Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HPLC Condition
2.3. Solubility Test
2.4. Characterization of Various Vehicle Compositions
2.5. Particle Size Measurement
2.6. Differential Scanning Calorimetry (DSC)
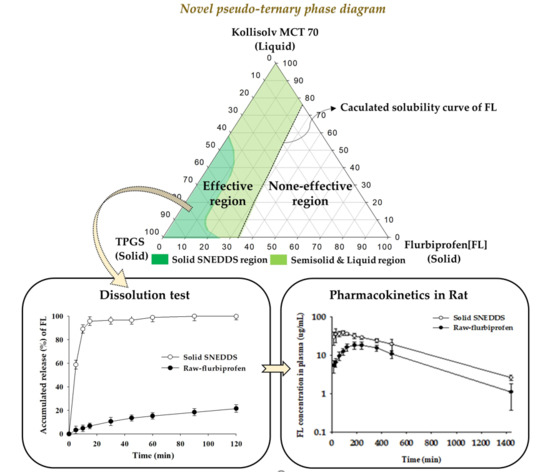
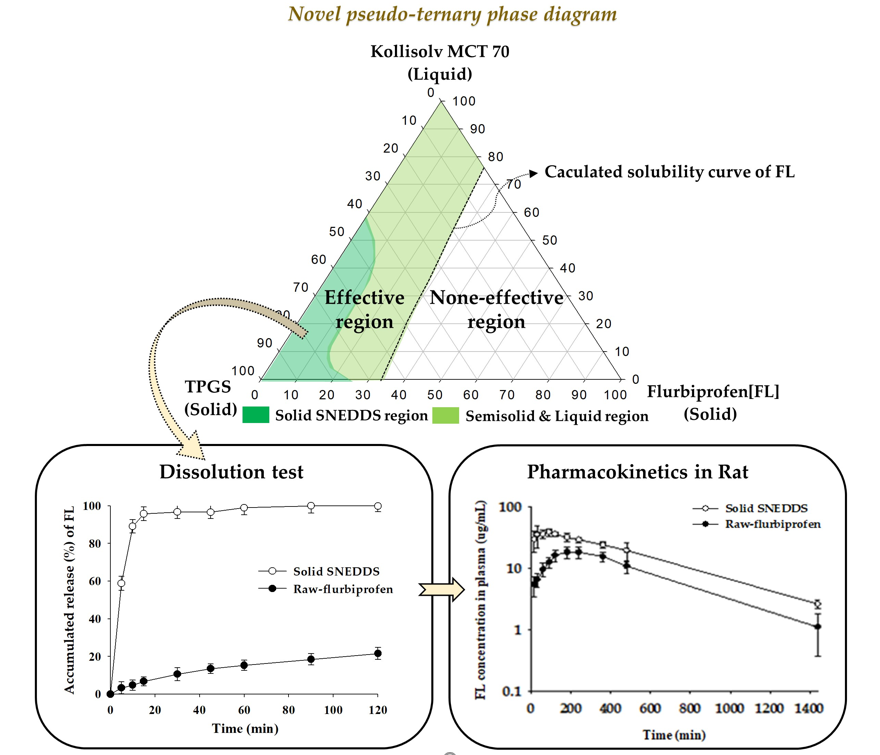
2.7. Construction of Pseudo-Ternary Phase Diagram
- C = predetermined composition (%) of Kollisolv MCT 70. D = predetermined composition (%) of TPGS. C + D + E = 100%.
