Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Preparation
2.2. RNA Preparation
2.3. PacBio cDNA Library Construction and Sequencing
2.4. Illumina cDNA Library Construction and Sequencing
2.5. Quality Filtering and Error Correction of PacBio Long Reads
2.6. Analysis of Detecting Coding Sequence (CDS), Simple Sequence Repeat (SSR), and Long Non-Coding RNA (lncRNA) Features
2.7. Functional Annotation
3. Results
3.1. General Properties of Single-Molecule Long-Reads
3.2. Acquisition of High-Quality Sequences and Error Correction of Long-Reads Using Illumina Data
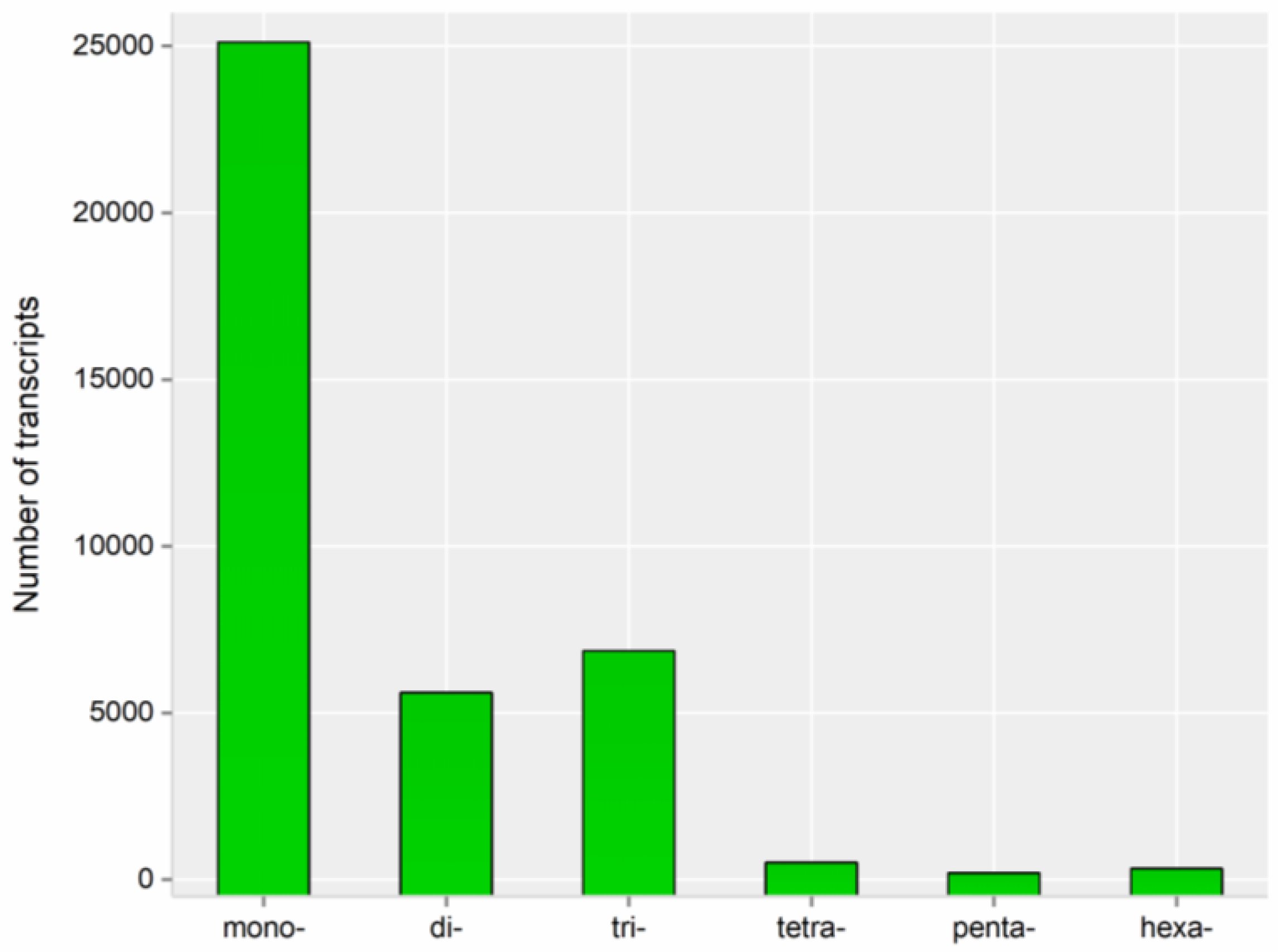
3.3. Predictions of ORFs, SSRs, and lncRNAs
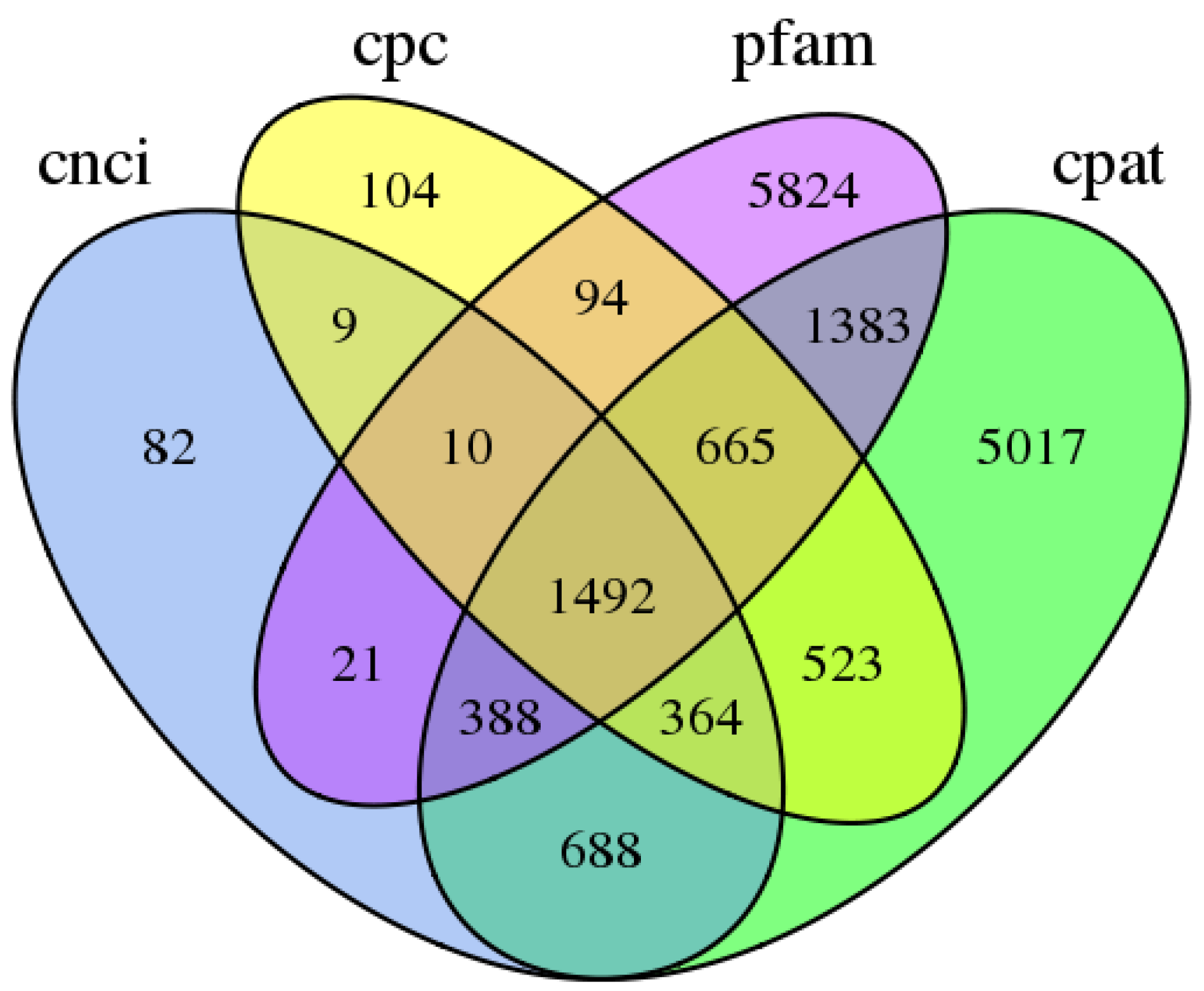
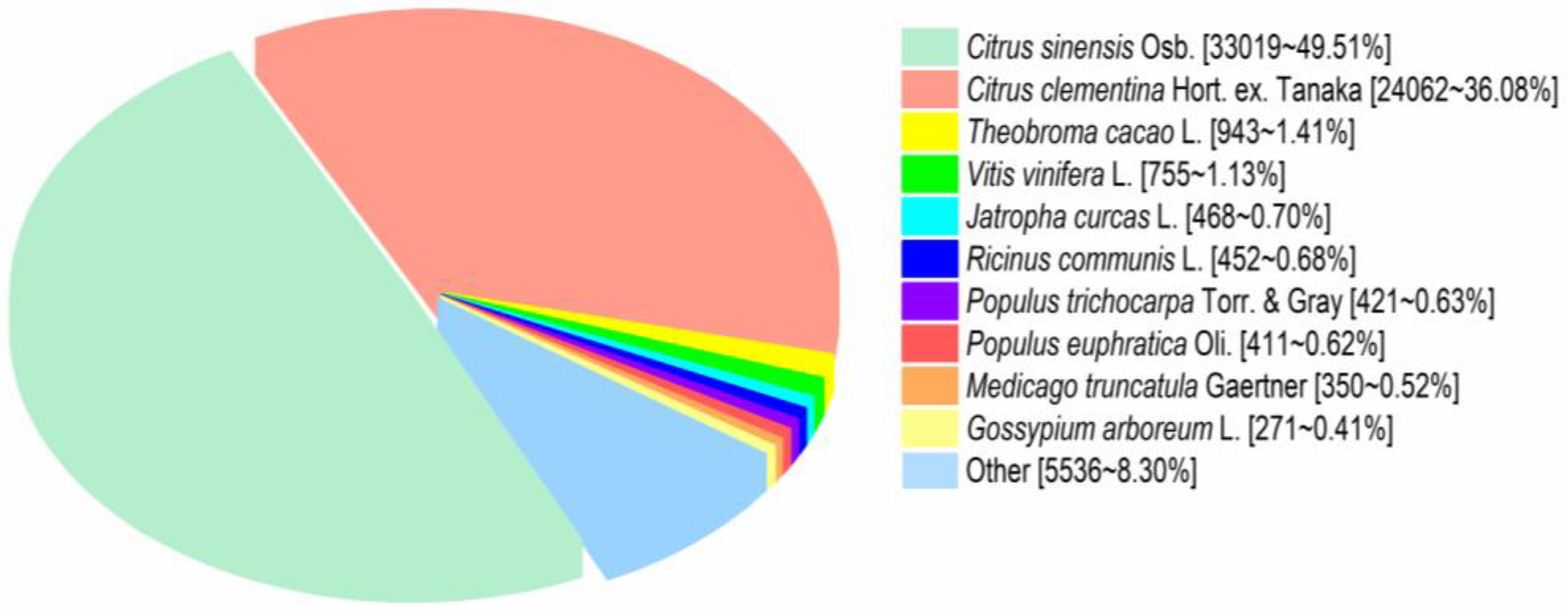
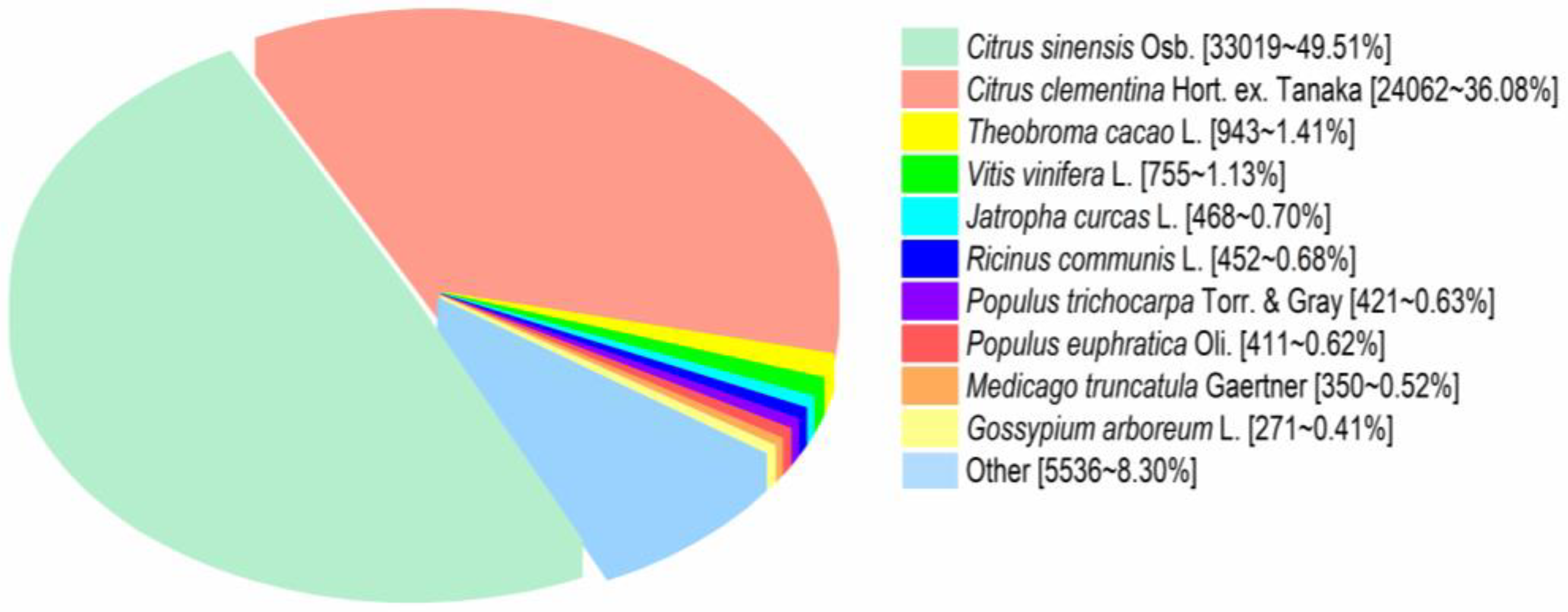
3.4. Functional Annotation of Transcripts
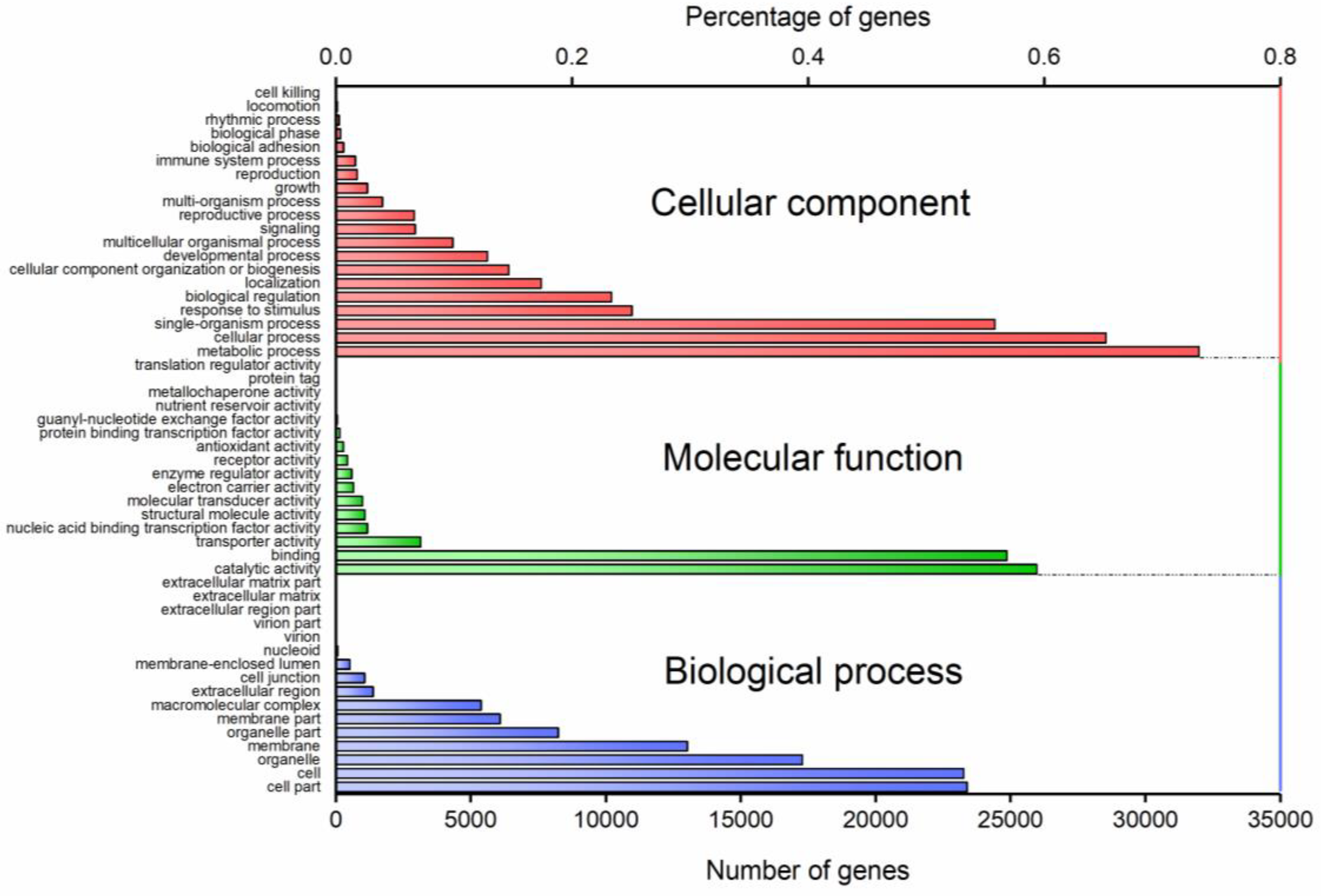
3.5. Gene Ontology (GO) Annotation
3.6. EggNOG Annotation
3.7. Analysis of KEGG Pathways and Gene Annotation Information
4. Discussion
4.1. Evaluation of SMRT Sequencing Quality
4.2. Application of lncRNA and NR Annotation
4.3. Excavation of KEGG Annotation Pathways Gene Annotation Information in Z. bungeanum
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xiang, L.; Liu, Y.; Xie, C.; Li, X.; Yu, Y.; Ye, M.; Chen, X. The chemical and genetic characteristics of Szechuan pepper (Zanthoxylum bungeanum and Z. armatum) cultivars and their suitable habitat. Front. Plant Sci. 2016, 7, 467. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Deng, Y.; Xie, H.; Yao, C.; Cai, C.; Pan, S.; Wang, Y. Antinociceptive and anti-inflammatory activities of ethyl acetate fraction from Zanthoxylum armatum in mice. Fitoterapia 2011, 82, 347–351. [Google Scholar] [CrossRef]
