On the Usefulness of Two Small-Scale In Vitro Setups in the Evaluation of Luminal Precipitation of Lipophilic Weak Bases in Early Formulation Development


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Physicochemical and Pharmacokinetic Properties of Model Drugs
2.2.2. Preparation of Level II FaSSIF V2 × 10 Concentrated for Biphasic Dissolution Experiments
2.2.3. Preparation of Level II FaSSIF V2 × 4 Concentrated for D-P Experiments
2.2.4. Dose Selection
2.2.5. Biphasic Dissolution Test
2.2.6. Biphasic Emulsification Risk Investigation
2.2.7. D-P Experiments
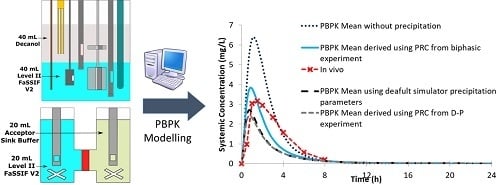
2.2.8. PBPK Modelling
3. Results and Discussion
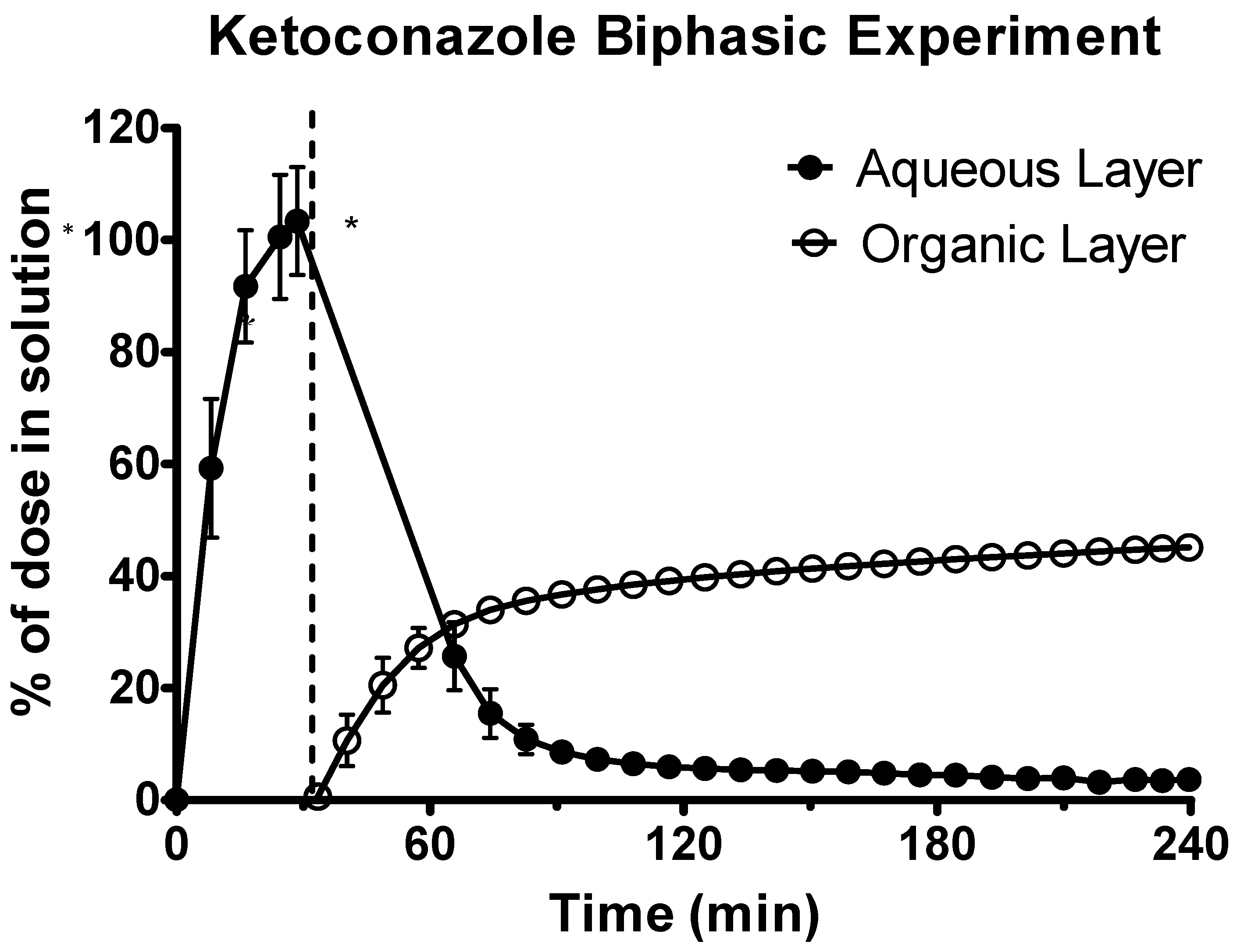
3.1. Data on the Emulsification Risk in the Biphasic Experiments
3.2. Data from the Biphasic Experiments
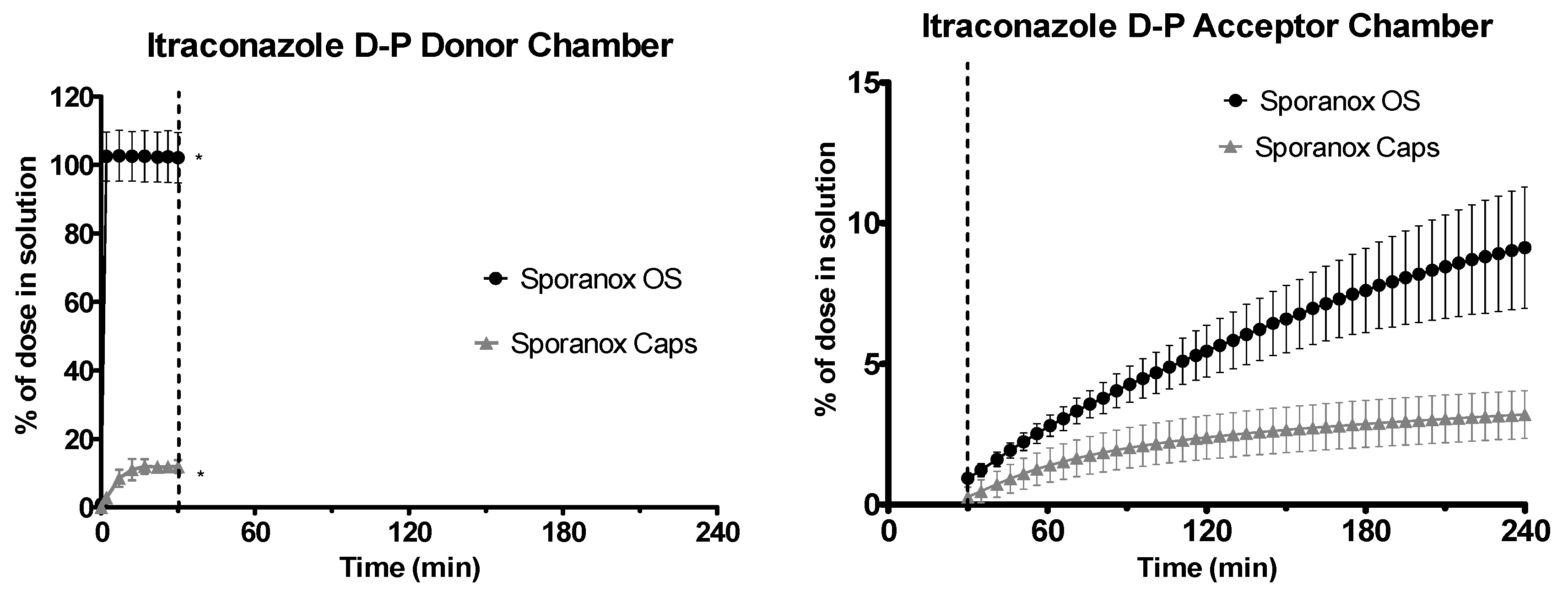
3.3. Data from the D-P Experiments
3.4. Limitations of UV Probes
3.5. PBPK Modelling
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Dwyer, P.J.; Litou, C.; Box, K.J.; Dressman, J.B.; Kostewicz, E.S.; Kuentz, M.; Reppas, C. In vitro methods to assess drug precipitation in the fasted small intestine—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 536–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.; Reppas, C. In-vitro simulation of luminal conditions for evaluation of performance of oral drug products: Choosing the appropriate test media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.J.; Pygall, S.R.; Cooper, V.B.; Mann, J.C. Overcoming sink limitations in dissolution testing: A review of traditional methods and the potential utility of biphasic systems. J. Pharm. Pharmacol. 2012, 64, 1549–1559. [Google Scholar] [CrossRef]
- Frank, K.J.; Locher, K.; Zecevic, D.E.; Fleth, J.; Wagner, K.G. In vivo predictive mini-scale dissolution for weak bases: Advantages of pH-shift in combination with an absorptive compartment. Eur. J. Pharm. Sci. 2014, 61, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.; Borghardt, J.M.; Frank, K.J.; Kloft, C.; Wagner, K.G. Evolution of a mini-scale biphasic dissolution model: Impact of model parameters on partitioning of dissolved API and modelling of in vivo-relevant kinetics. Eur. J. Pharm. Biopharm. 2016, 105, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Gabriëls, M.; Plaizier-Vercammen, J. Design of a dissolution system for the evaluation of the release rate characteristics of artemether and dihydroartemisinin from tablets. Int. J. Pharm. 2004, 274, 245–260. [Google Scholar] [CrossRef]
- Grundy, J.S.; Anderson, K.E.; Rogers, J.A.; Foster, R.T. Studies on dissolution testing of the nifedipine gastrointestinal therapeutic system. I. Description of a two-phase in vitro dissolution test. J. Control. Release 1997, 48, 1–8. [Google Scholar] [CrossRef]
- Hoa, N.T.; Kinget, R. Design and evaluation of two-phase partition-dissolution method and its use in evaluating artemisinin tablets. J. Pharm. Sci. 1996, 85, 1060–1063. [Google Scholar]
