dispersions. AAPS Pharmscitech 2018, 19, 1971–1984. [Google Scholar] [CrossRef] [PubMed]
- Blaabjerg, L.I.; Grohganz, H.; Lindenberg, E.; Löbmann, K.; Müllertz, A.; Rades, T. The influence of polymers on the supersaturation potential of poor and good glass formers. Pharmaceutics 2018, 10, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Ho, C. Amorphous solid dispersions: Utilization and challenges in drug discovery and development. J. Pharm. Sci. 2015, 104, 3237–3258. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef]
- Sawicki, E. Solid Dispersions in Oncology: A Solution to Solubility-Limited Oral Drug Absorption. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2017. [Google Scholar]
- Démuth, B.; Nagy, Z.K.; Balogh, A.; Vigh, T.; Marosi, G.; Verreck, G.; Van Assche, I.; Brewster, M.E. Downstream processing of polymer-based amorphous solid dispersions to generate tablet formulations. Int. J. Pharm. 2015, 486, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Supersaturation and crystallization: Non-equilibrium dynamics of amorphous solid dispersions for oral drug delivery. Expert Opin. Drug Deliv. 2017, 14, 735–743. [Google Scholar] [CrossRef]
- Davis, M.T.; Potter, C.B.; Walker, G.M. Downstream processing of a ternary amorphous solid dispersion: The impacts of spray drying and hot melt extrusion on powder flow, compression and dissolution. Int. J. Pharm. 2018, 544, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Doreth, M.; Hussein, M.A.; Priemel, P.A.; Grohganz, H.; Holm, R.; de Diego, H.L.; Rades, T.; Löbmann, K. Amorphization within the tablet: Using microwave irradiation to form a glass solution in situ. Int. J. Pharm. 2017, 519, 343–351. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Rades, T. The role of configurational entropy in amorphous systems. Pharmaceutics 2010, 2, 224–244. [Google Scholar] [CrossRef]
- Qian, F.; Huang, J.; Hussain, M.A. Drug-polymer solubility and miscibility: Stability consideration and practical challenges in amorphous solid dispersion development. J. Pharm. Sci. 2010, 99, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pang, H.; Guo, Z.; Lin, L.; Dong, Y.; Li, G.; Lu, M.; Wu, C. Interactions between drugs and polymers influencing hot melt extrusion. J. Pharm. Pharmacol. 2014, 66, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Priemel, P.A.; Grohganz, H.; Rades, T. Unintended and in situ amorphisation of pharmaceuticals. Adv. Drug Deliv. Rev. 2016, 100, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, M.; Jia, L.; Chen, M.; Du, S.; Gong, J. Investigation of Drug-polymer Miscibility, Molecular Interaction and Their Effects on the Physical Stabilities and Dissolution Behaviors of Norfloxacin Amorphous Solid Dispersions. Cryst. Growth Des. 2020, 20, 2952–2964. [Google Scholar] [CrossRef]
- Rams-Baron, M.; Jachowicz, R.; Boldyreva, E.; Zhou, D.; Jamroz, W.; Paluch, M. Amorphous Drug Formulation. In Amorphous Drugs: Benefits and Challenges, 1st ed.; Springer: New York, NY, USA, 2018; pp. 159–223. [Google Scholar] [CrossRef]
- Greco, S.; Authelin, J.; Leveder, C.; Segalini, A. A practical method to predict physical stability of amorphous solid dispersions. Pharm. Res. 2012, 29, 2792–2805. [Google Scholar] [CrossRef]
- Rahman, M.; Ozkan, S.; Lester, J.; Farzana, I.; Bi, V.; Dürig, T. Plasticizer Compatibility and Thermal and Rheological Properties of PlasdoneTM povidone and copovidone polymers for Hot-melt Extrusion Applications. In Proceedings of the Annual Meeting of the American Association of Pharmaceutical Scientists, Chicago, IL, USA, 14–18 October 2012; pp. 1–7. [Google Scholar]
- Mohapatra, S.; Samanta, S.; Kothari, K.; Mistry, P.; Suryanarayanan, R. Effect of polymer molecular weight on the crystallization behavior of indomethacin amorphous solid dispersions. Cryst. Growth Des. 2017, 17, 3142–3150. [Google Scholar] [CrossRef]