2.8. Powder X-ray Diffraction (PXRD)
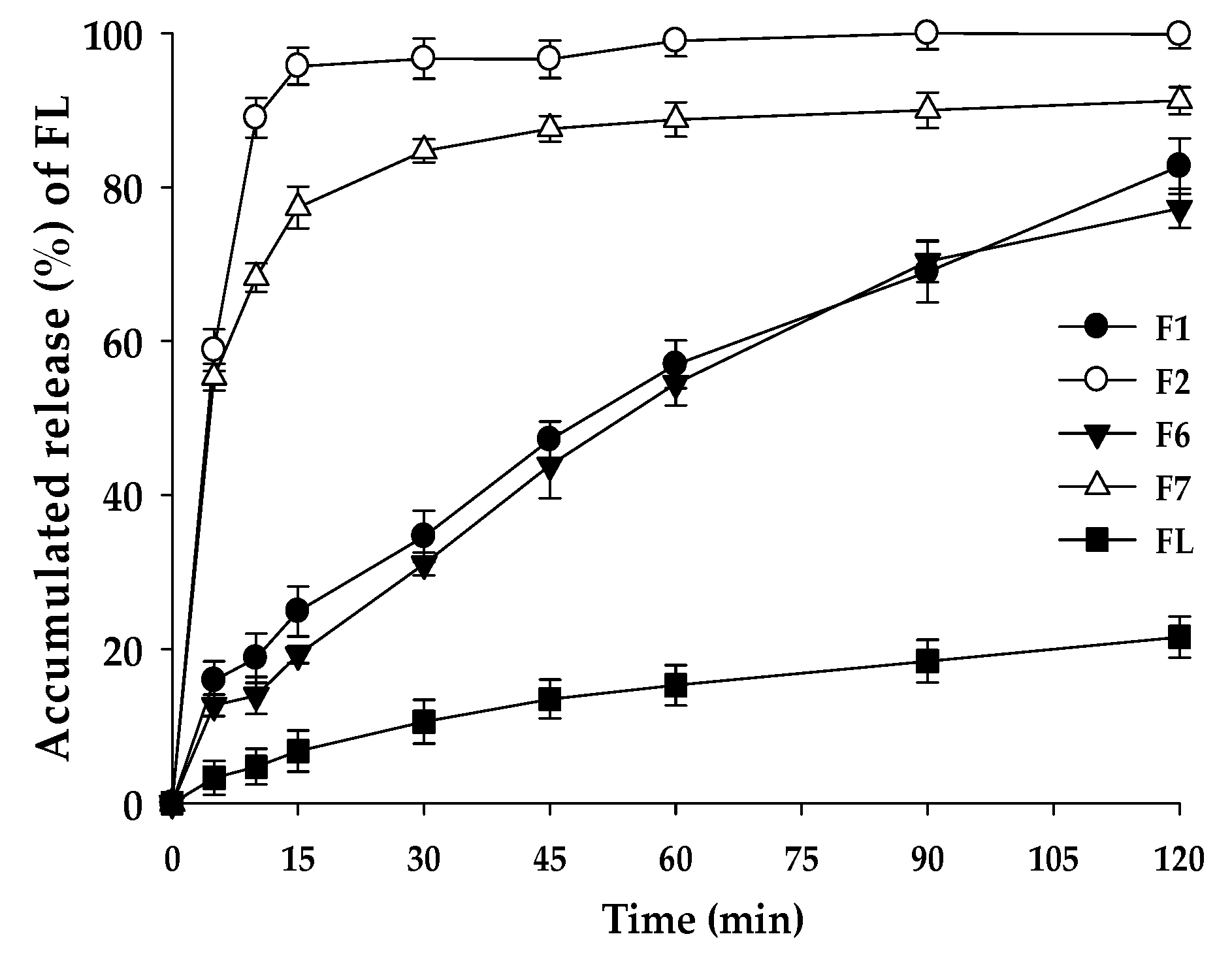
2.9. Dissolution Test
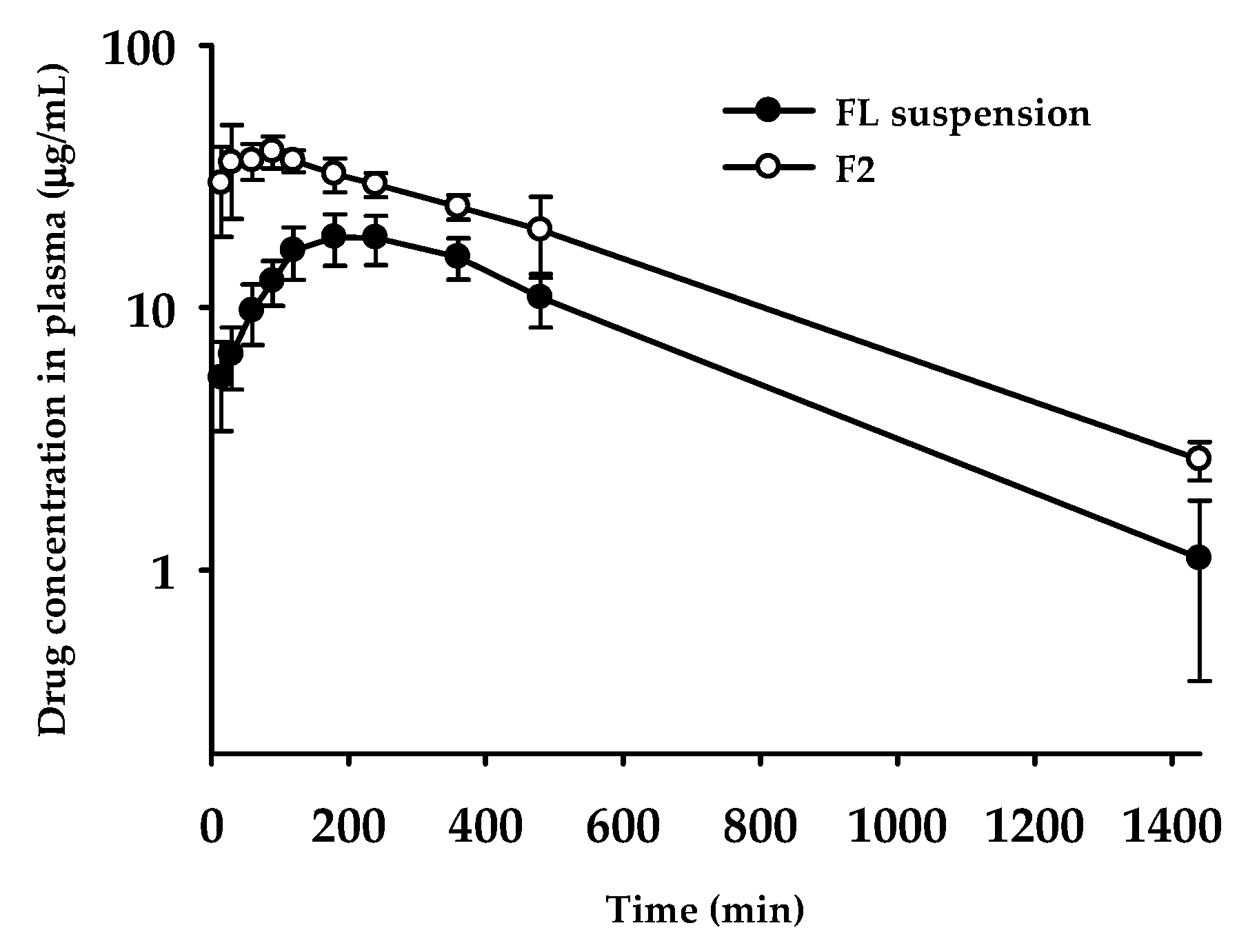
2.10. Oral Pharmacokinetic Study in Rats
2.11. Data Analysis
3. Results and Discussion
3.1. Solubility Test
3.2. Characterization of Various Vehicle Compositions
3.3. Construction of Pseudo-Ternary Phase Diagram
3.4. Particle Size and Peak Melting Temperature
3.5. Differential Scanning Calorimetry (DSC)
3.6. Powder X-ray Diffraction (PXRD)
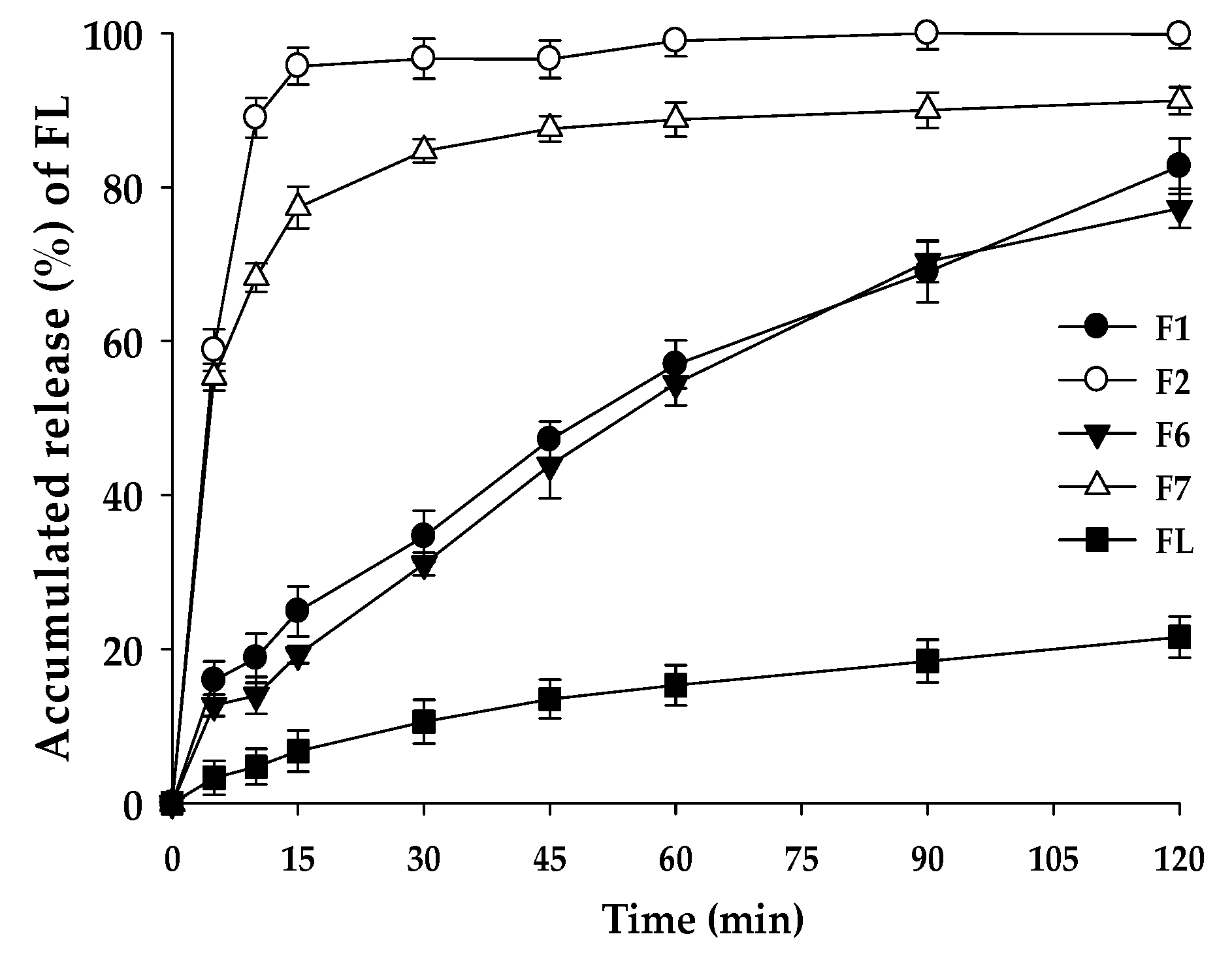
3.7. Dissolution Test
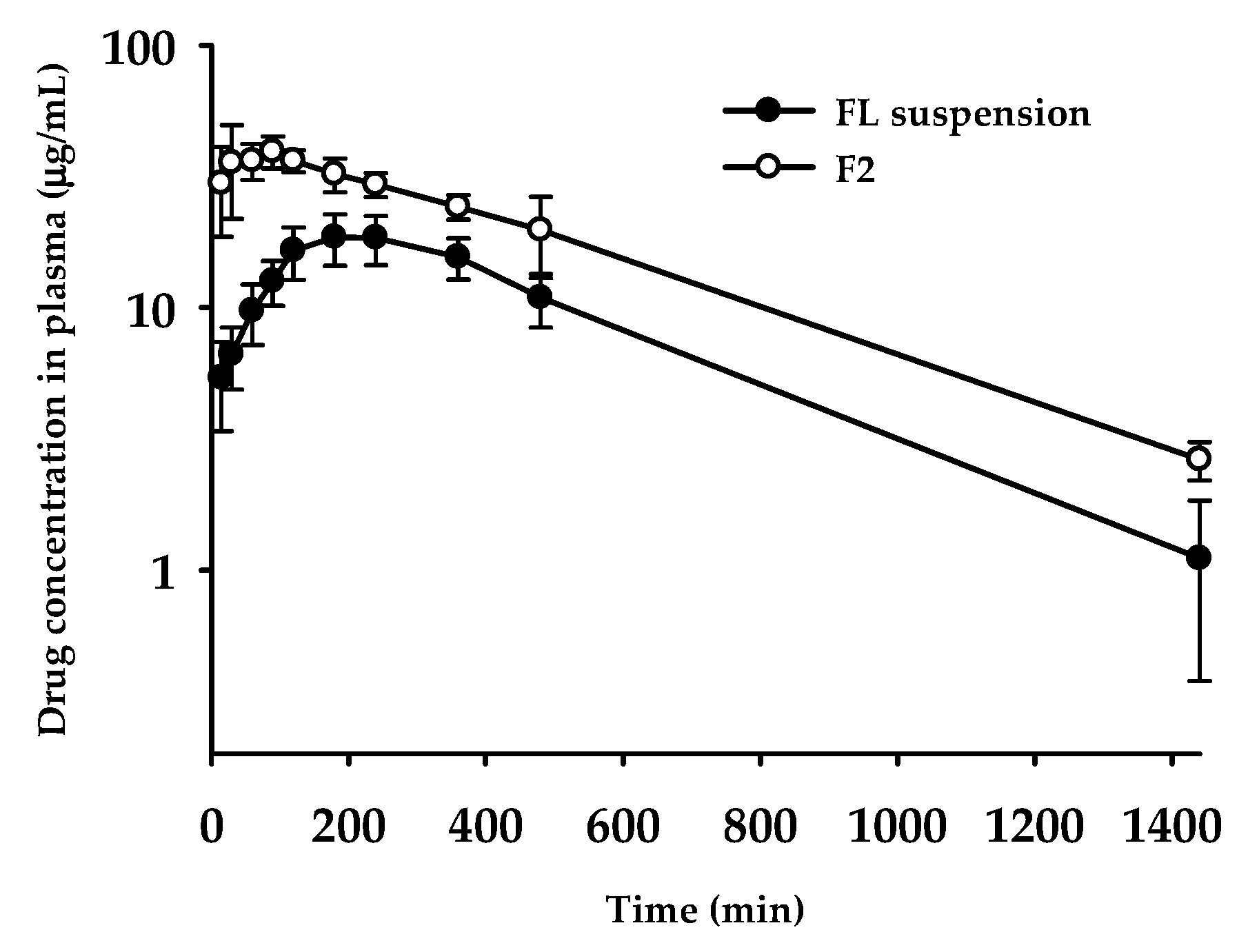
3.8. Oral Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fukumoto, A.; Tajima, K.; Hori, M.; Toda, Y.; Kaku, S.; Matsumoto, H. Analgesic effect of S (+)-flurbiprofen plaster in a rat model of knee arthritis: Analysis of gait and synovial fluid prostaglandin E2 levels. J. Pharm. Pharmacol. 2018, 70, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M. Clinical Pharmacokinetics of Flurbiprofen and its Enantiomers. Clin. Pharmacokinet. 1995, 28, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Perioli, L.; Ambrogi, V.; Nauta, L.D.; Nocchetti, M.; Rossi, C. Effects of hydrotalcite-like nanostructured compounds on biopharmaceutical properties and release of BCS class II drugs: The case of flurbiprofen. Appl. Clay Sci. 2011, 51, 407–413. [Google Scholar] [CrossRef]
- Junaid, D.; Ahmad, K.; Jallat, K.; Amjad, K.; Majid, G.K. Studies on self-nanoemulsifying drug delivery system of flurbiprofen employing long, medium and short chain triglycerides. Pak. J. Pharm. 2017, 30, 601–606. [Google Scholar]
