
Metabarcoding of Bacteria Associated with the Acute Oak Decline Syndrome in England

 ,
,
Abstract
:1. Introduction
2. Experimental Section
2.1. Sampling
2.2. Bacterial Communities Associated with Oak Tissue
2.3. Data Analysis
2.4. Nucleotide Sequence Accession Numbers
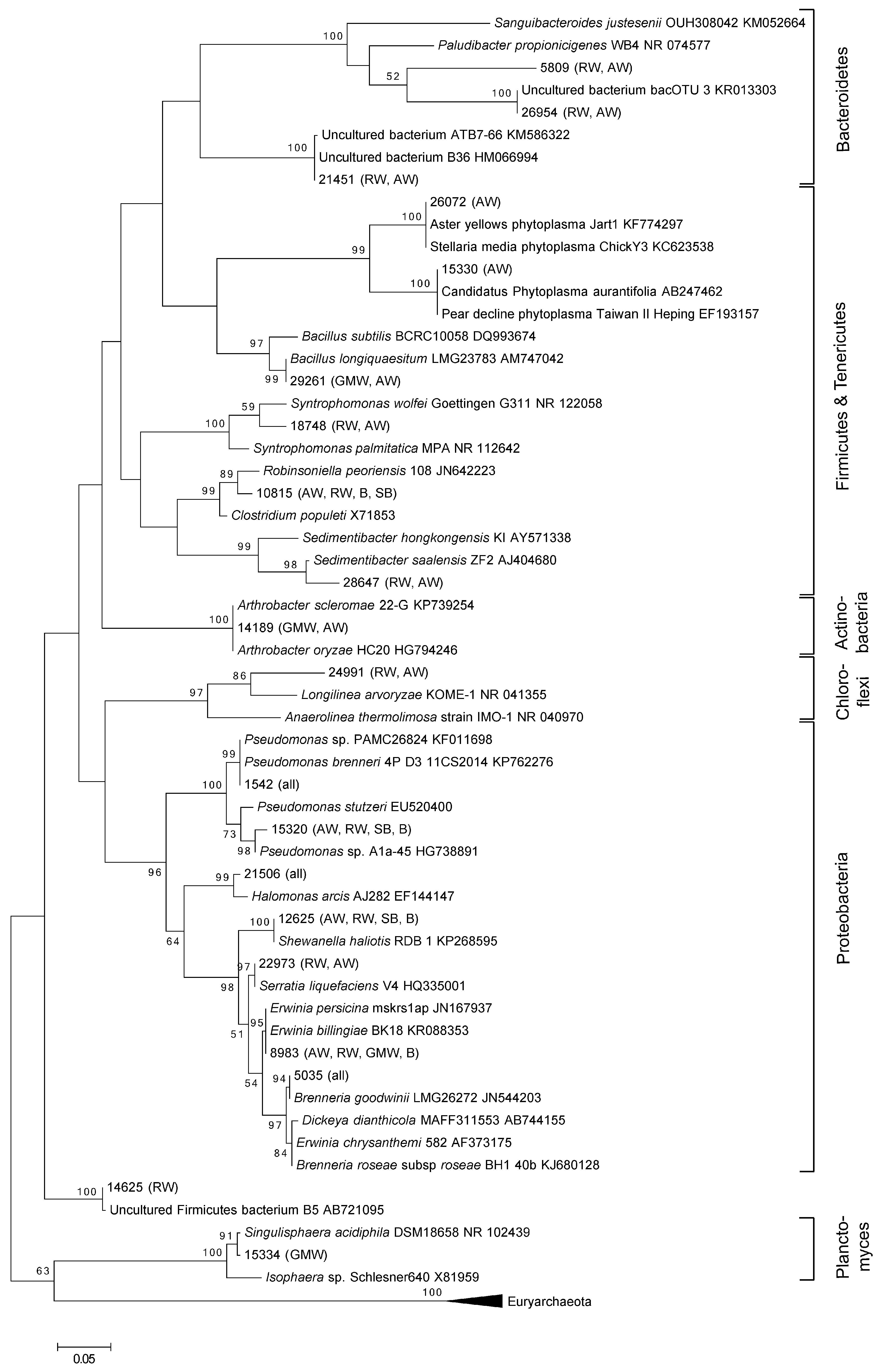
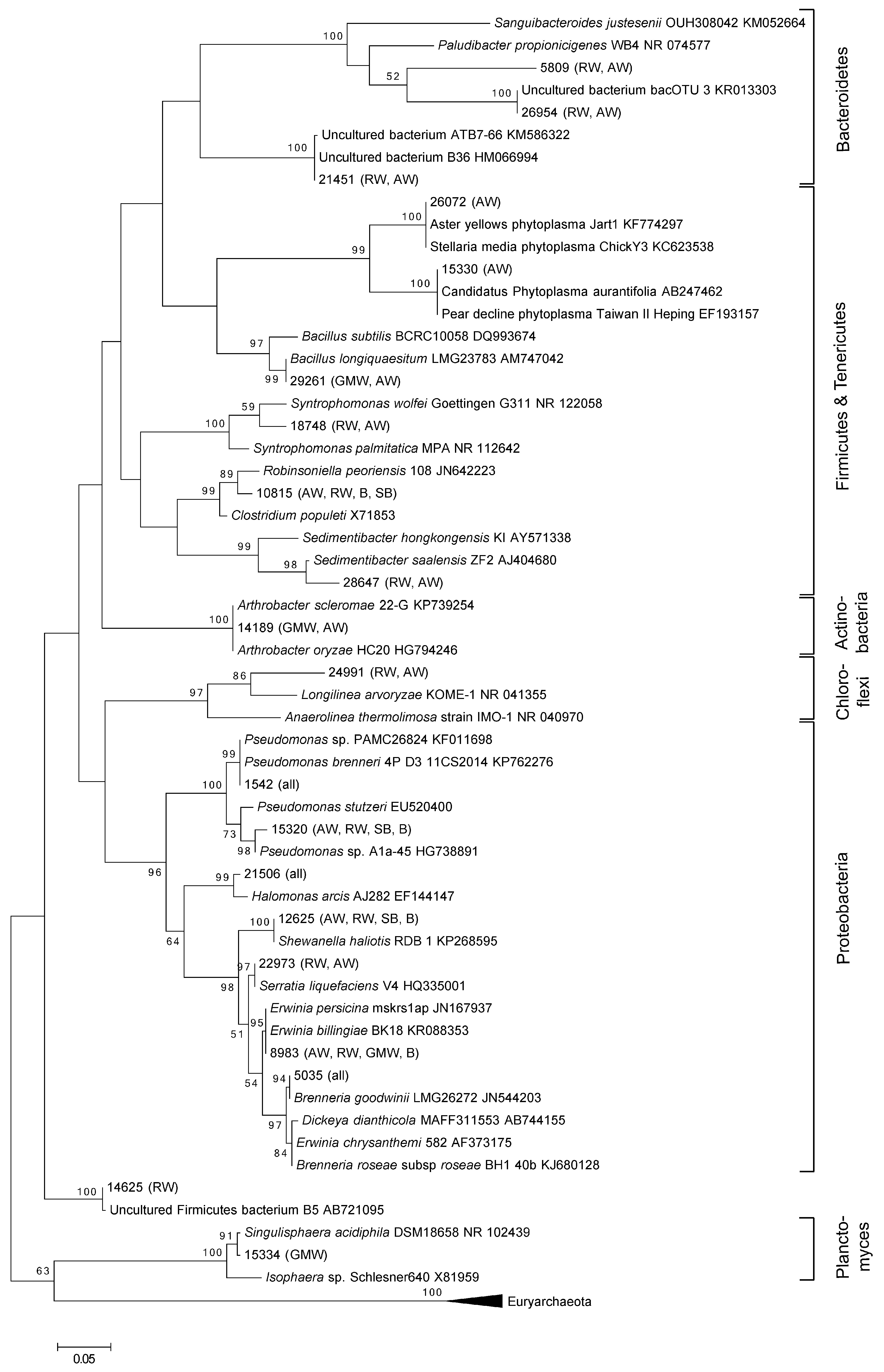
3. Results and Discussion
3.1. Total Oak Microbiome
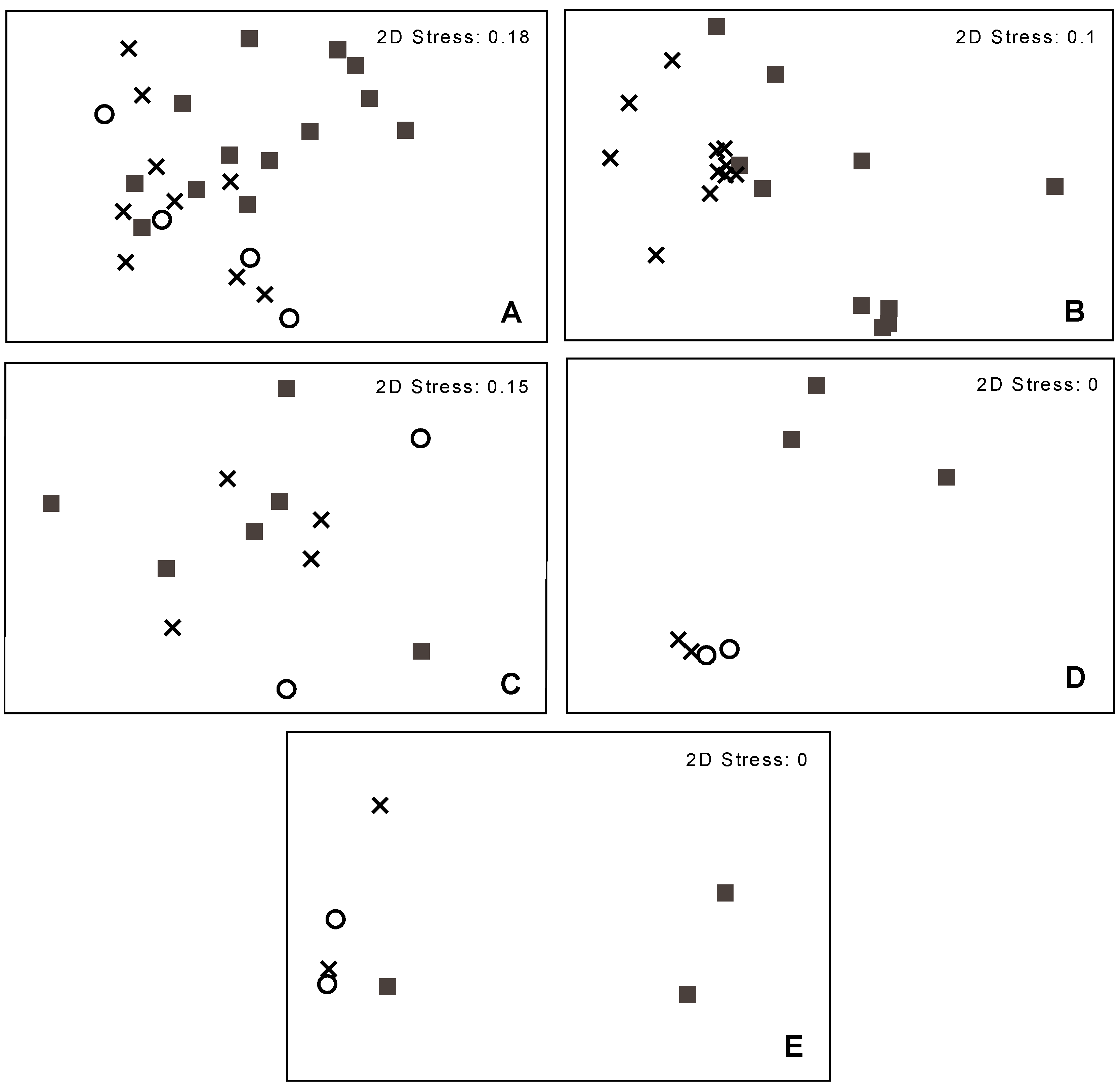
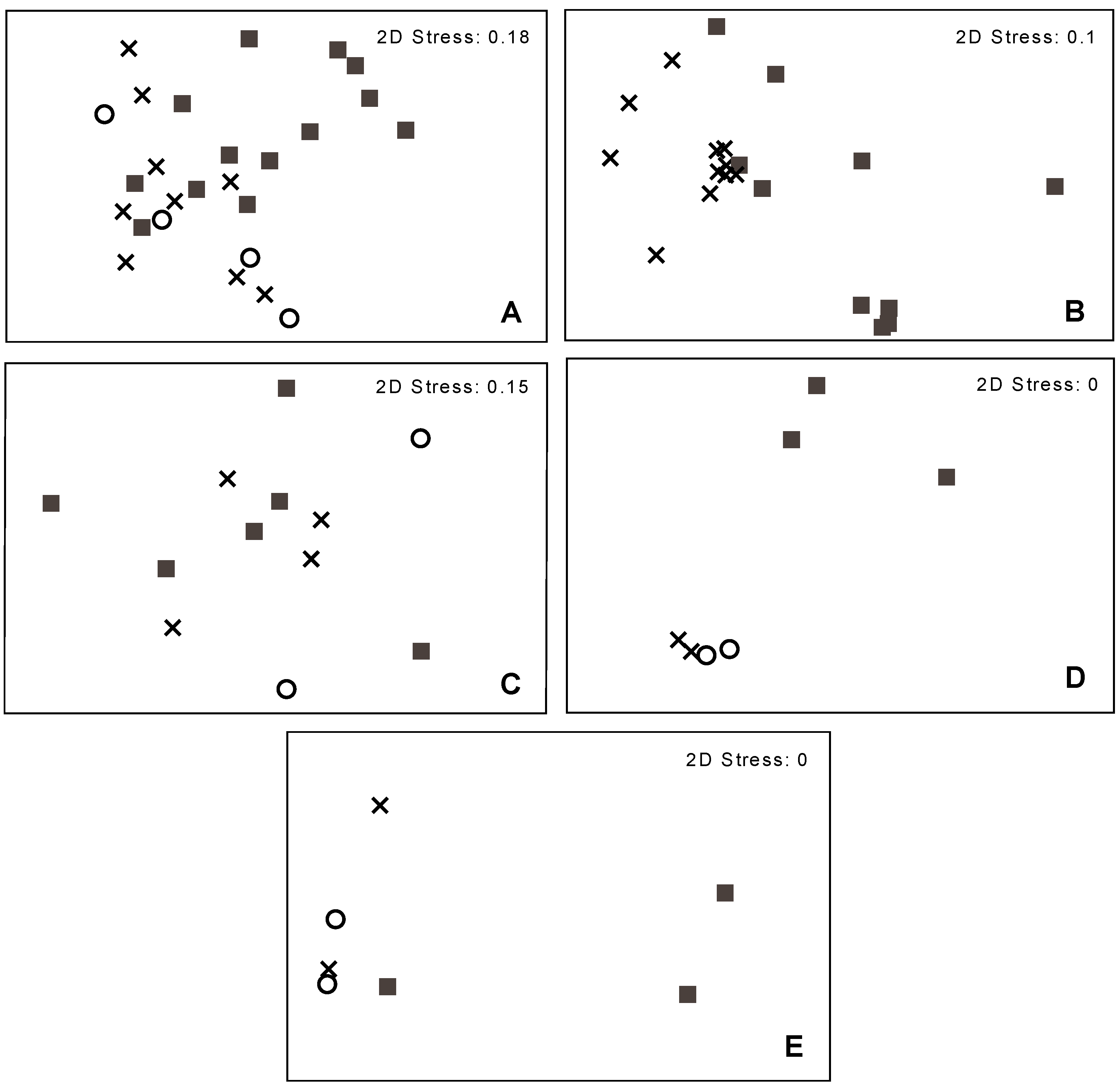
3.2. Bacterial Community Structure in Relation to the Tree’s Health Status
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Denman, S.; Brown, N.; Kirk, S.; Jeger, M.; Webber, J. A description of the symptoms of Acute Oak Decline in Britain and a comparative review on causes of similar disorders on oak in Europe. Forestry 2014, 87, 535–551. [Google Scholar] [CrossRef]
- Brown, N.; Jeger, M.; Kirk, S.; Xu, X.; Denman, S. Spatial and temporal patterns in symptom expression within eight woodlands affected by Acute Oak Decline. For. Ecol. Manag. 2016, 360, 97–109. [Google Scholar] [CrossRef]
- Denman, S.; Webber, J.F. Oak declines—New definitions and new episodes in Britain. Q. J. For. 2009, 103, 285–290. [Google Scholar]
- Andrews, J.H.; Harris, R.F. The Ecology and Biogeography of Microorganisms on Plant Surfaces. Annu. Rev. Phytopathol. 2000, 38, 145–180. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Junker, R.R.; Keller, A. Microhabitat heterogeneity across leaves and flower organs promotes bacterial diversity. FEMS Microbiol. Ecol. 2015, 91. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.R.; González Peña, A.; White, J.R.; Pettengill, J.B.; Li, C.; Allard, S.; Rideout, S.; Allard, M.; Hill, T.; Evans, P.; et al. Baseline survey of the anatomical microbial ecology of an important food plant: Solanum lycopersicum (tomato). BMC Microbiol. 2013, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. Unraveling the plant microbiome: Looking back and future perspectives. Front. Microbiol. 2014, 5, 1–7. [Google Scholar]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Km, A.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contracts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [PubMed]
- Weinert, N.; Piceno, Y.; Ding, G.-C.; Meincke, R.; Heuer, H.; Berg, G.; Schloter, M.; Andersen, G.; Smalla, K. PhyloChip hybridization uncovered an enormous bacterial diversity in the rhizosphere of different potato cultivars: Many common and few cultivar-dependent taxa. FEMS Microbiol. Ecol. 2011, 75, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Leff, J.W.; Del Tredici, P.; Friedman, W.E.; Fierer, N. Spatial structuring of bacterial communities within individual Ginkgo biloba trees. Environ. Microbiol. 2015, 17, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Finkel, O.M.; Burch, A.Y.; Lindow, S.E.; Post, A.F.; Belkin, S. Geographical location determines the population structure in phyllosphere microbial communities of a salt-excreting desert tree. Appl. Environ. Microbiol. 2011, 77, 7647–7655. [Google Scholar] [CrossRef] [PubMed]
- Lambais, M.R.; Crowley, D.E.; Cury, J.C.; Büll, R.C.; Rodrigues, R.R. Bacterial diversity in tree canopies of the atlantic forest. Science 2006, 312, 1917. [Google Scholar] [CrossRef] [PubMed]
- Kembel, S.W.; O’Connor, T.K.; Arnold, H.K.; Hubbell, S.P.; Wright, S.J.; Green, J.L. Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc. Natl. Acad. Sci. USA 2014, 111, 13715–13720. [Google Scholar] [CrossRef] [PubMed]