- Wang, B.; Wang, D.; Zhao, S.; Huang, X.; Zhang, J.; Lv, Y.; Liu, X.; Lv, G.; Ma, X. Evaluate the ability of PVP to inhibit crystallization of amorphous solid dispersions by density functional theory and experimental verify. Eur. J. Pharm. Sci. 2017, 96, 45–52. [Google Scholar] [CrossRef]
- Xie, T.; Taylor, L.S. Effect of temperature and moisture on the physical stability of binary and ternary amorphous solid dispersions of celecoxib. J. Pharm. Sci. 2017, 106, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Marsac, P.J.; Konno, H.; Rumondor, A.C.; Taylor, L.S. Recrystallization of nifedipine and felodipine from amorphous molecular level solid dispersions containing poly (vinylpyrrolidone) and sorbed water. Pharm. Res. 2008, 25, 647–656. [Google Scholar] [CrossRef]
- Lehmkemper, K.; Kyeremateng, S.O.; Heinzerling, O.; Degenhardt, M.; Sadowski, G. Impact of polymer type and relative humidity on the long-term physical stability of amorphous solid dispersions. Mol. Pharm. 2017, 14, 4374–4386. [Google Scholar] [CrossRef]
- Német, Z.; Sztatisz, J.; Demeter, A. Polymorph transitions of bicalutamide: A remarkable example of mechanical activation. J. Pharm. Sci. 2008, 97, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nollenberger, K.; Albers, J.; Qi, S. Molecular implications of drug-polymer solubility in understanding the destabilization of solid dispersions by milling. Mol. Pharm. 2014, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Van den Mooter, G. The role of the carrier in the formulation of pharmaceutical solid dispersions. Part II: Amorphous carriers. Expert Opin. Drug Deliv. 2016, 13, 1681–1694. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.K.; Balogh, A.; Démuth, B.; Pataki, H.; Vigh, T.; Bence, S.; Molnár, K.; Schmidt, B.T.; Horák, P.; Marosi, G. High speed electrospinning for scaled-up production of amorphous solid dispersion of itraconazole. Int. J. Pharm. 2015, 480, 137–142. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Solid state properties and drug release behavior of co-amorphous indomethacin-arginine tablets coated with Kollicoat® Protect. Eur. J. Pharm. Biopharm. 2017, 119, 150–160. [Google Scholar] [CrossRef]
- Tiwari, R.V.; Patil, H.; Repka, M.A. Contribution of hot-melt extrusion technology to advance drug delivery in the 21st century. Expert Opin. Drug Deliv. 2016, 13, 451–464. [Google Scholar] [CrossRef]
- Das, A.K.; Bhanja, S.; Srilakshmi, N. Formulation and evaluation of quetiapine immediate release film coated tablets. Asian J. Pharm. Clin. Res. 2013, 6, 107–112. [Google Scholar]
- Waters, L.J.; Bedford, S.; Parkes, G.M. Controlled microwave processing applied to the pharmaceutical formulation of ibuprofen. AAPS Pharmscitech 2011, 12, 1038–1043. [Google Scholar] [CrossRef]
- Moneghini, M.; Bellich, B.; Baxa, P.; Princivalle, F. Microwave generated solid dispersions containing Ibuprofen. Int. J. Pharm. 2008, 361, 125–130. [Google Scholar] [CrossRef]
- Ker, J.; Sr, I.S.; Kofler, B. Alternative solvent-free preparation methods for felodipine surface solid dispersions. Drug Dev. Ind. Pharm. 1998, 24, 359–363. [Google Scholar] [CrossRef]
- Doreth, M.; Löbmann, K.; Priemel, P.; Grohganz, H.; Taylor, R.; Holm, R.; de Diego, H.L.; Rades, T. Influence of PVP molecular weight on the microwave assisted in situ amorphization of indomethacin. Eur. J. Pharm. Biopharm. 2018, 122, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Edinger, M.; Knopp, M.M.; Kerdoncuff, H.; Rantanen, J.; Rades, T.; Löbmann, K. Quantification of microwave-induced amorphization of celecoxib in PVP tablets using transmission Raman spectroscopy. Eur. J. Pharm. Sci. 2018, 117, 62–67. [Google Scholar] [CrossRef]