- Li, D.X.; Han, M.J.; Balakrishnan, P.; Yan, Y.D.; Oh, D.H.; Joe, J.H.; Seo, Y.; Kim, J.O.; Park, S.M.; Yong, C.S.; et al. Enhanced oral bioavailability of flurbiprofen by combined use of micelle solution and inclusion compound. Arch. Pharm. Res. 2010, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Din, F.U.; Mustapha, O.; Kim, D.W.; Rashid, R.; Park, J.H.; Choi, J.Y.; Ku, S.K.; Yong, C.S.; Kim, J.O.; Choi, H.G. Novel dual-reverse thermosensitive solid lipid nanoparticle-loaded hydrogel for rectal administration of flurbiprofen with improved bioavailability and reduced initial burst effect. Eur. J. Pharm. Biopharm. 2015, 94, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Rudrangi, S.R.; Kaialy, W.; Ghori, M.U.; Trivedi, V.; Snowden, M.J.; Alexander, B.D. Solid-state flurbiprofen and methyl-b-cyclodextrin inclusion complexes prepared using a single-step, organic solvent-free supercritical fluid process. Eur. J. Pharm. Biopharm. 2016, 104, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oktay, A.N.; Karakucuk, A.; Ilbasmis, T.S.; Celebi, N. Dermal flurbiprofen nanosuspensions: Optimization with design ofexperiment approach and in vitro evaluation. Eur. J. Pharm. Sci. 2018, 122, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Vithani, K.; Hawley, A.; Jannin, V.; Pouton, C.; Boyd, B.J. Solubilisation behavior of poorly water-soluble drugs during digestion of solid SMEDDS. Eur. J. Pharm. Biopharm. 