- Lindow, S.E.; Brandl, M.T. MINIREVIEW: Microbiology of the phyllosphere. Appl. Environ. Microbiol. 2003, 69, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Bodenhausen, N.; Bortfeld-Miller, M.; Ackermann, M.; Vorholt, J.A. A synthetic community approach reveals plant genotypes affecting the Phyllosphere Microbiota. PLoS Genet. 2014, 10, e1004283. [Google Scholar] [CrossRef] [PubMed]
- Whipps, J.M.; Hand, P.; Pink, D.; Bending, G.D. Phyllosphere microbiology with special reference to diversity and plant genotype. J. Appl. Microbiol. 2008, 105, 1744–1755. [Google Scholar] [CrossRef] [PubMed]
- Barbaroux, C.; Bréda, N. Contrasting distribution and seasonal dynamics of carbohydrate reserves in stem wood of adult ring-porous sessile oak and diffuse-porous beech trees. Tree Physiol. 2002, 22, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Cai, J.; Duan, C.-Q.; Reeves, M.; He, F. A Review of polyphenolics in oak woods. Int. J. Mol. Sci. 2015, 16, 6978–7014. [Google Scholar] [CrossRef] [PubMed]
- Doussot, F.; Pardon, P.; Dedier, J.; De Jeso, B. Individual, species and geographic origin influence on cooperage oak extractible content (Quercus robur L. and Quercus petraea Liebl.). Analusis 2000, 28, 960–965. [Google Scholar] [CrossRef]
- De Simón, B.F.; Hernández, T.; Cadahía, E.; Dueñas, M.; Estrella, I. Phenolic compounds in a Spanish red wine aged in barrels made of Spanish, French and American oak wood. Eur. Food Res. Technol. 2003, 216, 150–156. [Google Scholar]
- Prida, A.; Puech, J.L. Influence of geographical origin and botanical species on the content of extractives in American, French, and East European oak woods. J. Agric. Food Chem. 2006, 54, 8115–8126. [Google Scholar] [CrossRef] [PubMed]
- Biosca, E.G.; González, R.; López-López, M.J.; Soria, S.; Montón, C.; Pérez-Laorga, E.; López, M.M. Isolation and characterization of Brenneria quercina, causal agent for bark canker and drippy nut of Quercus spp. in Spain. Phytopathology 2003, 93, 485–492. [Google Scholar] [CrossRef] [PubMed]
- González, R.; López-López, M.J.; Biosca, E.G.; López, F.; Santiago, R.; López, M.M. First report of bacterial deep bark canker of walnut caused by Brenneria (Erwinia) rubrifaciens in Europe. Plant Dis. 2002, 86, 696. [Google Scholar] [CrossRef]
- Maes, M.; Huvenne, H.; Messens, E. Brenneria salicis, the bacterium causing watermark disease in willow, resides as an endophyte in wood. Environ. Microbiol. 2009, 11, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Dujesiefken, D.; Stobbe, H.; Moreth, U.; Kehr, R.; Schröder, T. Pseudomonas syringae pv. aesculi associated with horse chestnut bleeding canker in Germany. For. Pathol. 2008, 38, 124–128. [Google Scholar]
- Webber, J.F.; Parkinson, N.M.; Rose, J.; Stanford, H.; Cook, R.T.A.; Elphinstone, J.G. Isolation and identification of Pseudomonas syringae pv. aesculi causing bleeding canker of horse chestnut in the UK. Plant Pathol. 2008, 57, 368. [Google Scholar] [CrossRef]