- Shi, Y.; Gao, P.; Gong, Y.; Ping, H. Application of a biphasic test for characterization of in vitro drug release of immediate release formulations of celecoxib and its relevance to in vivo absorption. Mol. Pharm. 2010, 7, 1458–1465. [Google Scholar] [CrossRef]
- Xu, H.; Shi, Y.; Vela, S.; Marroum, P.; Gao, P. Developing Quantitative In Vitro–In Vivo Correlation for Fenofibrate Immediate-Release Formulations with the Biphasic Dissolution-Partition Test Method. J. Pharm. Sci. 2018, 107, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Tsume, Y.; Igawa, N.; Drelich, A.J.; Amidon, G.E.; Amidon, G.L. The Combination of GIS and Biphasic to Better Predict In Vivo Dissolution of BCS Class IIb Drugs, Ketoconazole and Raloxifene. J. Pharm. Sci. 2018, 107, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Vela, S.; Shi, Y.; Marroum, P.; Gao, P. In Vitro Characterization of Ritonavir Drug Products and Correlation to Human in Vivo Performance. Mol. Pharm. 2017, 14, 3801–3814. [Google Scholar] [CrossRef] [PubMed]
- Jede, C.; Wagner, C.; Kubas, H.; Weber, C.; Weitschies, W. In-line derivative spectroscopy as a promising application to a small-scale in vitro transfer model in biorelevant supersaturation and precipitation testing. J. Pharm. Pharmacol. 2018, 70, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, S.; O’Dwyer, P.J.; Box, K.J.; Imanidis, G.; Reppas, C.; Kuentz, M. European Journal of Pharmaceutical Sciences Biphasic drug release testing coupled with di ff using wave spectroscopy for mechanistic understanding of solid dispersion performance. Eur. J. Pharm. Sci. 2019, 137, 105001. [Google Scholar] [CrossRef]
- Tsinman, K.; Tsinman, O.; Lingamaneni, R.; Zhu, S.; Riebesehl, B.; Grandeury, A.; Juhnke, M.; Van Eerdenbrugh, B. Ranking Itraconazole Formulations Based on the Flux through Artificial Lipophilic Membrane. Pharm. Res. 2018, 35, 161. [Google Scholar] [CrossRef]
- Stewart, A.M.; Grass, M.E.; Brodeur, T.J.; Goodwin, A.K.; Morgen, M.M.; Friesen, D.T.; Vodak, D.T. Impact of Drug-Rich Colloids of Itraconazole and HPMCAS on Membrane Flux in Vitro and Oral Bioavailability in Rats. Mol. Pharm. 2017, 14, 2437–2449. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.Z.X.; Ho, M.-C.D.; Gemski, C.K.; Chuang, B.-C.; Liao, M.; Xia, C.Q. Utilizing In Vitro Dissolution-Permeation Chamber for the Quantitative Prediction of pH-Dependent Drug-Drug Interactions with Acid-Reducing Agents: A Comparison with Physiologically Based Pharmacokinetic Modeling. AAPS J. 2016, 18, 1512–1523. [Google Scholar] [CrossRef]
- Bevernage, J.; Brouwers, J.; Clarysse, S.; Vertzoni, M.; Tack, J.A.N.; Annaert, P.; Augustijns, P. Drug Supersaturation in Simulated and Human Intestinal Fluids Representing Different Nutritional States. J. Pharm. Sci. 2010, 99, 4525–4534. [Google Scholar] [CrossRef]