2018, 130, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Nasr, A.; Gardouh, A.; Ghorab, M. Novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of olmesartan medoxomil: Design, formulation, pharmacokinetic and bioavailability evaluation. Pharmaceutics 2016, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Kulkarni, S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev. Ind. Pharm. 2014, 40, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Yang, S. Biowaiver extension potential and IVIVC for BCS Class II drugs by formulation design: Case study for cyclosporine self-microemulsifying formulation. Arch. Pharm. Res. 2010, 33, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Grove, M.; Mullertz, A.; Nielsen, J.; Pedersen, G. Bioavailability of seocalcitol II: Development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci. 2006, 28, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Lorand, K.; Fruzsina, R.; Alexandra, B.; Szilvia, V.; Bela, O.; Laszlo, G.; Piroska, S.; Marla, A. Kinetic analysis of the toxicity of pharmaceutical excipients Cremophor EL and RH40 on endothelial and epithelial cells. J. Pharm. Sci. 2013, 102, 1173–1181. [Google Scholar]
- Dokania, S.; Joshi, A.K. Self-microemulsifying drug delivery system (SMEDDS)—Challenges and road ahead. Drug Deliv. 2015, 22, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.H.; Oh, D.H.; Oh, Y.K.; Yong, C.S.; Choi, H.G. Effects of solid carriers on the crystalline properties, dissolution and bioavailability of flurbiprofen in solid self-nanoemulsifying drug delivery system (solid SNEDDS). Eur. J. Pharm. Biopharm. 