Mapping Fusiform Rust Resistance Genes within a Complex Mating Design of Loblolly Pine




 , and
, and 
Abstract
:1. Introduction
2. Experimental Section
2.1. Plant Material and Disease Phenotyping
2.2. Identification of Markers Using BayesCπ
2.3. Association Testing Using BAMD
2.4. Single Marker Regression Analyses and Mapping of Significant SNPs
3. Results
3.1. SNP Effect Size and Number Differs for Gall Score and Gall Length
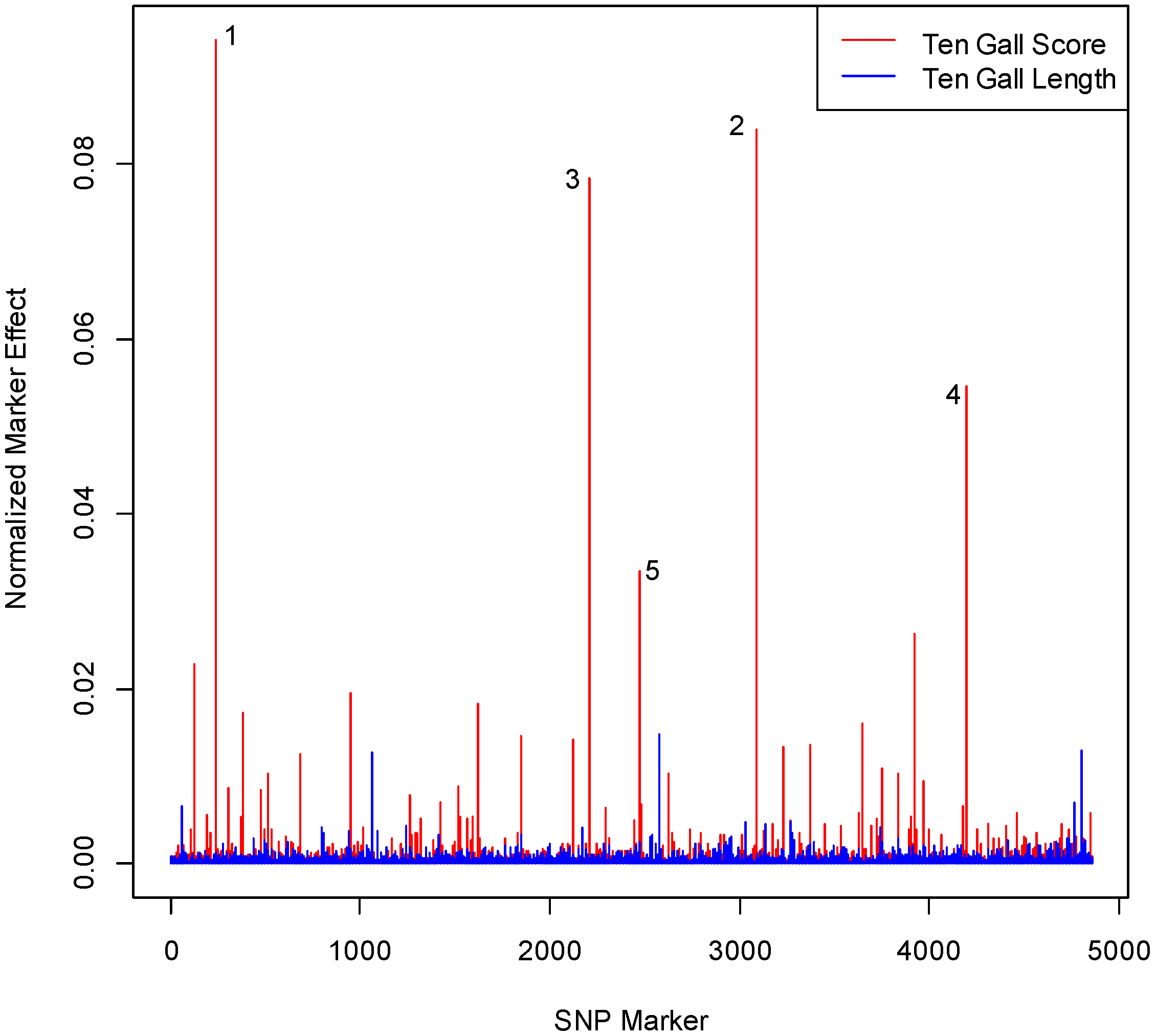
3.2. SNPs Differ in One Gall and 10 Gall Experiments

{kind=link}
{kind=link}
{kind=link}
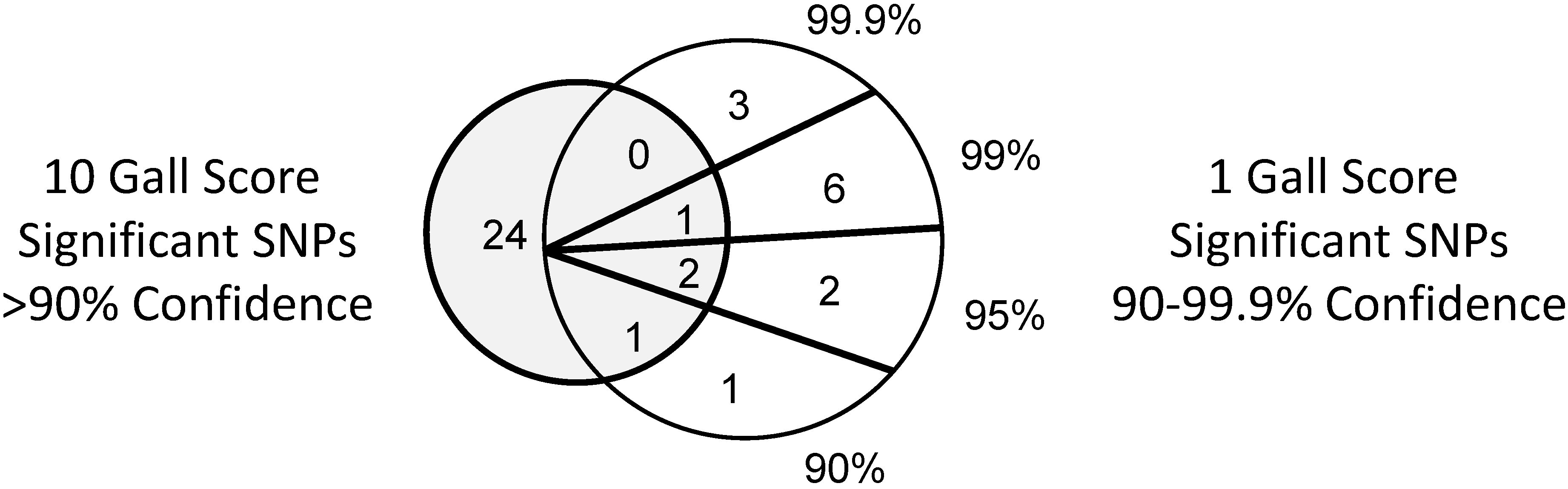
| Inoculum | Phenotype | SNPs at Confidence Interval (%) | |||
|---|---|---|---|---|---|
| 90–94.9 | 95–98.9 | 99–99.8 | >99.9 | ||
| 1 gall | Gall score | 2 | 4 | 7 | 3 |
| 10 gall | 7 | 10 | 4 | 7 | |
| 1 gall | Gall length | 2 | 2 | 0 | 0 |
| 10 gall | 1 | 4 | 3 | 2 | |
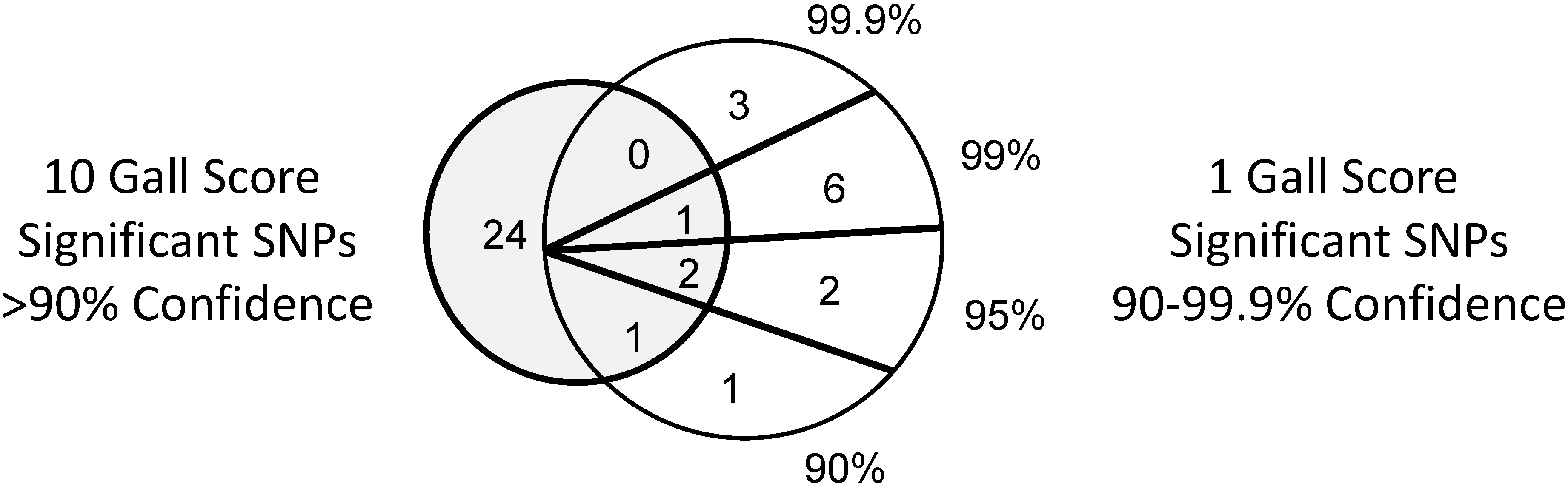
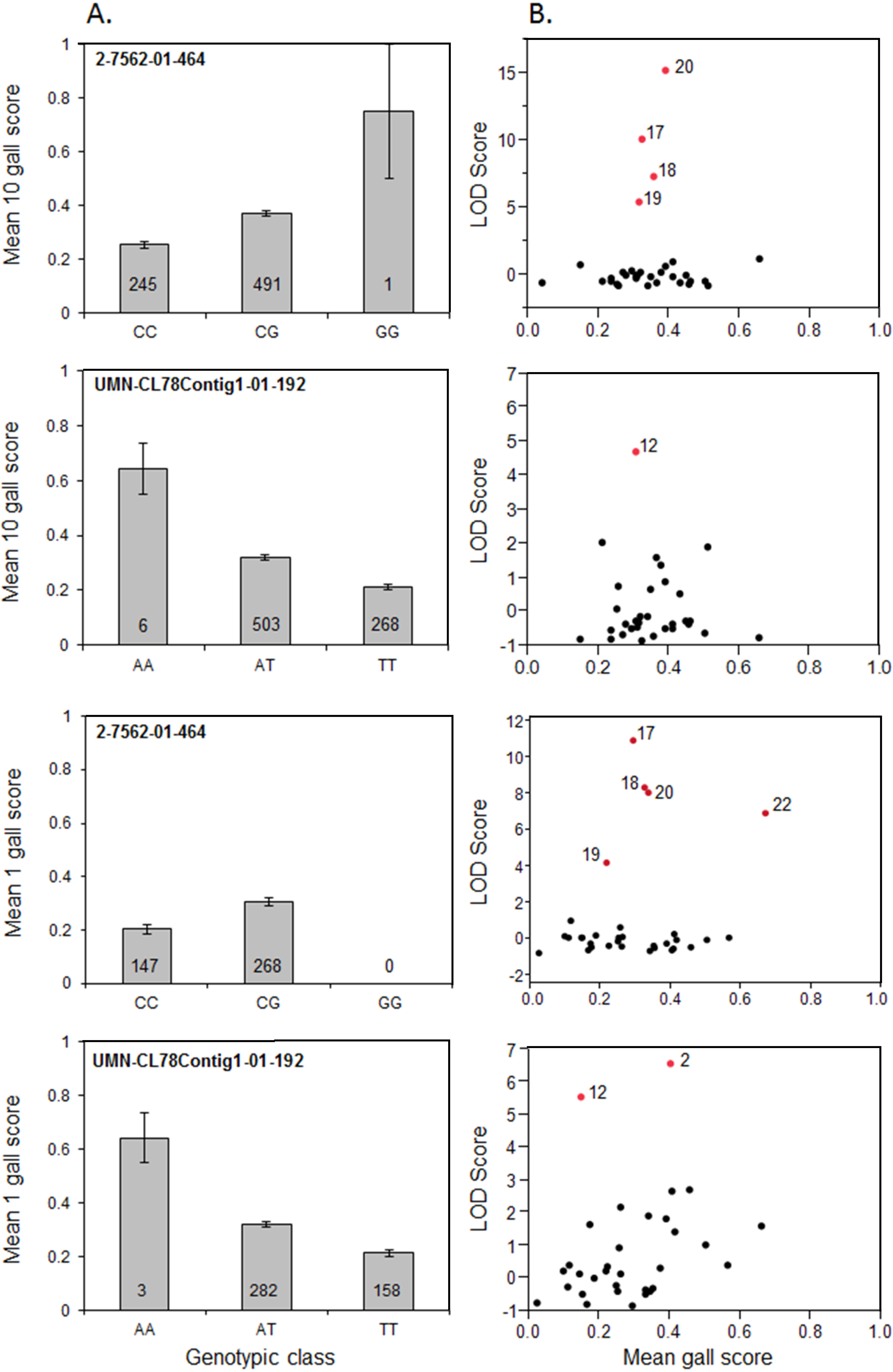
3.3. Significant SNP Markers are Associated with Resistance
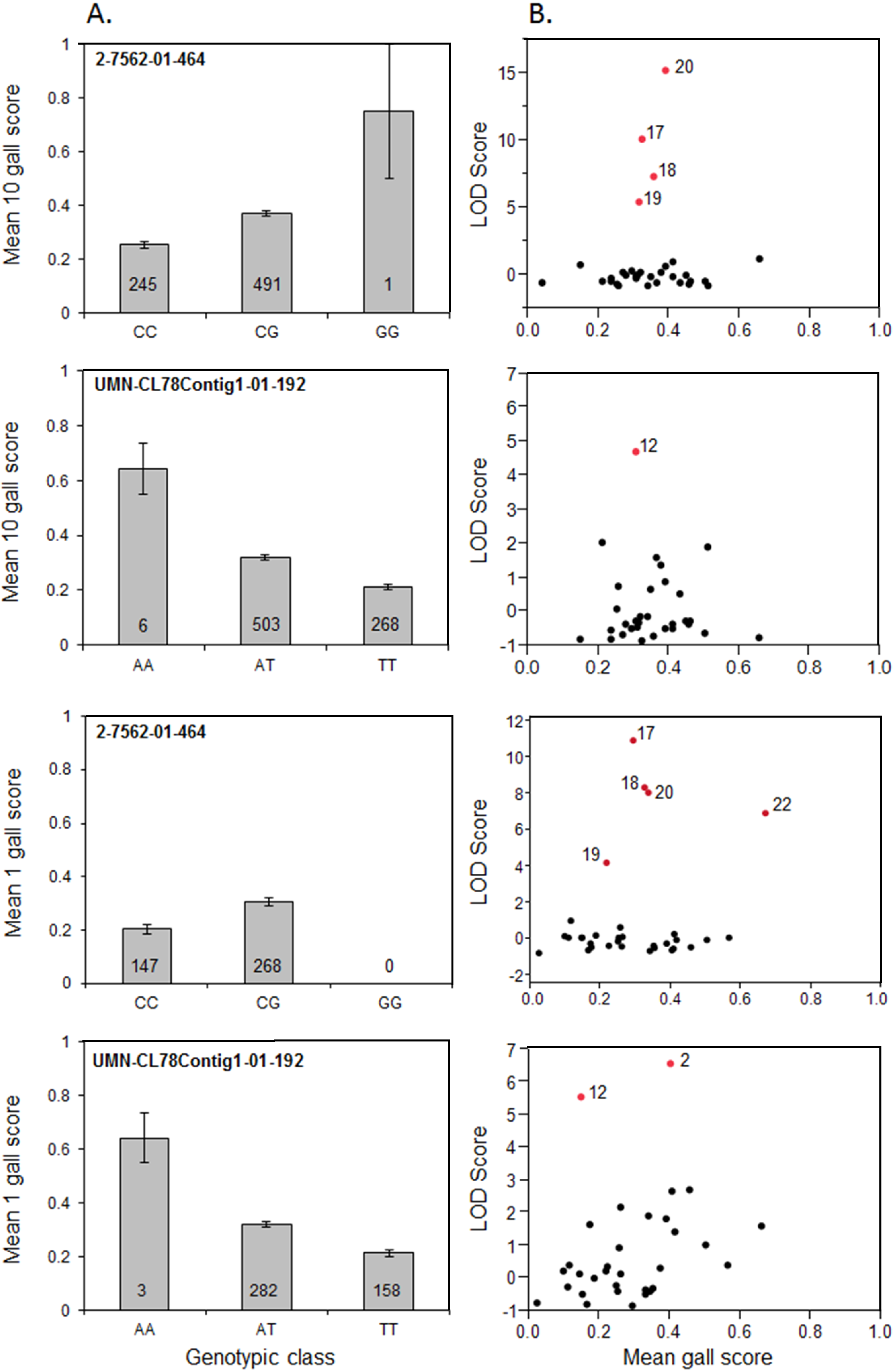
| Locus | LG | Parent | Families | Avr Source | Significant SNP (LOD 1Gall/ 10 Gall) | Genetic interpretation |
|---|---|---|---|---|---|---|
| Fr1 | 2 | 17 | 400, 401, 416, 436, 442 | 1 Gall/10 Gall | 2-7562-01-464 (10.9/10.0) | Parent 17 known Fr1/fr1, 1/10 gall known avirulent to Fr1 |
| 12 | 419, 428, 446, 466, 472 | 1 Gall/10 Gall | UMN-CL78 Contig1-01-192 (5.5/4.7) | |||
| Parents 12, 20 may segregate for Fr1 or non-Fr1 at locus, 1/10 gall avirulent | ||||||
| 20 | 408, 429, 430, 457, 461, 476, 477 | 1 Gall/10 Gall | 2-7562-01-464 (8.9/15.1) | |||