- Lei, X.; Cheng, S.; Peng, H.; He, Q.; Zhu, H.; Xu, M.; Wang, Q.; Liu, L.; Zhang, C.; Zhou, Q.; et al. Anti-inflammatory effect of Zanthoxylum bungeanum-cake-separated moxibustion on rheumatoid arthritis rats. Afr. J. Tradit. Complement. Altern. Med. 2016, 13, 45–52. [Google Scholar] [CrossRef]
- Barkatullah; Ibrar, M.; Muhammad, N.; Khan, A.; Khan, S.A.; Zafar, S.; Jan, S.; Riaz, N.; Ullah, Z.; Farooq, U.; et al. Pharmacognostic and phytochemical studies of Zanthoxylum armatum DC. Pak. J. Pharm. Sci. 2017, 30, 429–438. [Google Scholar] [PubMed]
- Alam, F.; Saqib, Q.N.U.; Ashraf, M. Zanthoxylum armatum DC extracts from fruit, bark and leaf induce hypolipidemic and hypoglycemic effects in mice- in vivo and in vitro study. BMC Complement. Altern. Med. 2018, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Cao, W.; Zhou, X.; Cao, W.; Xie, Y.; Wang, S. Anti-thrombotic effects of alpha-linolenic acid isolated from Zanthoxylum bungeanum Maxim seeds. BMC Complement. Altern. Med. 2014, 14, 348. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Park, Y.; Li, Y.; Xu, H.; Lee, J.; Kim, Y.; Jang, S.; Park, S.; Lee, Y.; Ahn, J.; et al. Biological safety and B cells activation effects of Zanthoxylum schinifolium. Mol. Cell. Toxicol. 2011, 7, 157–162. [Google Scholar] [CrossRef]
