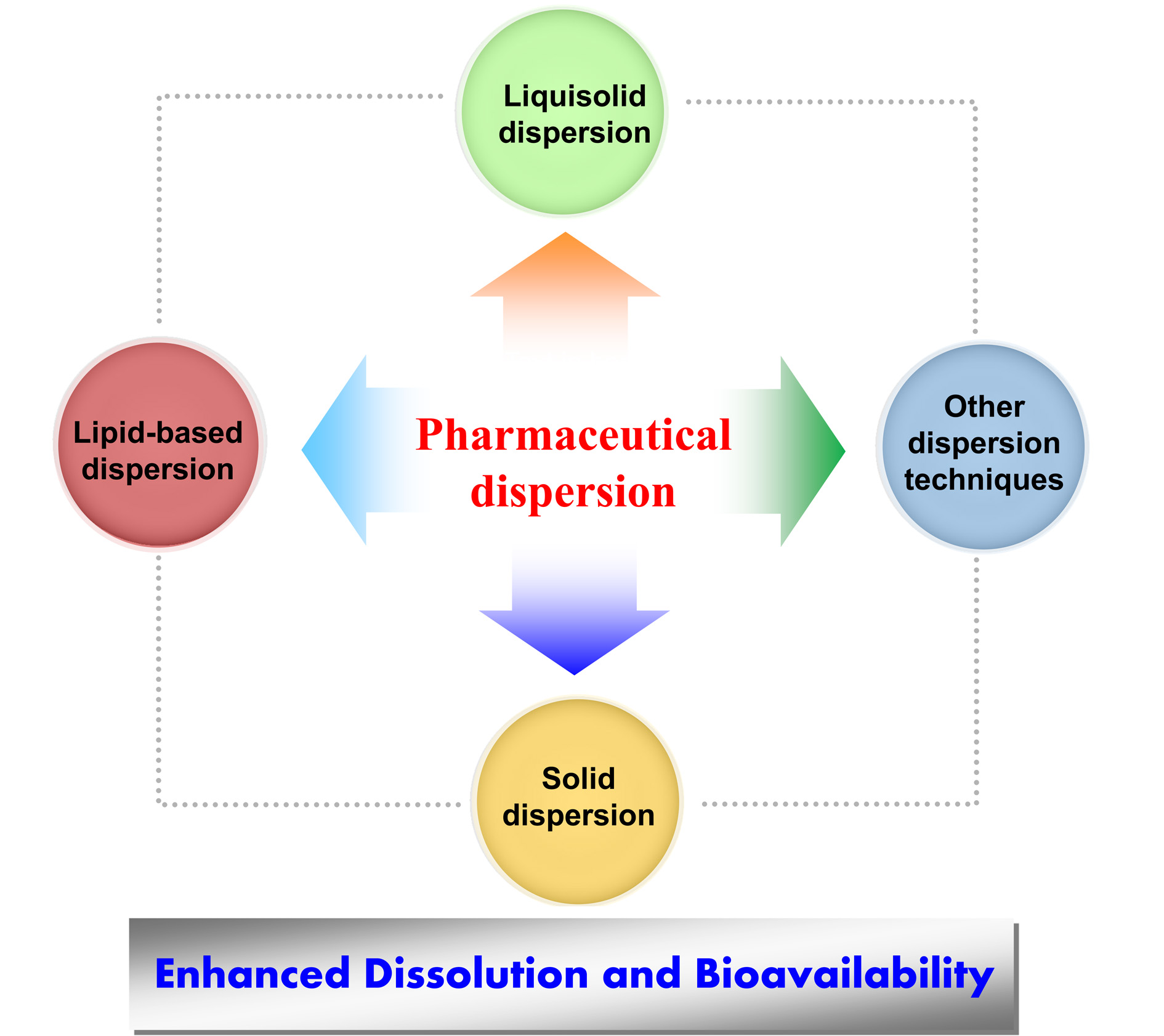
Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs



Abstract
: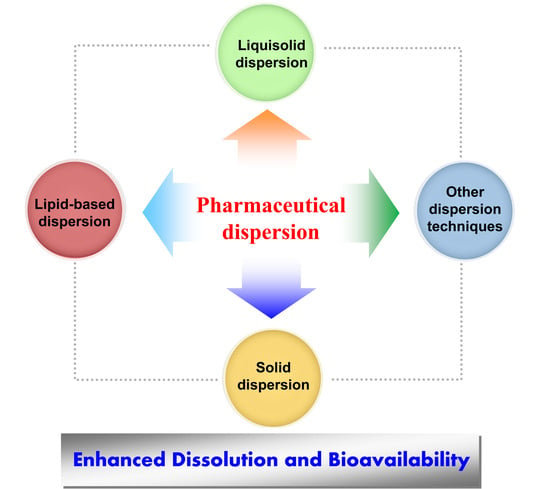
1. Introduction
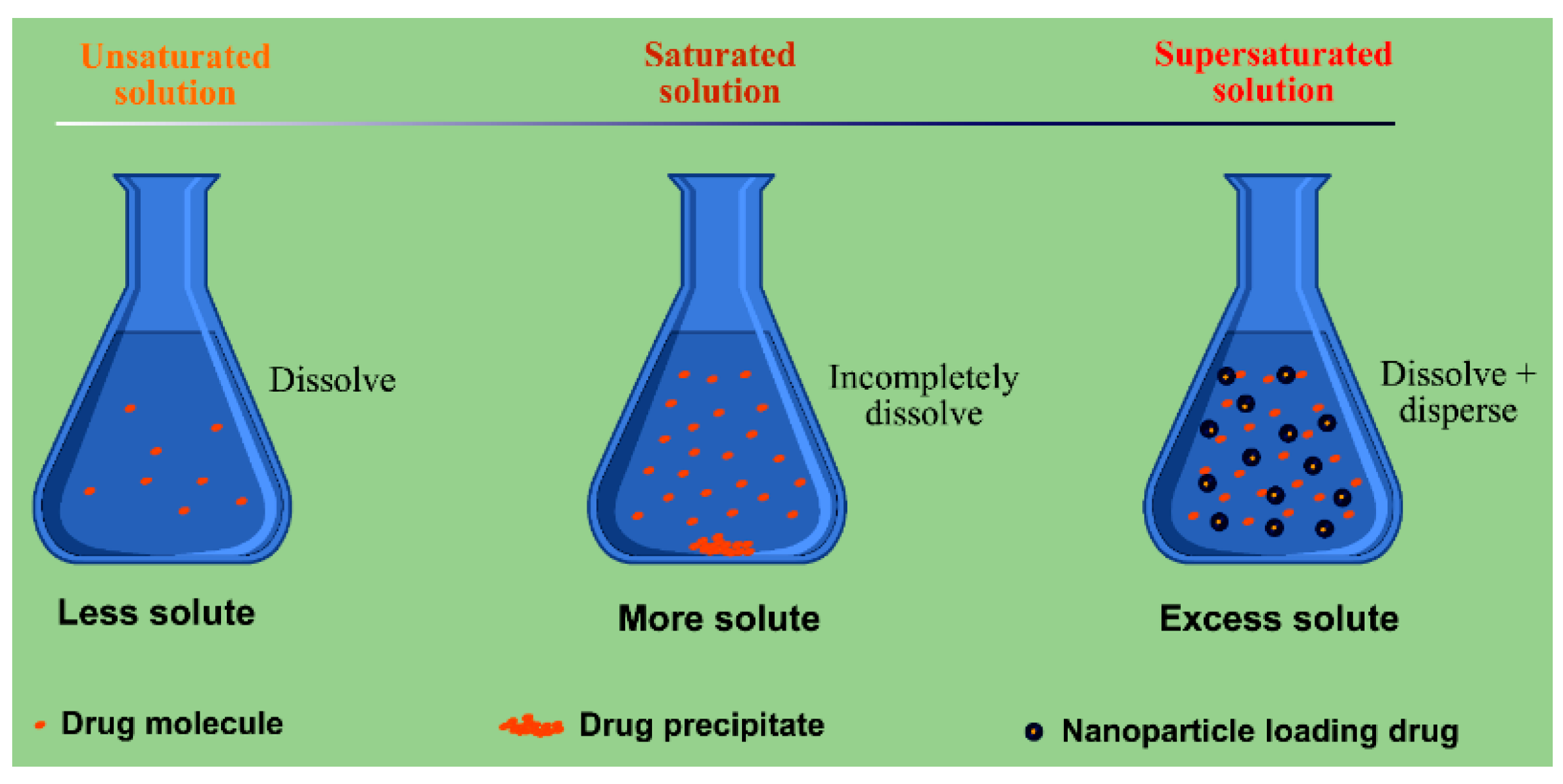
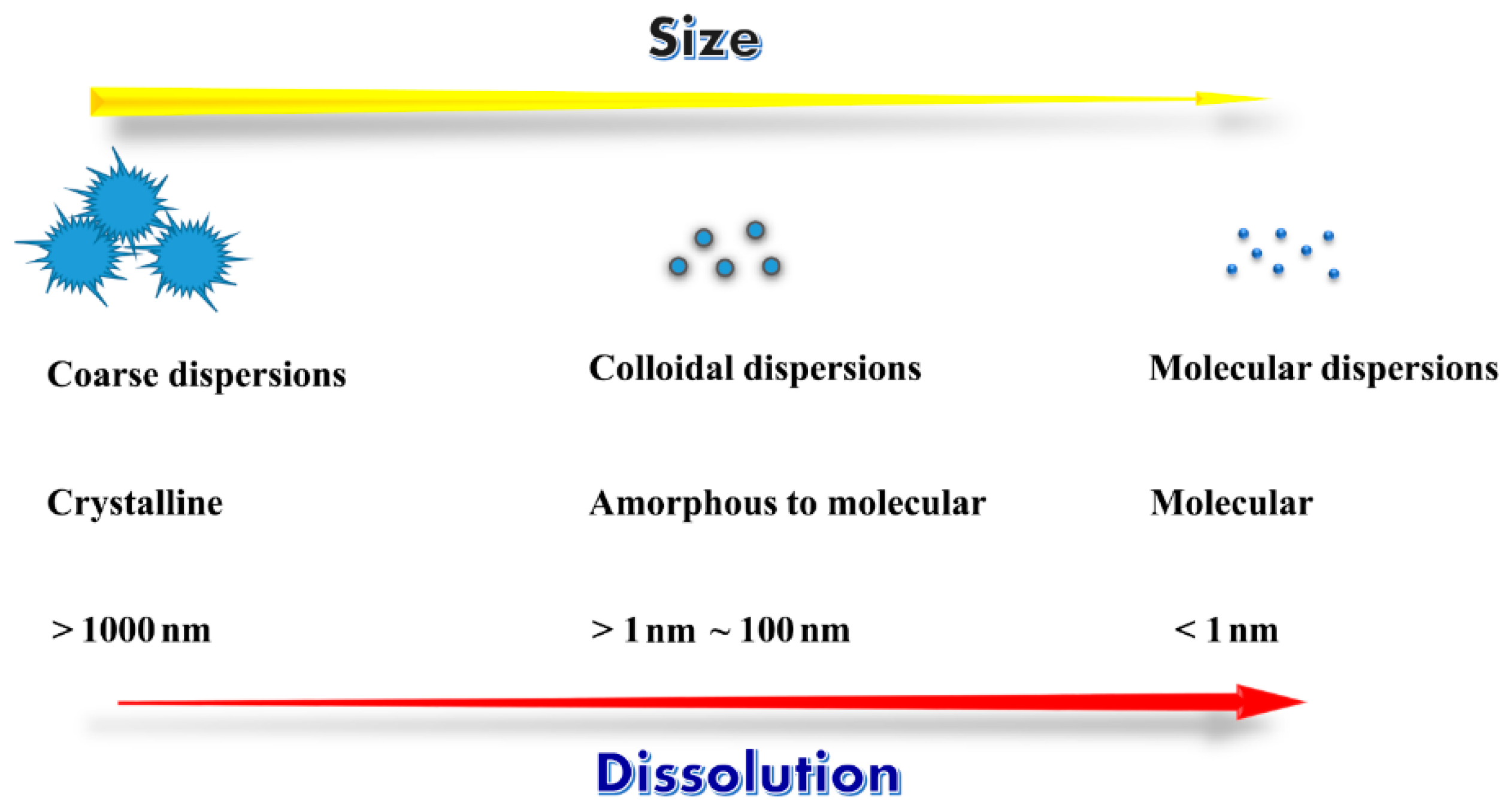
2. Interrelation between Dispersion and Drug Dissolution/Absorption
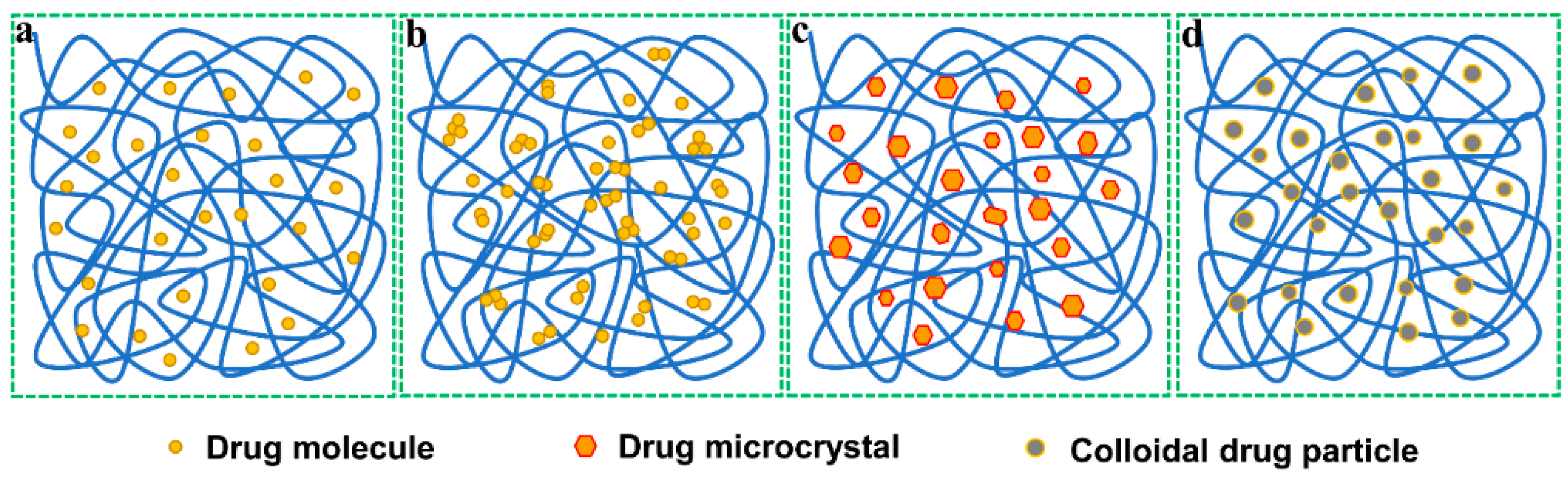
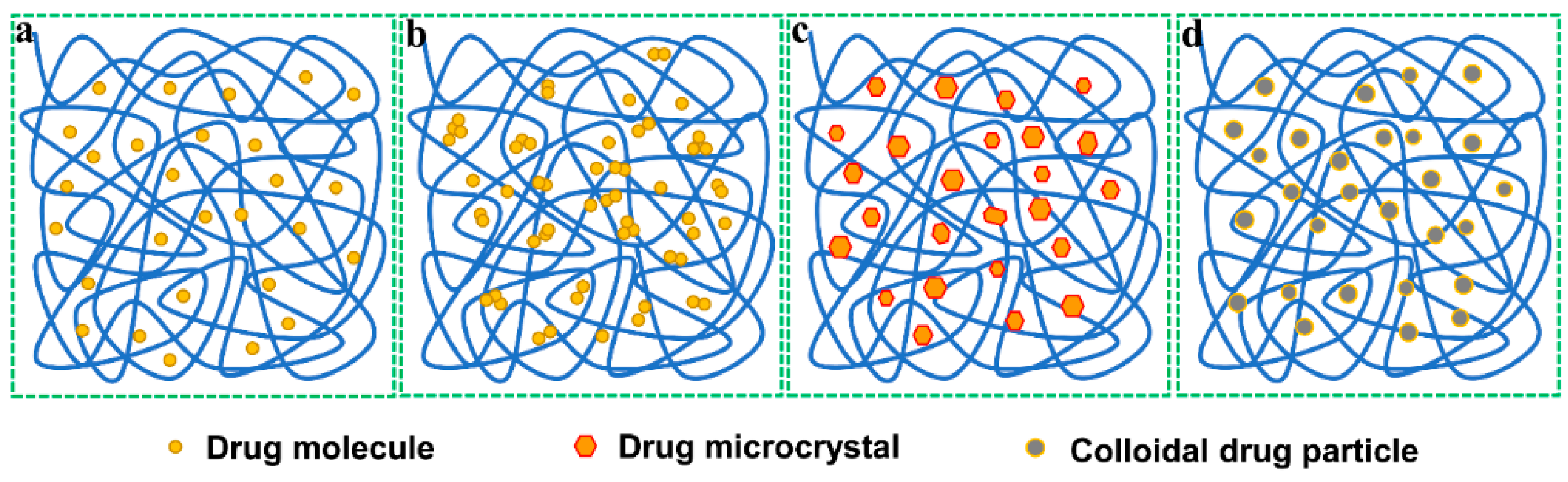
3. Solid Dispersion Technique
3.1. Solid Dispersions
3.2. Carrier Excipients of SDs
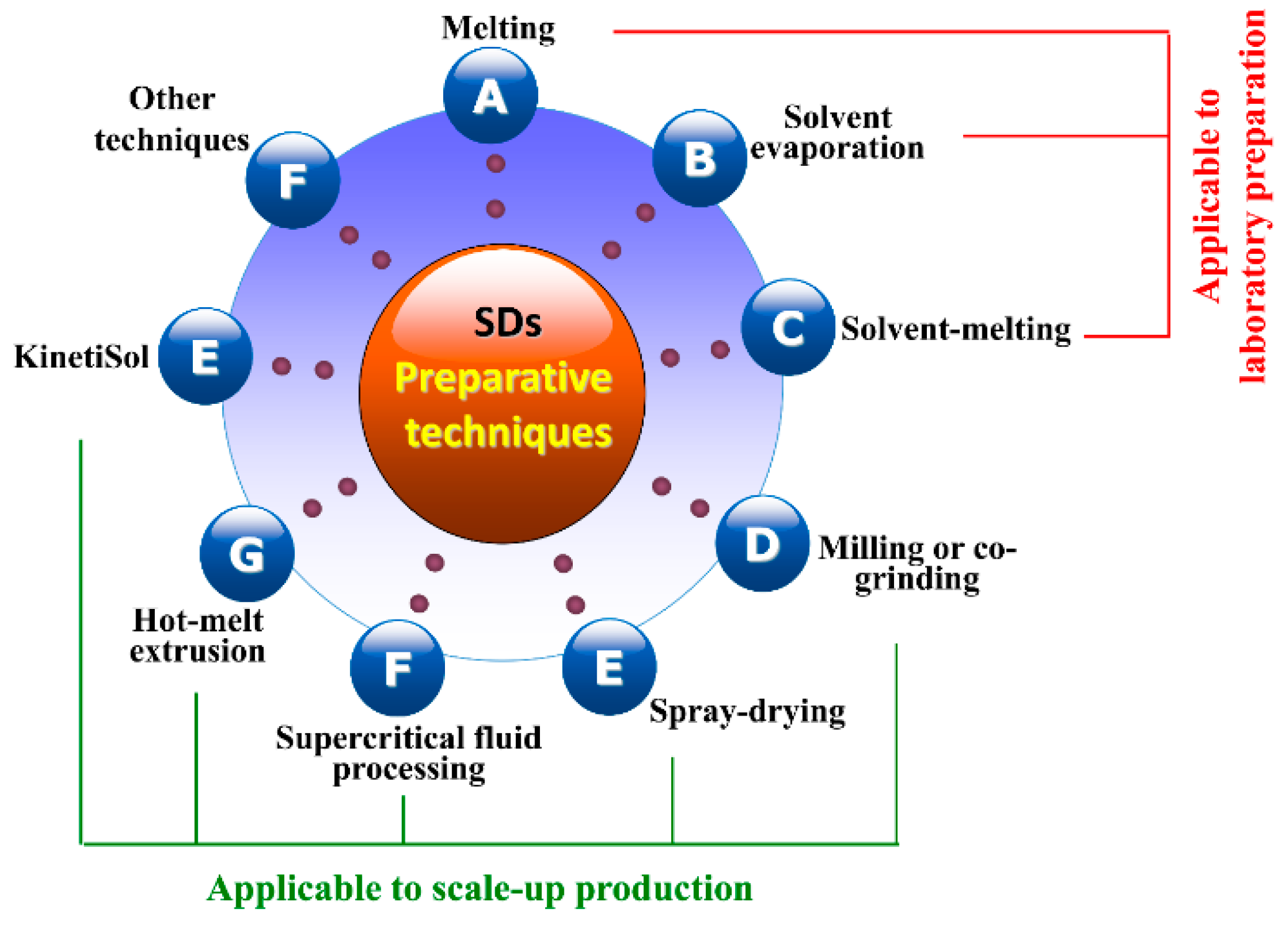
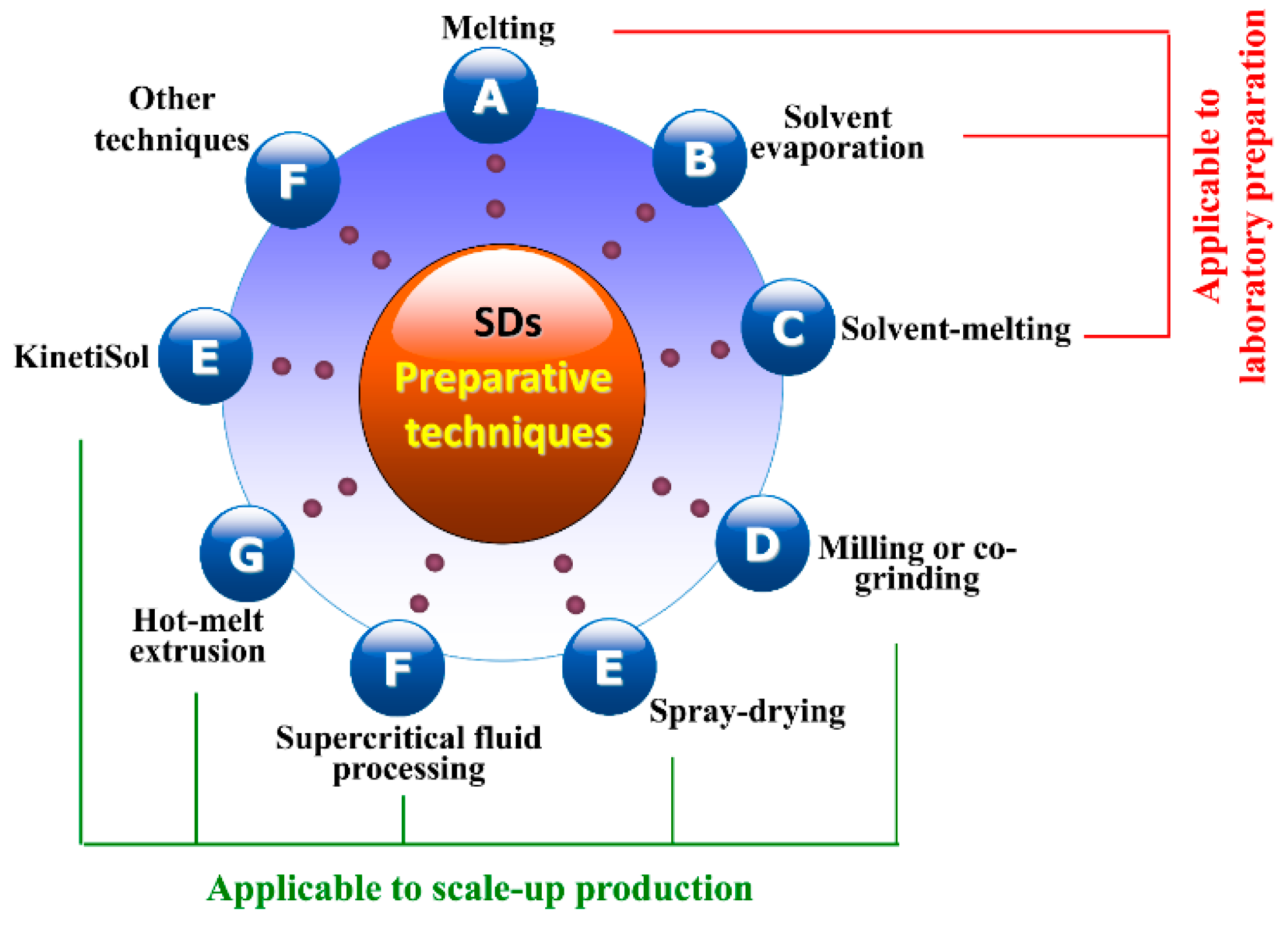
3.3. Preparative Processes of SDs
3.3.1. Melting Method
3.3.2. Solvent Evaporation Method
3.3.3. Solvent-Melting Method
3.3.4. Milling Method
3.3.5. Spray-Drying Method
3.3.6. Supercritical Fluid Processing
3.3.7. Hot-Melt Extrusion
3.3.8. KinetiSol® Technique
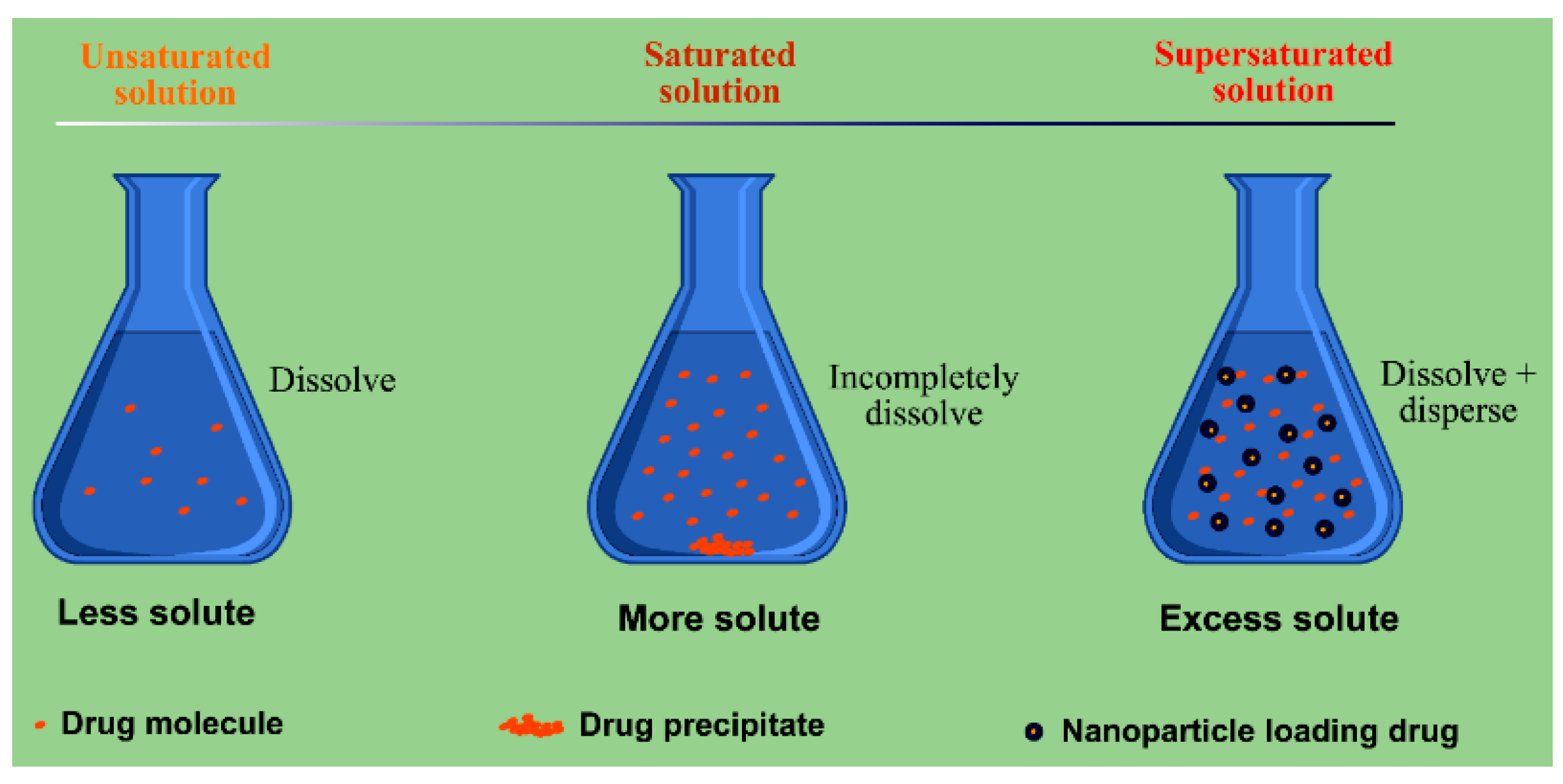
3.4. SDs-Based Dissolution and Bioavailability Enhancement
4. Lipid Dispersion Technique
4.1. Lipid Dispersions Accomplished by Lipid Nanoparticles
4.2. Commonly Used Lipid Dispersion Systems
4.2.1. Solid Lipid Nanoparticles
4.2.2. Nanostructured Lipid Carriers
4.2.3. Nanoemulsions
4.2.4. Liposomes and Phytosomes
4.3. Lipid Excipients
4.4. Lipid Nanocarriers-Based Enhancement of Bioavailability
4.5. Translation of Liquid Lipid Dispersions into Solid Formulations
4.5.1. Freeze Drying
4.5.2. Spray Drying
4.5.3. Self-Emulsifying
4.5.4. Developing Liquisolid Hybrid Formulations
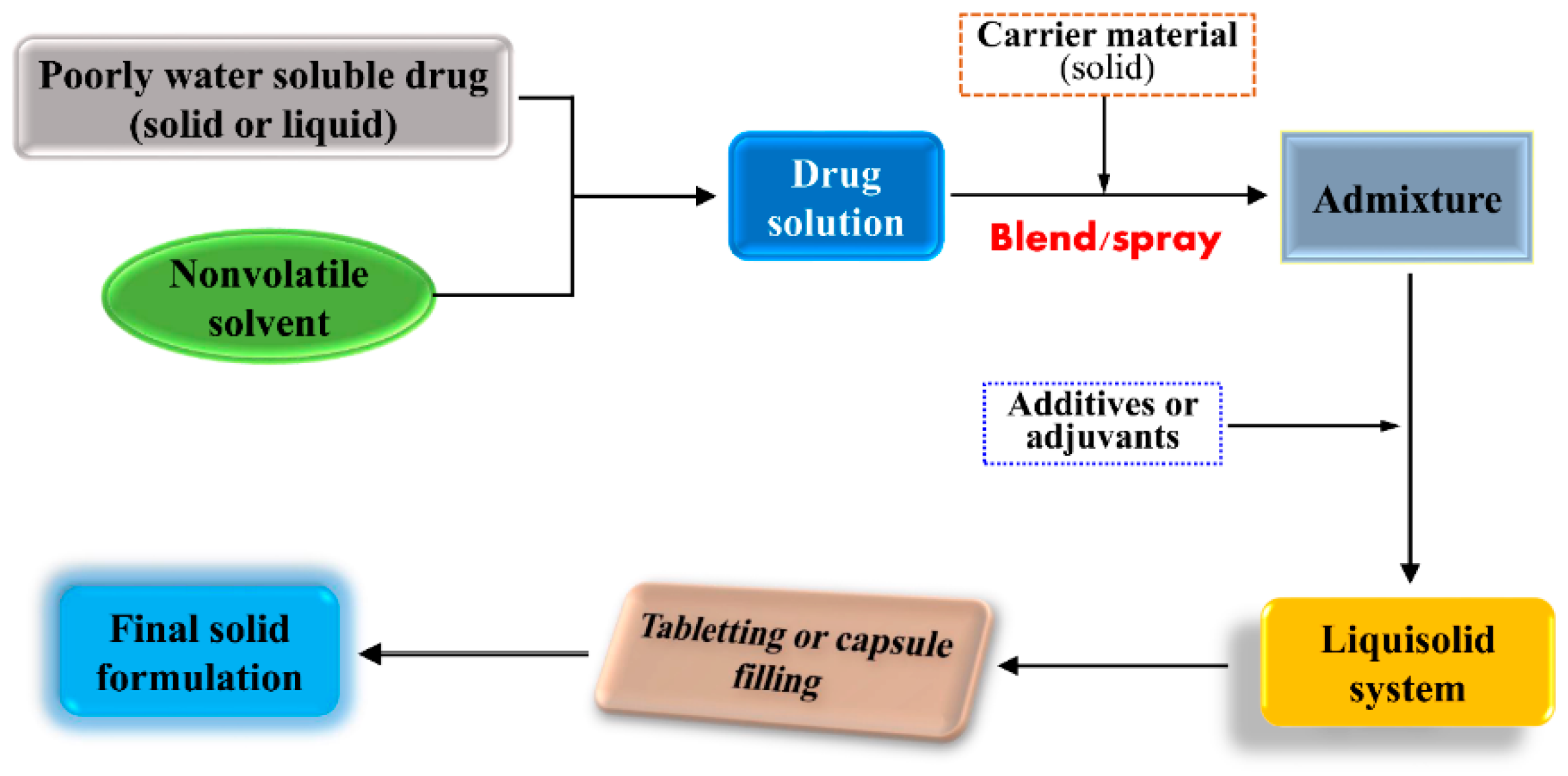
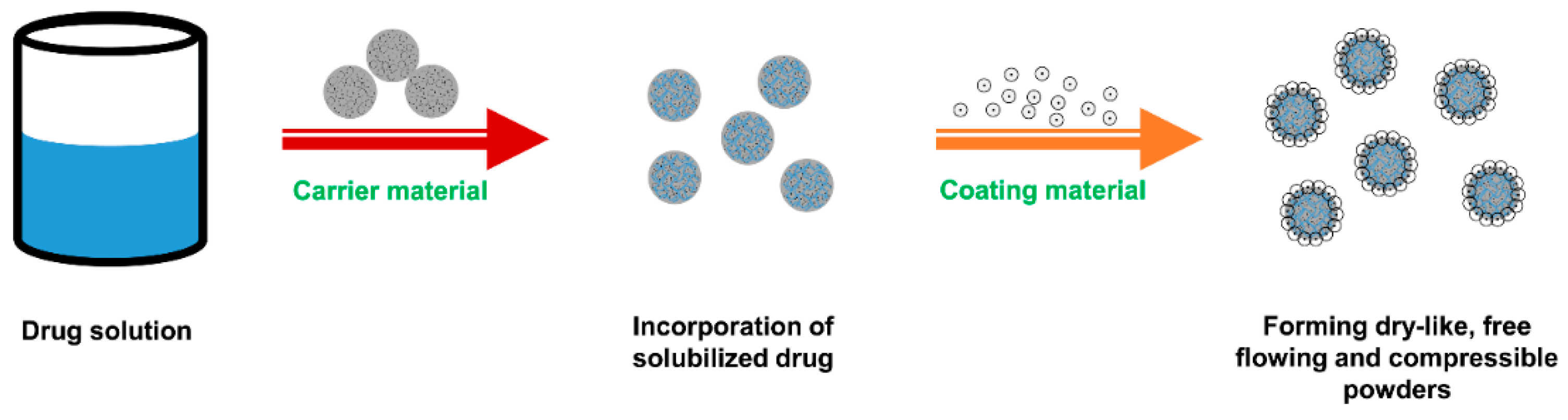
5. Liquisolid Dispersion Technique
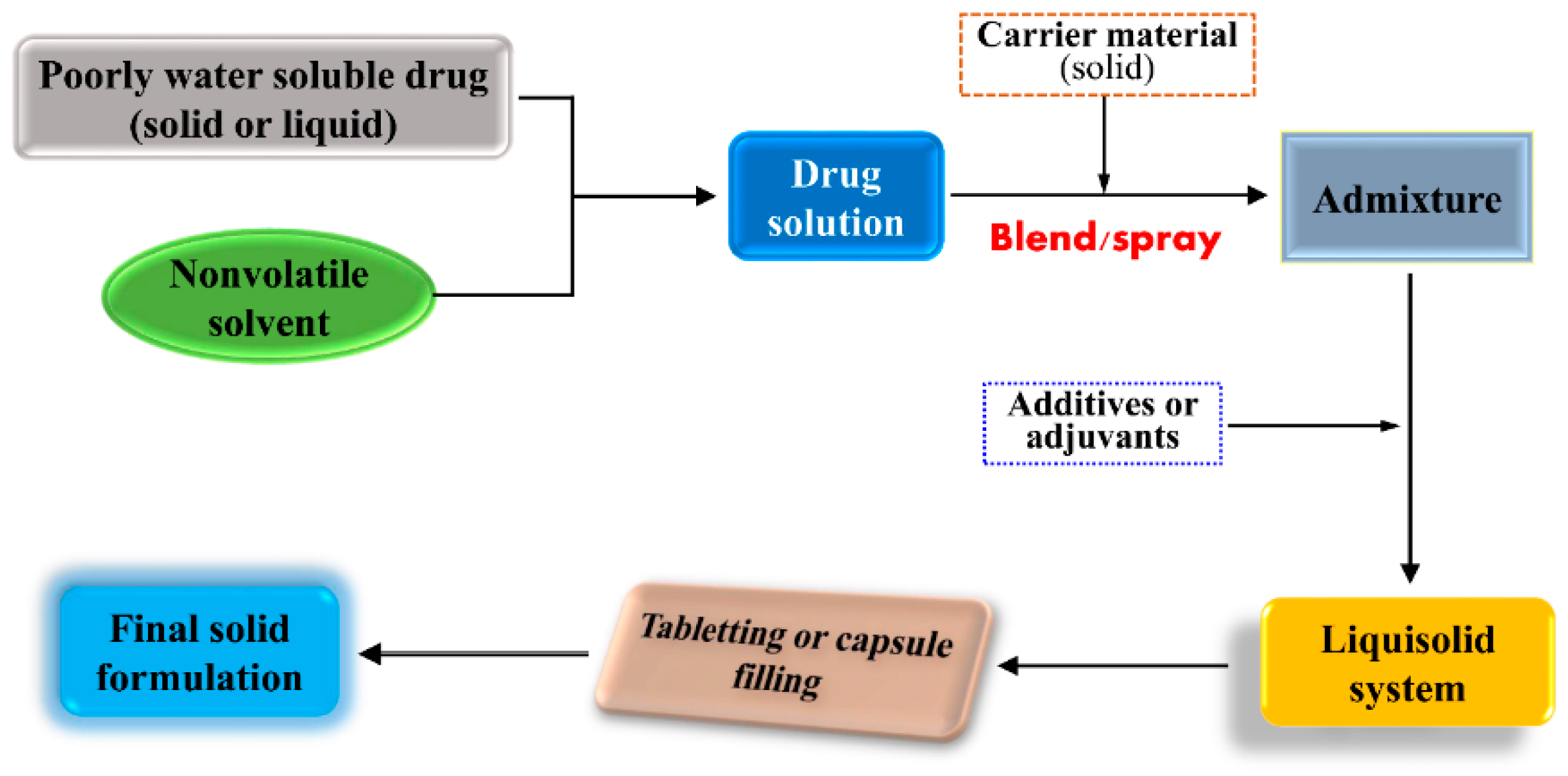
5.1. Overview of Liquisolid System
5.2. Formulation Components of Liquisolid System
5.3. Preparation of Liquisolid Compacts
5.4. Liquisolid System-Based Enhancement of Dissolution and Bioavailability
6. Other Dispersion Techniques
6.1. Co-Precipitation Technique
6.2. Concomitant Crystallization Technique
6.3. Inclusion Complexation Technique
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Goke, K.; Lorenz, T.; Repanas, A.; Schneider, F.; Steiner, D.; Baumann, K.; Bunjes, H.; Dietzel, A.; Finke, J.H.; Glasmacher, B.; et al. Novel strategies for the formulation and processing of poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2018, 126, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Leleux, J.; Williams, R.O., 3rd. Recent advancements in mechanical reduction methods: Particulate systems. Drug Dev. Ind. Pharm. 2014, 40, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Scholz, P.; Keck, C.M. Nanocrystals: From raw material to the final formulated oral dosage form—A review. Curr. Pharm. Des. 2015, 21, 4217–4228. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ho, C.; Yang, D.; Chen, J.; Orton, E. Measurement and accurate interpretation of the solubility of pharmaceutical salts. J. Pharm. Sci. 2017, 106, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, T.; Lan, Y.; Wu, B.; Shi, Z. Nanosuspensions containing oridonin/hp-beta-cyclodextrin inclusion complexes for oral bioavailability enhancement via improved dissolution and permeability. AAPS PharmSciTech 2016, 17, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Ezawa, T.; Inoue, Y.; Murata, I.; Takao, K.; Sugita, Y.; Kanamoto, I. Characterization of the dissolution behavior of piperine/cyclodextrins inclusion complexes. AAPS PharmSciTech 2018, 19, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Gadade, D.D.; Pekamwar, S.S. Pharmaceutical cocrystals: Regulatory and strategic aspects, design and development. Adv. Pharm. Bull. 2016, 6, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Cagel, M.; Tesan, F.C.; Bernabeu, E.; Salgueiro, M.J.; Zubillaga, M.B.; Moretton, M.A.; Chiappetta, D.A. Polymeric mixed micelles as nanomedicines: Achievements and perspectives. Eur. J. Pharm. Biopharm. 2017, 113, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Vo, C.L.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Vranikova, B.; Gajdziok, J. Liquisolid systems and aspects influencing their research and development. Acta Pharm. 2013, 63, 447–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalepu, S.; Nekkanti, V. Improved delivery of poorly soluble compounds using nanoparticle technology: A review. Drug Deliv. Transl. Res. 2016, 6, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Kwok, P.C.; Chan, H.K. Nanotechnology versus other techniques in improving drug dissolution. Curr. Pharm. Des. 2014, 20, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.S.; He, W.; Yin, L. A Smart paclitaxel-disulfiram nanococrystals for efficient mdr reversal and enhanced apoptosis. Pharm. Res. 2018, 35, 77. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Gala, U.; Chauhan, H. Classification of solid dispersions: Correlation to (i) stability and solubility (ii) preparation and characterization techniques. Drug Dev. Ind. Pharm. 2015, 41, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Fong, S.Y.; Bauer-Brandl, A.; Brandl, M. Oral bioavailability enhancement through supersaturation: An update and meta-analysis. Expert Opin. Drug Deliv. 2017, 14, 403–426. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.C.; Carbone, C.; Souto, E.B. Beyond liposomes: Recent advances on lipid based nanostructures for poorly soluble/poorly permeable drug delivery. Prog. Lipid Res. 2017, 68, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Wang, H.; Wu, B.; Zhang, X. Lipid nanoparticles for the delivery of active natural medicines. Curr. Pharm. Des. 2017, 23, 6705–6713. [Google Scholar] [CrossRef] [PubMed]
- Gajdziok, J.; Vranikova, B. Enhancing of drug bioavailability using liquisolid system formulation. Ceska Slov. Farm. 2015, 64, 55–66. [Google Scholar] [PubMed]
- Azharshekoufeh, B.L.; Shokri, J.; Adibkia, K.; Javadzadeh, Y. Liquisolid technology: What it can do for NSAIDs delivery? Colloids Surf. B Biointerfaces 2015, 136, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, N.; Wu, B.; Lu, Y.; Guan, T.; Wu, W. Physical characterization of lansoprazole/PVP solid dispersion prepared by fluid-bed coating technique. Powder Technol. 2008, 182, 480–485. [Google Scholar] [CrossRef]
- Arthur, A.N.; Willis, R.W. The rate of dissolution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar]
- Ghadi, R.; Dand, N. BCS class IV drugs: Highly notorious candidates for formulation development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Harde, H.; Das, M.; Jain, S. Solid lipid nanoparticles: An oral bioavailability enhancer vehicle. Expert Opin. Drug Deliv. 2011, 8, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, X.; Lian, R.; Zheng, S.; Yin, Z.; Lu, Y.; Wu, W. Enhanced dissolution and oral bioavailability of aripiprazole nanosuspensions prepared by nanoprecipitation/homogenization based on acid-base neutralization. Int. J. Pharm. 2012, 438, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Marano, S.; Barker, S.A.; Raimi-Abraham, B.T.; Missaghi, S.; Rajabi-Siahboomi, A.; Craig, D.Q.M. Development of micro-fibrous solid dispersions of poorly water-soluble drugs in sucrose using temperature-controlled centrifugal spinning. Eur. J. Pharm. Biopharm. 2016, 103, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.V., Jr.; Yanchick, V.A.; Maness, D.D. Dissolution rates of corticosteroids utilizing sugar glass dispersions. J. Pharm. Sci. 1977, 66, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Madgulkar, A.; Bandivadekar, M.; Shid, T.; Rao, S. Sugars as solid dispersion carrier to improve solubility and dissolution of the BCS class II drug: Clotrimazole. Drug Dev. Ind. Pharm. 2016, 42, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.M.; Videa, M.; Lopez Silva, T.; Castro, S.; Caballero, A.; Lara-Diaz, V.J.; Castorena-Torres, F. Two-phase amorphous-amorphous solid drug dispersion with enhanced stability, solubility and bioavailability resulting from ultrasonic dispersion of an immiscible system. Eur. J. Pharm. Biopharm. 2017, 119, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Nayak, A.K.; Mohanty, B.; Panda, S. Solubility and dissolution enhancement of etoricoxib by solid dispersion technique using sugar carriers. ISRN Pharm. 2011, 2011, 819765. [Google Scholar] [CrossRef] [PubMed]