| 2 | 406, 412, 423, 433, 435, 454, 469 | 1 Gall | UMN-CL78 Contig1-01-192 (6.5/1.9) | |||
| Parents 2, 22 may segregate for non-Fr1 at locus, 1 gall avirulent | ||||||
| 22 | 403, 408, 420, 451 | 1 Gall | 2-7562-01-464 (6.6/1.2) | |||
| Fr8 | - | 32 | 404, 461 | - | - | Parent 32 known Fr8/fr8, neither 1/10 gall appear avirulent to Fr8 |
| 10 | 28 | 402, 421, 433, 452, 453, 475 | 1 Gall | CL3925Contig1 -03-163 (6.4/-0.6) | ||
| Parent 28 may segregate for non-Fr8 at locus, 1 gall avirulent | ||||||
| Fr- | 11 | 23 | 420, 443, 448, 455, 467, 473 | 10 Gall | 0-13348-01-203 (2.8/4.1) | Parent 23 known unnamed Fr resistance gene (Fr-) 10 gall appears avirulent to Fr- |
| 4 | 415, 437, 469 | 10 Gall | 0-13348-01-203 (2.7/7.0) | |||
| Parent 4 may segregate for Fr- or non-Fr- at locus, 10 gall avirulent | ||||||
| 2 | 406, 412, 423, 433, 435, 454, 469 | 1 Gall/10 Gall | 0-2040-01-149 (6.8/5.2) | |||
| Parent 2 may segregate for non-Fr- at locus, 1/10 gall avirulent |
4. Discussion
4.1. SNPs Mapped to Fr1 (LG2)
4.2. SNPs Mapped to Fr8 (LG10)
4.3. SNPs Mapped to LG11
4.4. Genetic Architecture of Rust Resistance
4.5. Identifying Fr Genes: Paths Forward
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dwinell, L.D. Biology of Fusiform Rust, Management of Fusiform Rust in Southern Pines; Dinus, R.J., Schmidt, R.A., Eds.; University of Florida: Gainesville, FL, USA, 1977; p. 163. [Google Scholar]
- Cubbage, F.; Pye, J.; Holmes, T.; Wagner, J. An economic analysis of fusiform rust protection research. South. J. App. For. 2000, 24, 77–85. [Google Scholar]