2012, 80, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Bedi, N.; Tiwary, AK. Enhancing solubility of poorly aqueous soluble drugs: Critical appraisal of techniques. J. Pharm. Investig. 2017, 48, 509–526. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming lipid-based oral drug delivery systems into solid dosage forms: An overview of solid carriers, physicochemical properties, and biopharmaceutical performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Fang, Z.; Jung, H.Y.; Yoon, J.H.; Hong, S.S.; Maeng, H.J. Synthesis of gemcitabine-threonine amide prodrug effective on pancreatic cancer cells with improved pharmacokinetic properties. Molecules 2018, 23, 2608. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Black, W.C.; Ouimet, N.; Ouellet, M.; Percival, M.D. Structure-based design of COX-2 selectivity into flurbiprofen. Bioorg. Med. Chem. Lett. 1999, 9, 307–312. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasiya, A.; Singh, M.; Upadhyay, S.C.; Mukherjee, R.; Khar, R.K. Exemestane loaded self-microemulsifying drug delivery system (SMEDDS): Development and optimization. AAPS PharmSciTech 2008, 9, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Khadka, P.; Ro, J.; Kim, H.; Kim, I.; Kim, J.; Kim, H.; Cho, J.; Yun, G.; Lee, J. Pharmaceutical particle technologies: An approach to improve drug solubility, dissolution