Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review




Abstract
: Preceramic polymers, i.e., polymers that are converted into ceramics upon heat treatment, have been successfully used for almost 40 years to give advanced ceramics, especially belonging to the ternary SiCO and SiCN systems or to the quaternary SiBCN system. One of their main advantages is the possibility of combining the shaping and synthesis of ceramics: components can be shaped at the precursor stage by conventional plastic-forming techniques, such as spinning, blowing, injection molding, warm pressing and resin transfer molding, and then converted into ceramics by treatments typically above 800 °C. The extension of the approach to a wider range of ceramic compositions and applications, both structural and thermo-structural (refractory components, thermal barrier coatings) or functional (bioactive ceramics, luminescent materials), mainly relies on modifications of the polymers at the nano-scale, i.e., on the introduction of nano-sized fillers and/or chemical additives, leading to nano-structured ceramic components upon thermal conversion. Fillers and additives may react with the main ceramic residue of the polymer, leading to ceramics of significant engineering interest (such as silicates and SiAlONs), or cause the formation of secondary phases, significantly affecting the functionalities of the polymer-derived matrix.1. Introduction
Preceramic polymers, especially in the form of organo-silicon compounds (e.g., polymers based on a backbone of Si atoms containing also C, O, N, B and H atoms), have been widely recognized for the last 40 years as an extremely powerful tool for the production of advanced ceramics. Their key advantage over conventional (powder) synthesis procedures is represented by the possibility of adopting plastic-forming techniques (e.g., fiber spinning, foaming, warm pressing, extrusion, injection molding or resin transfer molding) to generate shaped components, later transformed into the desired ceramic parts (usually defined as polymer-derived ceramics or PDCs) by thermal treatment (pyrolysis) above ~800 °C, typically in a non-oxidative atmosphere (nitrogen or argon) [1–3].
The ceramics obtained from polymeric precursors usually feature a chemical composition not achievable by other techniques: as an example, silicones (polymers with a -Si-O- backbone) yield, upon firing in a non-oxidative atmosphere, an amorphous SiCO (silicon oxycarbide) residue, which can be considered as a silica glass modified by the presence of nano-sized domains based on SiC (Si atoms surrounded by four bridging carbon atoms instead of two oxygen atoms), together with Si atoms bonded to a varying number of C atoms and C clusters (“free carbon”) [4,5]. Such nano-domains transform into distinct phases upon treatment above 1200 °C. A similar microstructural evolution is found for another well investigated class of preceramic polymers, i.e., polysilazanes, yielding a SiCN (silicon carbonitride) ceramic containing nano-sized N- and C-rich areas at low pyrolysis temperature and Si3N4 and SiC nano-sized regions after heating at high temperature. Several efforts have been devoted to the control and understanding of the phase separation occurring in the ceramic residue with increasing pyrolysis temperature, with consequent (partial) crystallization, as well as to precisely describe the microstructure of the PDCs at various stages during pyrolysis [1,2,6].
A distinctive drawback of PDC technology is the poor control of shrinkage and structural integrity of the products of the polymer-to-ceramic transformation. The transformation implies the elimination of the organic moieties typical of a polymer (e.g., methyl or phenyl groups attached to the Si atoms), with consequent significant gas release (in the form of methane, benzene and hydrogen) and shrinkage (the density goes from ~0.8–1.2 g/cm3, typical values for a polymer, to ~2.2 g/cm3, a standard value for amorphous Si-based ceramics) [1,7]. Gas release not only leads to the formation of unwanted/uncontrolled porosity, but also causes a substantial cracking of monolithic pieces [7,8]. Thin-walled components (e.g., fibers, microtubes or highly porous open-celled foams) represent a notable exception, due to the intrinsic short diffusion paths for the generated gases, leading to very limited internal pressure build-up [9].
Hot-pressing of pre-pyrolyzed material has been proposed as a solution for crack-free polymer-derived ceramics since the early applications of the technology [4], and recently, spark plasma sintering (SPS) has been successfully used to produce fully dense, nano-structured components [10–12]. Although successful, a (more or less sophisticated) hot pressing treatment needs to be employed on PDC powders at least already partially pyrolyzed, as the generation of decomposition gases during pressing would create problems for the equipment.