- Amerson, H.V.; Kubisiak, T.L.; Garcia, S.A.; Kuhlman, G.C.; Nelson, C.D.; McKeand, S.E.; Mullin, T.J.; Li, B. Interacting Genes in the Pine-Fusiform Rust Forest Pathosystem. In proceedings of the 28th Biennial Southern Forest Tree Improvement Conference, Raleigh, NC, USA, 21–23 June 2005; U.S. Department of Agriculture, Forest Service: Raleigh, NC, USA, 2005; p. 60. [Google Scholar]
- Kubisiak, T.; Anderson, C.; Amerson, H.; Smith, J.; Davis, J.; Nelson, C. A genomic map enriched for markers linked to Avr1 Cronartium. quercuum f.sp. fusiforme. Fungal Gen. Biol. 2011, 48, 266–274. [Google Scholar] [CrossRef]
- Kubisiak, T.L.; Amerson, H.V.; Nelson, C.D. Genetic interaction of the fusiform rust fungus with resistance gene fr1 in loblolly pine. Phytopathology 2005, 95, 376–380. [Google Scholar] [CrossRef]
- Wilcox, P.L.; Amerson, H.V.; Kuhlman, E.G.; Liu, B.H.; O’Malley, D.M.; Sederoff, R.R. Detection of a major gene for resistance to fusiform rust disease in loblolly pine by genomic mapping. Proc. Natl. Acad. Sci. USA 1996, 93, 3859–3864. [Google Scholar]
- Nelson, C.; Kubisiak, T.; Amerson, H. Unravelling and managing fusiform rust disease: A model approach for coevolved forest tree pathosystems. For. Pathol. 2010, 40, 64–72. [Google Scholar] [CrossRef]
- Amerson, H. North Carolina State University: Raleigh, NC, USA; Forests, 2014; unpublished.
- Kayihan, G.; Nelson, C.; Huber, D.; Amerson, H.; White, T.; Davis, J. Clonal evaluation for fusiform rust disease resistance: Effects of pathogen virulence and disease escape. Can. J. For. Res. Rev. 2010, 40, 1042–1050. [Google Scholar] [CrossRef]