- Lebeis, S.L.; Rott, M.; Dangl, J.L.; Schulze-Lefert, P. Culturing a plant microbiome community at the cross-Rhodes. New Phytol. 2012, 196, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.; Denman, S.; Kirk, S.; Venter, S.; Rodríguez-Palenzuela, P.; Coutinho, T. Description of Gibbsiella quercinecans gen. nov., sp. nov., associated with Acute Oak Decline. Syst. Appl. Microbiol. 2010, 33, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.L.; Cleenwerck, I.; Denman, S.; Venter, S.N.; Rodríguez-Palenzuela, P.; Coutinho, T.A.; De Vos, P. Proposal to reclassify Brenneria quercina (Hildebrand and Schroth 1967) Hauben et al. 1999 into a new genus, Lonsdalea gen. nov., as Lonsdalea quercina comb. nov., descriptions of Lonsdalea quercina subsp. quercina comb. nov., Lonsdalea quercina subsp. ib. Int. J. Syst. Evol. Microbiol. 2012, 62, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Denman, S.; Brady, C.; Kirk, S.; Cleenwerck, I.; Venter, S.; Coutinho, T.; De Vos, P. Brenneria goodwinii sp. nov., associated with acute oak decline in the UK. Int. J. Syst. Evol. Microbiol. 2012, 62, 2451–2456. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.; Hunter, G.; Kirk, S.; Arnold, D.; Denman, S. Gibbsiella greigii sp. nov., a novel species associated with oak decline in the USA. Syst. Appl. Microbiol. 2014, 37, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.; Hunter, G.; Kirk, S.; Arnold, D.; Denman, S. Rahnella victoriana sp. nov., Rahnella bruchi sp. nov., Rahnella woolbedingensis sp. nov., classification of Rahnella genomospecies 2 and 3 as Rahnella variigena sp. nov. and Rahnella inusitata sp. nov., respectively and emended description of the genus Rahnella. Syst. Appl. Microbiol. 2014, 37, 545–552. [Google Scholar] [PubMed]
- Brady, C.; Hunter, G.; Kirk, S.; Arnold, D.; Denman, S. Description of Brenneria roseae sp. nov. and two subspecies, Brenneria roseae subspecies roseae ssp. nov and Brenneria roseae subspecies americana ssp. nov. isolated from symptomatic oak. Syst. Appl. Microbiol. 2014, 37, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Peplies, J.; Kottmann, R.; Ludwig, W.; Glöckner, F.O. A standard operating procedure for phylogenetic inference (SOPPI) using (rRNA) marker genes. Syst. Appl. Microbiol. 2008, 31, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, J.; Singh, K.; Fern, A.; Kirton, E.S.; He, S.; Woyke, T.; Lee, J.; Chen, F.; Dangl, J.L.; Tringe, S.G. Primer and platform effects on 16S rRNA tag sequencing. Front. Microbiol. 2015, 6, 771. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.M.; Thomas, W.K. FlowClus: Efficiently filtering and denoising pyrosequenced amplicons. BMC Bioinform. 2015, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Rideout, J.R.; He, Y.; Navas-molina, J.A.; Walters, W.A.; Ursell, L.K.; Gibbons, S.M.; Chase, J.; Mcdonald, D.; Gonzalez, A.; Robbins-pianka, A.; et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ 2014, 2, e545. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.K.; Sahl, J.W.; Castoe, T.A.; Wagner, B.D.; Pollock, D.D.; Spear, J.R. Comparison of Normalization Methods for construction of large, multiplex amplicon pools for next-generation sequencing. Appl. Environ. Microbiol. 2010, 76, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Gihring, T.M.; Green, S.J.; Schadt, C.W. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 2012, 14, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.E.; Bates, S.T.; Caporaso, J.G.; Lauber, C.L.; Leff, J.W.; Knight, R.; Fierer, N. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 2013, 15, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Krych, L.; Hansen, C.H.F.; Hansen, A.K.; van den Berg, F.W.J.; Nielsen, D.S. Quantitatively different, yet qualitatively alike: A meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS ONE 2013, 8, e62578. [Google Scholar] [CrossRef] [PubMed]
