Paclitaxel Magnetic Core–Shell Nanoparticles Based on Poly(lactic acid) Semitelechelic Novel Block Copolymers for Combined Hyperthermia and Chemotherapy Treatment of Cancer

 ,
, 
 , ,
, ,  , and
, and 
Abstract
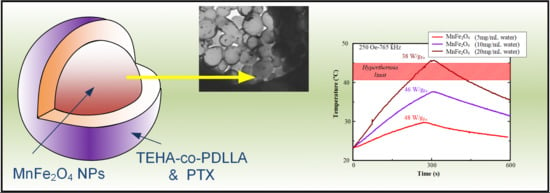
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Synthesis and Characterization of PDLLA and TEHA-co-PDLLA Copolymer
2.2.1. Synthesis of Biocompatible Polymers
2.2.2. Characterization of Biocompatible Block Polymers
2.3. Synthesis and Characterization of MnFe2O4 MNPs
2.3.1. Synthesis of MnFe2O4 MNPs
2.3.2. Characterization of MnFe2O4 MNPs
2.4. Preparation of Polymeric Nanoparticles and Core–Shell Magnetic Nanoparticles
2.5. Characterization of Nanoparticles and Core–Shell Magnetic Nanoparticles
2.6. Drug Release Studies
2.7. Cytotoxicity Studies
2.7.1. Caco-2 and hASCs Cell Culture
2.7.2. Sterilization of Nanoparticles
2.7.3. Measuring Cytotoxicity Levels with the MTT Test after 24 Hours Incubation
3. Results and Discussion
3.1. Characterization of Biocompatible Polymers
3.2. Characterization of MnFe2O4 MNPs
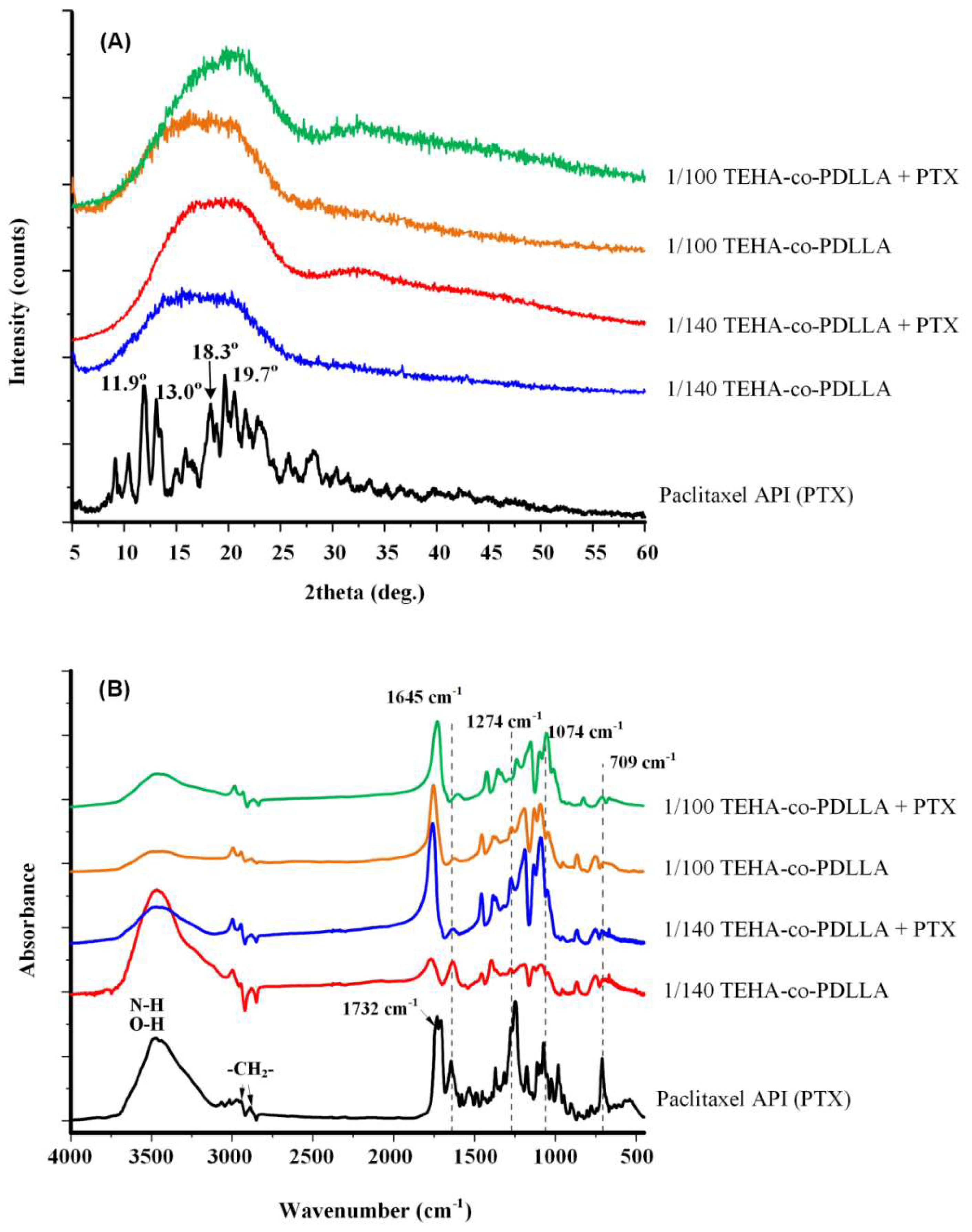
3.3. Characterization of Polymeric Nanoparticles
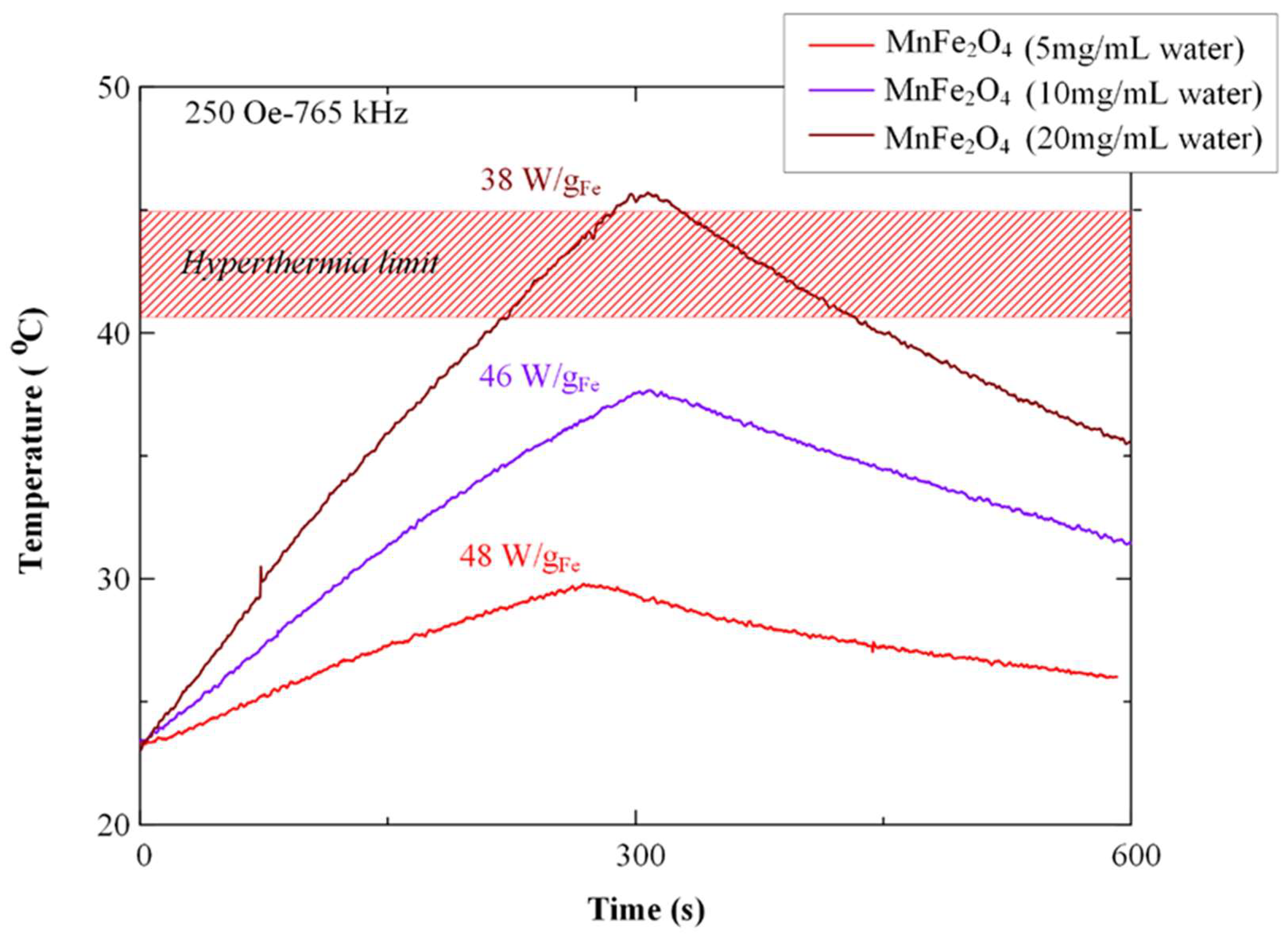
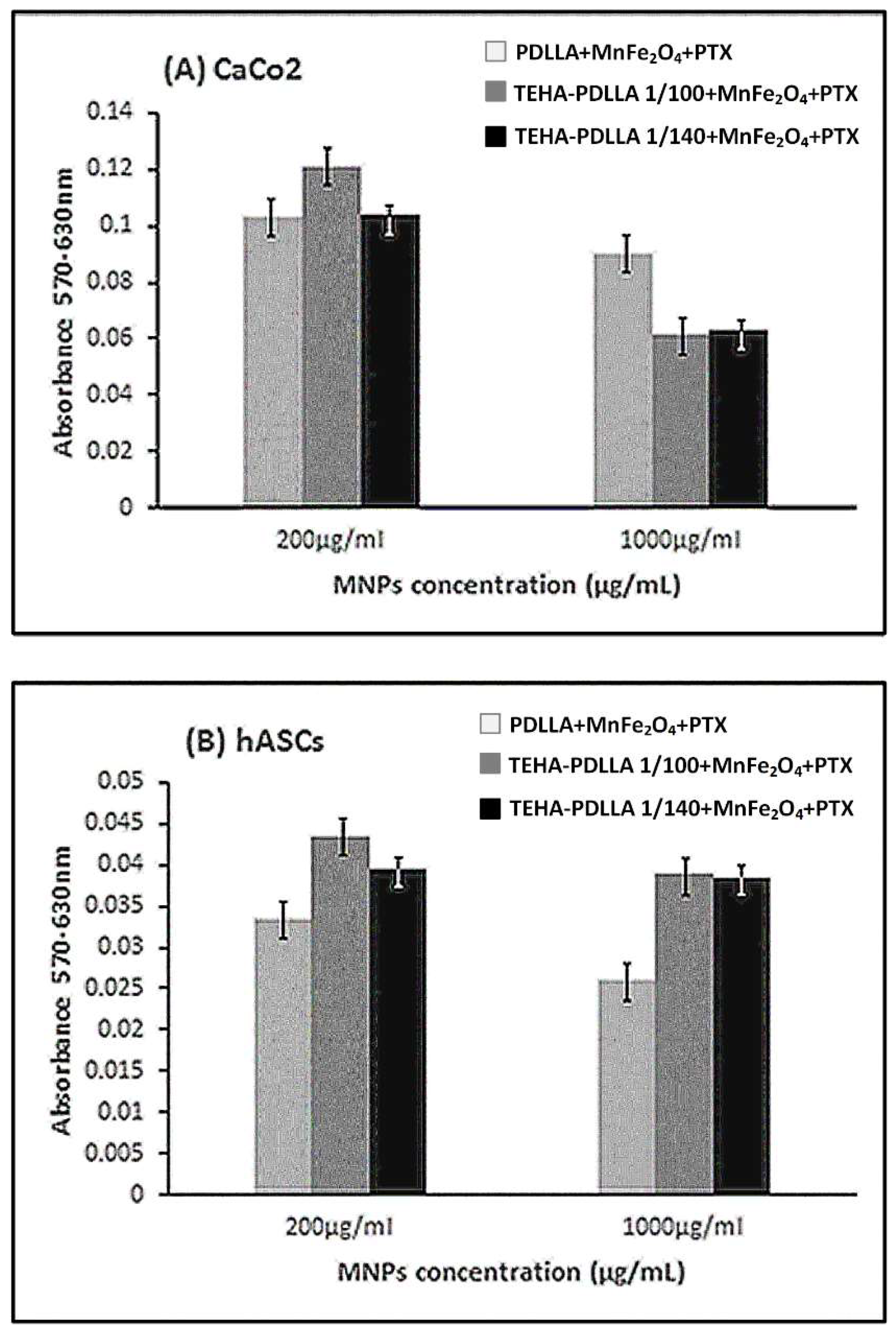
3.4. Characterization of Magnetic Core–Shell Drug-Loaded Nanoparticles
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A



References
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.L.; Zhang, Y.F.; Gao, F.; Li, G.L.; He, Y.; Peng, M.L.; Fan, H.M. Magnetic nanoparticles based cancer therapy: Current status and applications. Sci. China Life Sci. 2018, 61, 400–414. [Google Scholar] [CrossRef]