- Boehringer Ingelheim Limited. Persantin Ampoules 10 mg/2 mL Solution for Infusion. Professional Leaflet; Boehringer Ingelheim: Berks, UK, 2015. [Google Scholar]
- Huang, Y.; Colaizzi, J.L.; Bierman, R.H.; Woestenborghs, R.; Heykants, J. Pharmacokinetics and Dose Proportionality of Ketoconazole in Normal Volunteers. Antimicrob. Agents Chemother. 1986, 30, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Heykants, J.; Van Peer, A.; Van de Velde, V.; Van Rooy, P.; Meuldermans, W.; Lavrijsen, K.; Woestenborghs, R.; Van Cutsem, J.; Cauwenbergh, G. The Clinical Pharmacokinetics of Itraconazole: An Overview. Mycoses 1989, 32, 67–87. [Google Scholar] [CrossRef]
- Ricevuti, G.; Mazzone, A.; Pasotii, D.; Uccelli, E.; Pasquali, F.; Gazzani, G.; Fregnan, G.B. Pharmacokinetics of dipyridamole-B-cyclodextrin complex in healthy volunteers after single and multiple doses. Eur. J. Drug Metab. Pharmacokinet. 1991, 16, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Daneshmend, T.K.; Warnock, D.W.; Ene, M.D.; Johnson, E.M.; Potten, M.R.; Richardson, M.D.; Williamson, P.J. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob. Agents Chemother. 1984, 25, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, J.; Geboers, S.; Mols, R.; Tack, J.; Augustijns, P. Gastrointestinal behavior of itraconazole in humans—Part 1: Supersaturation from a solid dispersion and a cyclodextrin-based solution. Int. J. Pharm. 2017, 525, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.M.; Schaefer, K.J.; Jamei, M.; Turner, D.B. Biopharmaceutic IVIVE Mechanistic Modeling of Single- and Two-Phase In Vitro Experiments to Obtain Drug-Specific Parameters for Incorporation Into PBPK Models. J. Pharm. Sci. 2019, 108, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Skolnik, S.; Lin, X.; Wang, J.; Chen, X.-H.; He, T.; Zhang, B. Towards Prediction of In Vivo Intestinal Absorption Using a 96-Well Caco-2 Assay. J. Pharm. Sci. 2010, 99, 3246–3265. [Google Scholar] [CrossRef]
- Hintz, R.J.; Johnson, K.C. The effect of particle size distribution on dissolution rate and oral absorption. Int. J. Pharm. 1989, 51, 9–17. [Google Scholar] [CrossRef]
- Nielsen-Kudsk, F.; Pedersen, A.K. Pharmacokinetics of Dipyridamole. Acta Pharmacol. Toxicol. 1979, 44, 391–399. [Google Scholar] [CrossRef]
- Martínez-Jordá, R.; Rodriguez-Sasiain, J.M.; Suárez, E.; Calvo, R. Serum binding of ketoconazole in health and disease. Int. J. Clin. Pharmacol. Res. 1990, 10, 271–276. [Google Scholar]
- Pathak, S.M.; Ruff, A.; Kostewicz, E.S.; Patel, N.; Turner, D.B.; Jamei, M. Model-Based Analysis of Biopharmaceutic Experiments to Improve Mechanistic Oral Absorption Modeling: An Integrated in Vitro in Vivo Extrapolation Perspective Using Ketoconazole as a Model Drug. Mol. Pharm. 2017, 14, 4305–4320. [Google Scholar] [CrossRef] [Green Version]
