
On the Sr1−xBaxFeO2F Oxyfluoride Perovskites: Structure and Magnetism from Neutron Diffraction and Mössbauer Spectroscopy



Abstract
:
1. Introduction
2. Results
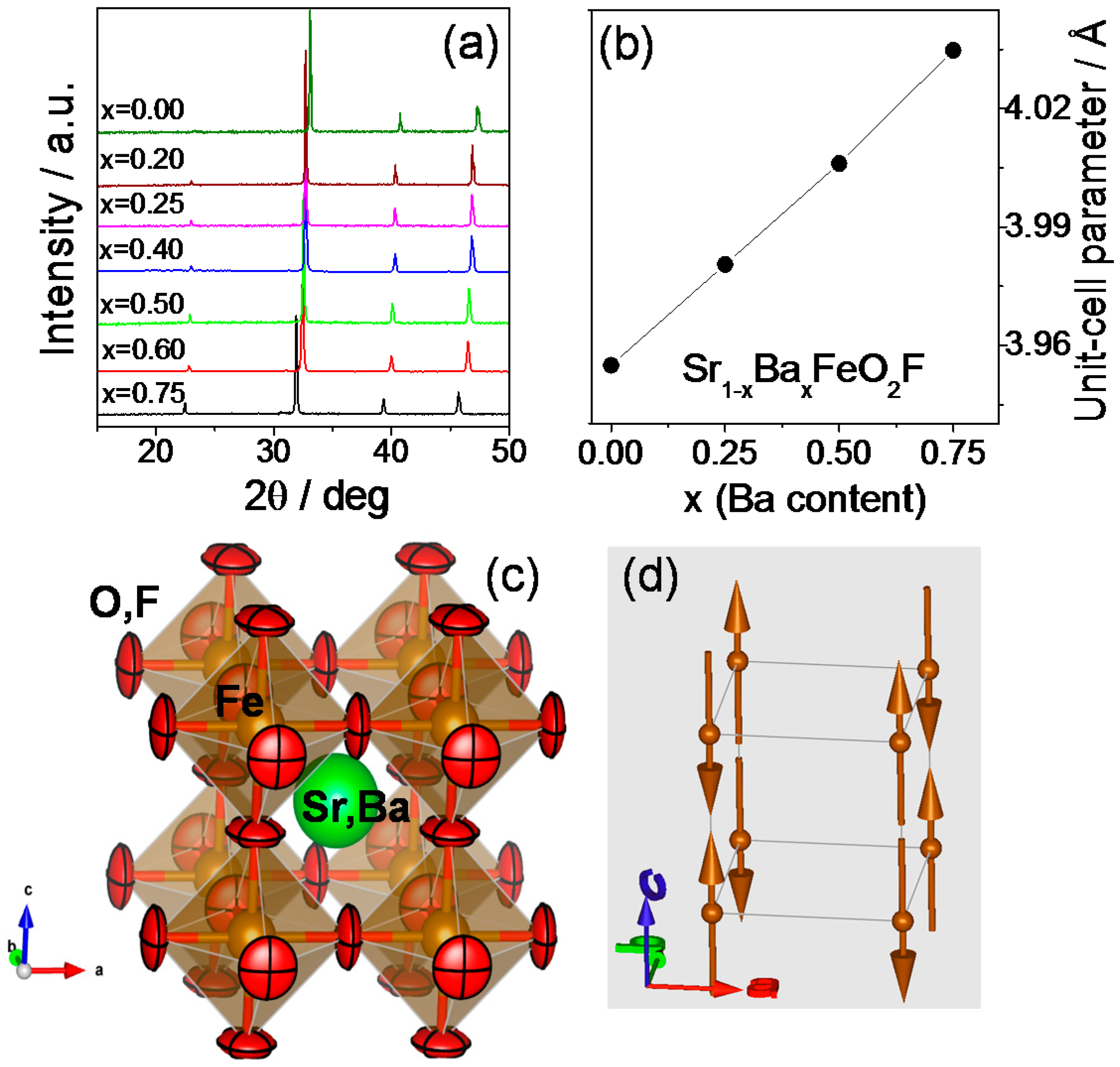
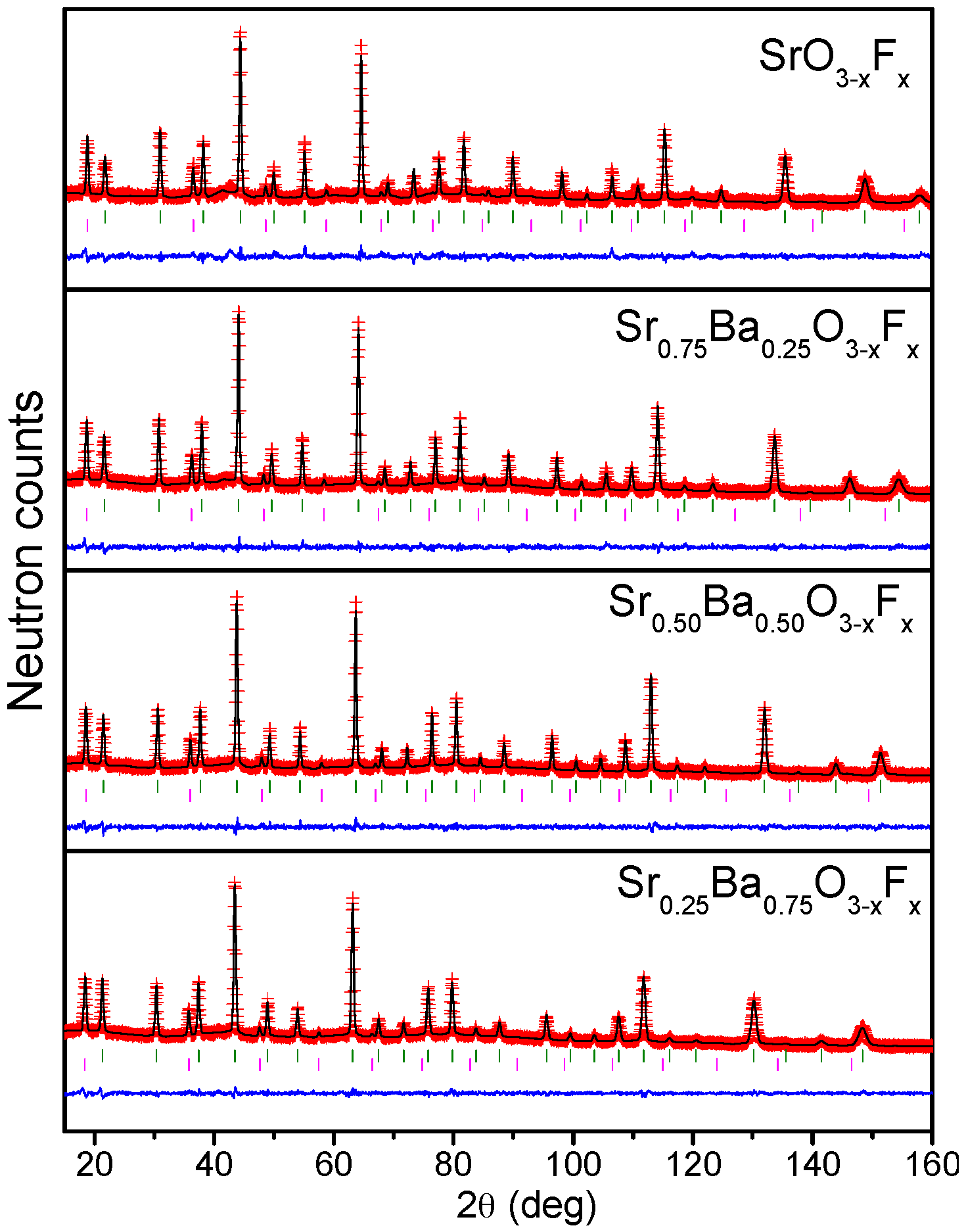
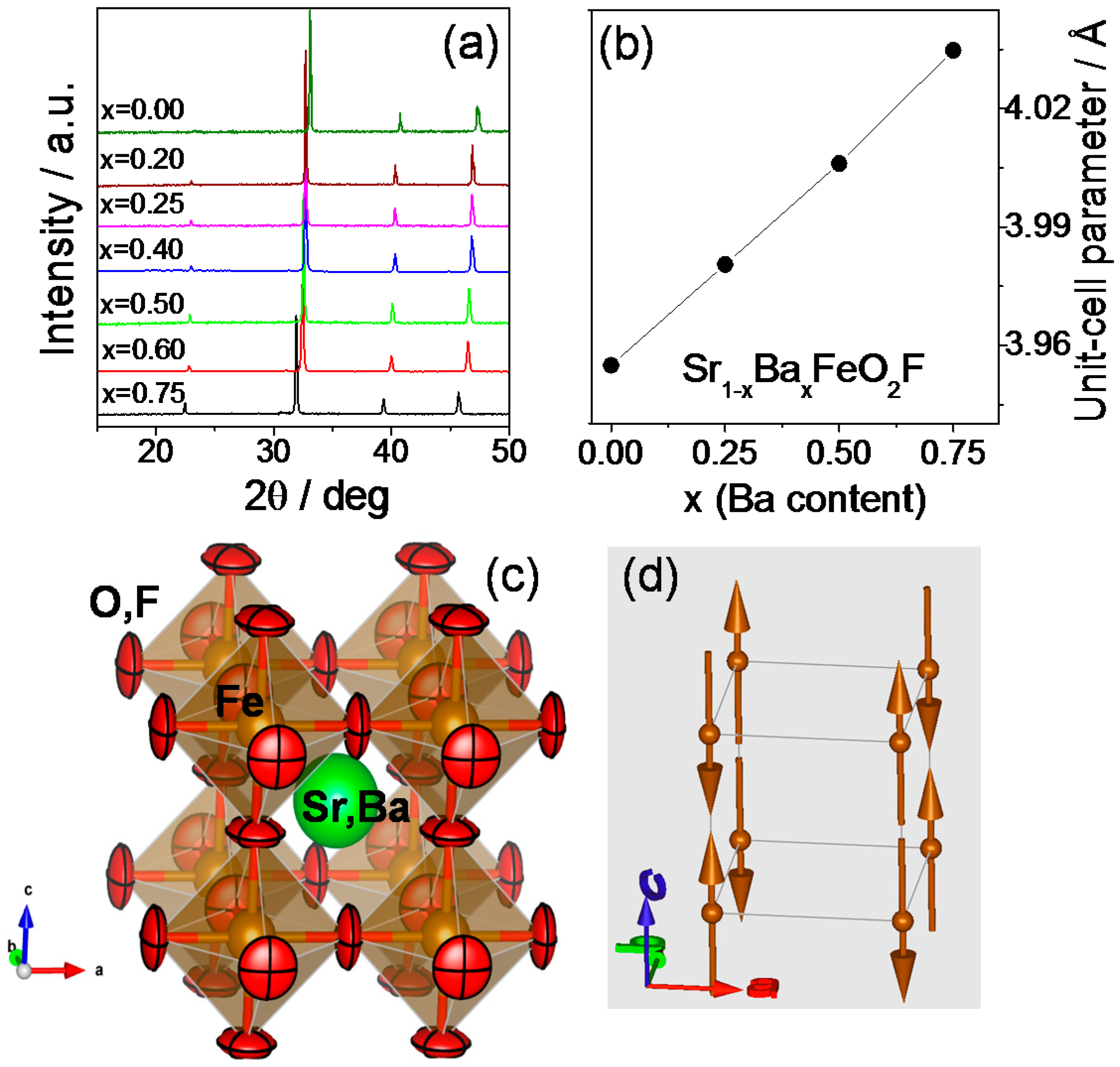
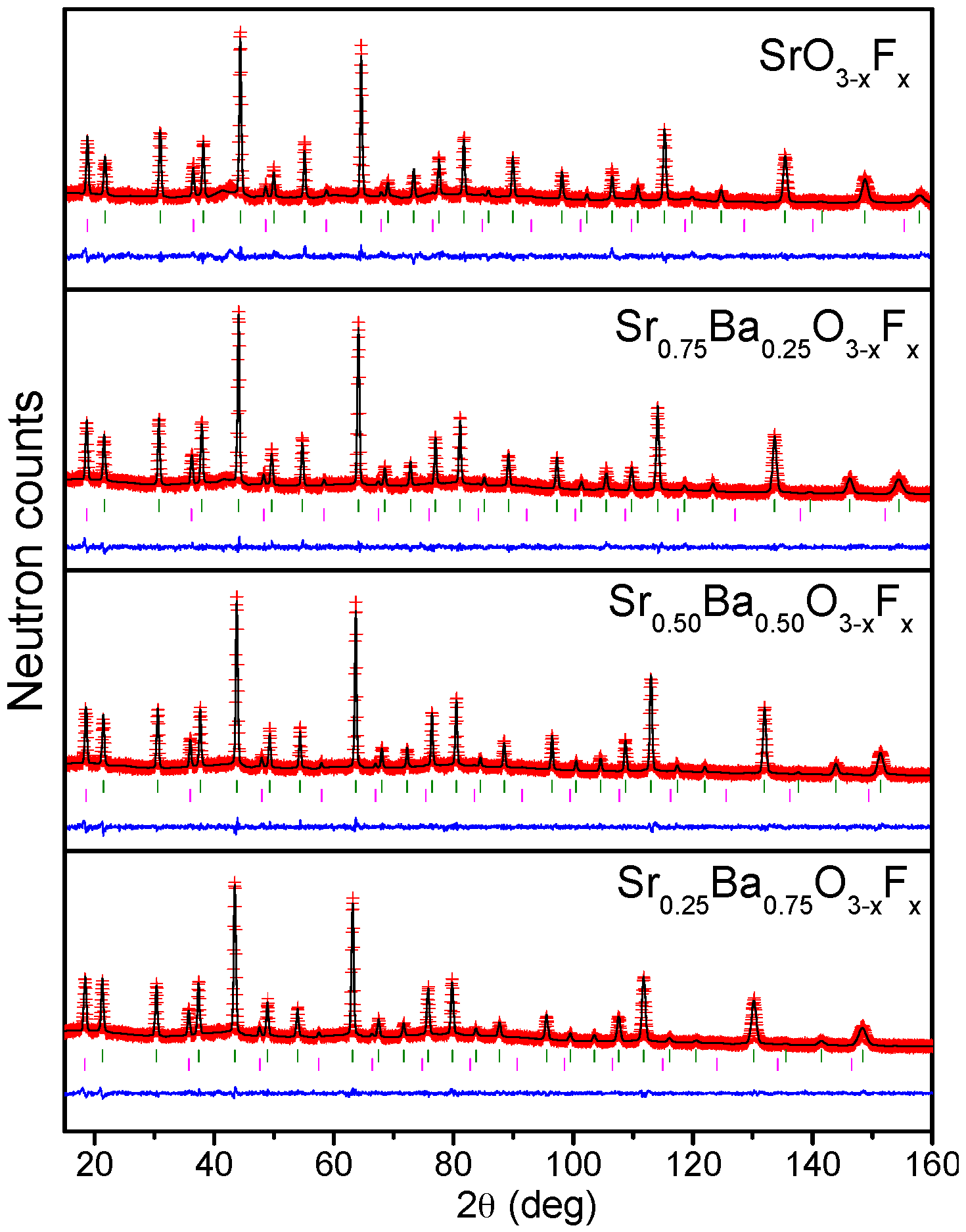
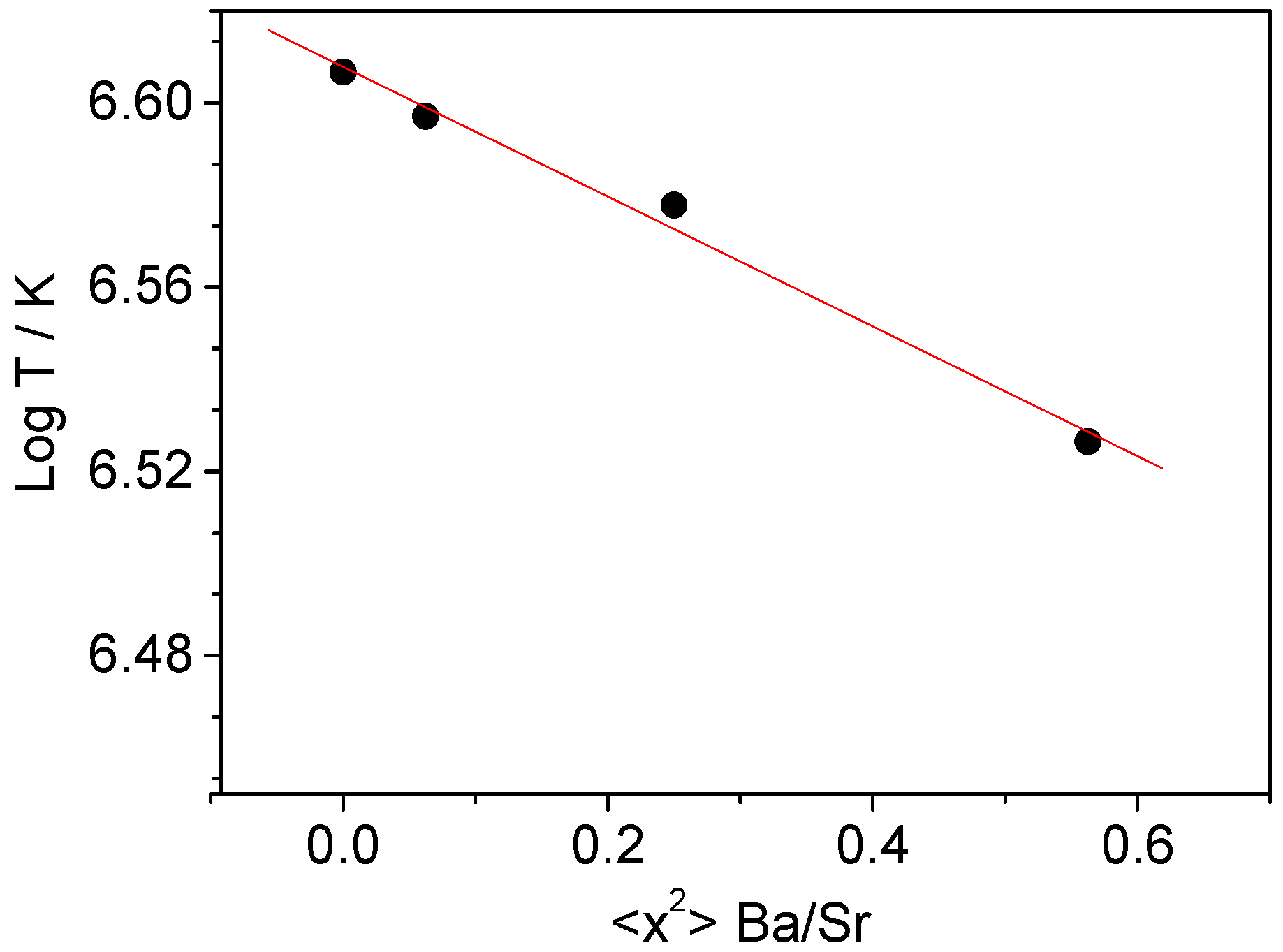
2.1. Crystallographic Characterization
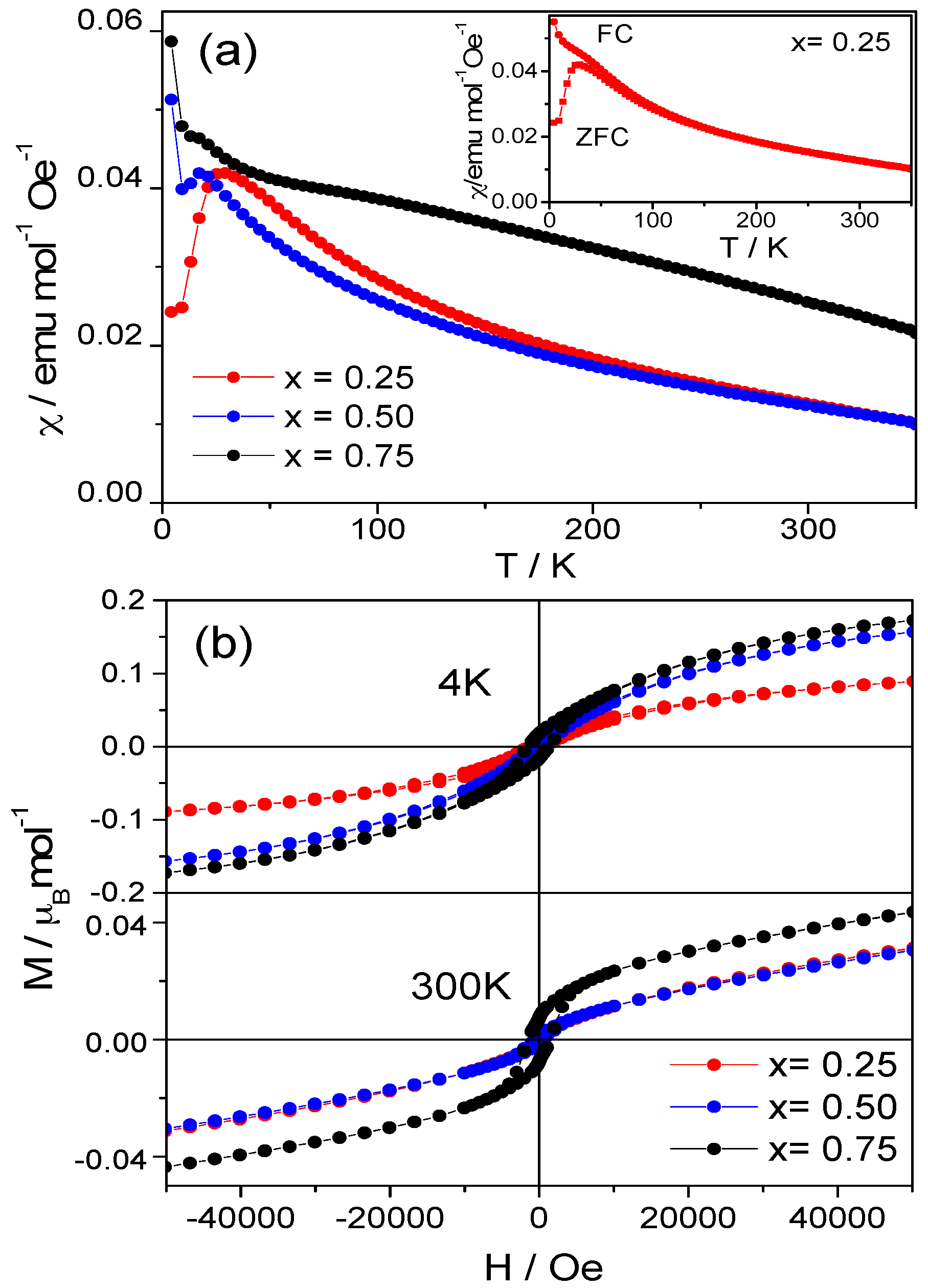
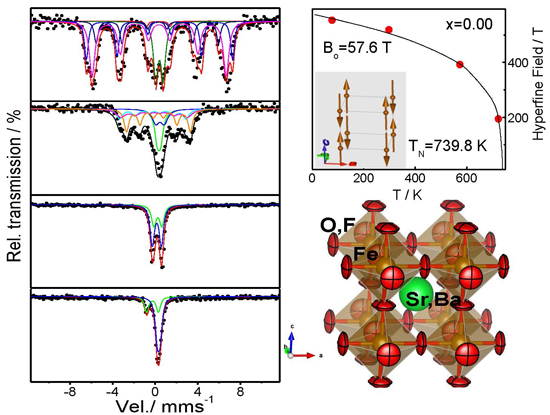
2.2. Magnetic Measurements
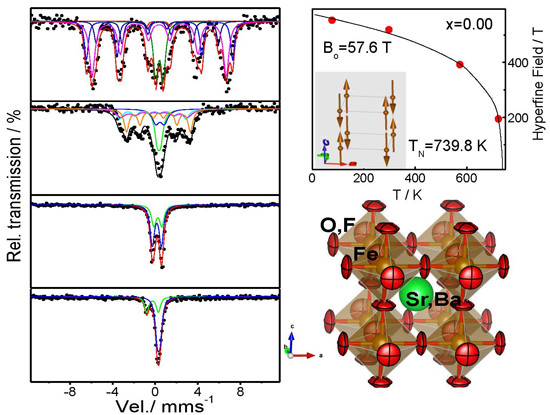
2.3. Determination of the Magnetic Structure
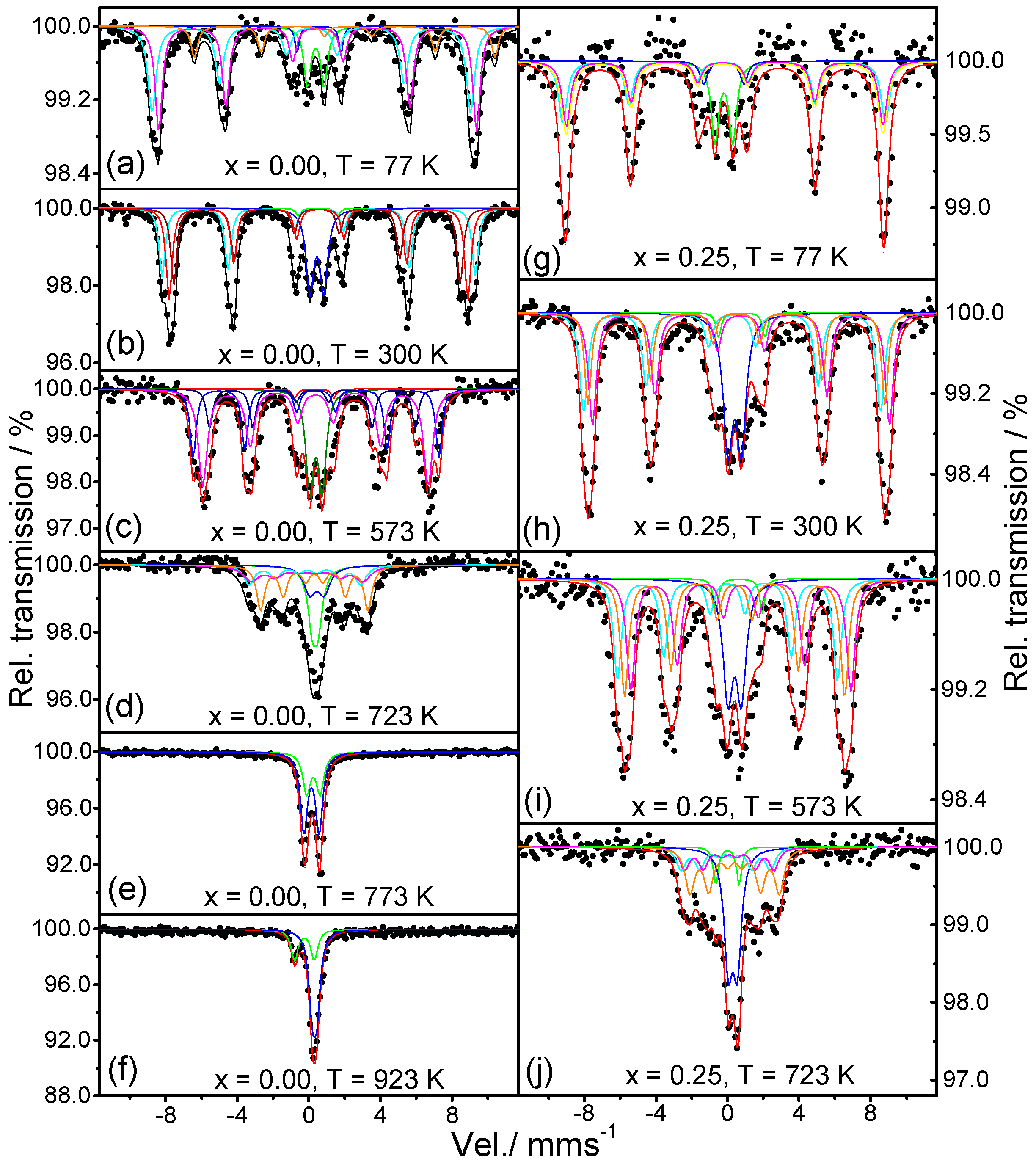
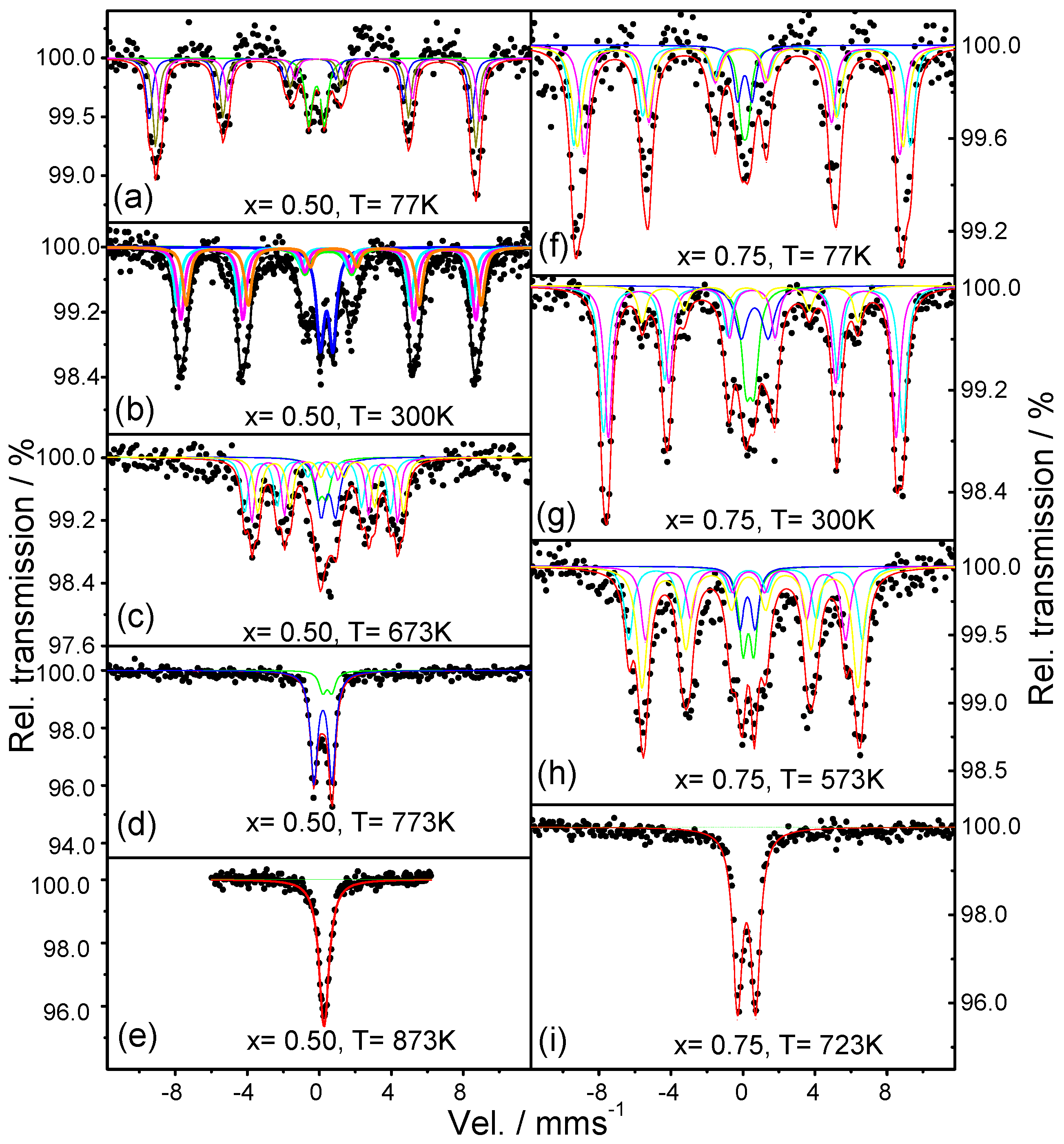
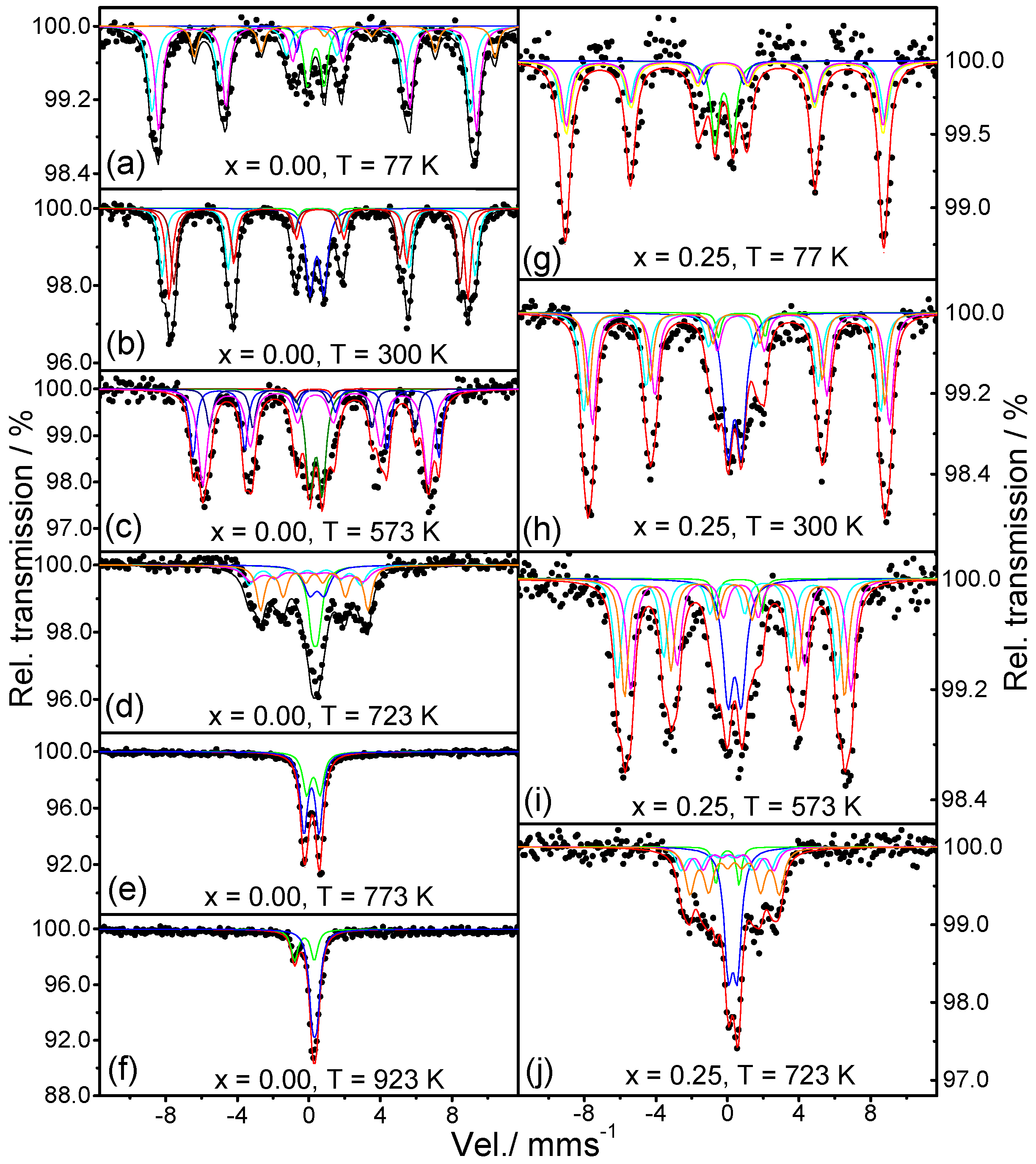
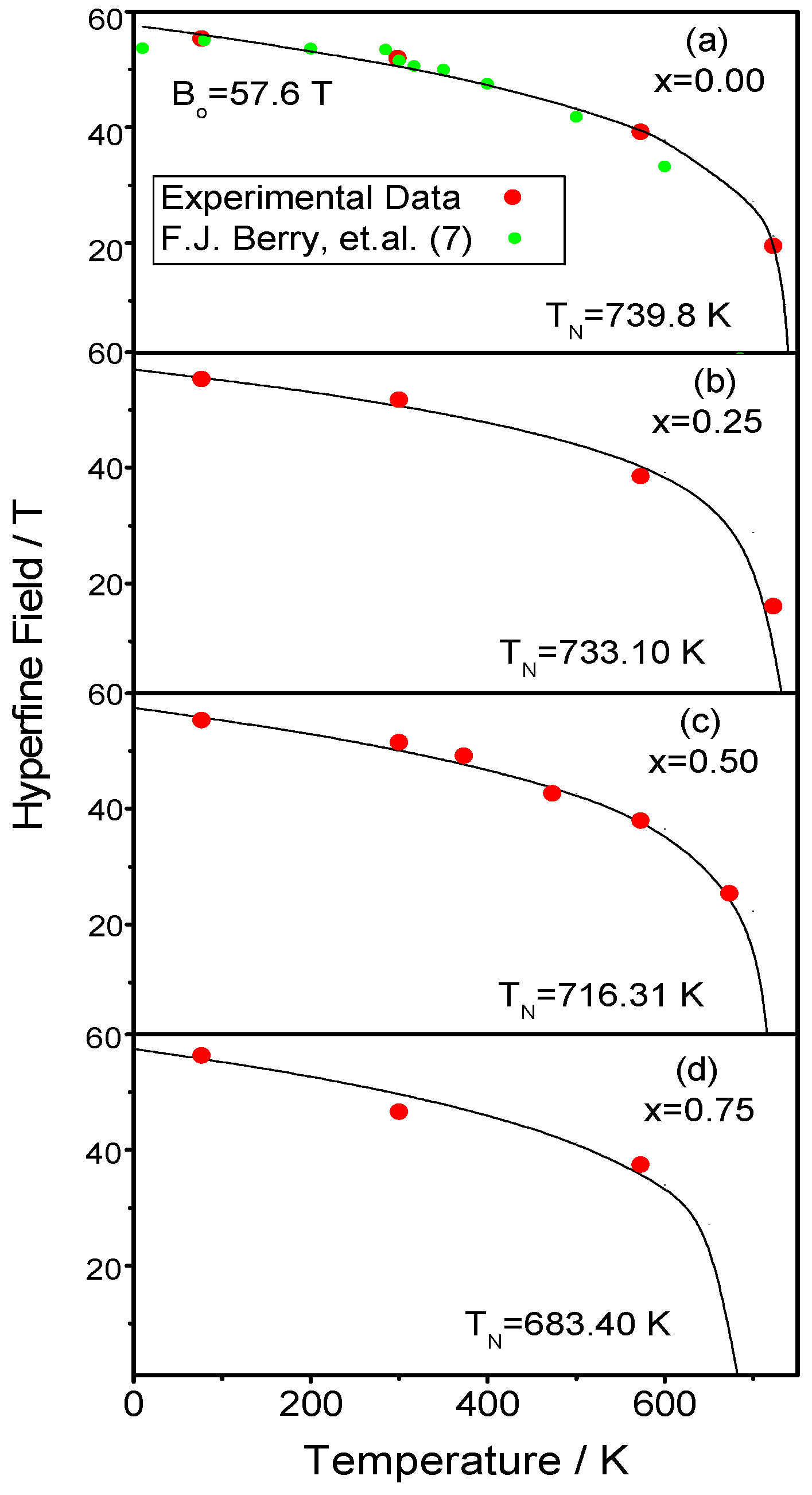
2.4. Mossbauer Analysis
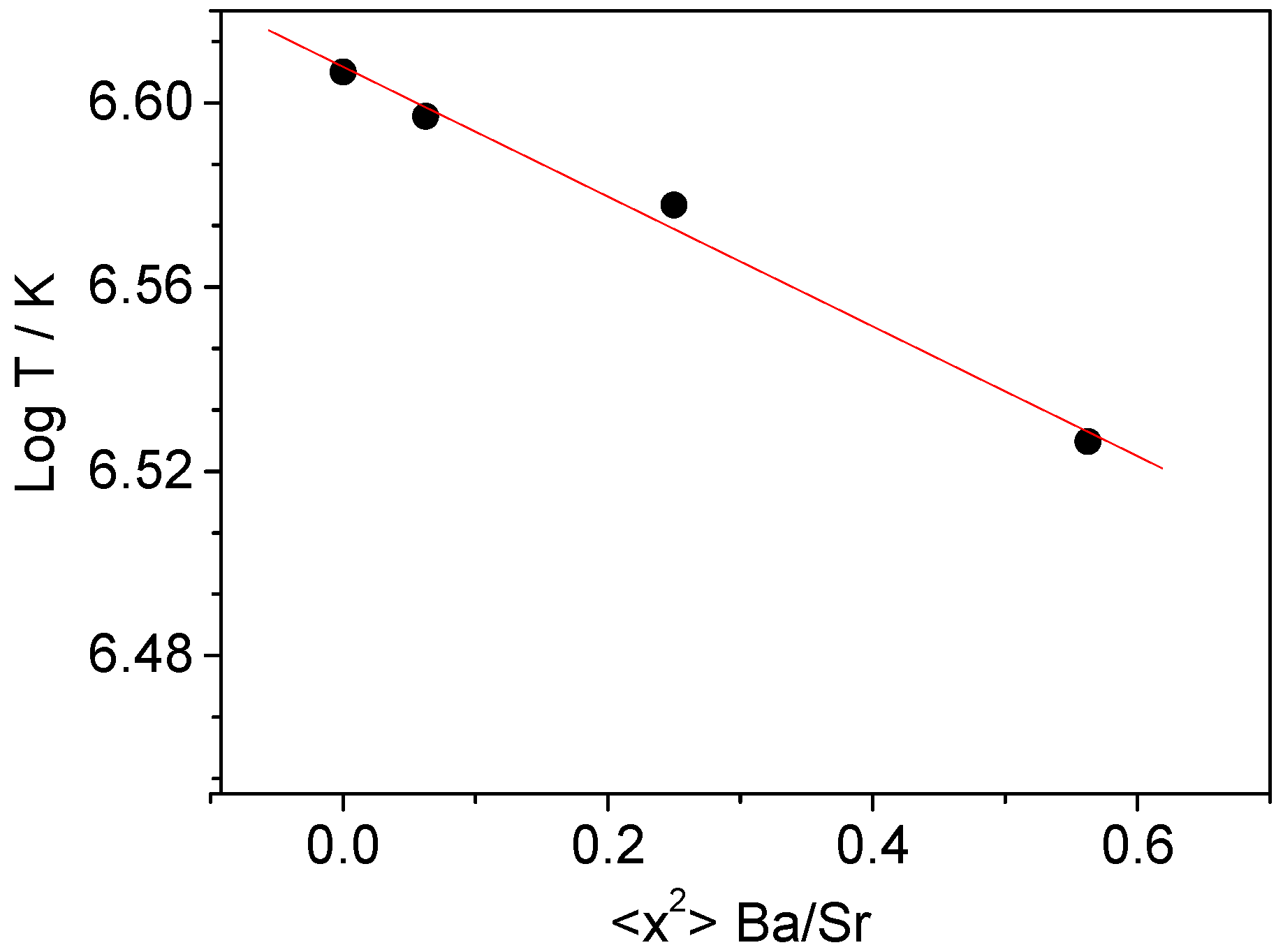
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. X-ray Diffraction and Neutron Diffraction Data
4.3. DC Magnetic Susceptibilities