- Spirou, S.V.; Costa Lima, S.A.; Bouziotis, P.; Vranjes-Djuric, S.; Efthimiadou, E.; Laurenzana, A.; Barbosa, A.I.; Garcia-Alonso, I.; Jones, C.; Jankovic, D.; et al. Recommendations for in vitro and in vivo testing of magnetic nanoparticle hyperthermia combined with radiation therapy. Nanomaterials 2018, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Shen, Z.; Yu, L.; Wei, M.; Li, Y. Manipulating nanoparticle transport within blood flow through external forces: An exemplar of mechanics in nanomedicine. Proc. Soc. A: Math. Phys. Eng. Sci. 2018, 474, 20170845. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Chen, H.; Hu, K.; Delahunty, I.; Gao, S.; Xie, J. Surface impact on nanoparticle-based magnetic resonance imaging contrast agents. Theranostics 2018, 8, 2521–2548. [Google Scholar] [CrossRef]
- DiStasio, N.; Lehoux, S.; Khademhosseini, A.; Tabrizian, M. The Multifaceted Uses and Therapeutic Advantages of Nanoparticles for Atherosclerosis Research. Materials 2018, 11, 754. [Google Scholar] [CrossRef]
- Farina, N.H.; Zingiryan, A.; Vrolijk, M.A.; Perrapato, S.D.; Ades, S.; Stein, G.S.; Lian, J.B.; Landry, C.C. Nanoparticle-based targeted cancer strategies for non-invasive prostate cancer intervention. J. Cell. Physiol. 2018, 233, 6408–6417. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Kraft, J.C.; Yu, D.; Ho, R.J.Y. Recent developments of nanotherapeutics for targeted and long-acting, combination hiv chemotherapy. Eur. J. Pharm. Biopharm. 2019, 138, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, Y.; Li, J.; Hang, Y.; Oupicky, D. Promise of chemokine network-targeted nanoparticles in combination nucleic acid therapies of metastatic cancer. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 11, e1528. [Google Scholar] [CrossRef] [PubMed]
- Fenton, O.S.; Olafson, K.N.; Pillai, P.S.; Mitchell, M.J.; Langer, R. Advances in biomaterials for drug delivery. Adv. Mater. 2018, e1705328. [Google Scholar] [CrossRef]
- Ganipineni, L.P.; Danhier, F.; Preat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef]