- Cristofoletti, R.; Patel, N.; Dressman, J.B. Differences in Food Effects for 2 Weak Bases With Similar BCS Drug-Related Properties: What Is Happening in the Intestinal Lumen? J. Pharm. Sci. 2016, 105, 2712–2722. [Google Scholar] [CrossRef] [Green Version]
- Matsui, K.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. The Evaluation of In Vitro Drug Dissolution of Commercially Available Oral Dosage Forms for Itraconazole in Gastrointestinal Simulator with Biorelevant Media. J. Pharm. Sci. 2016, 105, 2804–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, J.; Neeskens, P.; Tollenaere, J.P.; Van Remoortere, P.; Brewster, M.E. Characterization of the interaction of 2-hydroxypropyl-β-cyclodextrin with itraconazole at pH 2, 4, and 7. J. Pharm. Sci. 2002, 91, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.-M.; Cheymol, G. Optimisation of Itraconazole Therapy Using Target Drug Concentrations. Clin. Pharmacokinet. 1998, 35, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Hens, B.; Pathak, S.M.; Mitra, A.; Patel, N.; Liu, B.; Patel, S.; Jamei, M.; Brouwers, J.; Augustijns, P.; Turner, D.B. In Silico Modeling Approach for the Evaluation of Gastrointestinal Dissolution, Supersaturation, and Precipitation of Posaconazole. Mol. Pharm. 2017, 14, 4321–4333. [Google Scholar] [CrossRef] [PubMed]
- Owen, T. Sample handling and measurement. In Fundamentals of Modern UV-Visible Spectroscopy: A Primer; Agilent Technologies: Waldbronn, Germany, 2000; pp. 55–78. [Google Scholar]
- Mudie, D.M.; Shi, Y.; Ping, H.; Gao, P.; Amidon, G.L.; Amidon, G.E. Mechanistic analysis of solute transport in an in vitro physiological two-phase dissolution apparatus. Biopharm. Drug Dispos. 2012, 33, 378–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, C.M.; Tang, K.; Chokshi, H.; Fotaki, N. Surface Dissolution UV Imaging for Investigation of Dissolution of Poorly Soluble Drugs and Their Amorphous Formulation. AAPS PharmSciTech 2019, 20, 113. [Google Scholar] [CrossRef] [Green Version]
- Alvebratt, C.; Cheung, O.; Strømme, M.; Bergström, C.A.S. A Modified In Situ Method to Determine Release from a Complex Drug Carrier in Particle-Rich Suspensions. AAPS PharmSciTech 2018, 19, 2859–2865. [Google Scholar] [CrossRef]
- Psachoulias, D.; Vertzoni, M.; Goumas, K.; Kalioras, V.; Beato, S.; Butler, J.; Reppas, C. Precipitation in and supersaturation of contents of the upper small intestine after administration of two weak bases to fasted adults. Pharm. Res. 2011, 28, 3145–3158. [Google Scholar] [CrossRef]
- Hens, B.; Brouwers, J.; Corsetti, M.; Augustijns, P. Supersaturation and Precipitation of Posaconazole Upon Entry in the Upper Small Intestine in Humans. J. Pharm. Sci. 2016, 105, 2677–2684. [Google Scholar] [CrossRef] [Green Version]
- Kourentas, A.; Vertzoni, M.; Barmpatsalou, V.; Augustijns, P.; Beato, S.; Butler, J.; Holm, R.; Ouwerkerk, N.; Rosenberg, J.; Tajiri, T.; et al. The BioGIT System: A Valuable In Vitro Tool to Assess the Impact of Dose and Formulation on Early Exposure to Low Solubility Drugs after Oral Administration. AAPS J. 2018, 20, 71. [Google Scholar] [CrossRef]