- Huber, D.; Amerson, H. Performance of the loblolly pine fusiform rust disease resistance gene (Fr1) in a slashXloblolly pine hybrid family. Tree Genet. Genomes 2011, 7, 535–540. [Google Scholar] [CrossRef]
- Thomas, C.M.; Vos, P.; Zabeau, M.; Jones, D.A.; Norcott, K.A.; Chadwick, B.P.; Jones, J.D. Identification of amplified restriction fragment polymorphism (AFLP) markers tightly linked to the tomato Cf-9 gene for resistance to Cladosporium. fulvum. Plant J. 1995, 8, 785–794. [Google Scholar]
- Dodds, P.N.; Lawrence, G.J.; Catanzariti, A.M.; Teh, T.; Wang, C.I.; Ayliffe, M.A.; Kobe, B.; Ellis, J.G. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. USA 2006, 103, 8888–8893. [Google Scholar] [CrossRef]
- Bahri, B.; Kaltz, O.; Leconte, M.; de Vallavieille-Pope, C.; Enjalbert, J. Tracking costs of virulence in natural populations of the wheat pathogen, Puccinia. striiformis f.sp. tritici. BMC Evol. Biol. 2009, 9, 26. [Google Scholar] [CrossRef]
- McHale, L.K.; Truco, M.J.; Kozik, A.; Wroblewski, T.; Ochoa, O.E.; Lahre, K.A.; Knapp, S.J.; Michelmore, R.W. The genomic architecture of disease resistance in lettuce. Theor. Appl. Genet. 2009, 118, 565–580. [Google Scholar] [CrossRef]
- Eckert, A.; Pande, B.; Ersoz, E.; Wright, M.; Rashbrook, V.; Nicolet, C.; Neale, D. High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet. Genomes 2009, 5, 225–234. [Google Scholar] [CrossRef]