- Manzano, M.; Vallet-Regí, M. Mesoporous silica nanoparticles in nanomedicine applications. J. Mater. Sci. Mater. Med. 2018, 29, 65. [Google Scholar] [CrossRef]
- Villegas, M.R.; Baeza, A.; Vallet-Regí, M. Nanotechnological Strategies for Protein Delivery. Molecules 2018, 23, 1008. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhail, A.S.; Allen, C. Block copolymer micelles for delivery of cancer therapy: Transport at the whole body, tissue and cellular levels. J. Control. Release 2009, 138, 214–223. [Google Scholar] [CrossRef]
- Zensi, A.; Begley, D.; Pontikis, C.; Legros, C.; Mihoreanu, L.; Wagner, S.; Büchel, C.; Von Briesen, H.; Kreuter, J. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J. Control. Release 2009, 137, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Giustini, A.J.; Petryk, A.A.; Cassim, S.M.; Tate, J.A.; Baker, I.; Hoopes, P.J. Magnetic nanoparticle hyperthermia in cancer treatment. Nano Life 2010, 1, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Kettering, M.; Grau, I.; Pömpner, N.; Stapf, M.; Gajda, M.; Teichgräber, U.; Hilger, I. Means to increase the therapeutic efficiency of magnetic heating of tumors. Biomed. Eng./Biomed. Tech. 2015, 60, 505–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T. Cancer hyperthermia using magnetic nanoparticles. Biotechnol. J. 2011, 6, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.; Takano, Y.; Winterford, C.; Gobe, G. The Role of Apoptosis in the Response of Cells and Tumours to Mild Hyperthermia. Int. J. Radiat. Biol. 1991, 59, 489–501. [Google Scholar] [CrossRef]
- Gordon, R.T.; Hines, J.R.; Gordon, D. Intracellular hyperthermia. A biophysical approach to cancer treatment via intracellular temperature and biophysical alterations. Med. Hypotheses 1979, 5, 83–102. [Google Scholar] [CrossRef]
- Kufe, D.W.; Holland, J.F.; Frei, E.; Society, A.C. Cancer Medicine 6; BC Decker: New York, NY, USA, 2003. [Google Scholar]