- Duret, C.; Wauthoz, N.; Sebti, T.; Vanderbist, F.; Amighi, K. Solid dispersions of itraconazole for inhalation with enhanced dissolution, solubility and dispersion properties. Int. J. Pharm. 2012, 428, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Broekhuis, J.; Grasmeijer, N.; Tonnis, W.; Ketolainen, J.; Frijlink, H.W.; Hinrichs, W.L.J. Efficient production of solid dispersions by spray drying solutions of high solid content using a 3-fluid nozzle. Eur. J. Pharm. Biopharm. 2018, 123, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Thenmozhi, K.; Yoo, Y.J. Enhanced solubility of piperine using hydrophilic carrier-based potent solid dispersion systems. Drug Dev. Ind. Pharm. 2017, 43, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.P. Glass formation in barbiturates and solid dispersion systems of barbiturates with citric acid. J. Pharm. Sci. 1978, 67, 1606–1610. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M.; Ali, R. Effervescence assisted fusion technique to enhance the solubility of drugs. AAPS PharmSciTech 2015, 16, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Chawla, G.; Bansal, A.K. Improved dissolution of a poorly water soluble drug in solid dispersions with polymeric and non-polymeric hydrophilic additives. Acta Pharm. 2008, 58, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.P.; Gaikwad, N.J. Preparation and characterization of gliclazide-polyethylene glycol 4000 solid dispersions. Acta Pharm. 2009, 59, 57–65. [Google Scholar] [PubMed]
- Venkateskumar, K.; Parasuraman, S.; Gunasunderi, R.; Sureshkumar, K.; Nayak, M.M.; Shah, S.A.A.; Khoo, K.; Kai, H.W. Acyclovir-polyethylene glycol 6000 binary dispersions: Mechanistic insights. AAPS PharmSciTech 2017, 18, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.D.; Arantes, V.T.; Resende, J.A.; Speziali, N.L.; de Oliveira, R.B.; Vianna-Soares, C.D. Preparation and characterization of solid dispersion of simvastatin. Drug Dev. Ind. Pharm. 2010, 36, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Nguyen, J.H.; Becker, C.; Francke, N.M.; Jorgensen, E.B.; Holm, P.; Holm, R.; Mu, H.; Rades, T.; Langguth, P. Influence of polymer molecular weight on in vitro dissolution behavior and in vivo performance of celecoxib:PVP amorphous solid dispersions. Eur. J. Pharm. Biopharm. 2016, 101, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.; Tran, T.H.; Kim, S.Y.; Woo, K.B.; Choi, Y.J.; Choi, H.G.; Yong, C.S.; Kim, J.O. Preparation and characterization of fast dissolving flurbiprofen and esomeprazole solid dispersion using spray drying technique. Int. J. Pharm. 2016, 502, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Riekes, M.K.; Kuminek, G.; Rauber, G.S.; de Campos, C.E.; Bortoluzzi, A.J.; Stulzer, H.K. HPMC as a potential enhancer of nimodipine biopharmaceutical properties via ball-milled solid dispersions. Carbohydr. Polym. 2014, 99, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Cho, W.; Cha, K.H.; Ahn, J.; Han, K.; Hwang, S.J. Solubilization of the poorly water soluble drug, telmisartan, using supercritical anti-solvent (SAS) process. Int. J. Pharm. 2013, 441, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Rashid, R.; Kim, D.W.; Din, F.U.; Mustapha, O.; Yousaf, A.M.; Park, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.G. Effect of hydroxypropylcellulose and Tween 80 on physicochemical properties and bioavailability of ezetimibe-loaded solid dispersion. Carbohydr. Polym. 2015, 130, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Sarode, A.; Wang, P.; Cote, C.; Worthen, D.R. Low-viscosity hydroxypropylcellulose (HPC) grades SL and SSL: Versatile pharmaceutical polymers for dissolution enhancement, controlled release, and pharmaceutical processing. AAPS PharmSciTech 2013, 14, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, N.; Ishise, S.; Miyata, H.; Danjo, K. Application of nilvadipine solid dispersion to tablet formulation and manufacturing using crospovidone and methylcellulose as dispersion carriers. Chem. Pharm. Bull. (Tokyo) 2004, 52, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Song, C.K.; Yoon, I.S.; Kim, D.D. Poloxamer-based solid dispersions for oral delivery of docetaxel: Differential effects of F68 and P85 on oral docetaxel bioavailability. Int. J. Pharm. 2016, 507, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Balata, G.F.; Zidan, A.S.; Abourehab, M.A.; Essa, E.A. Rapid disintegrating tablets of simvastatin dispersions in polyoxyethylene-polypropylene block copolymer for maximized disintegration and dissolution. Drug Des. Dev. Ther. 2016, 10, 3211–3223. [Google Scholar] [CrossRef] [PubMed]
- Karolewicz, B.; Nartowski, K.; Pluta, J.; Gorniak, A. Physicochemical characterization and dissolution studies of acyclovir solid dispersions with Pluronic F127 prepared by the kneading method. Acta Pharm. 2016, 66, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorajana, A.; Kit, W.W.; Dua, K. Characterization and solubility study of norfloxacin-polyethylene glycol, polyvinylpyrrolidone and carbopol 974p solid dispersions. Recent Patent. Drug Deliv. Formul. 2015, 9, 167–182. [Google Scholar] [CrossRef]
- Meng, F.; Meckel, J.; Zhang, F. Investigation of itraconazole ternary amorphous solid dispersions based on povidone and Carbopol. Eur. J. Pharm. Sci. 2017, 106, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhu, S.J.; Zhou, Y.; Wei, Y.P.; Pei, Y.Y. Enhancement of dissolution of cyclosporine A using solid dispersions with polyoxyethylene (40) stearate. Pharmazie 2006, 61, 681–684. [Google Scholar] [PubMed]
- Parmar, K.R.; Shah, S.R.; Sheth, N.R. Studies in dissolution enhancement of ezetimibe by solid dispersions in combination with a surface adsorbent. Dissolut. Technol. 2011, 18, 55–61. [Google Scholar] [CrossRef]
- Daravath, B.; Tadikonda, R.R.; Vemula, S.K. Formulation and pharmacokinetics of gelucire solid dispersions of flurbiprofen. Drug Dev. Ind. Pharm. 2015, 41, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Panda, T.; Das, D.; Panigrahi, L. Formulation development of solid dispersions of bosentan using Gelucire 50/13 and Poloxamer 188. J. Appl. Pharm. Sci. 2016, 027–033. [Google Scholar] [CrossRef]
- Bali, D.E.; Osman, M.A.; El Maghraby, G.M. Enhancement of dissolution rate and intestinal stability of clopidogrel hydrogen sulfate. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, L.; Yang, P.; Wenslow, R.M., Jr.; Tan, B.; Zhang, H.; Deng, Z. Physicochemical characterization of felodipine-kollidon VA64 amorphous solid dispersions prepared by hot-melt extrusion. J. Pharm. Sci. 2013, 102, 1915–1923. [Google Scholar] [CrossRef] [PubMed]
- Knopp, M.M.; Nguyen, J.H.; Mu, H.; Langguth, P.; Rades, T.; Holm, R. Influence of copolymer composition on in vitro and in vivo performance of celecoxib-PVP/VA amorphous solid dispersions. AAPS J. 2016, 18, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Hiramatsu, T.; Suzuki, R.; Okamoto, R.; Shibagaki, K.; Fujita, K.; Takahashi, C.; Kawashima, Y.; Yamamoto, H. Improvement in the water solubility of drugs with a solid dispersion system by spray drying and hot-melt extrusion with using the amphiphilic polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer and d-mannitol. Eur. J. Pharm. Sci. 2018, 111, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, C.; Kelly, A.L.; Gough, T.; Jadhav, V.; Singh, K.K.; Paradkar, A. Application of hot melt extrusion for improving bioavailability of artemisinin a thermolabile drug. Drug Dev. Ind. Pharm. 2018, 44, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Swain, R.P.; Subudhi, B.B. Effect of semicrystalline polymers on self-emulsifying solid dispersions of nateglinide: In vitro and in vivo evaluation. Drug Dev. Ind. Pharm. 2018, 44, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; Kabra, D.; Pawar, Y.B.; Tikoo, K.; Bansal, A.K. Contribution of formulation and excipients towards enhanced permeation of curcumin. Arzneimittelforschung 2012, 62, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Grymonpre, W.; Bostijn, N.; Herck, S.V.; Verstraete, G.; Vanhoorne, V.; Nuhn, L.; Rombouts, P.; Beer, T.; Remon, J.P.; Vervaet, C. Downstream processing from hot-melt extrusion towards tablets: A quality by design approach. Int. J. Pharm. 2017, 531, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, K.; Obi, N. Studies on absorption of eutectic mixtures. I. A comparison of the behavior of eutectic mixtures of sulphathiazole and that of ordinary sulphathiazole in man. Chem. Pharm. Bull. 1961, 9, 866–872. [Google Scholar] [CrossRef]