- Doreth, M.; Löbmann, K.; Grohganz, H.; Holm, R.; De Diego, H.L.; Rades, T.; Priemel, P.A. Glass solution formation in water-In situ amorphization of naproxen and ibuprofen with Eudragit®E PO. J. Drug Deliv. Sci. Technol. 2016, 34, 32–40. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Undesired co-amorphisation of indomethacin and arginine during combined storage at high humidity conditions. Int. J. Pharm. 2018, 544, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation of arginine with indomethacin or furosemide during immersion in an acidic medium-A proof of concept study. Eur. J. Pharm. Biopharm. 2018, 133, 151–160. [Google Scholar] [CrossRef]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. In situ co-amorphisation in coated tablets-The combination of carvedilol with aspartic acid during immersion in an acidic medium. Int. J. Pharm. 2019, 558, 357–366. [Google Scholar] [CrossRef]
- Thostenson, E.T.; Chou, T. Microwave processing: Fundamentals and applications. Compos. Part A Appl. Sci. Manuf. 1999, 30, 1055–1071. [Google Scholar] [CrossRef]
- Atuonwu, J.C.; Tassou, S.A. Energy issues in microwave food processing: A review of developments and the enabling potentials of solid-state power delivery. Crit. Rev. Food Sci. 2019, 59, 1392–1407. [Google Scholar] [CrossRef]
- Risman, P.O.; Wäppling-Raaholt, B. Retro-modelling of a dual resonant applicator and accurate dielectric properties of liquid water from -20°C to +100°C. Meas. Sci. Technol. 2007, 18, 959. [Google Scholar] [CrossRef]
- Shah, R. Microwave-assisted Production of Solid Lipid Nanoparticles. Ph.D. Thesis, Swinburne University of Technology, Melbourne, Australia, 2016. [Google Scholar]
- Moneghini, M.; De Zordi, N. Microwave Applications in Pharmaceutical Dosage Form Development; Future Science and Newlands Press: London, UK, 2014; pp. 195–208. [Google Scholar] [CrossRef]
- Bonde, M.N.; Sohani, A.C.; Daud, A.S.; Sapkal, N.P. Microwave: An emerging trend in pharmaceutical processes and formulations. Int. J. Pharm. Technol. 2011, 3, 3499–3520. [Google Scholar]
- Kappe, C.O.; Stadler, A.; Dallinger, D. Microwaves in Organic and Medicinal Chemistry, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2012; pp. 11–12. [Google Scholar] [CrossRef]
- Jones, D.A.; Lelyveld, T.P.; Mavrofidis, S.D.; Kingman, S.W.; Miles, N.J. Microwave heating applications in environmental engineering—A review. Resour. Conserv. Recycl. 2002, 34, 75–90. [Google Scholar] [CrossRef]
- Haque, K.E. Microwave energy for mineral treatment processes—A brief review. Int. J. Miner. Process. 1999, 57, 1–24. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Galan, A.; Calinescu, I.; Trifan, A.; Winkworth-Smith, C.; Calvo-Carrascal, M.; Dodds, C.; Binner, E. New insights into the role of selective and volumetric heating during microwave extraction: Investigation of the extraction of polyphenolic compounds from sea buckthorn leaves using microwave-assisted extraction and conventional solvent extraction. Chem. Eng. Process. Process Intensif. 2017, 116, 29–39. [Google Scholar] [CrossRef]
- Tripathi, M.; Sahu, J.N.; Ganesan, P.; Monash, P.; Dey, T.K. Effect of microwave frequency on dielectric properties of oil palm shell (OPS) and OPS char synthesized by microwave pyrolysis of OPS. J. Anal. Appl. Pyrol. 2015, 112, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Bergese, P.; Colombo, I.; Gervasoni, D.; Depero, L.E. Microwave generated nanocomposites for making insoluble drugs soluble. Mater. Sci. Eng. C 2003, 23, 791–795. [Google Scholar] [CrossRef]
- Wong, T.W. Use of microwave in processing of drug delivery systems. Curr. Drug Deliv. 2008, 5, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Moneghini, M.; Zingone, G.; De Zordi, N. Influence of the microwave technology on the physical-chemical properties of solid dispersion with Nimesulide. Powder Technol. 2009, 195, 259–263. [Google Scholar] [CrossRef]