4.4. Mössbauer Spectroscopy
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aller, L.H.; Appenzeller, I.; Baschek, B.; Butler, K.; De Loore, C.; Duerbeck, H.W.; El Eid, M.F.; Fink, H.H.; Herczeg, T.; Richtler, T.; et al. Landolt-Börnstein: Numerical Data and Functional Relationship in Science and Technology; Springer: Berlin, Germany, 1996. [Google Scholar]
- Aguado, F.; Rodriguez, F.; Hirai, S.; Walsh, J.N.; Lennie, A.; Redfern, A.T. High-pressure behaviour of KMF3 perovskites. High Press. Res. 2008, 28, 539–544. [Google Scholar] [CrossRef]
- Kugel, K.I.; Khomskii, D.I. The Jahn-Teller effect and magnetism: Transition metal compounds. Sov. Phys. Uspekhi 1982, 25, 231–256. [Google Scholar] [CrossRef]
- Al-Mamouri, M.; Edwards, P.P.; Greaves, C.; Slaski, M. Synthesis and superconducting properties of the strontium copper oxy-fluoride Sr2CuO2F2+δ. Nature 1994, 369, 382–384. [Google Scholar] [CrossRef]
- Blakely, C.K.; Davis, J.D.; Bruno, S.R.; Kraemer, S.K.; Zhu, M.; Ke, X.; Bi, W.; Alp, E.E.; Poltavets, V.V. Multistep synthesis of the SrFeO2F perovskite oxyfluoride via the SrFeO2 infinite-layer intermediate. J. Fluor. Chem. 2014, 159, 8–14. [Google Scholar] [CrossRef]
- Thompson, C.M.; Blakely, C.K.; Flacau, R.; Greedan, J.E.; Poltavets, V.V. Structural and magnetic behavior of the cubic oxyfluoride SrFeO2F studied by neutron diffraction. J. Solid State Chem. 2014, 219, 173–178. [Google Scholar] [CrossRef]
- Berry, F.J.; Ren, X.; Heap, R.; Slater, P.R.; Thomas, M.F. Fluorination of perovskite-related SrFeO3−δ. Solid State Commun. 2005, 134, 621–624. [Google Scholar] [CrossRef]
- Heap, R.; Slater, P.R.; Berry, F.J.; Helgason, O.; Wright, A.J. Synthesis and structural determination of the new oxide fluoride BaFeO2F. Solid State Commun. 2007, 141, 467–470. [Google Scholar] [CrossRef]
- Malek, A.; Edwards, P.P.; Greaves, C.; Slater, P.R.; Slaski, M. Synthesis and structure of the calcium copper oxyfluoride, Ca2CuO2F2+δ. J. Mater. Chem. 1995, 5, 913–916. [Google Scholar]