- Filippousi, M.; Papadimitriou, S.A.; Bikiaris, D.N.; Pavlidou, E.; Angelakeris, M.; Zamboulis, D.; Tian, H.; Van Tendeloo, G. Novel core–shell magnetic nanoparticles for Taxol encapsulation in biodegradable and biocompatible block copolymers: Preparation, characterization and release properties. Int. J. Pharm. 2013, 448, 221–230. [Google Scholar] [CrossRef]
- Hervault, A.; Thanh, N.T.K. Magnetic nanoparticle-based therapeutic agents for thermo-chemotherapy treatment of cancer. Nanoscale 2014, 6, 11553–11573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, P.; Giri, J.; Rieken, F.; Koch, C.; Mykhaylyk, O.; Döblinger, M.; Banerjee, R.; Bahadur, D.; Plank, C. Targeted temperature sensitive magnetic liposomes for thermo-chemotherapy. J. Control. Release 2010, 142, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Wust, P.; Hildebrandt, B.; Sreenivasa, G.; Rau, B.; Gellermann, J.; Riess, H.; Félix, R.; Schlag, P. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002, 3, 487–497. [Google Scholar] [CrossRef]
- Rong, H.; Xiaogang, Y.; Jun, S.; Feng, G.; Bifeng, P.; Daxiang, C. Core/shell fluorescent magnetic silica-coated composite nanoparticles for bioconjugation. Nanotechnology 2007, 18, 315601. [Google Scholar]
- Salehizadeh, H.; Hekmatian, E.; Sadeghi, M.; Kennedy, K. Synthesis and characterization of core-shell Fe3O4-gold-chitosan nanostructure. J. Nanobiotechnol. 2012, 10, 3. [Google Scholar] [CrossRef]
- Verma, N.K.; Crosbie-Staunton, K.; Satti, A.; Gallagher, S.; Ryan, K.B.; Doody, T.; McAtamney, C.; MacLoughlin, R.; Galvin, P.; Burke, C.S.; et al. Magnetic core-shell nanoparticles for drug delivery by nebulization. J. Nanobiotechnol. 2013, 11, 1. [Google Scholar] [CrossRef]
- Lee, J.-H.; Huh, Y.-M.; Jun, Y.-W.; Seo, J.-W.; Jang, J.-T.; Song, H.-T.; Kim, S.; Cho, E.-J.; Yoon, H.-G.; Suh, J.-S.; et al. Artificially engineered magnetic nanoparticles for ultra-sensitive molecular imaging. Nat. Med. 2006, 13, 95–99. [Google Scholar] [CrossRef]