- Ruff, A.; Fiolka, T.; Kostewicz, E.S. Prediction of Ketoconazole absorption using an updated in vitro transfer model coupled to physiologically based pharmacokinetic modelling. Eur. J. Pharm. Sci. 2017, 100, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hens, B.; Talattof, A.; Paixão, P.; Bermejo, M.; Tsume, Y.; Löbenberg, R.; Amidon, G.L. Measuring the Impact of Gastrointestinal Variables on the Systemic Outcome of Two Suspensions of Posaconazole by a PBPK Model. AAPS J. 2018, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Berben, P.; Stappaerts, J.; Vink, M.J.A.; Domínguez-vega, E.; Somsen, G.W.; Brouwers, J.; Augustijns, P. Linking the concentrations of itraconazole and 2-hydroxypropyl-β-cyclodextrin in human intestinal fluids after oral intake of Sporanox®. Eur. J. Pharm. Biopharm. 2018, 132, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, E.; Thörn, H.; Tannergren, C. In Silico Modeling of Gastrointestinal Drug Absorption: Predictive Performance of Three Physiologically Based Absorption Models. Mol. Pharm. 2016, 13, 1763–1778. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Vertzoni, M.; Reppas, C. Physiologically based absorption modeling of salts of weak bases based on data in hypochlorhydric and achlorhydric biorelevant media. AAPS PharmSciTech 2018, in press. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter (Units) | Values Used | Refs/Comments |
|---|---|---|
| Physchem and Blood Binding Parameters | ||
| Mol wt (g/mol) | 504.6 | |
| Log Po:w | 3.97 | In house experimental database |
| Compound type | Monoprotic base | In house experimental database |
| pKa | 6.07 | In house experimental database |
| Fraction unbound in plasma | 0.002 | Pathak et al. [25] |
| Blood plasma ratio | 0.56 | Predicted in Simcyp |
| Fraction unbound in enterocyte | 1 | Simcyp compound file |
| Drug Absorption Parameters (ADAM Model) | ||
| Apparent Caco-2 cell permeability (×10−6 cm/s) apical pH 7.4 - basolateral pH 7.4 | 11.24 | Skolnik et al. [26] |
| Calibrator compounds Caco-2 cell permeability (×10−6 cm/s) | Cimetidine 1.64 Propranolol 21.29 Verapamil 22.68 Metoprolol 17.74 | Skolnik et al. [26] |
| PredictedPeff,man (×10−4 cm/s) | 2.90 | Predicted in Simcyp using Caco-2 cell permeability |
| Aqueous (Aq) intrinsic solubility (mg/mL) | 0.0065 | Calculated used pH solubility profile |
| Solubility Factor (SF) | 1081 | Estimated in Simcyp |
| Diffusion Layer Model (DLM) scalar | 1 | Simcyp default value |
| Particle density (g/mL) | 1.2 | Default Simcyp value |
| Particle size distribution | Monodispersed | Default Simcyp value |
| Particle radius (µm) | 10 | Default Simcyp value |
| Log bile micellar: buffer partition coefficient (Log Km:w) neutral | 4.65 | Estimated in Simcyp in vitro analysis (SIVA) toolkit |