- Slater, P.R.; Gover, R.K.B. Synthesis and structure of the new oxide fluoride Sr2TiO3F2 from the low temperature fluorination of Sr2TiO4: An example of a staged fluorine substitution/insertion reaction. J. Mater. Chem. 2002, 12, 291–294. [Google Scholar] [CrossRef]
- Slater, P.R. Poly(vinylidene fluoride) as a reagent for the synthesis of K2NiF4-related inorganic oxide fluorides. J. Fluor. Chem. 2002, 117, 43–45. [Google Scholar] [CrossRef]
- Clemens, O.; Haberkorn, R.; Slater, P.R.; Beck, H.P. Synthesis and characterisation of the SrxBa1−xFeO3−y system and the fluorinated phases SrxBa1−xFeO2F. Solid State Sci. 2010, 12, 1455–1463. [Google Scholar] [CrossRef]
- Berry, F.J.; Moore, E.A.; Ren, X.; Helgason, Ö.; Thomas, M.F.; Shim, S. Iron-57 Mössbauer spectroscopic study of fluorinated strontium orthoferrite. Hyperfine Interact. 2008, 185, 111–114. [Google Scholar] [CrossRef]
- Helgason, Ö. Mössbauer spectroscopy of perovskite-related oxide fluorides of composition Ba0.5Sr0.5FeO2F at elevated temperatures. Hyperfine Interact. 2008, 184, 143–146. [Google Scholar] [CrossRef]
- Takeda, T.; Kanno, R.; Kawamoto, Y.; Takano, M.; Kawasaki, S.; Kamiyama, T.; Izumi, F. Metal-semiconductor transition, charge disproportionation, and low-temperature structure of Ca1−xSrxFeO3 synthesized under high-oxygen pressure. Solid State Sci. 2000, 2, 673–687. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radio and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Physica B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Clemens, O.; Berry, F.J.; Wright, A.J.; Knight, K.S.; Perez-Mato, J.M.; Igartua, J.M.; Slater, P.R. Reply to Structural and magnetic behavior of the cubic oxyfluoride SrFeO2F studied by neutron diffraction. J. Solid State Chem. 2015, 226, 326–331. [Google Scholar] [CrossRef]