- Mikhaylov, G.; Mikac, U.; Magaeva, A.A.; Itin, V.I.; Naiden, E.P.; Psakhye, I.; Babes, L.; Reinheckel, T.; Peters, C.; Zeiser, R.; et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat. Nanotechnol. 2011, 6, 594–602. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 2016, 11, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.L.; Jie, P.; Venkatraman, S.S. Block copolymer ‘stealth’ nanoparticles for chemotherapy: Interactions with blood cells in vitro. Adv. Funct. Mater. 2008, 18, 716–725. [Google Scholar] [CrossRef]
- Romberg, B.; Hennink, W.E.; Storm, G. Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 2008, 25, 55–71. [Google Scholar] [CrossRef]
- Van Butsele, K.; Jérôme, R.; Jérôme, C. Functional amphiphilic and biodegradable copolymers for intravenous vectorisation. Polymer 2007, 48, 7431–7443. [Google Scholar] [CrossRef] [Green Version]
- Rowinsky, E.K.; Donehower, R.C. The clinical pharmacology of paclitaxel (Taxol). Semin. Oncol. 1993, 20, 16–25. [Google Scholar] [PubMed]
- Oĝuz, T.; Meier, M.A.R. Fatty acid derived monomers and related polymers via thiol-ene (click) additions. Macromol. Rapid Commun. 2010, 31, 1822–1826. [Google Scholar]
- Nerantzaki, M.; Adam, K.-V.; Koliakou, I.; Skoufa, E.; Avgeropoulos, A.; Papageorgiou, G.Z.; Bikiaris, D.N. Novel castor oil-derived block copolymers as promising candidates for biological applications: Biorelevant and biocompatible. Macromol. Chem. Phys. 2017, 218, 1700305. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- Nerantzaki, M.; Skoufa, E.; Adam, K.-V.; Nanaki, S.; Avgeropoulos, A.; Kostoglou, M.; Bikiaris, D. Amphiphilic Block Copolymer Microspheres Derived from Castor Oil, Poly(ε-carpolactone), and Poly(ethylene glycol): Preparation, Characterization and Application in Naltrexone Drug Delivery. Materials 2018, 11, 1996. [Google Scholar] [CrossRef]