The pioneering work of Greil [7–9,13–16] provided a fundamental solution for obtaining crack-free, almost dense (porosity typically below 15 vol%), strong monoliths with a single ceramization step, based on a more or less pronounced modification of the chemistry of PDCs. He demonstrated, in particular, the impact of two types of solid additives, or fillers:
Inert, or passive, fillers, which are ceramic powders that do not react with the ceramic residue from the preceramic polymer, the decomposition gases or the heating atmosphere [17]. Such fillers simply dilute the preceramic polymer, therefore decreasing the amount of gas generated and the associated volume shrinkage, reducing the likelihood of forming macroscopic cracks during processing. The final ceramic has a modified chemistry in the sense that a polymer-derived matrix is accompanied by secondary phases;
Active fillers, i.e., metallic or intermetallic powders that react, during pyrolysis, with the decomposition gases generated during heating, or the heating atmosphere or (less frequently) with the ceramic residue from the preceramic polymer [7,18]. The fillers are normally quite coarse, in the (several) micron range, for handling and safety reasons, as very small metallic particles may exhibit pyrophoricity. Typical products of the chemical reactions are carbides, nitrides or silicide phases, with a significant impact on the overall shrinkage. In fact, the metal-to-ceramic transformation generally occurs with a large volume expansion, due to a large density decrease, which compensates for the shrinkage associated with the ceramic conversion of polymers. Solid particles and the in situ reaction with the filler reduce the amount of gas generated and the local gas pressure in the part, respectively, therefore enabling the fabrication of near-net shape, bulk, uncracked ceramic components [7].
Even when large amounts (several tens of a percent) of fillers are introduced, the viscous flow of preceramic polymers is always sufficiently high to enable the use of the abovementioned plastic forming technologies. Moreover, the use of these fillers not only allows one to produce bulk polymer-derived ceramics, but they can also provide the possibility of manufacturing ceramics with functional properties. While metals (tungsten, niobium, molybdenum, iron alloy particles) that form carbides lead to components with high hardness and wear resistance [19–22], silicides may provide magnetic and electrical functionalities (for instance, silicone resins [23] may be easily converted into magnetic ceramics or into electrically conductive ceramics, by the addition of iron silicide or MoSi2 powders, respectively) [24,25]. In some cases, the addition of metal oxide particles, reduced by the carbon in the preceramic polymer during pyrolysis in an inert atmosphere, can also lead to the formation of metal clusters that afford additional functional properties, but with a limited impact on the composition of the main ceramic residue [26].
Modern nanotechnology offers a wide range of nano-sized particles, which allows for a substantial updating of the concept of preceramic polymers and fillers. In particular, oxide particles, if nano-sized, are far more interesting than micro-sized ones. Nano-sized oxide particles, contrary to micro-sized ones, have a distinctive character in that they are capable of reacting directly with the ceramic residue deriving from the heat treatment of a preceramic polymer, either in inert atmosphere [27] or, more interestingly, in air, i.e., upon transformation of Si-based polymers into pure silica [28–36]. Ceramics containing carbides and nitrides in an oxycarbide matrix are replaced, as products, by advanced oxide ceramics, developed in conditions of excellent phase purity. Mullite and mullite-based ceramics, from silicone resins containing γ-Al2O3 nanoparticles [28,29,31,33,34], can be seen as the starting point of this advancement in the original concept of active fillers. Micro-sized particles are far less reactive: for instance, corundum (or Al alloy) particles were also used in order to produce mullite after firing in air, but phase pure components could be obtained only after heating at very high temperatures (>1600 °C) [37,38]. The quasi-molecular mixing of the amorphous silica deriving from the thermal decomposition of the preceramic polymer in air with the oxide nano-sized fillers, on the one hand, is the reason for the very favorable reaction kinetics measured [31]. The formation of a multitude of nuclei, on the other hand, promotes a fine-grained microstructure [39], while secondary phases, if present, are also distributed at the nano-scale.
An alternative way to polymer-derived nano-structured ceramics, mainly for functional applications, is provided by the direct chemical modification of preceramic polymers using alkoxides or other chemical precursors (liquid or gaseous). This was proposed long ago for polycarbosilane, which has been modified with Ti [40–47], Al [48,49], Zr [50–52], Fe [53], Ta [54] or B [55,56], and has also been exploited for silazanes, for the realization of silicon carbonitrides containing boron (of particular interest for their exceptional high temperature properties) [57–66], early transition metals (e.g., Ti, Zr, Hf) [67–70], other transition metals (e.g., Ni, Fe, Co, Pd), aluminum [71] or others [72]. Silicone resins have also been modified using chemical precursors for metals [73–78], although the work has been devoted mainly to the fabrication of SiCO-based nanocomposites, heat treating the samples in inert atmosphere and not in air. Boron- or aluminum-modified silicones have also been produced [79–83]. It should be noted that silicon oxycarbides modified with main group or transition metals (SiMOC) have also been synthesized via pyrolysis of sol-gel, metal-modified precursors [84–87].