- Leff, J.W.; Fierer, N. Bacterial communities associated with the surfaces of fresh fruits and vegetables. PLoS ONE 2013, 8, e59310. [Google Scholar] [CrossRef] [PubMed]
- Prober, S.M.; Leff, J.W.; Bates, S.T.; Borer, E.T.; Firn, J.; Harpole, W.S.; Lind, E.M.; Seabloom, E.W.; Adler, P.B.; Bakker, J.D.; et al. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol. Lett. 2015, 18, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qu, Y.; Ma, Q.; Zhang, Z.; Li, D.; Wang, J.; Shen, W.; Shen, E.; Zhou, J. Illumina MiSeq sequencing reveals diverse microbial communities of activated sludge systems stimulated by different aromatics for indigo biosynthesis from indole. PLoS ONE 2015, 10, e0125732. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.R.; Snowberg, L.K.; Gregory Caporaso, J.; Knight, R.; Bolnick, D.I. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015, 9, 2515–2526. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, Y.-H.; Deng, Y.; Shi, Z.; Zhou, B.Y.; Xue, K.; Wu, L.; He, Z.; Yang, Y. Random sampling process leads to overestimation of β-diversity of microbial communities. mBio 2013, 4, e00324-13. [Google Scholar] [CrossRef] [PubMed]
- De Cárcer, D.A.; Denman, S.E.; McSweeney, C.; Morrison, M. Evaluation of subsampling-based normalization strategies for tagged high-throughput sequencing data sets from gut microbiomes. Appl. Environ. Microbiol. 2011, 77, 8795–8798. [Google Scholar] [CrossRef] [PubMed]
- Lallias, D.; Hiddink, J.G.; Fonseca, V.G.; Gaspar, J.M.; Sung, W.; Neill, S.P.; Barnes, N.; Ferrero, T.; Hall, N.; Lambshead, P.J.D.; et al. Environmental metabarcoding reveals heterogeneous drivers of microbial eukaryote diversity in contrasting estuarine ecosystems. ISME J. 2015, 9, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, K.M.B.; Krych, L.; Sørensen, D.B.; Pang, W.; Nielsen, D.S.; Josefsen, K.; Hansen, L.H.; Sørensen, S.J.; Hansen, A.K. Gut microbiota composition is correlated to grid floor induced stress and behavior in the BALB/c mouse. PLoS ONE 2012, 7, e46231. [Google Scholar]
- Ren, G.; Zhang, H.; Lin, X.; Zhu, J.; Jia, Z. Response of leaf endophytic bacterial community to elevated CO2 at different growth stages of rice plant. Front. Microbiol. 2015, 6, 855. [Google Scholar] [CrossRef] [PubMed]
- Touceda-González, M.; Brader, G.; Antonielli, L.; Ravindran, V.B.; Waldner, G.; Friesl-Hanl, W.; Corretto, E.; Campisano, A.; Pancher, M.; Sessitsch, A. Combined amendment of immobilizers and the plant growth-promoting strain Burkholderia phytofirmans PsJN favours plant growth and reduces heavy metal uptake. Soil Biol. Biochem. 2015, 91, 140–150. [Google Scholar] [CrossRef]
- Elshahed, M.S.; Youssef, N.H.; Spain, A.M.; Sheik, C.; Najar, F.Z.; Sukharnikov, L.O.; Roe, B.A.; Davis, J.P.; Schloss, P.D.; Bailey, V.L.; et al. Novelty and uniqueness patterns of rare members of the soil biosphere. Appl. Environ. Microbiol. 2008, 74, 5422–5428. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Casamayor, E.O.; Kirchman, D.L.; Lovejoy, C. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. USA 2009, 106, 22427–22432. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Van Dorst, J.; Bissett, A.; Palmer, A.S.; Brown, M.; Snape, I.; Stark, J.S.; Raymond, B.; McKinlay, J.; Ji, M.; Winsley, T.; et al. Community fingerprinting in a sequencing world. FEMS Microbiol. Ecol. 2014, 89, 316–330. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Gobet, A.; Pommier, T. Two decades of describing the unseen majority of aquatic microbial diversity. Mol. Ecol. 2012, 21, 1878–1896. [Google Scholar] [CrossRef] [PubMed]