- Panagi, Z.; Beletsi, A.; Evangelatos, G.; Livaniou, E.; Ithakissios, D.; Avgoustakis, K. Effect of dose on the biodistribution and pharmacokinetics of PLGA and PLGA–mPEG nanoparticles. Int. J. Pharm. 2001, 221, 143–152. [Google Scholar] [CrossRef]
- Georgiadou, V.; Makris, G.; Papagiannopoulou, D.; Vourlias, G.; Dendrinou-Samara, C. Octadecylamine Mediated Versatile Coating of CoFe2O4 NPs for the Sustained Release of Anti-inflammatory Drug Naproxen and in vivo Target Selectivity. ACS Appl. Mater. Interfaces 2016, 8, 9345–9360. [Google Scholar] [CrossRef]
- Beletsi, A.; Leontiadis, L.; Klepetsanis, P.; Ithakissios, D.; Avgoustakis, K. Effect of preparative variables on the properties of poly(dl-lactide-co-glycolide)–methoxypoly(ethyleneglycol) copolymers related to their application in controlled drug delivery. Int. J. Pharm. 1999, 182, 187–197. [Google Scholar] [CrossRef]
- Nerantzaki, M.; Prokopiou, L.; Bikiaris, D.N.; Patsiaoura, D.; Chrissafis, K.; Klonos, P.; Kyritsis, A.; Pissis, P. In situ prepared poly(DL-lactic acid)/silica nanocomposites: Study of molecular composition, thermal stability, glass transition and molecular dynamics. Thermochim. Acta 2018, 669, 16–29. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, B.; Yang, G.; Gauthier, M. Poly(Lactic Acid)-Based Biomaterials: Synthesis, Modification and Applications. In Biomedical Science, Engineering and Technology; Ghista, D.N., Ed.; IntechOpen: London, UK, 2012. [Google Scholar] [Green Version]
- Papageorgiou, G.; Achilias, D.; Nanaki, S.; Beslikas, T.; Bikiaris, D.; Achilias, D. PLA nanocomposites: Effect of filler type on non-isothermal crystallization. Thermochim. Acta 2010, 511, 129–139. [Google Scholar] [CrossRef]