The metal precursors have been added for various purposes, ranging from the crosslinking of the preceramic polymer, to the control of the crystallization of the ceramic residue, from the improvement of the high temperature stability to the affording of functional properties or to the formation of catalytic particles. From the microstructural point of view, these additions have been incorporated into the Si-based ceramic residue, forming new crystalline phases, or have modified the amorphous Si-based network of the ceramic residue or have phase-separated creating new carbide, nitride, silicide, oxide or metallic crystalline phases. In some cases, the additions have allowed the preceramic polymers to retain their plastic shaping capability, while in other cases, the increase of the degree of crosslinking prevents the viscous flow of the polymer.
Finally, we must mention that also nano-sized fillers can be inert or passive. In fact, carbon nanotubes [88–90], carbon nanofibers [91] or graphene [92] (also via graphene oxide [93]) have been recently added to preceramic polymers and acted as non-reactive fillers.
The purpose of the present paper is to give an up-to-date overview of all strategies for the modification of preceramic polymers at the nano-scale, focusing on the latest developments. We will first address the chemical modifications of polymers, then discuss passive nano-sized fillers and, finally, summarize the main findings relative to the introduction of (mainly oxide) nano-sized active fillers, which are the object of specific studies conducted by our research group. We will present some case studies, with the aim of highlighting the flexibility of the approach for the manufacturing of a wide range of advanced ceramics.
2. Chemical Modification of the Polymer Structure
The phase separation in the ceramic residue is a fundamental issue of polymer-derived ceramics [1–3,6,77]. Amorphous SiCO and SiCN residues from polysiloxanes and polysilazanes typically start decomposing by carbothermal reactions, causing the development of SiC and Si3N4 crystals, at temperatures above ~1250 °C. The chemical modification of the polymers by the sol-gel technique, with metal alkoxides [3], represents an excellent opportunity to extend the temperature stability of the amorphous residues. As an example, Al-containing alkoxide compounds, such as alumatrane (C6H12NO3Al), added to commercial silicones, led to a SiAlOC residue, stable up to 1300 °C [79]. The potential of the additives, however, does not simply rely on avoiding phase separation, but also on allowing controlled crystallization with the development of new phases. The mixing of the polymer and additive occurs at a molecular level, so that the modified polymers act as a single-phase precursors for ceramic nano-composites. Above 1300 °C, the SiAlOC residue yields nano-crystalline mullite (3Al2O3·2SiO2), in addition to SiC, with positive effects on the structural integrity of the polymer-derived ceramic, which could be exploited for the fabrication of ceramic micro electro-mechanical systems (MEMS) [79].
Figure 1 illustrates the typical strategy for the preparation of polymer-derived ceramic nano-composites (PDC-NCs) [73]. We can note that the process can be applied to a vast range of metal (M) alkoxides, attached to polymer chains by means of condensation reactions, involving the -OH side groups. Ionescu et al. [74] recently provided thermo-dynamic models useful for predicting the conversion of SiMOC residues into nano-composites; in fact, a SiCO matrix may embed metal oxides (MOx), silicides (MSix), carbides (MCx) or metals. The stability of MOx against carbothermal reduction is essential for understanding the further evolution into silicates, on the one hand, or silicides and carbides, on the other. Lu and Mn, as an example, lead to stable oxides, in turn favoring the formation of silicate phases.