- Goodman, L.A. On the estimation of the number of classes in a population. Ann. Math. Stat. 1949, 20, 572–579. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Sapp, M.; Schwaderer, A.S.; Wiltshire, K.H.; Hoppe, H.-G.; Gerdts, G.; Wichels, A. Species-specific bacterial communities in the phycosphere of microalgae? Microb. Ecol. 2007, 53, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Delmas, J.; Breysse, F.; Devulder, G.; Flandrois, J.-P.; Chomarat, M. Rapid identification of Enterobacteriaceae by sequencing DNA gyrase subunit B encoding gene. Diagn. Microbiol. Infect. Dis. 2006, 55, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-T.; Lee, F.-L.; Tai, C.-J.; Kasai, H. Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA-DNA hybridization in the Bacillus subtilis group. Int. J. Syst. Evol. Microbiol. 2007, 57, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Lundin, D.; Severin, I.; Logue, J.B.; Östman, Ö.; Andersson, A.F.; Lindström, E.S. Which sequencing depth is sufficient to describe patterns in bacterial α-and β-diversity? Environ. Microbiol. Rep. 2012, 4, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Idris, R.; Trifonova, R.; Puschenreiter, M.; Wenzel, W.W.; Sessitsch, A. Bacterial communities associated with flowering plants of the Ni hyperaccumulator Thlaspi goesingense. Appl. Environ. Microbiol. 2004, 70, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, A.E.; Simmons, S.J. Plant control of phyllosphere diversity: Genotype interactions with ultraviolet-B radiation. In Microbial Ecology of Aerial Plant Surfaces; Bailey, M., Lilley, A., Timms-Wilson, T., Spencer-Phillips, P., Eds.; CAB International: Wallingford, UK, 2006; pp. 223–238. [Google Scholar]
- Rasche, F.; Marco-Noales, E.; Velvis, H.; van Overbeek, L.S.; López, M.M.; van Elsas, J.D.; Sessitsch, A. Structural characteristics and plant-beneficial effects of bacteria colonizing the shoots of field grown conventional and genetically modified T4-lysozyme producing potatoes. Plant Soil 2006, 289, 123–140. [Google Scholar] [CrossRef]
- Rasche, F.; Trondl, R.; Naglreiter, C.; Reichenauer, T.G.; Sessitsch, A. Chilling and cultivar type affect the diversity of bacterial endophytes colonizing sweet pepper (Capsicum anuum L.). Can. J. Microbiol. 2006, 52, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.P.; Chatterjee, A.K. The genus Erwinia: Enterobacteria pathogenic to plants and animals. Annu. Rev. Microbiol. 1972, 26, 389–426. [Google Scholar] [CrossRef] [PubMed]
- Gasparich, G.E. Spiroplasmas and phytoplasmas: Microbes associated with plant hosts. Biologicals 2010, 38, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-M.; Gundersen-Rindal, D.E.; Bertaccini, A. Phytoplasma: Ecology and genomic diversity. Phytopathology 1998, 88, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, P.R.; van Overbeek, L.S.; van Elsas, J.D. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 2008, 16, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Lalucat, J.; Bennasar, A.; Bosch, R.; García-Valdés, E.; Palleroni, N.J. Biology of Pseudomonas stutzeri. Microbiol. Mol. Biol. Rev. 2006, 70, 510–547. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Kim, Y.S.; Kim, S.D. Pseudomonas stutzeri YPL-1 genetic transformation and antifungal mechanism against Fusarium solani, an agent of plant root rot. Appl. Environ. Microbiol. 1991, 57, 510–516. [Google Scholar] [PubMed]