- Berry, F.J.; Coomer, F.C.; Hancock, C.; Helgason, Ö.; Moore, E.A.; Slater, P.R.; Wright, A.J.; Thomas, M.F. Structure and magnetic properties of the cubic oxide fluoride BaFeO2F. J. Solid State Chem. 2011, 184, 1361–1366. [Google Scholar] [CrossRef]
- Berry, F.J.; Heap, R.; Helgason, Ö.; Moore, E.A.; Shim, S.; Slater, P.R.; Thomas, M.F. Magnetic order in perovskite-related SrFeO2F. J. Phys. Condens. Matter 2008, 20, 215207. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Ruskov, T.; García, C.; Asenov, S.; Spirov, I.; Mönch, I.; Graff, A.; Kozhuharova, R.; Kozhuharova, R.; Leonhardt, A.; Muhl, T.; et al. Mossbauer transmission and back scattered conversion electron study of Fe nanowires encapsulated in multiwalled carbon nanotubes. J. Appl. Phys. 2004, 96, 7514–7518. [Google Scholar] [CrossRef]
- Shenoy, G.K.; Friedt, J.M.; Maleta, H.; Ruby, S.L. Curve fitting and the transmission integral: Warnings and suggestions. In Mössbauer Effect Methodology; Springer: New York, NY, USA, 1974; Volume 9, pp. 277–305. [Google Scholar]