- Maurya, D.; Belgamwar, V.; Tekade, A. Microwave induced solubility enhancement of poorly water soluble atorvastatin calcium. J. Pharm. Pharmacol. 2010, 62, 1599–1606. [Google Scholar] [CrossRef]
- Moneghini, M.; De Zordi, N.; Solinas, D.; Macchiavelli, S.; Princivalle, F. Characterization of solid dispersions of itraconazole and vitamin E TPGS prepared by microwave technology. Future Med. Chem. 2010, 2, 237–246. [Google Scholar] [CrossRef]
- Zawar, L.R.; Bari, S.B. Preparation, characterization and in vivo evaluation of antihyperglycemic activity of microwave generated repaglinide solid dispersion. Chem. Pharm. Bull. 2012, 60, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, P.H.; Belgamwar, V.S.; Patil, P.R.; Surana, S.J. Enhancement of solubility and dissolution rate of poorly water soluble raloxifene using microwave induced fusion method. Braz. J. Pharm. Sci. 2013, 49, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Zawar, L.; Bari, S. Microwave induced solid dispersion as a novel technique for enhancing dissolution rate of repaglinide. Adv. Pharmacol. Pharm. 2013, 1, 95–101. [Google Scholar] [CrossRef]
- Sonawane, R.; Bonde, P.; Chatap, V.; Bari, S.; Zawar, L. Preparation and Characterization of Microwave-assisted Candesartan Cilexetil Solid Dispersions. Res. Rev. J. Pharm. Sci. 2013, 4, 9–19. [Google Scholar]
- Shi, N.Q.; Lai, H.W.; Zhang, Y.; Feng, B.; Xiao, X.; Zhang, H.M.; Li, Z.Q.; Qi, X.R. On the inherent properties of Soluplus and its application in ibuprofen solid dispersions generated by microwave-quench cooling technology. Pharm. Dev. Technol 2016, 23, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Shakeel, F.; Ibrahim, M.; Elzayat, E.; Altamimi, M.; Shazly, G.; Mohsin, K.; Alkholief, M.; Alsulays, B.; Alshetaili, A.; et al. Influence of the microwave technology on solid dispersions of mefenamic acid and flufenamic acid. PLoS ONE 2017, 12, e0182011. [Google Scholar] [CrossRef] [Green Version]
- Alshehri, S.; Imam, S.S.; Altamimi, M.A.; Hussain, A.; Shakeel, F.; Elzayat, E.; Mohsin, K.; Ibrahim, M.; Alanazi, F. Enhanced dissolution of luteolin by solid dispersion prepared by different methods: Physicochemical characterization and antioxidant activity. ACS Omega 2020, 5, 6461–6471. [Google Scholar] [CrossRef] [Green Version]
- Radacsi, N.; Stefanidis, G.D.; Szabó Révész, P.; Ambrus, R. Analysis of niflumic acid prepared by rapid microwave-assisted evaporation. J. Pharm. Biomed. 2014, 98, 16–21. [Google Scholar] [CrossRef]
- Abreu-Villela, R.; Adler, C.; Caraballo, I.; Kuentz, M. Electron microscopy/energy dispersive X-ray spectroscopy of drug distribution in solid dispersions and interpretation by multifractal geometry. J. Pharm. Biomed. 2018, 150, 241–247. [Google Scholar] [CrossRef]
- Otsuka, M.; Maeno, Y.; Fukami, T.; Inoue, M.; Tagami, T.; Ozeki, T. Solid dispersions of efonidipine hydrochloride ethanolate with improved physicochemical and pharmacokinetic properties prepared with microwave treatment. Eur. J. Pharm. Biopharm. 2016, 108, 25–31. [Google Scholar] [CrossRef]
- Sridhar, I.; Doshi, A.; Joshi, B.; Wankhede, V.; Doshi, J. Solid dispersions: An approach to enhance solubility of poorly water soluble drug. J. Sci. Innov. Res. 2013, 2, 685–694. [Google Scholar]
- Papadimitriou, S.A.; Bikiaris, D.; Avgoustakis, K. Microwave induced enhancement of the dissolution rate of poorly water soluble tibolone from poly (ethylene glycol) solid dispersions. J. Appl. Polym. Sci. 2008, 108, 1249–1258. [Google Scholar] [CrossRef]
- Barba, A.A.; Dalmoro, A.; D’Amore, M. Microwave assisted drying of cellulose derivative (HPMC) granular solids. Powder Technol. 2013, 237, 581–585. [Google Scholar] [CrossRef]