The most significant investigations that can be found in the literature concern, besides Al-alkoxides, Zr- and Hf-containing compounds. Figure 1 refers to the separation of a hafnia-rich phase, in the form of nano-sized droplets, from SiHfOC heated at 1100 °C; a further heating, at 1300 °C, leads to nano-sized tetragonal hafnia crystals (a diameter of 5 nm) [73,74]. Zr- and Hf-containing compounds are interesting, above all, for the excellent high-temperature stability of the related nano-composites [76,78,94]; zirconia and hafnia, being particularly resistant against carbothermal reduction, may form silicates, such as zircon (ZrSiO4) and hafnon (HfSiO4), stable up to 1600 °C. The same choice of additives was found to be suitable also for use at moderate temperatures (up to 250 °C), but in hydrothermal conditions. SiZrOC and SiHfOC are more resistant in this environment than pure SiCO (in turn, more resistant than SiC, being comparable to Si3N4), owing to a synergistic effect. While zirconia and hafnia have a relatively low, but appreciable, solubility in water under the testing conditions, the SiCO matrix protects the dispersed phases from the water-induced tetragonal-to-monoclinic transformation [95].
The dispersed phases, in PDC-NCs, may be interesting also for advanced applications at room temperature. As an example, Hojamberdiev et al. [97], exploiting the poor stability of iron oxide towards carbothermal reduction, successfully formed Fe-silicides, i.e., Fe3Si and Fe5Si3, from a commercial silicone modified with an iron alkoxide (Fe(AcAc)3) after heat treatments in Ar below or above 1300 °C. Fe-silicides were developed according to a multi-step reaction, involving: (i) separation of FeOx in a SiCO matrix; (ii) reduction into Fe3C in SiCO; (iii) formation of a Fe-Si-C alloy; and (iv) separation of Fe3Si and graphitic carbon, or Fe5Si3 and β-SiC. The specific iron-containing phases led to monoliths behaving as soft magnets, which could be applied in magnetic data storage devices.
Hf-containing compounds can also be used for modifying silazanes [69,70]. In this case, the molecular structure was found to significantly affect the high-temperature behavior with respect to decomposition and crystallization phenomena. In particular, the high homogeneity achievable in polysilazane-derived ceramics favors high temperature stability, whereas hafnia-rich zones, in cyclotrisilazane-derived ceramics, promotes a pronounced decomposition.
3. Nano-Sized Passive Fillers
A few papers report the incorporation of carbon nanotubes, carbon fibers and graphene as nano-sized passive fillers in ceramic matrix, in order to induce or improve mechanical, thermal, electrical or rheological properties. While conventional powder-based ceramic processing has been shown to disrupt the integrity of the fillers, as well as to be not effective in their homogeneous dispersion, the polymer precursor-based approach offers several advantages for the purpose of producing ceramic matrix composites [88,90]. First, the availability of common polymer processing techniques allows one to obtain a homogeneous dispersion of a nano-sized filler in a preceramic polymer and helps to avoid the formation of agglomerates; then, the typical low pyrolysis temperatures employed can minimize the damage of the nano-structures [98]; moreover, complex shapes can be realized in small scales for microelectromechanical devices, such as MEMS [99].
Considering the brittle nature of ceramics, one of the most interesting applications of such fillers is their effect on the fracture toughness. From this point of view, Katsuda et al. [88] reinforced precursor-derived Si-C-N ceramics with multi-walled carbon nanotubes (MWCNTs). They demonstrated that an amount of 1–2 wt% MWCNTs produces a remarkable increase in the fracture toughness of the ceramic matrix, due to pulling-out and bridging phenomena. However, the authors highlighted that the success of this reinforcement technique is essentially linked to the nature of the nanotubes: amorphous MWCNTs were found to degrade upon thermolysis, and so, they were not effective in the toughening mechanism. Moreover, in their study, they revealed that the addition of MWCNTs up to 2 wt% does not influence the basic material properties of the matrix, such as the Young modulus, the Poisson’s ratio, the coefficient of thermal expansion and the bulk density [88].
A further promising peculiarity of nanotubes is that their use can be suggested as a new way to tailor the rheology and the processability of spun preceramic polymer fibers, as described in the study by Kokott et al. [89], concerning the fabrication of SiCN-fibers by spinning the ABSE polycarbosilazane. Originally, the mentioned polymer needs thermal and catalytic treatments to increase its molecular weight in order to become suitable for the melt-spinning process. According to the authors, this can be avoided by the addition of MWCNTs: their study showed that only 1 wt% of MWCNTs can significantly improve the preceramic polymer spinnability. The key point for achieving this is producing a very good dispersion of the nanotubes: agglomerates cause a lack of stability in the spinning process and, besides, irregular fiber diameters and rough surfaces. Moreover, Kokott et al. pointed out that also the tensile strength of the green fibers is increased with higher fractions of MWCNTs, thus facilitating their handling [89]. An example of fiber embedding with well-dispersed MWCNTs is shown in Figure 2.