- Vamvakidis, K.; Katsikini, M.; Sakellari, D.; Paloura, E.C.; Kalogirou, O.; Dendrinou-Samara, C. Reducing the inversion degree of MnFe2O4 nanoparticles through synthesis to enhance magnetization: Evaluation of their 1 H NMR relaxation and heating efficiency. Dalton Trans. 2014, 43, 12754–12765. [Google Scholar] [CrossRef] [PubMed]
- Vamvakidis, K.; Sakellari, D.; Angelakeris, M.; Dendrinou-Samara, C. Size and compositionally controlled manganese ferrite nanoparticles with enhanced magnetization. J. Nanopart. 2013, 15, 1743. [Google Scholar] [CrossRef]
- Liggins, R.T.; Hunter, W.L.; Burt, H.M. Solid-State Characterization of Paclitaxel. J. Pharm. Sci. 1997, 86, 1458–1463. [Google Scholar] [CrossRef]
- Avgoustakis, K. Effect of copolymer composition on the physicochemical characteristics, in vitro stability, and biodistribution of PLGA–mPEG nanoparticles. Int. J. Pharm. 2003, 259, 115–127. [Google Scholar] [CrossRef]
- Avgoustakis, K. PLGA–mPEG nanoparticles of cisplatin: In vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control. Release 2002, 79, 123–135. [Google Scholar] [CrossRef]
- Bigham, A.; Foroughi, F.; Motamedi, M.; Rafienia, M. Multifunctional nanoporous magnetic zinc silicate-ZNFE2O4 core–shell composite for bone tissue engineering applications. Ceram. Int. 2018, 44, 11798–11806. [Google Scholar] [CrossRef]
- Hildebrandt, B. The cellular and molecular basis of hyperthermia. Crit. Rev. Oncol. 2002, 43, 33–56. [Google Scholar] [CrossRef]
- Goldstein, L.S.; Dewhirst, M.W.; Repacholi, M.; Kheifets, L. Summary, conclusions and recommendations: Adverse temperature levels in the human body. Int. J. Hyperth. 2003, 19, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. Int. J. Pharm. 2006, 321, 1–11. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
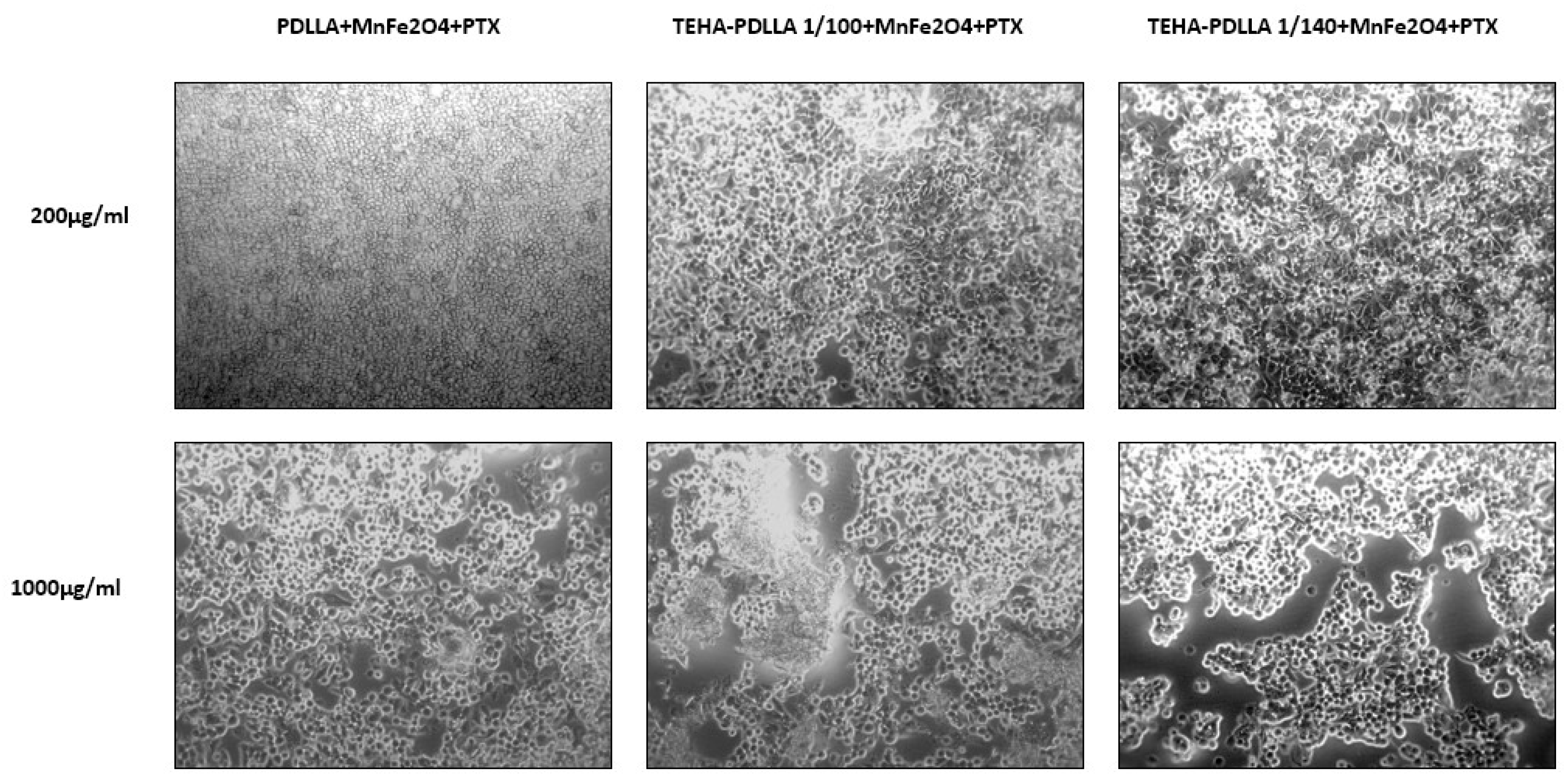{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Molar Ratio TEHA:d,l-lactide | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|