| Log Km:w ion | 4.07 | Estimated in SIVA |
| Particle diffusion layer thickness (heff) prediction | Hintz–Johnson method | Default Simcyp method [27] |
| Critical Supersaturation Ratio (CSR) | 13.33 | Calculated from experimental data, this study |
| Precipitation Rate Constant (PRC) (1/h) | 1.08 (from biphasic exp)/2.02 (from D-P exp) | Calculated from experimental data, this study |
| Secondary PRC (sPRC) (1/h) | N/A | |
| Monomer diffusion coeff (10−4 cm2/min) | 3.70 | Predicted in Simcyp |
| Micelle diffusion coeff (10−4 cm2/min) | 0.78 | Default Simcyp value |
| Distribution Parameters | ||
| Model | Full PBPK Model | |
| Method | Method 3 | |
| Tissue-plasma partition coefficient (Kp) scalar | 1 | Pathak et al. [25] |
| Steady State Volume of Distribution (Vss) (L/kg) | 0.41 | Predicted within Simcyp |
| Elimination Parameters | ||
| Intravenous clearance (CLiv) (L/h) | 12 | Persantin® Ampoules 10 mg/2 mL solution for infusion product information [19] |
| Renal clearance (L/h) | 0 | Nielsen-Kudsk and Pedersen [28] |
| Parameter (Units) | Values Used | Refs/Comments |
|---|---|---|
| Physchem and Blood Binding Parameters | ||
| Mol wt (g/mol) | 531.4 | |
| Log Po:w | 3.84 | In house experimental database |
| Compound type | Diprotic base | In house experimental database |
| pKa | 3.16, 6.13 | In house experimental database |
| Fraction unbound in plasma | 0.029 | Martinez-Jorda et al. [29] |
| Blood plasma ratio | 0.62 | Simcyp inhibitor compound file |
| Fraction unbound in enterocyte | 0.06 | Simcyp inhibitor compound file |
| Drug Absorption Parameters (ADAM Model) | ||
| Apparent Caco-2 cell permeability (×10−6 cm/s) apical pH 7.4 - basolateral pH 7.4 | 15.95 | Skolnik et al. [26] |
| Calibrator compounds Caco-2 cell permeability (×10−6 cm/s) | Cimetidine 1.64 Propranolol 21.29 Verapamil 22.68 Metoprolol 17.74 | Skolnik et al. [26] |
| PredictedPeff,man (×10−4 cm/s) | 3.70 | Predicted in Simcyp using Caco-2 cell permeability |
| Aq intrinsic solubility (mg/mL) | 0.0064 | Back calculated using pH solubility data |
| Solubility factor | 2167 | Estimated in SIVA |
| DLM scalar | 1 | Simcyp default value |
| Particle density (g/mL) | 1.2 | Simcyp default value |
| Particle size distribution | Monodispersed | Default Simcyp |
| Particle radius (µm) | 12 | Pathak et al. [30] |
| Log Km:w neutral | 4.30 | Estimated in SIVA |
| Log Km:w ion | 4.28 | Estimated in SIVA |
| Particle heff prediction | Hintz–Johnson method | Default Simcyp method [27] |
| CSR | 16.93 | Calculated from experimental data, this study |
| PRC (1/h) | 2.02 (from biphasic exp)/6.14 (from D-P exp) | Calculated from experimental data, this study |
| sPRC (1/h) | N/A | |
| Monomer diffusion coeff (10−4 cm2/min) | 3.62 | Predicted in Simcyp |
| Micelle diffusion coeff (10−4 cm2/min) | 0.78 | Default Simcyp Value |