- Kim, S.; Seo, H.; Al Mahmud, H.; Islam, M.I.; Lee, B.E.; Cho, M.L.; Song, H.Y. In vitro activity of collinin isolated from the leaves of Zanthoxylum schinifolium against multidrug- and extensively drug-resistant mycobacterium tuberculosis. Phytomedicine 2018, 46, 104–110. [Google Scholar] [CrossRef]
- Cai, X.; Mu, X.; Zhang, Z.; Hua, Z.; Huang, Y.; Sun, Q. Polyembryony and multiple seedlings in the apomictic plants. Acta Bot. Sin. 1997, 39, 590–595. [Google Scholar]
- Liu, Y. Apomixis in Zanthoxylum bungeanum and Z. simulans. J. Genet. Genomics 1987, 14, 107–113. [Google Scholar]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genomics 2017, 18, 395. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 17, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- Ning, G.; Cheng, X.; Luo, P.; Liang, F.; Wang, Z.; Yu, G.; Li, X.; Wang, D.; Bao, M. Hybrid sequencing and map finding (HySeMaFi): Optional strategies for extensively deciphering gene splicing and expression in organisms without reference genome. Sci. Rep. 2017, 7, 43793. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Ye, C.Y.; Xia, X.; Yin, W. De novo sequencing and transcriptome analysis of the desert shrub, Ammopiptanthus mongolicus, during cold acclimation using Illumina/Solexa. BMC Genomics 2013, 14, 488. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Raxwal, V.K.; Joshi, B.; Jagannath, A.; Katiyar-Agarwal, S.; Goel, S.; Kumar, A.; Agarwal, M. De novo transcriptome profiling of cold-stressed siliques during pod filling stages in Indian mustard (Brassica juncea L.). Front. Plant Sci. 2015, 6, 932. [Google Scholar] [CrossRef]