- Cotta, M.A.; Whitehead, T.R.; Falsen, E.; Moore, E.; Lawson, P.A. Robinsoniella peoriensis gen. nov., sp. nov., isolated from a swine-manure storage pit and a human clinical source. Int. J. Syst. Evol. Microbiol. 2009, 59, 150–155. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Sample | Sequences after Denoising | Symptom Status of Tree | Symptom Status of Tissue | Tissue | Site | Barcode Sequence |
|---|---|---|---|---|---|---|
| GMW2 | 3293 | Healthy | Healthy | Inner bark | GMW | ACGAGTGCGT |
| GMW3 | 3586 | Healthy | Healthy | Sapwood | GMW | ACGCTCGACA |
| GMW6 | 4255 | AOD | Symptomatic | Inner bark | GMW | AGACGCACTC |
| GMW7 | 3138 | AOD | Symptomatic | Sapwood | GMW | AGCACTGTAG |
| GMW9 | 3498 | AOD | Symptomatic | Gallery | GMW | ATCAGACACG |
| GMW11 | 3509 | AOD | Non-symptomatic | Inner bark | GMW | ATATCGCGAG |
| GMW12 | 3829 | AOD | Non-symptomatic | Sapwood | GMW | CGTGTCTCTA |
| GMW6 * | 3893 | AOD | Symptomatic | Inner bark | GMW | CTCGCGTGTC |
| GMW7 * | 3798 | AOD | Symptomatic | Sapwood | GMW | TAGTATCAGC |
| GMW9 * | 3847 | AOD | Symptomatic | Gallery | GMW | TCTCTATGCG |
| GMW11 * | 3975 | AOD | Non-symptomatic | Inner bark | GMW | TGATACGTCT |
| GMW12 * | 3689 | AOD | Non-symptomatic | Sapwood | GMW | TACTGAGCTA |
| R1 | 1138 | Healthy | Healthy | Outer bark | RW | ACGAGTGCGT |
| R2 | 1061 | Healthy | Healthy | Inner bark | RW | ACGCTCGACA |
| R3 | 826 | Healthy | Healthy | Sapwood | RW | AGACGCACTC |
| R4 | 1019 | Healthy | Healthy | Heartwood | RW | AGCACTGTAG |
| R5 | 633 | AOD | Symptomatic | Outer bark | RW | ATCAGACACG |
| R6 | 727 | AOD | Symptomatic | Inner bark | RW | ATATCGCGAG |
| R7 | 887 | AOD | Symptomatic | Sapwood | RW | CGTGTCTCTA |
| R8 | 968 | AOD | Symptomatic | Heartwood | RW | CTCGCGTGTC |
| R9 | 910 | AOD | Symptomatic | Gallery | RW | TAGTATCAGC |
| R10 | 980 | AOD | Non-symptomatic | Outer bark | RW | TCTCTATGCG |
| R11 | 7569 | AOD | Non-symptomatic | Inner bark | RW | TGATACGTCT |
| R12 | 8490 | AOD | Non-symptomatic | Sapwood | RW | TACTGAGCTA |
| R13 | 8088 | AOD | Non-symptomatic | Heartwood | RW | ACGAGTGCGT |
| R14 | 7795 | AOD | Symptomatic | Outer bark | RW | ACGCTCGACA |
| R15 | 7440 | AOD | Symptomatic | Inner bark | RW | AGACGCACTC |
| R16 | 7969 | AOD | Symptomatic | Sapwood | RW | AGCACTGTAG |
| R17 | 4444 | AOD | Symptomatic | Heartwood | RW | ATCAGACACG |
| R18 | 10658 | AOD | Symptomatic | Gallery | RW | ATATCGCGAG |
| R19 | 7535 | AOD | Non-symptomatic | Outer bark | RW | CGTGTCTCTA |
| R20 | 6931 | AOD | Non-symptomatic | Inner bark | RW | CTCGCGTGTC |
| R21 | 7303 | AOD | Non-symptomatic | Sapwood | RW | TAGTATCAGC |
| R22 | 6873 | AOD | Non-symptomatic | Heartwood | RW | TCTCTATGCG |
| R23 * | 5395 | AOD | Symptomatic | Outer bark | RW | TGATACGTCT |
| R24 * | 9518 | AOD | Symptomatic | Inner bark | RW | TACTGAGCTA |
| R25 * | 8223 | AOD | Symptomatic | Sapwood | RW | ACGAGTGCGT |
| R26 * | 6742 | AOD | Symptomatic | Heartwood | RW | ACGCTCGACA |
| R27 * | 10902 | AOD | Symptomatic | Gallery | RW | AGACGCACTC |
| R28 * | 7134 | AOD | Non-symptomatic | Outer bark | RW | AGCACTGTAG |
| R29 * | 4855 | AOD | Non-symptomatic | Inner bark | RW | ATCAGACACG |