| Distribution Parameters | ||
| Model | Full PBPK Model | |
| Method | Method 2 | |
| Kp scalar | 0.012 | Cristofoletti et al. [31] |
| Vss (L/kg) | 0.2 | Predicted within Simcyp |
| Elimination Parameters | ||
| CLiv (L/h) | 14.4 | Huang et al. [20] |
| Renal clearance (L/h) | 0.15 | Pathak et al. [30] |
| Parameter (Units) | Values Used | Refs/Comments |
|---|---|---|
| Physchem and Blood Binding Parameters | ||
| Mol wt (g/mol) | 704.64 | |
| Log Po:w | 5.66 | FDA Label Sporanox |
| Compound type | Monoprotic base | In house experimental database |
| pKa | 3.87 | In house experimental database |
| Fraction unbound in plasma | 0.002 | FDA Label Sporanox |
| Blood plasma ratio | 0.58 | Simcyp inhibitor compound file |
| Fraction unbound in enterocyte | 0.016 | Simcyp inhibitor compound file |
| Drug Absorption Parameters (ADAM Model) | ||
| MCDK II (×10−6 cm/s) | 57.1 | Simcyp inhibitor compound file |
| Calibrator compounds MCDK II permeability (×10−6 cm/s) | Cimetidine 2.00 Propranolol 49.60 Verapamil 59.10 | Simcyp inhibitor compound file |
| PredictedPeff,man (×10−4 cm/s) | 9.85 | Predicted in Simcyp using MCDK cell permeability |
| Aq intrinsic solubility (mg/mL) | 8.94E-05 | Back calculated using pH solubility data |
| Solubility factor | 222.37 | Estimated in SIVA |
| DLM scalar | 1 | Simcyp default value |
| Particle density (g/mL) | 1.2 | Simcyp default value |
| Particle size distribution | Monodispersed | Simcyp default value |
| Particle radius (µm) | 10 | Simcyp default value |
| Log Km:w neutral | 5.62 | Predicted in SIVA |
| Log Km:w ion | 5.48 | Predicted in SIVA |
| Particle heff prediction | Hintz–Johnson method | Default Simcyp method [27] |
| CSR | 44.97 | Calculated from experimental data, this study |
| PRC (1/h) | 1.98 (OS), 2.11 (capsules) | Calculated from experimental data, this study |
| sPRC (1/h) | N/A | |
| Monomer diffusion coeff (10−4 cm2/s) | 3.17 | Predicted within Simcyp |
| Micelle diffusion coeff (10−4 cm2/s) | 0.78 | Default Simcyp Value |
| Solid state 2 (Capsules only) | ||
| Aq. intrinsic solubility (mg/ml) | 0.001 | Matsui et al. [32] |
| DLM scalar | 0.69 | Estimated in SIVA from dissolution profile |
| Excipient Mediated Solubility (OS only) | ||
| Binding constant (M−1) | 1654 (K1:1), 13 (K1:2) | Peeters et al. [33] |
| Binding constant (M−1) stomach | 9895 (K1:1), 23 (K1:2) | Peeters et al. [33] |
| Distribution Parameters | ||
| Model | Full PBPK Model | |
| Method | Method 2 | |
| Kp scalar | 0.19 | Poirier et al. [34] |
| Vss (L/kg) | 11.06 | Predicted within Simcyp |
| Elimination Parameters | ||
| CLiv (L/h) | 22.9 | Heykants et al. [21] |
| Renal clearance (L/h) | 0 | Simcyp inhibitor compound file |
| PK Parameter | Ricevuti et al. ± SD | PBPK Using Experimental Biphasic InForm PRC Value ± SD (% PE) | PBPK Using Experimental D-P PRC Value ± SD (% PE) | PBPK Using Default Simulator Precipitation Values ± SD (% PE) | PBPK with no Precipitation ± SD (% PE) |