- Alhayali, A.; Tavellin, S.; Velaga, S. Dissolution and precipitation behavior of ternary solid dispersions of ezetimibe in biorelevant media. Drug Dev. Ind. Pharm. 2017, 43, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, K.M.; Barbosa, R.M.; Vargas, F.G.A.; de Azevedo, E.P.; Lins, A.; Camara, C.A.; Aragao, C.F.S.; Moura, T.; Raffin, F.N. Development of solid dispersions of beta-lapachone in PEG and PVP by solvent evaporation method. Drug Dev. Ind. Pharm. 2017, 44, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, Q.; Chen, Z.; Zheng, J.; Xu, H.; Li, J.; Liu, H. Preparation and characterization of emulsified solid dispersions containing docetaxel. Arch. Pharm. Res. 2011, 34, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Nokhodchi, A.; Talari, R.; Valizadeh, H.; Jalali, M.B. An investigation on the solid dispersions of chlordiazepoxide. Int. J. Biomed. Sci. 2007, 3, 211–216. [Google Scholar] [PubMed]
- Weuts, I.; Kempen, D.; Verreck, G.; Decorte, A.; Heymans, K.; Peeters, J.; Brewster, M.; Van den Mooter, G. Study of the physicochemical properties and stability of solid dispersions of loperamide and PEG6000 prepared by spray drying. Eur. J. Pharm. Biopharm. 2005, 59, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Naylor, A.; Lewis, A.L.; Ilium, L. Supercritical fluid-mediated methods to encapsulate drugs: Recent advances and new opportunities. Ther. Deliv. 2011, 2, 1551–1565. [Google Scholar] [CrossRef] [PubMed]
- Hezave, A.Z.; Aftab, S.; Esmaeilzadeh, F. Micronization of creatine monohydrate via Rapid Expansion of Supercritical Solution (RESS). J. Supercrit. Fluids 2010, 55, 316–324. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Liu, T.; Zhao, L.; Zhao, J.; Feng, N. Development and in-vivo assessment of the bioavailability of oridonin solid dispersions by the gas anti-solvent technique. Int. J. Pharm. 2011, 411, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Daintree, L.S.; Ding, S.; Ledger, D.M.; Wang, B.; Zhao, W.; Qi, J.; Wu, W.; Han, J. Itraconazole solid dispersion prepared by a supercritical fluid technique: Preparation, in vitro characterization, and bioavailability in beagle dogs. Drug Des. Dev. Ther. 2015, 9, 2801–2810. [Google Scholar] [PubMed]
- LaFountaine, J.S.; McGinity, J.W.; Williams, R.O., 3rd. Challenges and strategies in thermal processing of amorphous solid dispersions: A review. AAPS PharmSciTech 2016, 17, 43–55. [Google Scholar] [CrossRef] [PubMed]
- LaFountaine, J.S.; Jermain, S.V.; Prasad, L.K.; Brough, C.; Miller, D.A.; Lubda, D.; McGinity, J.W.; Williams, R.O., 3rd. Enabling thermal processing of ritonavir-polyvinyl alcohol amorphous solid dispersions by KinetiSol® Dispersing. Eur. J. Pharm. Biopharm. 2016, 101, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Rumondor, A.C.; Dhareshwar, S.S.; Kesisoglou, F. Amorphous solid dispersions or prodrugs: Complementary strategies to increase drug absorption. J. Pharm. Sci. 2016, 105, 2498–2508. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Wang, Y.; Gong, T.; Sun, X.; Zhang, Z.R. Dissolution and bioavailability enhancement of alpha-asarone by solid dispersions via oral administration. Drug Dev. Ind. Pharm. 2017, 43, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Thiry, J.; Kok, M.G.; Collard, L.; Frere, A.; Krier, F.; Fillet, M.; Evrard, B. Bioavailability enhancement of itraconazole-based solid dispersions produced by hot melt extrusion in the framework of the Three Rs rule. Eur. J. Pharm. Sci. 2017, 99, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Maeno, Y.; Fukami, T.; Inoue, M.; Tagami, T.; Ozeki, T. Solid dispersions of efonidipine hydrochloride ethanolate with improved physicochemical and pharmacokinetic properties prepared with microwave treatment. Eur. J. Pharm. Biopharm. 2016, 108, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhang, X.; Zhang, T.; Wang, H.; Wu, B. Design and evaluation of injectable niclosamide nanocrystals prepared by wet media milling technique. Drug Dev. Ind. Pharm. 2015, 41, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Salituro, G.M.; Lee, K.J.; Bak, A.; Leung, D.H. Modulating drug release and enhancing the oral bioavailability of torcetrapib with solid lipid dispersion formulations. AAPS PharmSciTech 2015, 16, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dong, X.; Lu, Y.; Qi, J.; Zhao, W.; Wu, W. Bioimaging of nanoparticles: The crucial role of discriminating nanoparticles from free probes. Drug Discov. Today 2017, 22, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.H.; Mader, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Muchow, M.; Maincent, P.; Muller, R.H. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev. Ind. Pharm. 2008, 34, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, Z.; Zhang, K.; Yang, G.; Wang, Z.; Zhao, J.; Hu, R.; Feng, N. Ethyl oleate-containing nanostructured lipid carriers improve oral bioavailability of trans-ferulic acid ascompared with conventional solid lipid nanoparticles. Int. J. Pharm. 2016, 511, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.M.; de Fatima Reis, C.; Maione-Silva, L.; Anjos, J.L.; Alonso, A.; Serpa, R.C.; Marreto, R.N.; Lima, E.M.; Taveira, S.F. Impact of lipid dynamic behavior on physical stability, in vitro release and skin permeation of genistein-loaded lipid nanoparticles. Eur. J. Pharm. Biopharm. 2014, 88, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Poonia, N.; Kharb, R.; Lather, V.; Pandita, D. Nanostructured lipid carriers: Versatile oral delivery vehicle. Future Sci. OA 2016, 2, Fso135. [Google Scholar] [CrossRef] [PubMed]
- Aboalnaja, K.O.; Yaghmoor, S.; Kumosani, T.A.; McClements, D.J. Utilization of nanoemulsions to enhance bioactivity of pharmaceuticals, supplements, and nutraceuticals: Nanoemulsion delivery systems and nanoemulsion excipient systems. Expert Opin. Drug Deliv. 2016, 13, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xiang, C.; Wang, P.; Yin, Y.; Hou, Y. Biocompatible nanoemulsions based on hemp oil and less surfactants for oral delivery of baicalein with enhanced bioavailability. Int. J. Nanomed. 2017, 12, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Gursoy, R.N.; Benita, S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, J.; Lu, Y.; He, W.; Li, X.; Wu, W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomedicine 2014, 10, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Xie, Q.; Wang, H.; Ma, Z.; Wu, B.; Zhang, X. Selenium nanoparticles as versatile carriers for oral delivery of insulin: Insight into the synergic antidiabetic effect and mechanism. Nanomedicine 2017, 13, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Franze, S.; Bruneau, A.; Alami, M.; Denis, S.; Nicolas, V.; Lesieur, S.; Legrand, F.X.; Barratt, G.; Messaoudi, S.; et al. Formulation and in vitro efficacy of liposomes containing the Hsp90 inhibitor 6BrCaQ in prostate cancer cells. Int. J. Pharm. 2016, 499, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.O.F.; Fernandes, R.S.; Oda, C.M.R.; Lopes, S.C.; Townsend, D.M.; Cardoso, V.N.; Oliveira, M.C.; Leite, E.A.; Rubello, D.; de Barros, A.L.B. Paclitaxel-loaded folate-coated long circulating and pH-sensitive liposomes as a potential drug delivery system: A biodistribution study. Biomed. Pharmacother. 2017, 97, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Daeihamed, M.; Dadashzadeh, S.; Haeri, A.; Akhlaghi, M.F. Potential of liposomes for enhancement of oral drug absorption. Curr. Drug Deliv. 2017, 14, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Gupta, B.P.; Thakur, N.; Jain, R.; Banweer, J.; Jain, D.K.; Jain, S. Phytosome: A novel drug delivery system for herbal medicine. Int. J. Pharm. Sci. Drug Res. 2010, 2, 224–228. [Google Scholar]