- Cranshaw, T.E. The deduction of the best values of the parameters from Mossbauer spectra. J. Phys. E 1974, 7, 122–124. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| x | 0 | 0.25 | 0.50 | 0.75 |
| a (Å) | 3.95500(7) | 3.98055(6) | 4.00610(5) | 4.03476(6) |
| V (Å3) | 61.864(2) | 63.071(2) | 64.293(1) | 65.683(2) |
| Sr/Ba 1b (½ ½ ½) | ||||
| B (Å2) | 0.80(4) | 0.81(3) | 0.87(3) | 0.84(3) |
| Fe 1a (0 0 0) | ||||
| B (Å2) | 1.62(4) | 1.89(3) | 2.29(3) | 2.68(3) |
| μB | 3.63(4) | 3.50(3) | 3.37(3) | 3.40(2) |
| O/F 3d (0 0 ½) | ||||
| B (Å2) | 2.36(4) * | 1.55(3) | 1.30(3) | 1.10(2) |
| Occupancy O/F | 1 | 1 | 0.98(1) | 0.96(1) |
| Main bond distances (Å) | ||||
| Sr-O/F (×12) | 2.79661(4) | 2.81467(3) | 2.83274(2) | 2.85301(3) |
| Fe-O/F (×6) | 1.97750(4) | 1.99028(3) | 2.00305(2) | 2.01738(3) |
| Reliability factors ** | ||||
| χ2 | 1.78 | 1.39 | 1.33 | 1.35 |