|---|---|---|---|---|---|
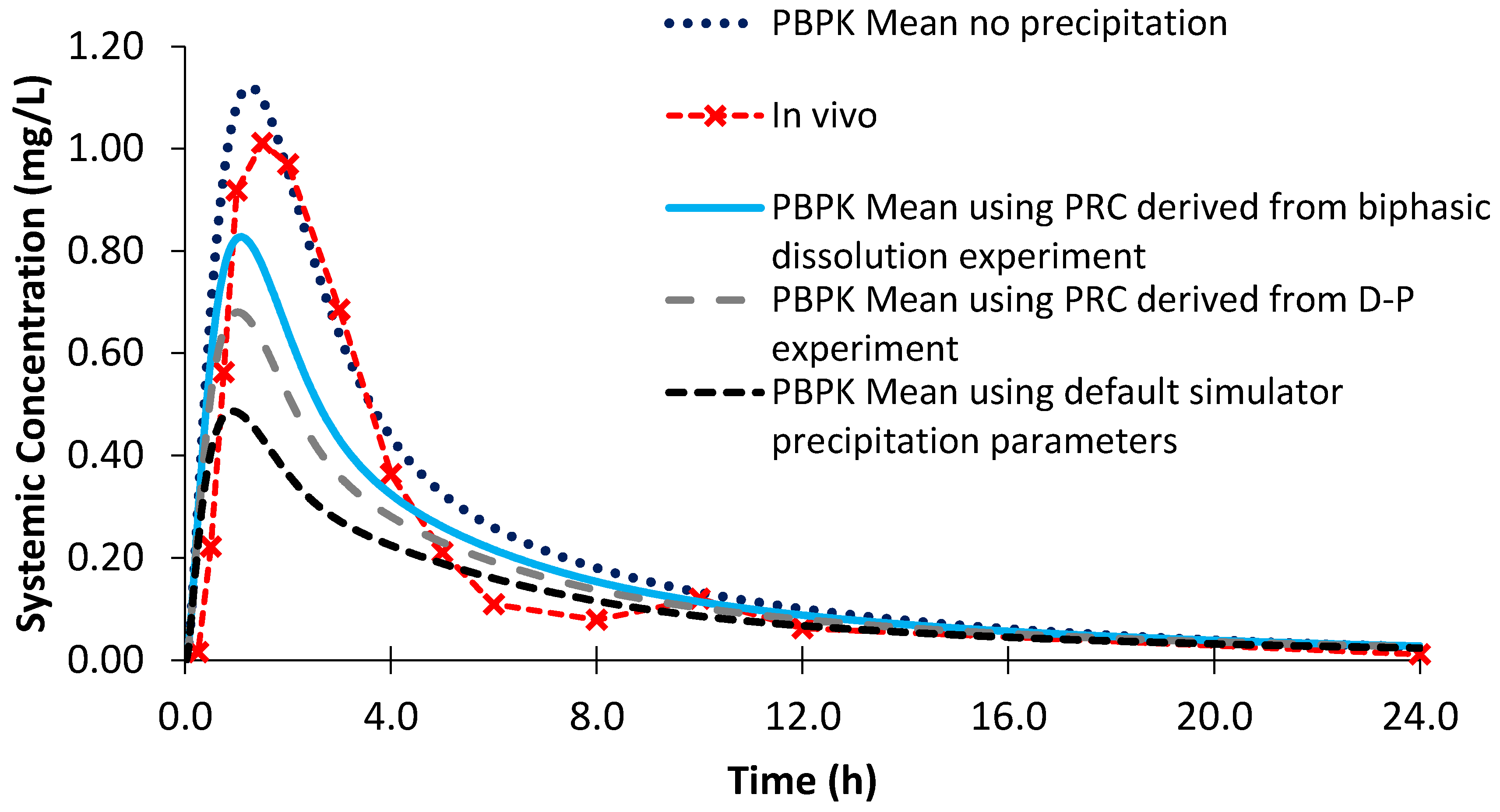
| AUC (mg/L h) | 4.13 ± 0.52 | 4.12 ± 1.49 (0.24%) | 3.58 ± 1.32 (13.32%) | 2.81 ± 1.13 (31.96%) | 5.28 ± 1.98 (27.85%) |
| Cmax (mg/L) | 0.93 ± 0.13 | 0.83 ± 0.21 (10.75%) | 0.71 ± 0.18 (23.66%) | 0.49 ± 0.15 (47.31%) | 1.14 ± 0.29 (22.58%) |
| PK Parameter. | Daneshmend et al. ± SD | PBPK Using Biphasic Experimental Biphasic InForm PRC Value ± SD (% PE) | PBPK Using Experimental D-P PRC Value ± SD (% PE) | PBPK Default Simulator Precipitation Values ± SD (% PE) | PBPK with no Precipitation ± SD (% PE) |
|---|---|---|---|---|---|
| AUC (mg/L h) | 12.9 ± 1.50 | 10.49 ± 4.37 (18.68%) | 7.08 ± 3.27 (45.12%) | 7.44 ± 3.41 (42.33%) | 17.93 ± 7.87 (38.99%) |
| Cmax (mg/L) | 4.36 ± 0.54 | 4.01 ± 1.23 (8.03%) | 2.52 ± 0.99 (42.20%) | 2.80 ± 1.00 (35.78%) | 6.53 ± 2.06 (49.77%) |
| PK Parameter | Brouwers et al. ± SD | PBPK Using Experimental Biphasic InForm PRC and CSR Values ± SD (% PE) | PBPK Using Experimental D-P PRC Value ± SD (% PE) | PBPK Using Default Simulator Precipitation Values ± SD (% PE) | PBPK with no Precipitation ± SD (% PE) |
|---|---|---|---|---|---|
| AUC (mg/L h) | 3.64 | 3.32 ± 1.12 (8.79%) | D-P PRC value not available * | 2.18 ± 0.78 (40.11%) | 3.52 ± 1.17 (3.30 %) |
| Cmax (mg/L) | 0.54 | 0.84 ± 0.23 (55.56%) | 0.37 ± 0.14 (31.48%) | 1.10 ± 0.28 (103.70%) |
| PK Parameter | Brouwers et al. ± SD | PBPK Using Experimental Biphasic InForm PRC Value ± SD (% PE) | PBPK Using Experimental D-P PRC Value ± SD (% PE) | PBPK Using Default Simulator Precipitation Values ± SD (% PE) | PBPK with No Precipitation ± SD (% PE) |
|---|---|---|---|---|---|
| AUC (mg/L h) | 0.87 | 0.67 ± 0.35 (22.99%) | D-P PRC value not available * | 0.58 ± 0.31 (33.33%) | 0.70 ± 0.36 (19.54%) |
| Cmax (mg/L) | 0.084 | 0.089 ± 0.045 (5.95%) | 0.076 ± 0.037 (9.52%) | 0.092 ± 0.047 (9.52%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Dwyer, P.J.; Imanidis, G.; Box, K.J.; Reppas, C. On the Usefulness of Two Small-Scale In Vitro Setups in the Evaluation of Luminal Precipitation of Lipophilic Weak Bases in Early Formulation Development. Pharmaceutics 2020, 12, 272. https://doi.org/10.3390/pharmaceutics12030272
O’Dwyer PJ, Imanidis G, Box KJ, Reppas C. On the Usefulness of Two Small-Scale In Vitro Setups in the Evaluation of Luminal Precipitation of Lipophilic Weak Bases in Early Formulation Development. Pharmaceutics. 2020; 12(3):272. https://doi.org/10.3390/pharmaceutics12030272
Chicago/Turabian StyleO’Dwyer, Patrick J., Georgios Imanidis, Karl J. Box, and Christos Reppas. 2020. "On the Usefulness of Two Small-Scale In Vitro Setups in the Evaluation of Luminal Precipitation of Lipophilic Weak Bases in Early Formulation Development" Pharmaceutics 12, no. 3: 272. https://doi.org/10.3390/pharmaceutics12030272