and bioavailability. Asian J. Pharm. Sci. 2014, 9, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Tadros, T.; Izquierdo, P.; Esquena, J.; Solans, C. Formation and stability of nano-emulsions. Adv. Colloid Interface Sci. 2004, 108–109, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Kim, J. Physicochemical characterization of solid dispersion of furosemide with TPGS. Int. J. Pharm. 2003, 251, 79–84. [Google Scholar] [CrossRef]
- Singh, A.; Chaurasiya, A.; Awasthi, A.; Mishra, G.; Asati, D.; Khar, RK.; Mukherjee, R. Oral bioavailability enhancement of exemestane from self-microemulsifying drug delivery system (SMEDDS). AAPS PharmSciTech 2009, 10, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Kyatanwar, A.U.; Jadhav, K.R.; Kadam, V.J. Self micro-emulsifying drug delivery system (SMEDDS). J. Pharm. Res. 2010, 3, 75–83. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition (Weight Ratio) | Physical State at 25 °C | Particle Size (nm, mean ± S.D., n = 3) | Peak Melting Temperature (°C, n = 1) | ||
|---|---|---|---|---|---|
| Kollisolv MCT 70 | Gelucire 44/14 | 1/3 | Solid | 354.42 ± 21.82 | 41.68 |
| 2/2 | Solid | 445.73 ± 34.30 | 41.22 | ||
| 3/1 | Solid | 571.91 ± 40.48 | 40.99 | ||
| Kolliphor HS 15 | 1/3 | Solid | 129.40 ± 2.31 | 27.31 | |
| 2/2 | Liquid | 196.35 ± 18.73 | No peak | ||
| 3/1 | Liquid | 490.63 ± 63.16 | No peak | ||
| TPGS | 1/3 | Solid | 132.12 ± 10.51 | 36.96 | |
| 2/2 | Solid | 278.95 ± 26.25 | 36.58 | ||
| 3/1 | Solid | 396.63 ± 21.34 | 36.37 | ||