It should be noted that the achievement of a homogenous dispersion inside the ceramic matrix is the main issue concerning the use of carbon nanotubes as reinforcing fillers for polymer precursors. Such a dispersion is particularly difficult to obtain for SWCNTs, because of the strong cohesive force between individual tubes [90]. Li et al. investigated a novel chemical modification method involving the radical addition of an organosilicon compound to the sidewall of SWCNTs, aimed at improving the dispersion of the carbon nanotubes in a Si-C-N matrix derived from preceramic polymers. Simultaneously, the chemical modification could also be designed to provide a suitable interfacial bonding to ensure efficient load transfer from the matrix to the nanotubes [90].
Carbon-based reinforcement can also be embedded in a preceramic matrix in the shape of fibers, to enhance electrical and mechanical properties. Shibuya et al. [91] investigated the effect of vapor-grown carbon fibers (VGCFs) on the electrical resistivity before and after the pyrolysis of a preceramic methyl silicone resin (MSR), used as a precursor to a silicon oxycarbide ceramic (SiCO). Being that the ceramics obtained by pyrolysis were subjected to a large weight loss and shrinkage, resulting in the fracture of the bulk component during the heat treatment, sacrificial poly(methyl methacrylate) (PMMA) microbeads were incorporated to obtain microcellular VGCFs/ceramic composites, a well-established technique to eliminate cracking problems and produce highly porous components [101–103]. The bending stress and the elastic modulus of the VGCFs-reinforced ceramic were maximized when adding the PMMA microbeads in an amount of 50 wt% with respect to MSR. An optimum combination of low density, high electrical conductivity and good mechanical properties was achieved operating with 10 wt% VGCFs and 50 wt% PMMA microbeads with respect to MSR [91].
Bulk ceramics containing nano-sized reinforcements can also be fabricated from preceramic polymers when employing suitable techniques for sintering and consolidating the material, which are the crucial points for achieving dense products. For instance, graphene is a very attractive nano-sized filler, considering its unique mechanical properties that make it one of the strongest materials available, as well as its super-electrical and very good thermal properties [104–107]. This novel approach was recently followed by Rahman et al. [92], who fabricated graphene-reinforced silicon carbide nano-composites by incorporating graphene nano-platelets in polycarbosilane, followed by pyrolysis, grinding into fine powders and sintering by SPS. The SPS technique was necessary to consolidate the final product, thus obtaining a nano-crystalline bulk component. The mechanical properties of the reinforced samples were affected by graphene inclusions, and an increase in strength and microhardness was observed. The ceramic microstructure was also affected by the nano-platelets, with graphene inclusions found to restrict grain growth for samples processed at lower temperatures. Moreover, SiC with and without graphene inclusions showed a trend of increasing density with increasing processing temperatures, while porosity was found to decrease with increasing processing temperatures [92].
4. Nano-Sized Active Fillers
Table 1 summarizes the many types of silicate and oxynitride ceramics that have been so far produced by the authors, starting from preceramic polymers containing nano-sized active fillers. Preceramic polymers are typically dissolved in a suitable solvent (e.g., isopropanol or acetone), and filler powders are added, obtaining a diluted suspension (solid content 20 to 30 wt%). The mixtures are stirred magnetically for 15–20 min and then ultrasonicated for another 15–20 min in an ultrasonic bath; finally, the suspensions are dried overnight at 60 °C to produce composite (preceramic polymer + filler) powders, unless the fabrication procedure of the desired component requires the use of a fluid paste [108]. The composite powders (biphasic systems, unlike those determined by chemical modification) can then be subjected to shaping using several methods, including cold uniaxial pressing (pressure of 20 to 40 MPa), warm pressing (180 °C, 20 MPa), fused deposition, machining of warm/cold pressed blocks and coating (after dispersing the powders in water or when still in the dissolved state) [109]. The obtained shaped component is later subjected to thermal treatment to obtain the ceramic component. If the desired silicate is used in the form of powders, like in the case of inorganic phosphors for light-emitting diode (LED) devices, the thermal treatment can be applied directly to the composite powders [109]. It should be stressed that the production of silicate ceramics generally implies handling, drying and thermal treatment in air, i.e., they can be prepared very easily, by conversion of the preceramic polymers into reactive amorphous silica. On the contrary, oxynitride ceramics required more stringent processing conditions. If obtained from silicone polymers, only the thermal treatment must be conducted in nitrogen in order to achieve a silicon oxycarbide (SiCO) residue, instead of silica; if obtained from polysilazanes, yielding a silicone carbonitride (SiCN) residue, drying may also be quite critical. Precautions are necessary in order to avoid the reaction of polysilazanes with H2O (moisture from the atmosphere or impurity in polar solvents), which may reduce the ceramic yield (by decomposition of the polymers and the consequent release of ammonia), modify the composition of the precursor and promote oxygen contaminations in the ceramic residue. Dispersions therefore need to be produced using pure apolar solvents (n-hexane, toluene) under N2 or Ar atmosphere (by using the Schlenk technique) and, subsequently, directly dried and cured into a tube furnace (typically at 350 °C for 1 h) under flowing N2. The specific processing conditions adopted for each case can be found in the published literature [110].