- Chen, E.; Wei, D.; Shen, G.; Yuan, G.; Bai, P.; Wang, J. De novo characterization of the Dialeurodes citri transcriptome: Mining genes involved in stress resistance and simple sequence repeats (SSRs) discovery. Insect Mol. Biol. 2014, 23, 52–66. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hackl, T.; Hedrich, R.; Schultz, J.; Foester, F. Proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Muench, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166–e166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Biswas, M.; Nath, U.; Howlader, J.; Bagchi, M.; Natarajan, S.; Kayum, M.A.; Kim, H.-T.; Park, J.I.; Kang, J.-G.; Nou, S., III. Exploration and exploitation of novel SSR markers for candidate transcription factor genes in Lilium species. Genes 2018, 9, 97. [Google Scholar] [CrossRef]
- Powell, S.; Szklarczyk, D.; Trachana, K.; Roth, A.; Kuhn, M.; Muller, J.; Arnold, R.; Rattei, T.; Letunic, I.; Doerks, T.; et al. eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2011, 40, D284–D289. [Google Scholar] [CrossRef]
- Minoche, A.E.; Dohm, J.C.; Schneider, J.; Holtgräwe, D.; Viehöver, P.; Montfort, M.; Sörensen, T.R.; Weisshaar, B.; Himmelbauer, H. Exploiting single-molecule transcript sequencing for eukaryotic gene prediction. Genome Biol. 2015, 16, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Deng, F.; Jia, X.; Li, C.; Lai, S. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.-H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef]