| Lauroglycol 90 | Gelucire 44/14 | 1/3 | Solid | 533.96 ± 78.33 | 35.55 |
| 2/2 | Solid | 546.20 ± 50.48 | 34.92 | ||
| 3/1 | Solid | 592.21 ± 68.34 | 32.66 | ||
| Kolliphor HS 15 | 1/3 | Liquid | 228.95 ± 16.85 | No peak | |
| 2/2 | Liquid | 475.82 ± 35.43 | No peak | ||
| 3/1 | Liquid | 795.76 ± 104.22 | No peak | ||
| TPGS | 1/3 | Solid | 262.11 ± 30.47 | 36.70 | |
| 2/2 | Liquid | 556.73 ± 45.61 | No peak | ||
| 3/1 | Liquid | 598.68 ± 61.63 | No peak | ||
| Capmul MCM C8 | Gelucire 44/14 | 1/3 | Solid | 171.30 ± 27.38 | 33.28 |
| 2/2 | Liquid | 280.14 ± 56.94 | No peak | ||
| 3/1 | Liquid | 490.26 ± 62.36 | No peak | ||
| Kolliphor HS 15 | 1/3 | Liquid | 98.16 ± 2.63 | No peak | |
| 2/2 | Liquid | 134.21 ± 9.45 | No peak | ||
| 3/1 | Liquid | 312.86 ± 53.72 | No peak | ||
| TPGS | 1/3 | Solid | 133.47 ± 4.34 | 35.36 | |
| 2/2 | Liquid | 259.23 ± 9.10 | No peak | ||
| 3/1 | Liquid | 421.85 ± 48.53 | No peak | ||
| Formulation | Composition (Weight %) | Particle Size (nm, mean ± S.D., n = 3) | Peak Melting Temperature (°C, n = 1) | ||
|---|---|---|---|---|---|
| FL | Kollisolv MCT 70 | TPGS | |||
| F1 | 10 | 0 | 90 | 26.82 ± 2.25 | 33.59 |
| F2 | 10 | 10 | 80 | 13.74 ± 2.21 | 32.37 |
| F3 | 10 | 20 | 70 | 17.03 ± 2.31 | 31.52 |
| F4 | 10 | 30 | 60 | 31.42 ± 5.29 | 30.74 |
| F5 | 10 | 70 | 20 | 594.38 ± 113.15 | Liquid |
| F6 | 20 | 0 | 80 | 190.46 ± 48.37 | 26.13/30.72 |
| F7 | 20 | 10 | 70 | 186.81 ± 24.21 | Semi-solid |
| F8 | 30 | 0 | 70 | 245.24 ± 67.09 | Liquid |