| PDLLA | - | 147,600 | 470,500 | 3.2 |
| TEHA-co- PDLLA 1/5 | 1:5 | 12,200 | 29,700 | 2.4 |
| TEHA-co- PDLLA 1/50 | 1:50 | 29,000 | 107,000 | 3.7 |
| TEHA-co- PDLLA 1/70 | 1:70 | 45,800 | 170,900 | 3.7 |
| TEHA-co- PDLLA 1/140 | 1:140 | 86,200 | 231,000 | 2.7 |
| Polymer Used | Particle Size (nm) | ζ-Potential (mV) | Yield (%) | Entrapment Efficiency (%) |
|---|---|---|---|---|
| TEHA-co-PDLLA 1/100 | 111 ± 10 | -32 | 58.6 ± 2.8 | 6.23 |
| TEHA-co-PDLLA 1/140 | 124 ± 13 | -34 | 75.6 ± 4.2 | 5.61 |
| PDLLA | 140 ± 12 | -28 | 73 ± 1.7 | 5.28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christodoulou, E.; Nerantzaki, M.; Nanaki, S.; Barmpalexis, P.; Giannousi, K.; Dendrinou-Samara, C.; Angelakeris, M.; Gounari, E.; Anastasiou, A.D.; Bikiaris, D.N. Paclitaxel Magnetic Core–Shell Nanoparticles Based on Poly(lactic acid) Semitelechelic Novel Block Copolymers for Combined Hyperthermia and Chemotherapy Treatment of Cancer. Pharmaceutics 2019, 11, 213. https://doi.org/10.3390/pharmaceutics11050213
Christodoulou E, Nerantzaki M, Nanaki S, Barmpalexis P, Giannousi K, Dendrinou-Samara C, Angelakeris M, Gounari E, Anastasiou AD, Bikiaris DN. Paclitaxel Magnetic Core–Shell Nanoparticles Based on Poly(lactic acid) Semitelechelic Novel Block Copolymers for Combined Hyperthermia and Chemotherapy Treatment of Cancer. Pharmaceutics. 2019; 11(5):213. https://doi.org/10.3390/pharmaceutics11050213
Chicago/Turabian StyleChristodoulou, Evi, Maria Nerantzaki, Stavroula Nanaki, Panagiotis Barmpalexis, Kleoniki Giannousi, Catherine Dendrinou-Samara, Makis Angelakeris, Eleni Gounari, Antonis D. Anastasiou, and Dimitrios N. Bikiaris. 2019. "Paclitaxel Magnetic Core–Shell Nanoparticles Based on Poly(lactic acid) Semitelechelic Novel Block Copolymers for Combined Hyperthermia and Chemotherapy Treatment of Cancer" Pharmaceutics 11, no. 5: 213. https://doi.org/10.3390/pharmaceutics11050213