- Kong, S.H.; Lam, S.S.; Yek, P.N.Y.; Liew, R.K.; Ma, N.L.; Osman, M.S.; Wong, C.C. Self purging microwave pyrolysis: An innovative approach to convert oil palm shell into carbon rich biochar for methylene blue adsorption. J. Chem. Technol. Biotechnol. 2019, 94, 1397–1405. [Google Scholar] [CrossRef]
- Isaac, J.; Kaity, S.; Ganguly, S.; Ghosh, A. Microwave-induced solid dispersion technology to improve bioavailability of glipizide. J. Pharm. Pharmacol. 2013, 65, 219–229. [Google Scholar] [CrossRef]
- Shi, N.; Wang, S.; Zhang, Y.; Huo, J.; Wang, L.; Cai, J.; Li, Z.; Xiang, B.; Qi, X. Hot melt extrusion technology for improved dissolution, solubility and ‘‘spring-parachute’’ processes of amorphous self-micellizing solid dispersions containing BCS II drugs indomethacin and fenofibrate: Profiles and mechanisms. Eur. J. Pharm. Sci. 2019, 130, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Knopp, M.M.; Berthelsen, R.; Löbmann, K. Convection-induced vs. microwave radiation-induced in situ drug amorphization. Molecules 2020, 25, 1068. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Knopp, M.M.; Berthelsen, R.; Zeitler, J.A.; Löbmann, K. The influence of drug and polymer particle size on the in situ amorphization using microwave irradiation. Eur. J. Pharm. Biopharm. 2020, 149, 77–84. [Google Scholar] [CrossRef]
- Serajuddin, A.T. Solid dispersion of poorly water soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | API | Upstream Processes | Inducements of in situ Amorphization | Post-Amorphization Dosage Form | Impact of in situ Amorphization | Ref. |
|---|---|---|---|---|---|---|
| Dissolution-mediated in situ amorphization | Paracetamol, ibuprofen (IBU), ketoprofen and naproxen | Mixing (API–polyethylene oxide), compaction | Moisture uptake during storage (room temperature (RT), 94% relative humidity (RH)) of compacts for 3–4 weeks | Compact | NA | [26] |
| IBU | Dry mixing for 3 h (API–hydroxypropyl methyl cellulose) | Methanol or water spray followed by milling for 1–3 h; storage in saturated methanol vapor for 16 h; saturated methanol vapor followed by milling for 16 h | Powder | NA | [26] | |
| Indomethacin (IND) | Dry mixing for 3 h (API–hydroxypropyl methyl cellulose) | Saturated methanol vapor followed by milling for 16 h | ||||
| IND | Mixing (API–Eudragit® E), compaction | Immersion in pH 6.8 phosphate buffer for 1–3 h at 37 °C | Compact | Improved dissolution behavior when the swelling degree was high | [26] | |
| Naproxen, IBU | Mixing (API–Eudragit® E), compaction | Immersion in 50 mL purified water for 1 h | Compact | Improved dissolution behavior | [48] | |
| IND | Mixing (API–L-arginine), blending, compaction, coating (Kollicoat® Protect) | Moisture uptake during storage (RT, 75% RH), not completely amorphized even after 91 days | Coated compact | Good mechanical stability; improved bioavailability | [40] | |
| IND | Mixing (API–L-arginine) | Moisture uptake during storage (RT, 75% RH), not completely amorphized even after 101 days | Powder | Chemical degradation | [49] | |
| Furosemide, IND | Mixing (API–L-arginine), blending, compaction, coating (Eudragit® L) | Immersion in 0.1 M HCl for 10–30 min at 37 °C (co-amorphization); recrystallization after longer immersion times | Coated compact | L-arginine induced chemical degradation of IND (prevented by adding citric acid but at the expense of amorphization) | [50] | |
| Carvedilol | Mixing (API–L-aspartic acid), blending, compaction, coating (Eudragit® L) | Immersion in 0.1 M HCl for 45 min at 37 °C (co-amorphization) | Coated compact | Insufficient disintegration and poor drug release | [51] | |
| Vapor-mediated in situ amorphization | Aspirin, phenacetin | Mixing (API–magnesium aluminum silicate/activate carbon) | Storage at 25 °C with silica gel for 1–2 week; storage at 40 °C with reduced pressure for 2–8 h | Powder | NA | [26] |