As previously mentioned, a general trend is that oxide fillers react with the decomposition products of the preceramic polymers, producing the desired new phases; for silicates, when developed from silicones treated in air, the reaction is particularly simple, as follows:
where m/n expresses the molar ratio between a metal oxide (MxOy) and silica in the desired silicate. The molar ratio is obviously associated with a weight ratio (WR) between oxide and silica. Given the ceramic yield (CY) in air of a silicone, the desired silicate will be obtained operating with a weight ratio between oxide and polymer equal to WR·CY. If the metal oxide is provided starting from a carbonate or other compounds, like sulfates, nitrates, etc., the weight ratio between filler and polymer will be inferred from the WR·CY product, according to the yield in the metal oxide of the adopted compound. As an example, for wollastonite (CaO·SiO2), the CaO/SiO2 molar ratio is one, associated with WR = 0.93; considering the commercial silicone polymer MK (Wacker Chemie AG, see Table 1) as the silica source (a polymer with a particularly high ceramic yield; ~84% of the starting weight of the polymer is converted into silica [39], so that CY = 0.84), the CaO/silicone ratio will be WR·CY = 0.93 × 0.84 = 0.78; considering the yield in CaO of CaCO3 (0.56), we will obtain the final ratio of CaCO3/silicone = 0.78/0.56 = 1.40.
The small dimension of fillers allows for very favorable reaction kinetics, with the formation in most cases of fine-grained phase pure ceramics at low temperature [39], even starting from bi-phasic systems. The phase purity is particularly significant, if we consider that silicates, due to their characteristic (partially covalent) chemical bonding, generally feature a poor ionic interdiffusion [116] and no liquid phase (which could accelerate the diffusion) forms during the process of the conversion of preceramic polymers and the reaction with fillers. The limited dimensions of crystals of the desired phase favors the incorporation of secondary phases; in particular, ZrO2 nano-particles form zirconia agglomerates that do not exceed the critical size for tetragonal-to-monoclinic transformation upon cooling, thus posing the conditions for an effective transformation toughening (with ceramics composites achieving a fracture toughness, KC, of 6.5 MPa·m0.5) [34].
While phase purity and limited grain size are easy to achieved, densification remains an important issue. The poor interdiffusion, if positive for limiting the grain growth, has a negative impact on density. Dense zones are generally surrounded by a number of submicron pores, which could be ascribed to the gas evolution upon ceramic transformation, as well as incomplete sintering. The porosity in pure mullite from MK silicone and nano-sized γ-Al2O3, as shown in Figure 3, exceeds 20%. The introduction of ZrO2, in order to prepare zirconia-toughened mullite, is advantageous mainly for the dilution of the transforming mass; the higher is the content secondary oxide, acting as an inert filler, the lower is the amount of polymer, with lower gas evolution. For composites with 30 vol% ZrO2, the densification, however, is still poor.
An important improvement, especially for pure mullite, comes from the introduction of further fillers. As an example, TiO2 nano-particles promote the densification by decreasing the viscosity (above 1200 °C) of the amorphous silica provided by the polymer; in other words, silica is modified by the incorporation of titania, and transient viscous sintering (as reported by other authors [117,118]) may occur before the nucleation of the mullite phase. Figure 3 clearly shows that the relative density of pure mullite goes from ~77% to 85%; the effect is less and less important, with increasing ZrO2 addition, consistent with the reduction of silicone and, consequently, of silicone-derived silica. Improvements in the densification, associated with the presence of TiO2 nano-particles, were observed also for other systems, such as zircon (ZrSiO4) [111] and, more recently, forsterite (Mg2SiO4). The mixing of titania with silica, in forsterite, is testified by the development of a solid solution, e.g., Mg2Si0.9Ti0.1O4, at only 1100 °C [112].