| R30 * | 7838 | AOD | Non-symptomatic | Sapwood | RW | ATATCGCGAG |
| R31 * | 7314 | AOD | Non-symptomatic | Heartwood | RW | CGTGTCTCTA |
| A1 | 4168 | Healthy | Healthy | Outer bark | AW | CTCGCGTGTC |
| A2 | 3320 | Healthy | Healthy | Inner bark | AW | TAGTATCAGC |
| A3 | 4221 | Healthy | Healthy | Sapwood | AW | TCTCTATGCG |
| A4 | 3228 | Healthy | Healthy | Heartwood | AW | TGATACGTCT |
| A5 | 4448 | AOD | Symptomatic | Outer bark | AW | TACTGAGCTA |
| A6 | 10358 | AOD | Symptomatic | Inner bark | AW | ACGAGTGCGT |
| A7 | 3664 | AOD | Symptomatic | Sapwood | AW | ACGCTCGACA |
| A8 | 2067 | AOD | Symptomatic | Heartwood | AW | AGACGCACTC |
| A9 | 2528 | AOD | Symptomatic | Gallery | AW | AGCACTGTAG |
| A10 | 3556 | AOD | Non-symptomatic | Outer bark | AW | ATCAGACACG |
| A11 | 9465 | AOD | Non-symptomatic | Inner bark | AW | ATATCGCGAG |
| A12 | 3186 | AOD | Non-symptomatic | Sapwood | AW | CGTGTCTCTA |
| A13 | 2698 | AOD | Non-symptomatic | Heartwood | AW | CTCGCGTGTC |
| A14 | 5970 | AOD | Symptomatic | Outer bark | AW | TAGTATCAGC |
| A15 | 4036 | AOD | Symptomatic | Inner bark | AW | TCTCTATGCG |
| A16 | 3969 | AOD | Symptomatic | Sapwood | AW | TGATACGTCT |
| A17 | 9214 | AOD | Symptomatic | Heartwood | AW | TACTGAGCTA |
| A18 | 5015 | AOD | Symptomatic | Gallery | AW | ACGAGTGCGT |
| A19 | 4404 | AOD | Non-symptomatic | Outer bark | AW | ACGCTCGACA |
| A20 | 3172 | AOD | Non-symptomatic | Inner bark | AW | AGACGCACTC |
| A21 | 5008 | AOD | Non-symptomatic | Sapwood | AW | AGCACTGTAG |
| A22 | 1524 | AOD | Non-symptomatic | Heartwood | AW | ATCAGACACG |
| A23 * | 4870 | AOD | Symptomatic | Outer bark | AW | ATATCGCGAG |
| A24 * | 7765 | AOD | Symptomatic | Inner bark | AW | CGTGTCTCTA |
| A25 * | 10540 | AOD | Symptomatic | Sapwood | AW | CTCGCGTGTC |
| A26 * | 6306 | AOD | Symptomatic | Heartwood | AW | TAGTATCAGC |
| A27 * | 5994 | AOD | Symptomatic | Gallery | AW | TCTCTATGCG |
| A28 * | 2996 | AOD | Non-symptomatic | Outer bark | AW | TGATACGTCT |
| A29 * | 4971 | AOD | Non-symptomatic | Inner bark | AW | TACTGAGCTA |
| A30 * | 6072 | AOD | Non-symptomatic | Sapwood | AW | ACGAGTGCGT |
| A31 * | 2730 | AOD | Non-symptomatic | Heartwood | AW | ACGCTCGACA |
| B2 | 12559 | Healthy | Healthy | Inner bark | BW | ACGAGTGCGT |
| B3 | 13716 | Healthy | Healthy | Sapwood | BW | ACGCTCGACA |
| B6 * | 7248 | AOD | Symptomatic | Inner bark | BW | AGACGCACTC |
| B7 * | 7907 | AOD | Symptomatic | Sapwood | BW | AGCACTGTAG |
| B9 * | 10338 | AOD | Symptomatic | Gallery | BW | ATCAGACACG |
| B11 * | 19265 | AOD | Non-symptomatic | Inner bark | BW | ATATCGCGAG |
| B12 * | 9824 | AOD | Non-symptomatic | Sapwood | BW | CGTGTCTCTA |
| SB2 | 12491 | Healthy | Healthy | Inner bark | SB | ACGAGTGCGT |
| SB3 | 7845 | Healthy | Healthy | Sapwood | SB | ACGCTCGACA |
| SB6 * | 6759 | AOD | Symptomatic | Inner bark | SB | AGACGCACTC |
| SB7 * | 9311 | AOD | Symptomatic | Sapwood | SB | AGCACTGTAG |
| SB11 * | 7031 | AOD | Non-symptomatic | Inner bark | SB | ATCAGACACG |
| SB12 * | 5127 | AOD | Non-symptomatic | Sapwood | SB | ATATCGCGAG |
| SB9 * | 12471 | AOD | Symptomatic | Gallery | SB | CGTGTCTCTA |
| Sample ID | No OTUs | Richness Chao1 | Shannon Diversity H′(log2) | PD † |
|---|---|---|---|---|
| GMW2 | 516 | 2499 | 7.86 | 52.57 |
| GMW3 | 495 | 2783 | 7.63 | 51.32 |
| GMW6 | 472 | 2170 | 7.47 | 50.73 |
| GMW7 | 505 | 2007 | 7.72 | 51.15 |
| GMW9 | 471 | 1609 | 7.65 | 49.65 |