| IBU | Mixing (API–silica) | Storage at 40 °C, 0% RH for 4–5 weeks; hydrophilic, small pore diameter silica was preferred | Powder | Potential instabilities due to drug molecules migration and subsequent interaction with excipients | [26] | |
| Diflunisal | Mixing (API–silica) | Storage at 80 °C, 0% RH for 2–3 weeks | ||||
| In situ amorphization during lipolysis | Cinnarizine, halofantrine and simvastatin | Preparing the self-microemulsifying drug delivery systems (SMEDDS) or the self-nanoemulsifying drug delivery systems (SNEDDS) | Immersion in lipolysis medium for approx. 1 h | SMEDDS or SNEDDS | Unintended partial amorphous precipitation. Lack of correlation of in vitro precipitation to the in vivo performance | [26] |
| Year | Microwave Instrument | Power (W) | Processing Time (min) | Microwave Absorbing Sample Holder | API | Carrier | Cooling Method | Drug Released % | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 2008 | CE297DN, Samsung | 600 | 6 | Beaker | IBU | β-CD; PVP/VA 64 (did not achieve fully amorphous) | NA | 90 (2 min) | [44] |
| 2009 | CE297DN, Samsung | 600 | 15; 9 | Beaker | Nimesulide | Gelucire® 50/13; Poloxamer 188 | NA | >90 (16 min); >90 (70 s) | [66] |
| 2010 | CATA-2R, Catalyst Systems, Pune, India | 590 | 3; 4; 5; 6 | Beaker | Atorvastatin calcium | PEG6000 | RT | 52; 57; 61; 64 (120 min) | [67] |
| 2010 | CE297DN-Samsung, Surrey, England | 600 | 10 | Beaker | Itraconazole | D-α-tocopheryl polyethylene glycol 1000 succinate | RT | >90 (2 min) | [68] |
| 2011 | Modified domestic microwave oven | Variable power | Around 30 | Crucible | IBU | Stearic acid; PVP 40 | RT | >60 (20 min) | [43] |
| 2012 | CATA-2R, Catalyst Systems, Pune, India | 600 | 3; 4; 5; 6 | Glass beaker | Repaglinide | Poloxamer 188 | NA | 68; 73; 80; 82 (60 min) | [69] |
| 2013 | CATA-2R, Catalyst Systems, Pune, India | 590 | 3; 4; 5; 6 | Beaker | Raloxifene | HPMC E5 LV | RT | 50; 57; 60; 65 (120 min) | [70] |
| 2013 | CATA-2R, Catalyst Systems, Pune, India | 600 | 5 | Glass beaker | Repaglinide | PEG 6000 | NA | 86 (120 min) | [71] |
| 2013 | Catalyst systems, Pune, India | 440 | NA | NA | Glipizide | PEG 4000 | Ice bath | NA | [83] |
| 2014 | CATA-2R, Catalyst Systems, Pune, India | 560 | NA | Glass beaker | Candesartan Cilexetil | PEG 6000; HPMC E5 | NA | > 90 (5 min) | [72] |
| 2016 | P70F23P-G5(SO), Glanze | 550 | 10 | Crucible | IBU | Soluplus® | Liquid nitrogen (−196 °C) | NA | [73] |
| 2017 | ME0113M1, Samsung | 900 | Different time of interval | Glass beaker | Mefenamic acid; flufenamic acid | Pluronic F127®; Eudragit EPO®; PEG 4000; Gelucire 50/13 | RT, with one exception under −80 °C | 80 (40 min) for optimum prescriptions | [74] |
| 2019 | P70F23P-G5(SO), Galz, Guangzhou, China | 700 | 2 | Porcelain dish | IND; fenofibrate | Soluplus® | Liquid nitrogen at −196 °C | NA | [84] |
| 2020 | ME0113M1, Samsung | 500 | NA | Glass beaker | Luteolin | PEG 4000 | NA | >60 (20 min) | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiang, W.; Löbmann, K.; McCoy, C.P.; Andrews, G.P.; Zhao, M. Microwave-Induced In Situ Amorphization: A New Strategy for Tackling the Stability Issue of Amorphous Solid Dispersions. Pharmaceutics 2020, 12, 655. https://doi.org/10.3390/pharmaceutics12070655
Qiang W, Löbmann K, McCoy CP, Andrews GP, Zhao M. Microwave-Induced In Situ Amorphization: A New Strategy for Tackling the Stability Issue of Amorphous Solid Dispersions. Pharmaceutics. 2020; 12(7):655. https://doi.org/10.3390/pharmaceutics12070655
Chicago/Turabian StyleQiang, Wei, Korbinian Löbmann, Colin P. McCoy, Gavin P. Andrews, and Min Zhao. 2020. "Microwave-Induced In Situ Amorphization: A New Strategy for Tackling the Stability Issue of Amorphous Solid Dispersions" Pharmaceutics 12, no. 7: 655. https://doi.org/10.3390/pharmaceutics12070655