- Kayihan, G.; Huber, D.; Morse, A.; White, T.; Davis, J. Genetic dissection of fusiform rust and pitch canker disease traits in loblolly pine. Theor. App. Genet. 2005, 110, 948–958. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.; Kizilkaya, K.; Garrick, D. Extension of the bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Resende, M.; Munoz, P.; Resende, M.; Garrick, D.; Fernando, R.; Davis, J.; Jokela, E.; Martin, T.; Peter, G.; Kirst, M. Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 2012, 190, 1503–1510. [Google Scholar] [CrossRef]
- Li, Z.; Gopal, V.; Li, X.; Davis, J.M.; Casella, G. Simultaneous SNP identification in association studies with missing data. Ann. App. Statist. 2012, 6, 432–456. [Google Scholar] [CrossRef]
- Echt, C.S.; Saha, S.; Krutovsky, K.V.; Wimalanathan, K.; Erpelding, J.E.; Liang, C.; Nelson, C.D. An annotated genetic map of loblolly pine based on microsatellite and cDNA markers. BMC Genet. 2011, 12, 17. [Google Scholar]
- Munoz, P.R.; Resende, M.F.R.; Huber, D.A.; Quesada, T.; Resende, M.D.V.; Neale, D.V.; Wegrzyn, J.L.; Kirst, M.; Peter, G.F. Genomic relationship matrix for correcting pedigree errors in breeding populations: Impact on genetic parameters and genomic selection accuracy. Crop Sci. 2013. [Google Scholar] [CrossRef]
- Eckert, A.; van Heerwaarden, J.; Wegrzyn, J.; Nelson, C.; Ross-Ibarra, J.; Gonzalez-Martinez, S.; Neale, D. Patterns of population structure and environmental associations to aridity across the range of loblolly pine (Pinus taeda L., pinaceae). Genetics 2010, 185, 969–982. [Google Scholar] [CrossRef]
- Garrick, D.; Taylor, J.; Fernando, R. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet. Sel. Evol. 2009, 41. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- SAS Institute Inc. SAS/STAT® 9.3. User’s Guide, SAS Institute, Inc: Cary, NC, USA, 2011.