- Matias, D.; Rijo, P.; Reis, C.P. Phytosomes as biocompatible carriers of natural drugs. Curr. Med. Chem. 2017, 24, 568–589. [Google Scholar] [CrossRef] [PubMed]
- Gnananath, K.; Sri Nataraj, K.; Ganga Rao, B. Phospholipid complex technique for superior bioavailability of phytoconstituents. Adv. Pharm. Bull. 2017, 7, 35–42. [Google Scholar] [CrossRef] [PubMed]
- El-Gazayerly, O.N.; Makhlouf, A.I.; Soelm, A.M.; Mohmoud, M.A. Antioxidant and hepatoprotective effects of silymarin phytosomes compared to milk thistle extract in CCl4 induced hepatotoxicity in rats. J. Microencapsul. 2014, 31, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.; Shakeri, A.; Rashidi, B.; Jalili, A.; Banikazemi, Z.; Sahebkar, A. Phytosomal curcumin: A review of pharmacokinetic, experimental and clinical studies. Biomed. Pharmacother. 2017, 85, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Telange, D.R.; Patil, A.T.; Pethe, A.M.; Fegade, H.; Anand, S.; Dave, V.S. Formulation and characterization of an apigenin-phospholipid phytosome (APLC) for improved solubility, in vivo bioavailability, and antioxidant potential. Eur. J. Pharm. Sci. 2017, 108, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, H.; Holm, R.; Mullertz, A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int. J. Pharm. 2013, 453, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L. Lipid excipients and delivery systems for pharmaceutical development: A regulatory perspective. Adv. Drug Deliv. Rev. 2008, 60, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Park, Y.J. Improved antitumor efficacy of hyaluronic acid-complexed paclitaxel nanoemulsions in treating non-small cell lung cancer. Biomol. Ther. (Seoul) 2017, 25, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Najlah, M.; Kadam, A.; Wan, K.W.; Ahmed, W.; Taylor, K.M.; Elhissi, A.M. Novel paclitaxel formulations solubilized by parenteral nutrition nanoemulsions for application against glioma cell lines. Int. J. Pharm. 2016, 506, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hayes, D.G.; Chen, G.; Zhong, Q. Transparent dispersions of milk-fat-based nanostructured lipid carriers for delivery of beta-carotene. J. Agric. Food Chem. 2013, 61, 9435–9443. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Yang, C.R.; Yang, K.L.; Li, K.X.; Hu, H.Y.; Chen, D.W. Preparation and characterization of nanostructured lipid carriers loaded traditional Chinese medicine, zedoary turmeric oil. Drug Dev. Ind. Pharm. 2010, 36, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, T.; Ahmad, A.; Ahmed, A.; Ahmed, Z. Optimization of olive oil based O/W nanoemulsions prepared through ultrasonic homogenization: A response surface methodology approach. Food Chem. 2017, 229, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Koutelidakis, A.E.; Argyri, K.; Sevastou, Z.; Lamprinaki, D.; Panagopoulou, E.; Paximada, E.; Sali, A.; Papalazarou, V.; Mallouchos, A.; Evageliou, V.; et al. Bioactivity of epigallocatechin gallate nanoemulsions evaluated in mice model. J. Med. Food 2017, 20, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Burneo, N.; Busquets, M.A.; Estelrich, J. Magnetic Nanoemulsions: Comparison between nanoemulsions formed by ultrasonication and by spontaneous emulsification. Nanomaterials 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, S.I.; Sheikh, B.Y.; Taha, M.M.; How, C.W.; Abdullah, R.; Yagoub, U.; El-Sunousi, R.; Eid, E.E. Thymoquinone-loaded nanostructured lipid carriers: Preparation, gastroprotection, in vitro toxicity, and pharmacokinetic properties after extravascular administration. Int. J. Nanomed. 2013, 8, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Mikulcova, V.; Kasparkova, V.; Humpolicek, P.; Bunkova, L. Formulation, characterization and properties of hemp seed oil and its emulsions. Molecules 2017, 22, E700. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Liu, S.; Wang, Q.; Li, X. Nanoemulsions as novel oral carriers of stiripentol: Insights into the protective effect and absorption enhancement. Int. J. Nanomed. 2015, 10, 4937–4946. [Google Scholar]
- Panatieri, L.F.; Brazil, N.T.; Faber, K.; Medeiros-Neves, B.; von Poser, G.L.; Rott, M.B.; Zorzi, G.K.; Teixeira, H.F. Nanoemulsions containing a coumarin-rich extract from pterocaulon balansae (Asteraceae) for the treatment of ocular acanthamoeba keratitis. AAPS PharmSciTech 2017, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Ragelle, H.; Crauste-Manciet, S.; Seguin, J.; Brossard, D.; Scherman, D.; Arnaud, P.; Chabot, G.G. Nanoemulsion formulation of fisetin improves bioavailability and antitumour activity in mice. Int. J. Pharm. 2012, 427, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejri, A.; Khosravi, A.; Gharanjig, K.; Hejazi, M. Optimisation of the formulation of beta-carotene loaded nanostructured lipid carriers prepared by solvent diffusion method. Food Chem. 2013, 141, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.W.; Billa, N.; Roberts, C.R.; Burley, J.C. Surfactant effects on the physical characteristics of Amphotericin B-containing nanostructured lipid carriers. Colloids Surf. Physicochem. Eng. Aspects 2010, 372, 73–79. [Google Scholar] [CrossRef]
- Wang, L.; Luo, Q.; Lin, T.; Li, R.; Zhu, T.; Zhou, K.; Ji, Z.; Song, J.; Jia, B.; Zhang, C.; et al. PEGylated nanostructured lipid carriers (PEG-NLC) as a novel drug delivery system for biochanin A. Drug Dev. Ind. Pharm. 2015, 41, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Sood, J.; Sapra, B.; Tiwary, A.K. Microemulsion transdermal formulation for simultaneous delivery of valsartan and nifedipine: Formulation by design. AAPS PharmSciTech 2017, 18, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Ramesh, D. Design, optimization and evaluation of glipizide solid self-nanoemulsifying drug delivery for enhanced solubility and dissolution. Saudi Pharm. J. 2015, 23, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.L.; Botto, C.; Amore, E.; Emma, M.R.; Augello, G.; Craparo, E.F.; Cervello, M. Lipid nanocarriers containing sorafenib inhibit colonies formation in human hepatocarcinoma cells. Int. J. Pharm. 2015, 493, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, A.; Ponnusamy, C.; Kandasamy, P.; Krishnaswami, V.; Palanichamy, R.; Kandasamy, R.; Lakshmanan, M.; Natesan, S. Development and evaluation of camptothecin loaded polymer stabilized nanoemulsion: Targeting potential in 4T1-breast tumour xenograft model. Eur. J. Pharm. Sci. 2018, 116, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Griesser, J.; Burtscher, S.; Kollner, S.; Nardin, I.; Prufert, F.; Bernkop-Schnurch, A. Zeta potential changing self-emulsifying drug delivery systems containing phosphorylated polysaccharides. Eur. J. Pharm. Biopharm. 2017, 119, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.V.; Desai, H.H.; Thool, P.; Dalrymple, D.; Serajuddin, A.T.M. Development of self-microemulsifying drug delivery system for oral delivery of poorly water-soluble nutraceuticals. Drug Dev. Ind. Pharm. 2018, 44, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pan, W.; Gan, L.; Zhu, C.; Gan, Y.; Nie, S. Preparation of a dispersible PEGylate nanostructured lipid carriers (NLC) loaded with 10-hydroxycamptothecin by spray-drying. Chem. Pharm. Bull. 2008, 56, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Katiyar, S.S.; Kushwah, V.; Jain, S. Nanoemulsion loaded gel for topical co-delivery of clobitasol propionate and calcipotriol in psoriasis. Nanomedicine 2017, 13, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Shelat, P.; Lalwani, A. Statistical modeling, optimization and characterization of solid self-nanoemulsifying drug delivery system of lopinavir using design of experiment. Drug Deliv. 