- Ochogavía, A.; Galla, G.; Seijo, J.G.; González, A.M.; Bellucci, M.; Pupilli, F.; Barcaccia, G.; Albertini, E.; Pessino, S. Structure, target-specificity and expression of PN_LNC_N13, a long non-coding RNA differentially expressed in apomictic and sexual Paspalum notatum. Plant Mol. Biol. 2018, 96, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Zhang, X.; Wang, W.; Yuan, R.; Shen, F. Identification of Gossypium hirsutum long non-coding RNAs (lncRNAs) under salt stress. BMC Plant Biol. 2018, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Laporte, P.; Merchan, F.; Amor, B.B.; Wirth, S.; Crespi, M. Riboregulators in plant development. Biochem. Soc. Trans. 2007, 35, 1638–1642. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Luan, Y.; Jiang, N.; Bao, H.; Meng, J. Comparative transcriptome analysis between resistant and susceptible tomato allows the identification of lncRNA16397 conferring resistance to Phytophthora infestans by co-expressing glutaredoxin. Plant J. 2017, 89, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Solda, G.; Sunkin, S.M.; et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res. 2008, 18, 1433–1445. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Chen, L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.; Hao, B.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar] [CrossRef]
- Deng, Z.; Huang, S.; Ling, P.; Chen, C.; Yu, C.; Weber, C.A.; Moore, G.A.; Gmitter, F.G., Jr. Cloning and characterization of NBS-LRR class resistance-gene candidate sequences in citrus. Theor. Appl. Genet. 2000, 101, 814–822. [Google Scholar] [CrossRef]
- Diao, W.; Hu, Q.; Feng, S.; Li, W.; Xu, J. Chemical composition and antibacterial activity of the essential oil from Green Huajiao (Zanthoxylum schinifolium) against selected foodborne pathogens. J. Agr. Food Chem. 2013, 61, 6044–6049. [Google Scholar] [CrossRef] [PubMed]
- Trapp, O.; Seeliger, K.; Puchta, H. Homologs of Breast Cancer Genes in Plants. Front. Plant Sci. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Puchta, H.; Kobbe, D.; Wanieck, K.; Knoll, A.; Suer, S.; Focke, M.; Hartung, F. Role of human disease genes for the maintenance of genome stability in plants. In Induced Plant Mutations in the Genomics Era; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2009; pp. 129–132. [Google Scholar]
- Wang, S.; Durrant, W.E.; Song, J.; Spivey, N.W.; Dong, X. Arabidopsis BRCA2 and RAD51 proteins are specifically involved in defense gene transcription during plant immune responses. Proc. Natl. Acad. Sci. USA 2010, 107, 22716–22721. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cDNA Size | 1–2 kb | 2–3 kb | >3 kb | Total |
|---|---|---|---|---|
| No. of ROIs | 113,885 | 102,613 | 65,603 | 282,101 |
| No. of five primer reads | 62,689 | 56,205 | 40,479 | 159,373 |
| No. of three primer reads | 69,924 | 60,719 | 42,195 | 441,474 |
| No. of poly(A) reads | 68,221 | 60,120 | 42,172 | 170,513 |
| No. of filtered short reads | 15,681 | 8132 | 1886 | 25,699 |