- Munoz, P.; Peter, G. University of Florida: Gainesville, FL, USA, Unpublished Data.
- Li, B.; McKeand, S.; Weir, R. Tree improvement and sustainable forestry—Impact of two cycles of loblolly pine breeding in the USA. For. Genet. 1999, 6, 229–234. [Google Scholar]
- McKeand, S.; Bridgwater, F. A strategy for the third breeding cycle of loblolly pine in the southeastern US. Silvae Genet. 1998, 47, 223–234. [Google Scholar]
- Nelson, C.D.; Johnsen, K.H. Genomic and physiological approaches to advancing forest tree improvement. Tree Physiol. 2008, 28, 1135–1143. [Google Scholar] [CrossRef]
- Myburg, H.; Morse, A.; Amerson, H.; Kubisiak, T.; Huber, D.; Osborne, J.; Garcia, S.; Nelson, C.; Davis, J.; Covert, S.; et al. Differential gene expression in loblolly pine (Pinus taeda L.) challenged with the fusiform rust fungus, Cronartium. quercuum f.sp fusiforme. Physiol. Mol. Plant Pathol. 2006, 68, 79–91. [Google Scholar] [CrossRef]
- Cumbie, W.; Isik, F.; McKeand, S. Genetic improvement of sawtimber potential in loblolly pine. For. Sci. 2012, 58, 168–177. [Google Scholar]
- McKeand, S.E.; Jokela, E.J.; Huber, D.A.; Byram, T.D.; Allen, H.L.; Li, B.; Mullin, T.J. Performance of improved genotypes of loblolly pine across different soils, climates, and silvicultural inputs. For. Ecol. Manag. 2006, 227, 178–184. [Google Scholar] [CrossRef]
- Schmidt, R.; Powers, H.; Snow, G. Application of genetic-disease resistance for the control of fusiform rust in intensively managed southern pine. Phytopathology 1981, 71, 993–997. [Google Scholar] [CrossRef]