2016, 23, 3027–3042. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.V.; Seth, A.K.; Balaraman, R.; Aundhia, C.J.; Maheshwari, R.A.; Parmar, G.R. Nanostructured lipid carriers for oral bioavailability enhancement of raloxifene: Design and in vivo study. J. Adv. Res. 2016, 7, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, Y.S.; El-Massik, M.A.; Abdallah, O.Y. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: Design and optimization. Int. J. Pharm. 2009, 380, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Katare, O.P.; Singh, B. Optimized self nano-emulsifying systems of ezetimibe with enhanced bioavailability potential using long chain and medium chain triglycerides. Colloids Surf. B Biointerfaces 2012, 100, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Kamboj, S.; Rana, V. Quality-by-design based development of a self-microemulsifying drug delivery system to reduce the effect of food on Nelfinavir mesylate. Int. J. Pharm. 2016, 501, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Yang, G.; Ren, J.; Guo, T.; Du, Y.; Feng, N. Formulation design, preparation, and in vitro and in vivo characterizations of beta-Elemene-loaded nanostructured lipid carriers. Int. J. Nanomed. 2013, 8, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Mustapha, O.; Kim, K.S.; Shafique, S.; Kim, D.S.; Jin, S.G.; Seo, Y.G.; Youn, Y.S.; Oh, K.T.; Lee, B.J.; Park, Y.J.; et al. Development of novel cilostazol-loaded solid SNEDDS using a SPG membrane emulsification technique: Physicochemical characterization and in vivo evaluation. Colloids Surf. B Biointerfaces 2017, 150, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Drechsler, M.; Mariani, P.; Carducci, F.; Servadio, M.; Melancia, F.; Ratano, P.; Campolongo, P.; Trezza, V.; Cortesi, R.; et al. Lipid nanoparticles for administration of poorly water soluble neuroactive drugs. Biomed. Microdevices 2017, 19, 44. [Google Scholar] [CrossRef] [PubMed]
- Dawoud, M.Z.; Nasr, M. Comparison of drug release from liquid crystalline monoolein dispersions and solid lipid nanoparticles using a flow cytometric technique. Acta Pharm. Sin. B 2016, 6, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Brusewitz, C.; Schendler, A.; Funke, A.; Wagner, T.; Lipp, R. Novel poloxamer-based nanoemulsions to enhance the intestinal absorption of active compounds. Int. J. Pharm. 2007, 329, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.G.; Kim, D.W.; Cho, K.H.; Yousaf, A.M.; Kim, D.S.; Kim, J.H.; Kim, J.O.; Yong, C.S.; Choi, H.G. Preparation and pharmaceutical evaluation of new tacrolimus-loaded solid self-emulsifying drug delivery system. Arch. Pharm. Res. 2015, 38, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Thatipamula, R.; Palem, C.; Gannu, R.; Mudragada, S.; Yamsani, M. Formulation and in vitro characterization of domperidone loaded solid lipid nanoparticles and nanostructured lipid carriers. Daru 2011, 19, 23–32. [Google Scholar] [PubMed]
- Hsu, H.J.; Huang, R.F.; Kao, T.H.; Inbaraj, B.S.; Chen, B.H. Preparation of carotenoid extracts and nanoemulsions from Lycium barbarum L. and their effects on growth of HT-29 colon cancer cells. Nanotechnology 2017, 28, 135103. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Modi, S.R.; Bansal, A.K. Role of solid carriers in pharmaceutical performance of solid supersaturable SEDDS of celecoxib. Int. J. Pharm. 2015, 495, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, M.; Ibrahim, H.M.; Ahmed, M.A.; Shalaby, K.; Salama, A.; Hefesha, H. Atorvastatin-loaded nanostructured lipid carriers (NLCs): Strategy to overcome oral delivery drawbacks. Drug Deliv. 2017, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Zhou, C.L.; Chen, F.P.; Han, D.; Wang, C.Y.; Li, J.X.; Chi, Z.; Liu, C.G. Development of a carboxymethyl chitosan functionalized nanoemulsion formulation for increasing aqueous solubility, stability and skin permeability of astaxanthin using low-energy method. J. Microencapsul. 2017, 34, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Kamble, R.N.; Mehta, P.P.; Kumar, A. Efavirenz self-nano-emulsifying drug delivery system: In vitro and in vivo evaluation. AAPS PharmSciTech 2016, 17, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Safwat, S.; Ishak, R.A.H.; Hathout, R.M.; Mortada, N.D. Nanostructured lipid carriers loaded with simvastatin: Effect of PEG/glycerides on characterization, stability, cellular uptake efficiency and in vitro cytotoxicity. Drug Dev. Ind. Pharm. 2017, 43, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Cuomo, F.; Cofelice, M.; Venditti, F.; Ceglie, A.; Miguel, M.; Lindman, B.; Lopez, F. In-vitro digestion of curcumin loaded chitosan-coated liposomes. Colloids Surf. B. Biointerfaces 2018, 168, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Freag, M.S.; Saleh, W.M.; Abdallah, O.Y. Self-assembled phospholipid-based phytosomal nanocarriers as promising platforms for improving oral bioavailability of the anticancer celastrol. Int. J. Pharm. 2017, 535, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, T.; Zhou, X.; Liu, H.; Sun, H.; Ma, Z.; Wu, B. Enhancement of oral bioavailability of tripterine through lipid nanospheres: Preparation, characterization, and absorption evaluation. J. Pharm. Sci. 2014, 103, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lu, S.T.; Zhang, L.J.; Zhuo, R.X.; Xu, H.B.; Huang, S.W. Codelivery of doxorubicin and triptolide with reduction-sensitive lipid-polymer hybrid nanoparticles for in vitro and in vivo synergistic cancer treatment. Int. J. Nanomed. 2017, 12, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Wu, M.; Zhang, C.; Lin, X.; Wei, Z.; Zheng, Y.; Da, Z.; Zhang, Z.; Liu, X. Photoresponsive lipid-polymer hybrid nanoparticles for controlled doxorubicin release. Nanotechnology 2017, 28, 255101. [Google Scholar] [CrossRef] [PubMed]
- Valicherla, G.R.; Dave, K.M.; Syed, A.A.; Riyazuddin, M.; Gupta, A.P.; Singh, A.; Mitra, K.; Datta, D.; Gayen, J.R. Formulation optimization of Docetaxel loaded self-emulsifying drug delivery system to enhance bioavailability and anti-tumor activity. Sci. Rep. 2016, 6, 26895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaber, D.M.; Nafee, N.; Abdallah, O.Y. Myricetin solid lipid nanoparticles: Stability assurance from system preparation to site of action. Eur. J. Pharm. Sci. 2017, 109, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. Design and in vivo evaluation of solid lipid nanoparticulate systems of Olanzapine for acute phase schizophrenia treatment: Investigations on antipsychotic potential and adverse effects. Eur. J. Pharm. Sci. 2017, 104, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, L.M.; Maestrelli, F.; Di Cesare Mannelli, L.; Ghelardini, C.; Almeida, A.J.; Mura, P. Development of solid lipid nanoparticles as carriers for improving oral bioavailability of glibenclamide. Eur. J. Pharm. Biopharm. 2016, 102, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Hu, X.; Ma, Y.; Xie, Y.; Lu, Y.; Qi, J.; Xiang, L.; Li, F.; Wu, W. Lipids-based nanostructured lipid carriers (NLCs) for improved oral bioavailability of sirolimus. Drug Deliv. 2016, 23, 1469–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, M.; Samy, A.; Raslan, M.A.; Salama, A.; Said, R.A.; Abdelaziz, A.E.; El-Eraky, W.; El Awdan, S.; Viitala, T. Enhancement of bioavailability and pharmacodynamic effects of thymoquinone via nanostructured lipid carrier (NLC) formulation. AAPS PharmSciTech 2016, 17, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Meng, X.; Wang, Z.; Fan, A.; Wang, G.; Zhao, Y.; Tang, Y. Engineering hot-melt extruded solid dispersion for controlled release of hydrophilic drugs. Eur. J. Pharm. Sci. 2017, 100, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Lim, S.J.; Lee, M.K. Cellular uptake and antitumour activity of paclitaxel incorporated into trilaurin-based solid lipid nanoparticles in ovarian cancer. J. Microencapsul. 2013, 30, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, P.C.; Zhang, L.; Mullertz, A.; Nielsen, H.M.; Yang, M.; Mu, H. Solid lipid particles for oral delivery of peptide and protein drugs II--the digestion of trilaurin protects desmopressin from proteolytic degradation. Pharm. Res. 2014, 31, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; McClements, D.J.; Xiao, H. Improving oral bioavailability of nutraceuticals by engineered nanoparticle-based delivery systems. Curr. Opin. Food Sci. 2015, 2, 14–19. [Google Scholar] [CrossRef]