| GMW11 | 482 | 2030 | 7.64 | 49.96 |
| GMW12 | 459 | 2084 | 7.39 | 47.74 |
| GMW6 * | 454 | 1598 | 7.55 | 48.69 |
| GMW7 * | 469 | 2052 | 7.55 | 51.53 |
| GMW9 * | 512 | 3307 | 7.81 | 52.43 |
| GMW11 * | 480 | 2020 | 7.66 | 49.31 |
| GMW12 * | 465 | 2087 | 7.52 | 51.05 |
| R11 | 550 | 2607 | 8.03 | 68.33 |
| R12 | 543 | 2828 | 8.00 | 63.92 |
| R13 | 552 | 2695 | 8.02 | 65.34 |
| R14 | 604 | 2522 | 8.46 | 73.48 |
| R15 | 508 | 2053 | 7.87 | 65.01 |
| R16 | 227 | 855 | 5.34 | 32.01 |
| R17 | 279 | 774 | 6.12 | 41.90 |
| R18 | 121 | 447 | 2.16 | 21.76 |
| R19 | 388 | 1173 | 7.07 | 56.14 |
| R20 | 400 | 1392 | 7.35 | 53.29 |
| R21 | 388 | 1016 | 7.21 | 53.43 |
| R22 | 347 | 1053 | 7.05 | 50.83 |
| R23 * | 277 | 781 | 5.85 | 44.63 |
| R24 * | 128 | 351 | 2.37 | 26.69 |
| R25 * | 122 | 345 | 2.46 | 25.39 |
| R26 * | 349 | 995 | 7.00 | 50.94 |
| R27 * | 99 | 233 | 1.75 | 19.48 |
| R28 * | 404 | 1057 | 7.73 | 57.80 |
| R29 * | 348 | 903 | 7.25 | 54.40 |
| R30 * | 337 | 964 | 6.89 | 48.95 |
| R31 * | 329 | 872 | 6.84 | 49.47 |
| A1 | 23 | 32 | 2.01 | 6.05 |
| A2 | 95 | 169 | 3.68 | 16.46 |
| A3 | 54 | 101 | 3.03 | 11.63 |
| A4 | 204 | 341 | 5.98 | 37.49 |
| A6 | 103 | 180 | 3.79 | 16.79 |
| A7 | 59 | 107 | 2.96 | 11.77 |
| A8 | 180 | 317 | 5.28 | 28.59 |
| A9 | 114 | 194 | 3.83 | 19.84 |
| A11 | 28 | 46 | 2.22 | 7.97 |
| A12 | 132 | 229 | 4.00 | 25.72 |
| A13 | 188 | 349 | 5.12 | 25.59 |
| A14 | 185 | 313 | 6.23 | 25.04 |
| A15 | 46 | 60 | 3.27 | 10.11 |
| A16 | 144 | 306 | 4.68 | 22.85 |
| A17 | 126 | 344 | 3.71 | 20.06 |
| A18 | 151 | 494 | 4.60 | 27.46 |
| A19 | 107 | 222 | 4.16 | 16.44 |
| A20 | 32 | 56 | 2.40 | 7.43 |
| A21 | 104 | 282 | 3.61 | 18.39 |
| A23 * | 106 | 194 | 4.85 | 16.80 |
| A24 * | 167 | 303 | 5.69 | 21.11 |
| A25 * | 87 | 152 | 4.17 | 16.60 |
| A27 * | 158 | 417 | 4.77 | 28.99 |
| A28 * | 166 | 384 | 4.53 | 31.77 |
| A29 * | 63 | 119 | 2.86 | 15.65 |
| A30 * | 167 | 317 | 5.76 | 32.26 |
| B2 | 27 | 53 | 0.94 | 6.89 |
| B3 | 28 | 85 | 0.90 | 7.38 |
| B6 * | 58 | 94 | 2.69 | 10.66 |
| B7 * | 30 | 240 | 1.05 | 6.79 |
| B9 * | 57 | 135 | 2.65 | 11.00 |
| B11 * | 35 | 64 | 1.33 | 8.41 |
| B12 * | 15 | 20 | 0.74 | 4.13 |
| SB2 | 24 | 29 | 0.75 | 6.46 |
| SB3 | 29 | 53 | 0.96 | 7.59 |
| SB6 * | 158 | 424 | 5.06 | 20.85 |
| SB7 * | 182 | 426 | 5.38 | 23.60 |
| SB11 * | 18 | 63 | 0.55 | 3.99 |
| SB12 * | 28 | 52 | 0.88 | 8.79 |
| SB9 * | 69 | 102 | 3.92 | 13.02 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapp, M.; Lewis, E.; Moss, S.; Barrett, B.; Kirk, S.; Elphinstone, J.G.; Denman, S. Metabarcoding of Bacteria Associated with the Acute Oak Decline Syndrome in England. Forests 2016, 7, 95. https://doi.org/10.3390/f7050095
Sapp M, Lewis E, Moss S, Barrett B, Kirk S, Elphinstone JG, Denman S. Metabarcoding of Bacteria Associated with the Acute Oak Decline Syndrome in England. Forests. 2016; 7(5):95. https://doi.org/10.3390/f7050095
Chicago/Turabian StyleSapp, Melanie, Erin Lewis, Stephen Moss, Ben Barrett, Susan Kirk, John G. Elphinstone, and Sandra Denman. 2016. "Metabarcoding of Bacteria Associated with the Acute Oak Decline Syndrome in England" Forests 7, no. 5: 95. https://doi.org/10.3390/f7050095
APA StyleSapp, M., Lewis, E., Moss, S., Barrett, B., Kirk, S., Elphinstone, J. G., & Denman, S. (2016). Metabarcoding of Bacteria Associated with the Acute Oak Decline Syndrome in England. Forests, 7(5), 95. https://doi.org/10.3390/f7050095