- Schmidt, R.; Gramacho, K.; Miller, T.; Young, C. Components of partial resistance in the slash pine-fusiform rust pathosystem. Phytopathology 2000, 90, 1005–1010. [Google Scholar] [CrossRef]
- Poland, J.; Balint-Kurti, P.; Wisser, R.; Pratt, R.; Nelson, R. Shades of gray: The world of quantitative disease resistance. Trends Plant. Sci. 2009, 14, 21–29. [Google Scholar] [CrossRef]
- Ercolano, M.R.; Sanseverino, W.; Carli, P.; Ferriello, F.; Frusciante, L. Genetic and genomic approaches for r-gene mediated disease resistance in tomato: Retrospects and prospects. Plant Cell Rep. 2012, 31, 973–985. [Google Scholar] [CrossRef]
- Vleeshouwers, V.G.; Raffaele, S.; Vossen, J.H.; Champouret, N.; Oliva, R.; Segretin, M.E.; Rietman, H.; Cano, L.M.; Lokossou, A.; Kessel, G.; et al. Understanding and exploiting late blight resistance in the age of effectors. Annu. Rev. Phytopathol. 2011, 49, 507–531. [Google Scholar] [CrossRef]
- Xiao, W.; Yang, Q.; Wang, H.; Duan, J.; Guo, T.; Liu, Y.; Zhu, X.; Chen, Z. Identification and fine mapping of a major r gene to Magnaporthe oryzae in a broad-spectrum resistant germplasm in rice. Mol. Breed. 2012, 30, 1715–1726. [Google Scholar] [CrossRef]
- Vida, G.; Gal, M.; Uhrin, A.; Veisz, O.; Syed, N.; Flavell, A.; Wang, Z.; Bedo, Z. Molecular markers for the identification of resistance genes and marker-assisted selection in breeding wheat for leaf rust resistance. Euphytica 2009, 170, 67–76. [Google Scholar] [CrossRef]
- Panwar, P.; Jha, A.; Pandey, P.; Gupta, A.; Kumar, A. Functional markers based molecular characterization and cloning of resistance gene analogs encoding NBS-LRR disease resistance proteins in finger millet (Eleusine coracana). Mol. Biol. Rep. 2011, 38, 3427–3436. [Google Scholar] [CrossRef]