- Keck, C.M.; Kovacevic, A.; Muller, R.H.; Savic, S.; Vuleta, G.; Milic, J. Formulation of solid lipid nanoparticles (SLN): The value of different alkyl polyglucoside surfactants. Int. J. Pharm. 2014, 474, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, L.N.; Franz-Montan, M.; Breitkreitz, M.C.; Alcantara, A.C.; Castro, S.R.; Guilherme, V.A.; Barbosa, R.M.; de Paula, E. Nanostructured lipid carriers as robust systems for topical lidocaine-prilocaine release in dentistry. Eur. J. Pharm. Sci. 2016, 93, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumar, A.; Baskaran, R.; Jang, Y.S.; Oh, S.H.; Yoo, B.K. Quercetin-loaded solid lipid nanoparticle dispersion with improved physicochemical properties and cellular uptake. AAPS PharmSciTech 2017, 18, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Wu, P.C.; Huang, Y.B.; Chang, J.S.; Lin, C.L.; Tsai, Y.H.; Fang, J.Y. Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int. J. Pharm. 2012, 423, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergstrom, C.A.S.; Porter, C.J.H. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zhang, G.G.Z. Physical chemistry of supersaturated solutions and implications for oral absorption. Adv. Drug Deliv. Rev. 2016, 101, 122–142. [Google Scholar] [CrossRef] [PubMed]
- Shekhawat, P.B.; Pokharkar, V.B. Understanding peroral absorption: Regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm. Sin. B 2017, 7, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.; Yang, W.; Yu, M.; Sun, S.; Xie, B. Improved oral bioavailability and liver targeting of sorafenib solid lipid nanoparticles in rats. AAPS PharmSciTech 2018, 19, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, X.; Ye, Y.; Zhang, T.; Wang, H.; Ma, Z.; Wu, B. Nanostructured lipid carriers used for oral delivery of oridonin: An effect of ligand modification on absorption. Int. J. Pharm. 2015, 479, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, T.; Ye, Y.; Zhang, X.; Wu, B. Enhanced bioavailability of tripterine through lipid nanoparticles using broccoli-derived lipids as a carrier material. Int. J. Pharm. 2015, 495, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Zamudio, R.; Ochoa-Flores, A.A.; Soto-Rodriguez, I.; Garcia-Varela, R.; Garcia, H.S. Preparation, characterization and bioavailability by oral administration of O/W curcumin nanoemulsions stabilized with lysophosphatidylcholine. Food Funct. 2017, 8, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; He, X.; Zhang, Y.; Zhang, Y.; Chai, F.; Jiang, L.; Webster, T.J.; Zheng, C. A dabigatran etexilate phospholipid complex nanoemulsion system for further oral bioavailability by reducing drug-leakage in the gastrointestinal tract. Nanomedicine 2017. [Google Scholar] [CrossRef] [PubMed]
- Rushmi, Z.T.; Akter, N.; Mow, R.J.; Afroz, M.; Kazi, M.; de Matas, M.; Rahman, M.; Shariare, M.H. The impact of formulation attributes and process parameters on black seed oil loaded liposomes and their performance in animal models of analgesia. Saudi Pharm. J. 2017, 25, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, J.; Yu, Z.; Xie, Y.; He, H.; Qi, J.; Dong, X.; Lu, Y.; Zhao, W.; Wu, W. Environment-responsive aza-BODIPY dyes quenching in water as potential probes to visualize the in vivo fate of lipid-based nanocarriers. Nanomedicine 2015, 11, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Fan, W.; Jiang, S.; Ma, Y.; Lu, Y.; Qi, J.; Ahmad, E.; Dong, X.; Zhao, W.; Wu, W. Size-dependent translocation of nanoemulsions via oral delivery. ACS Appl. Mater. Interfaces 2017, 9, 21660–21672. [Google Scholar] [CrossRef] [PubMed]
- Heurtault, B.; Saulnier, P.; Pech, B.; Proust, J.E.; Benoit, J.P. Physico-chemical stability of colloidal lipid particles. Biomaterials 2003, 24, 4283–4300. [Google Scholar] [CrossRef]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming lipid-based oral drug delivery systems into solid dosage forms: An overview of solid carriers, physicochemical properties, and biopharmaceutical performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.D.; Lu, X.; Jay, M.; Dziubla, T.D. Optimization of the lyophilization process for long-term stability of solid-lipid nanoparticles. Drug Dev. Ind. Pharm. 2012, 38, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Freag, M.S.; Elnaggar, Y.S.; Abdallah, O.Y. Lyophilized phytosomal nanocarriers as platforms for enhanced diosmin delivery: Optimization and ex vivo permeation. Int. J. Nanomed. 2013, 8, 2385–2397. [Google Scholar]
- Wang, T.; Hu, Q.; Zhou, M.; Xue, J.; Luo, Y. Preparation of ultra-fine powders from polysaccharide-coated solid lipid nanoparticles and nanostructured lipid carriers by innovative nano spray drying technology. Int. J. Pharm. 2016, 511, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Hu, Q.; Zhou, M.; Xia, Y.; Nieh, M.P.; Luo, Y. Development of “all natural” layer-by-layer redispersible solid lipid nanoparticles by nano spray drying technology. Eur. J. Pharm. Biopharm. 2016, 107, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Yi, Y.; Yuan, H.; Han, J.; Zhang, X.; Xie, Y.; Lu, Y.; Qi, J.; Wu, W. Solidification of nanostructured lipid carriers (NLCs) onto pellets by fluid-bed coating: Preparation, in vitro characterization and bioavailability in dogs. Powder Technol. 2013, 247, 120–127. [Google Scholar] [CrossRef]
- Ali, M.E.; Lamprecht, A. Spray freeze drying as an alternative technique for lyophilization of polymeric and lipid-based nanoparticles. Int. J. Pharm. 2017, 516, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Chouhan, N.; Mittal, V.; Kaushik, D.; Khatkar, A.; Raina, M. Self emulsifying drug delivery system (SEDDS) for phytoconstituents: A review. Curr. Drug Deliv. 2015, 12, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Keohane, K.; Rosa, M.; Coulter, I.S.; Griffin, B.T. Enhanced colonic delivery of ciclosporin A self-emulsifying drug delivery system encapsulated in coated minispheres. Drug Dev. Ind. Pharm. 2016, 42, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.; Dhingani, A.; Garala, K.; Raval, M.; Sheth, N. Quality by design approach for oral bioavailability enhancement of irbesartan by self-nanoemulsifying tablets. Drug Deliv. 2014, 21, 412–435. [Google Scholar] [CrossRef] [PubMed]
- Nnamani, P.O.; Ugwu, A.A.; Ibezim, E.C.; Kenechukwu, F.C.; Akpa, P.A.; Ogbonna, J.N.; Obitte, N.C.; Odo, A.N.; Windbergs, M.; Lehr, C.M.; et al. Sustained-release liquisolid compact tablets containing artemether-lumefantrine as alternate-day regimen for malaria treatment to improve patient compliance. Int. J. Nanomed. 2016, 11, 6365–6378. [Google Scholar] [CrossRef] [PubMed]
- Spireas, S.; Sadu, S.; Grover, R. In vitro release evaluation of hydrocortisone liquisolid tablets. J. Pharm. Sci. 1998, 87, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Nokhodchi, A.; Hentzschel, C.M.; Leopold, C.S. Drug release from liquisolid systems: Speed it up, slow it down. Expert Opin. Drug Deliv. 2011, 8, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Kamble, P.R.; Shaikh, K.S.; Chaudhari, P.D. Application of liquisolid technology for enhancing solubility and dissolution of rosuvastatin. Adv. Pharm. Bull. 2014, 4, 197–204. [Google Scholar] [PubMed]
- Badawy, M.A.; Kamel, A.O.; Sammour, O.A. Use of biorelevant media for assessment of a poorly soluble weakly basic drug in the form of liquisolid compacts: In vitro and in vivo study. Drug Deliv. 2016, 23, 818–827. [Google Scholar] [PubMed]