| Rp (%) | 3.46 | 2.71 | 2.87 | 2.12 |
| Rwp (%) | 4.34 | 3.41 | 3.63 | 2.69 |
| Rexp (%) | 3.25 | 2.89 | 3.15 | 2.31 |
| RI (%) | 4.91 | 2.60 | 2.01 | 1.53 |
| Rmag (%) | 17.14 | 8.51 | 7.12 | 6.09 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Ramos, C.A.; Retuerto, M.; Alonso, J.A. On the Sr1−xBaxFeO2F Oxyfluoride Perovskites: Structure and Magnetism from Neutron Diffraction and Mössbauer Spectroscopy. Materials 2016, 9, 970. https://doi.org/10.3390/ma9120970
García-Ramos CA, Retuerto M, Alonso JA. On the Sr1−xBaxFeO2F Oxyfluoride Perovskites: Structure and Magnetism from Neutron Diffraction and Mössbauer Spectroscopy. Materials. 2016; 9(12):970. https://doi.org/10.3390/ma9120970
Chicago/Turabian StyleGarcía-Ramos, Crisanto A., María Retuerto, and José Antonio Alonso. 2016. "On the Sr1−xBaxFeO2F Oxyfluoride Perovskites: Structure and Magnetism from Neutron Diffraction and Mössbauer Spectroscopy" Materials 9, no. 12: 970. https://doi.org/10.3390/ma9120970