- Magbanua, Z.V.; Ozkan, S.; Bartlett, B.D.; Chouvarine, P.; Saski, C.A.; Liston, A.; Cronn, R.C.; Nelson, C.D.; Peterson, D.G. Adventures in the enormous: A 1.8 million clone BAC library for the 21.7 gb genome of loblolly pine. PLoS One 2011, 6, e16214. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quesada, T.; Resende Jr., M.F.R.; Muñoz, P.; Wegrzyn, J.L.; Neale, D.B.; Kirst, M.; Peter, G.F.; Gezan, S.A.; Nelson, C.D.; Davis, J.M. Mapping Fusiform Rust Resistance Genes within a Complex Mating Design of Loblolly Pine. Forests 2014, 5, 347-362. https://doi.org/10.3390/f5020347
Quesada T, Resende Jr. MFR, Muñoz P, Wegrzyn JL, Neale DB, Kirst M, Peter GF, Gezan SA, Nelson CD, Davis JM. Mapping Fusiform Rust Resistance Genes within a Complex Mating Design of Loblolly Pine. Forests. 2014; 5(2):347-362. https://doi.org/10.3390/f5020347
Chicago/Turabian StyleQuesada, Tania, Marcio F.R. Resende Jr., Patricio Muñoz, Jill L. Wegrzyn, David B. Neale, Matias Kirst, Gary F. Peter, Salvador A. Gezan, C. Dana Nelson, and John M. Davis. 2014. "Mapping Fusiform Rust Resistance Genes within a Complex Mating Design of Loblolly Pine" Forests 5, no. 2: 347-362. https://doi.org/10.3390/f5020347
APA StyleQuesada, T., Resende Jr., M. F. R., Muñoz, P., Wegrzyn, J. L., Neale, D. B., Kirst, M., Peter, G. F., Gezan, S. A., Nelson, C. D., & Davis, J. M. (2014). Mapping Fusiform Rust Resistance Genes within a Complex Mating Design of Loblolly Pine. Forests, 5(2), 347-362. https://doi.org/10.3390/f5020347