- Khames, A. Liquisolid technique: A promising alternative to conventional coating for improvement of drug photostability in solid dosage forms. Expert Opin. Drug Deliv. 2013, 10, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Pathak, K. Effect of hydrogen bond formation/replacement on solubility characteristics, gastric permeation and pharmacokinetics of curcumin by application of powder solution technology. Acta Pharm. Sin. B 2016, 6, 600–613. [Google Scholar] [CrossRef] [PubMed]
- Yehia, S.A.; El-Ridi, M.S.; Tadros, M.I.; El-Sherif, N.G. Enhancement of the oral bioavailability of fexofenadine hydrochloride via Cremophor® El-based liquisolid tablets. Adv. Pharm. Bull. 2015, 5, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Chella, N.; Narra, N.; Rama Rao, T. Preparation and characterization of liquisolid compacts for improved dissolution of telmisartan. J. Drug Deliv. 2014. [Google Scholar] [CrossRef] [PubMed]
- Elkordy, A.A.; Essa, E.A.; Dhuppad, S.; Jammigumpula, P. Liquisolid technique to enhance and to sustain griseofulvin dissolution: Effect of choice of non-volatile liquid vehicles. Int. J. Pharm. 2012, 434, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Khames, A. Investigation of the effect of solubility increase at the main absorption site on bioavailability of BCS class II drug (risperidone) using liquisolid technique. Drug Deliv. 2017, 24, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javadzadeh, Y.; Jafari-Navimipour, B.; Nokhodchi, A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine). Int. J. Pharm. 2007, 341, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kala, N.P.; Shastri, D.H.; Shelat, P.K. Design and Characterization of buccoadhesive liquisolid system of an antihypertensive drug. J. Drug Deliv. 2015, 2015, 574247. [Google Scholar] [CrossRef] [PubMed]
- Hentzschel, C.M.; Alnaief, M.; Smirnova, I.; Sakmann, A.; Leopold, C.S. Enhancement of griseofulvin release from liquisolid compacts. Eur. J. Pharm. Biopharm. 2012, 80, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Vranikova, B.; Gajdziok, J. Evaluation of sorptive properties of various carriers and coating materials for liquisolid systems. Acta Pol. Pharm. 2015, 72, 539–549. [Google Scholar] [PubMed]
- Prajapati, S.T.; Bulchandani, H.H.; Patel, D.M.; Dumaniya, S.K.; Patel, C.N. Formulation and evaluation of liquisolid compacts for olmesartan medoxomil. J. Drug Deliv. 2013. [Google Scholar] [CrossRef] [PubMed]
- Venkateswarlu, K.; Preethi, J.K.; Chandrasekhar, K.B. Enhancement of loperamide dissolution rate by liquisolid compact technique. Adv. Pharm. Bull. 2016, 6, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Hentzschel, C.M.; Sakmann, A.; Leopold, C.S. Suitability of various excipients as carrier and coating materials for liquisolid compacts. Drug Dev. Ind. Pharm. 2011, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xing, H.; Jiang, J.; Chen, X.; Yang, T.; Wang, D.; Ding, P. Liquisolid technique and its applications in pharmaceutics. Asian J. Pharm. Sci. 2017, 12, 115–123. [Google Scholar] [CrossRef]
- Javadzadeh, Y.; Musaalrezaei, L.; Nokhodchi, A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int. J. Pharm. 2008, 362, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, A.B.; Gonjari, I.D.; Hosmani, A.H. Liquisolid technology for dissolution rate enhancement or sustained release. Expert Opin. Drug Deliv. 2010, 7, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, A.B.; Gonjari, I.D.; Hosmani, A.H.; Dhabale, P.N. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Drug Discov. Ther. 2010, 4, 26–32. [Google Scholar] [PubMed]
- Spireas, S.S.; Jarowski, C.I.; Rohera, B.D. Powdered solution technology: Principles and mechanism. Pharm. Res. 1992, 9, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Aloorkar, N.H.; Mane, M.S.; Gaja, J.B. Liquisolid systems: A review. Int. J. Pharm. Sci. Nanotechnol. 2010, 3, 795–802. [Google Scholar]
- Gavali, S.M.; Pacharane, S.S.; Sankpal, S.V.; Jadhav, K.R.; Kadam, V.J. Liquisolid compact: A new technique for enhancement of drug dissolution. Int. J. Res. Pharm. Chem. 2011, 1, 705–713. [Google Scholar]
- Barmpalexis, P.; Grypioti, A.; Eleftheriadis, G.K.; Fatouros, D.G. Development of a new aprepitant liquisolid formulation with the aid of artificial neural networks and genetic programming. AAPS PharmSciTech 2018, 19, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Karmarkar, A.B. Effect of Ceolus KG-802 on the dissolution rate of fenofibrate liquisolid tablets: Preformulation and formulation development studies. Drug Discov. Ther. 2010, 4, 493–498. [Google Scholar] [PubMed]
- Saeedi, M.; Akbari, J.; Morteza-Semnani, K.; Enayati-Fard, R.; Sar-Reshteh-Dar, S.; Soleymani, A. Enhancement of dissolution rate of indomethacin: Using liquisolid compacts. Iran. J. Pharm. Sci. 2011, 10, 25–34. [Google Scholar]
- Vittal, G.V.; Deveswaran, R.; Bharath, S.; Basavaraj, B.; Madhavan, V. Formulation and characterization of ketoprofen liquisolid compacts by Box-Behnken design. Int. J. Pharm. Investig. 2012, 2, 150–156. [Google Scholar] [PubMed]
- El-Hammadi, M.; Awad, N. Investigating the use of liquisolid compacts technique to minimize the influence of pH variations on loratadine release. AAPS PharmSciTech 2012, 13, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Vaskula, S.; Vemula, S.K.; Bontha, V.K.; Garrepally, P. Liquisolid compacts: An approach to enhance the dissolution rate of nimesulide. J. Appl. Pharm. Sci. 2012, 2, 115–121. [Google Scholar]
- Akinlade, B.; Elkordy, A.A.; Essa, E.A.; Elhagar, S. Liquisolid systems to improve the dissolution of furosemide. Sci. Pharm. 2010, 78, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.S.; Pipaliya, R.M.; Surti, N. Liquisolid tablets for dissolution enhancement of a hypolipidemic drug. Indian J. Pharm. Sci. 2015, 77, 290–298. [Google Scholar] [PubMed]
- Chella, N.; Shastri, N.; Tadikonda, R.R. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm. Sin. B 2012, 2, 502–508. [Google Scholar] [CrossRef]
- Shin, S. Dissolution characteristics of furosemide-polymer coprecipitates. Arch. Pharm. Res. 1979, 2, 35–48. [Google Scholar] [CrossRef]
- Shin, S.-C.; Oh, I.-J.; Lee, Y.-B.; Choi, H.-K.; Choi, J.-S. Enhanced dissolution of furosemide by coprecipitating or cogrinding with crospovidone. Int. J. Pharm. 1998, 175, 17–24. [Google Scholar] [CrossRef]
- Planinsek, O.; Kovacic, B.; Vrecer, F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int. J. Pharm. 2011, 406, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, A.; Malamatari, M.; Kachrimanis, K. Pharmaceutical cocrystals: New solid phase modification approaches for the formulation of APIs. Pharmaceutics 2018, 10, E18. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodriguez-Hornedo, N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Wang, Y.; Wu, S.; Yu, B.; Shi, P.; Bian, L.; Zhang, D.; Hou, J.; Wang, J.; Gong, J. Two novel cocrystals of lamotrigine with isomeric bipyridines and in situ monitoring of the cocrystallization. Eur. J. Pharm. Sci. 2017, 110, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Sun, C.C. Improving dissolution rate of carbamazepine-glutaric acid cocrystal through solubilization by excess coformer. Pharm. Res. 2017, 35, 4. [Google Scholar] [CrossRef] [PubMed]
- Semalty, A. Cyclodextrin and phospholipid complexation in solubility and dissolution enhancement: A critical and meta-analysis. Expert Opin. Drug Deliv. 2014, 11, 1255–1272. [Google Scholar] [CrossRef] [PubMed]
- Mady, F.M.; Farghaly Aly, U. Experimental, molecular docking investigations and bioavailability study on the inclusion complexes of finasteride and cyclodextrins. Drug Des. Dev. Ther. 2017, 11, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Moya-Ortega, M.D.; Alvarez-Lorenzo, C.; Concheiro, A. Pharmacokinetics of cyclodextrins and drugs after oral and parenteral administration of drug/cyclodextrin complexes. J. Pharm. Pharmacol. 2016, 68, 544–555. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier Excipients | Example | Comments | Reference |
|---|---|---|---|