| F9 | 30 | 10 | 60 | 817.13 ± 128.77 | Liquid |
| Vehicle | * Solubility (w/w%) |
|---|---|
| Oils | |
| Kollisolv MCT 70 | 24.85 ± 0.20 |
| Lauroglycol 90 | 22.43 ± 0.92 |
| Capmul MCM C8 | 21.38 ± 0.95 |
| Solid or Semisolid Surfactants | |
| Gelucire 44/14 | 33.09 ± 1.44 |
| Kolliphor HS 15 | 31.97 ± 2.45 |
| TPGS | 34.28 ± 4.26 |
| Pharmacokinetic Parameters | FL Suspension | F2 |
|---|---|---|
| Tmax (min) | 228 ± 89 | 60.0 ± 34.6* |
| Cmax (μg /mL) | 19.6 ± 3.8 | 44.1 ± 3.4 |
| T1/2 (min) | 282 ± 73 | 340 ± 49 |
| AUClast (μg∙min/mL) | 12700 ± 2052 | 24200 ± 2886* |
| AUC∞ (μg∙min/mL) | 13200 ± 2279 | 25500 ± 2531* |
| MRT a (min) | 448 ± 76 | 462 ± 34 |
| Relative BA b (%) | - | 193 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, R.M.; Jang, D.-J.; Kim, Y.C.; Yoon, J.-H.; Min, K.A.; Maeng, H.-J.; Cho, K.H. Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats. Pharmaceutics 2018, 10, 247. https://doi.org/10.3390/pharmaceutics10040247
Kim RM, Jang D-J, Kim YC, Yoon J-H, Min KA, Maeng H-J, Cho KH. Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats. Pharmaceutics. 2018; 10(4):247. https://doi.org/10.3390/pharmaceutics10040247
Chicago/Turabian StyleKim, Rae Man, Dong-Jin Jang, Yu Chul Kim, Jin-Ha Yoon, Kyoung Ah Min, Han-Joo Maeng, and Kwan Hyung Cho. 2018. "Flurbiprofen-Loaded Solid SNEDDS Preconcentrate for the Enhanced Solubility, In-Vitro Dissolution and Bioavailability in Rats" Pharmaceutics 10, no. 4: 247. https://doi.org/10.3390/pharmaceutics10040247