| No. of full-length reads | 52,960 | 46,744 | 34,696 | 134,400 |
| No. of full-length non-chimeric reads | 52,785 | 46,629 | 34,660 | 134,074 |
| Full-length percentage (%) | 46.50 | 45.55 | 52.89 | 47.64 |
| Mean length of ROIs | 2000 | 2891 | 3567 | 2819 |
| Size | No. of Consensus Isoforms | Average Consensus Isoforms Read Length | No. of Polished High-Quality Isoforms | No. of Polished Low-Quality Isoforms | Percentage of Polished High-Quality Isoforms (%) |
|---|---|---|---|---|---|
| 0–1 kb | 1821 | 903 | 1608 | 213 | 88.30 |
| 1–2 kb | 26,917 | 1442 | 23,043 | 3874 | 85.61 |
| 2–3 kb | 24,913 | 2387 | 19,181 | 5732 | 76.99 |
| 3–6 kb | 21,823 | 3582 | 14,603 | 7220 | 66.92 |
| >6 kb | 499 | 8757 | 10 | 489 | 2.00 |
| Total | 75,973 | 3414 | 58,445 | 17,528 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, J.; Feng, S.; Liu, Y.; Zhao, L.; Tian, L.; Hu, Y.; Yang, T.; Wei, A. Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes. Forests 2018, 9, 765. https://doi.org/10.3390/f9120765
Tian J, Feng S, Liu Y, Zhao L, Tian L, Hu Y, Yang T, Wei A. Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes. Forests. 2018; 9(12):765. https://doi.org/10.3390/f9120765
Chicago/Turabian StyleTian, Jieyun, Shijing Feng, Yulin Liu, Lili Zhao, Lu Tian, Yang Hu, Tuxi Yang, and Anzhi Wei. 2018. "Single-Molecule Long-Read Sequencing of Zanthoxylum bungeanum Maxim. Transcriptome: Identification of Aroma-Related Genes" Forests 9, no. 12: 765. https://doi.org/10.3390/f9120765