| Saccharides | Sucrose Glucose Lactose Dextrose | Ordinary dispersibility; having potential effect on drug absorption. | [29,30,31,32,33] |
| Alcohols | Mannitol Sorbitol | Ordinary dispersibility; weak absorption-promoting effect. | [34,35] [36] |
| Organic acids | Citric acid Tartaric acid | Effervescent dispersion;Simple dispersing material, not applicable for acid-labile API. | [37,38] [39] |
| Polyethylene glycol | PEG 4000 PEG 6000 | High dispersibility; able to solubilize drug and delay aging of SDs. | [40] [41,42] |
| Polyvidone | PVP k15 PVP k30 | High dispersibility; able to inhibit recrystallization. | [42] [23,43,44] |
| Cellulose derivative | HPMC HPC MC | High dispersibility; less plasticity and hygroscopicity, easy to process. | [45,46] [47,48] [49] |
| Poly(oxyethylene–co-oxypropylene) | Poloxamer 188 Poloxamer 407 | High dispersibility; able to solubilize drug and having absorption-promoting effect. | [50,51] [52] |
| Carboxypolymethylene | Carbopol 947 Carbopol 907 | Ionic polymers; good dispersibility; rapid drug release in the intestine. | [53] [54] |
| Polyoxyethylene stearate | Polyoxyethylene (40) stearate | Fine dispersibility; contribute less to dissolution; used rarely. | [55] |
| Fatty acid macrogolglycerides | Gelucire 44/14 Gelucire 50/13 | Functional dispersing materials; either able to enhance dissolution or to promote drug absorption. | [56,57] [58,59] |
| Poly(vinylpyrrolidone-co-vinyl acetate) | PVP/VA | Fine dispersibility but low hygroscopicity; superior to PVP in function. | [60,61] |
| Poly(vinyl acetate-co-vinyl caprolactame-co-ethylene glycol) | Soluplus® | Novel dispersing material; excellent capability to form solid solution. | [62,63] |
| Lipid Excipient | Chemical | Carrier Type | Comments | Reference |
|---|---|---|---|---|
| Soybean oil | Long-chain triglycerides | Nanoemulsions; NLCs | Liquid, high biocompatibility, negligible physiological effect, solubilizing capacity a little weak. | [109,110,111,112] |
| Olive oil | Long-chain triglycerides | Nanoemulsions; NLCs | Liquid, good health benefits, containing more monounsaturated fatty acid, easy to emulsify. | [110,113,114,115,116] |
| Hemp oil | Medium/long-chain triglycerides blended with low-molecular-weight lipids | Nanoemulsions | Liquid; rich in essential fatty acids, having tocopherols, tocotrienols, phytosterols, phospholipids, etc., excellent hydrophilicity and self-emulsifiability. | [93,117] |
| Caprylic/capric triglycerides | Medium-chain triglycerides | Nanoemulsions; NLCs | Liquid, fine solubilizing capacity, good compatibility with other lipids, easy to emulsify. | [118,119,120,121,122,123] |
| Captex® series | Medium/short-chain triglycerides | Nanoemulsions; SEDDS; NLCs | liquid, fine solubilizing and emulsifying capacities, miscible with other lipids. | [124,125,126] |
| Capmul MCM | Medium-chain mono/diglycerides | Nanoemulsions; SEDDS; NLCs | Liquid, excellent solvent powder for many organic compounds, can use as emulsifier. | [127,128,129,130] |
| Capmul MCM C8 | Glyceryl monocaprylate | Nanoemulsions; SEDDS; NLCs | Liquid, properties similar to that of Capmul MCM. | [131,132,133] |
| Maisine TM 35-1 | Glyceryl monolinoleate | SEDDS | Liquid, solubilizer, bioavailability enhancer, oil phase in SEDDS. | [134,135,136,137] |
| PeceolTM | Glyceryl monooleate | SEDDS; NLCs; Cubosomes; | Liquid, lipid dispersion agent, oil-soluble surfactant, moisturizer. | [138,139,140] |
| Lauroglycol® 90 | Propylene glycol monolaurate | Nanoemulsions; SEDDS; NLCs | Liquid, water insoluble surfactant of SEDDS, solubilizer, bioavailability enhancer, skin penetration solubilizer enhancer. | [141,142,143] |
| CapryolTM series | Propylene glycol monocaprylate | Nanoemulsions; SEDDS; NLCs | Liquid, properties similar to that of Lauroglycol® 90. | [144,145,146] |
| Labrafil M 1944 CS | Oleoyl polyoxyl-6 glycerides | Nanoemulsions; SEDDS; NLCs | Liquid, water dispersible surfactant, able to self-emulsify, good miscibility with other lipids, bioavailability enhancer, solubilizer, co-emulsifier. | [147,148,149] |
| Lecithin | Phosphatidylcholine blended with a small amount of other lipid components. | Liposomes; phytosomes; sorts of lipid nanoparticles | Semi-solid, an amphiphilic lipid, used as vesicles-forming material, solubilizing, emulsifying, and stabilizing agents. | [150,151,152,153,154] |
| Gelucire® series | Lipid blends consisting of mono-, di-, or triglycerides and fatty acid macrogolglycerides | SEDDS; SLNs; NLCs | Semi-solid, non-ionic water soluble surfactant for solid/semi-solid dispersions and SEDDS, bioavailability enhancer, micelles-forming material, solubilizing and wetting agent. | [146,155,156] |
| Monostearin | Glyceryl monostearate | SLNs; NLCs | Solid, lipid matrix for SLNs and NLCs; thickening, solidifying and control release adjusting agent. | [133,157] |
| Precirol® ATO 5 | Glyceryl distearate | SLNs; NLCs | Solid, lipid matrix for SLNs and NLCs, hydrophobicity and melting point greater than glyceryl monostearate. | [158,159] |
| Compritol® 888 ATO | Glyceryl behenate | SLNs; NLCs; solid lipid dispersions | Solid, high melting point lipid, used for preparation of SLNs and NLCs, lipid matrix for sustained release, used as atomized powders. | [160,161,162] |
| Trilaurin | Glyceryl trilaurate | SLNs; NLCs; | Solid, lipid matrix for SLNs and NLCs, sustained release material, thickening agent. | [163,164,165] |
| Cetyl palmitate | Palmityl palmitate | SLNs; NLCs; | Solid, wax-like substance, used for preparation of SLNs and NLCs. | [166,167] |
| Tripalmitin | Glyceryl tripalmitate | SLNs; NLCs; | Solid, as lipid matrix of SLNs and NLCs, skin-conditioning agent. | [168,169] |
| Excipients Type | Characteristics | Function | Examples | Reference |
|---|---|---|---|---|
| Non-volatile solvent | Inert, water-miscible, compatible with drug candidate, excellent dissolving powder. | Non-volatile solvent acts as a solvent and a binding agent in a liquisolid system. | PEG series; glycerin; propylene glycol; polysorbate; Cremophor® EL; Transcutol HP; CapryolTM 90; 2-pyrrolidone; Labrasol, etc. | [195,196,197,198,199,200,201,202] |
| Carrier material | Porous, large specific surface area, sufficient adsorption ability, good flowability and compressibility. | Carrier material plays a fundamental role in forming the dry form of powders from liquid medication. | Microcrystalline cellulose (MCC, e.g., Avicel®, Ceolus®, Vivapur®, Emcocel®); lactose; mannitol; Magnesium Aluminometasilicate (Neusilin®); Dibasic calcium phosphate anhydrous (Fujculin®); | [196,197,203,204,205,206,207] |
| Coating material | Ultrafine and highly adsorptive particles, good flow-aided effect. | Coating material contributes to covering the wet surface of particles by adsorbing excess liquid to ensure a good flowability of powders. | Colloidal silicon dioxide (e.g., Aerosil®, Cab-O-Sil® M5); Neusilin®; Calcium Silicate (Florite®) | [195,196,204,208,209] |
| Other adjuvants | Disintegrant, lubricant, release modifiers, flavoring and coloring agents, etc. | The selected adjuvants can improve the quality of solid dosage forms. | Sodium starch glycolate (CMS-Na); crospovidone; L-HPC; PVP k25; PEG 6000; HPMC; Eudragit. | [22,210] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Xing, H.; Zhao, Y.; Ma, Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics 2018, 10, 74. https://doi.org/10.3390/pharmaceutics10030074
Zhang X, Xing H, Zhao Y, Ma Z. Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics. 2018; 10(3):74. https://doi.org/10.3390/pharmaceutics10030074
Chicago/Turabian StyleZhang, Xingwang, Huijie Xing, Yue Zhao, and Zhiguo Ma. 2018. "Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs" Pharmaceutics 10, no. 3: 74. https://doi.org/10.3390/pharmaceutics10030074
APA StyleZhang, X., Xing, H., Zhao, Y., & Ma, Z. (2018). Pharmaceutical Dispersion Techniques for Dissolution and Bioavailability Enhancement of Poorly Water-Soluble Drugs. Pharmaceutics, 10(3), 74. https://doi.org/10.3390/pharmaceutics10030074