The limited interdiffusion is a constraint also for the morphology of crystals. All polymer-derived mullite-based materials developed so far possess, in fact, equiaxed grains. A remarkable exception is represented by recently prepared mullite ceramics, clearly exhibiting anisotropic crystal growth, as illustrated in Figure 4. Such morphology is due to the introduction of small amounts of borax (hydrated sodium borate, 3 wt% referring to the weight of mullite) in silicone/γ-Al2O3 mixtures. The role of B2O3 in lowering the mullitization temperatures and in promoting the anisotropic growth of mullite crystals has been widely discussed in the literature [119]; the use of a borate, in the present case, was aimed at providing a low viscosity intergranular phase (in turn, stimulating anisotropic growth [120,121]), that could be dissolved easily by acid leaching (it should be noted that B3+ ions may be incorporated in mullite in relatively high amounts, while Na+ can be accommodated in the crystal structure only in limited quantities [122,123]). Figure 4 provides an example of mullite ceramic (fired at 1300 °C, for 1 h) after acid leaching; although quite preliminary, the image demonstrates the potential for the fabrication of highly porous bodies, composed of interlocking mullite fibers. Such cellular ceramics could provide an alternative to acicular mullite ceramics prepared by much more complicated thermo-chemical processing (e.g., controlled decomposition of fluoro-topaz [124]).
The density of polymer-derived mullite may be improved also by changing the silica source. In particular, with 50% of the silica provided by MK and 50% by another polymer, H62C (a liquid polymer, with a silica yield of 58% [39]), it was possible to achieve a relative density of 97% for pure mullite, as illustrated in Figure 3. In the authors’ opinion, this could be justified by the different molecular structure of the polymers, leading to amorphous silica with a different degree of network connectivity and a number of defects; in other words, a more defective silica network would lead to Si-O network fragments that can be more easily accommodated in the mullite structure. This hypothesis has not been yet verified by dedicated experiments (such as detailed Raman spectroscopy) and will constitute the focus of future work.
Dense mullite-based ceramics, e.g., with relative density from 93% to 97%, can be produced also by another refined approach, involving changes in the filler and in the firing atmosphere. Riedel et al. [27] used nano-sized γ-Al2O3 powders functionalized at the surface by octylsilane groups; mixed with MK polymer, such fillers proved to be more reactive, leading to the formation of a larger weight fraction of mullite crystals at lower processing temperatures (1300 °C) as compared to un-functionalized nano-γ-Al2O3 filler. Such improved reactivity is ascribed to the enhanced homogeneity of the distribution of alumina nano-particles in the starting polysiloxane system. Since the firing treatments were performed in nitrogen, MK converted into a silicon oxycarbide (SiCO) ceramic, rather than into silica; the interaction with functionalized nano-γ-Al2O3 determined the formation of SiC crystals, in the range of 1–8 nm, embedded in a mullite matrix, with crystals in the range of 60–160 nm. The obtained crack-free monoliths may be seen as a refinement of analogous SiC/mullite nano-composites from polymethylsiloxane gels filled with α-Al2O3 (fired at higher temperatures) [125] and serve as prototypes for complex shaped high temperature- and corrosion-resistant ceramic devices. Similar results were obtained by the same research group [126], by using MK coupled with aluminum nano-sized particle filler; the different reaction paths (involving the oxidation of Al nano-particles) led to ternary composites, with nano-sized α-Al2O3 as an additional phase together with SiC and mullite.
In some cases (e.g., in zircon, forsterite and cordierite ceramics [111–113]), the partial replacement of MK with H62C proved to be advantageous for avoiding extensive cracking of samples. Several factors may justify the increase of compactness obtained when substituting part of MK by H62C; besides a different molecular structure of the polymeric precursors (in turn, providing a different ability to relax structural rearrangements and eliminate gases during the pyrolysis step, without the local pressure accumulation phenomena), the substitution could be advantageous for:
the reduction of the gas release during polymer cross-linking, due to different cross-linking reactions occurring in the two polymers;
the application of a crosslinking step (30 min at 250 °C) before powder compaction, which involves a certain degree of shrinkage, which is consequently absent in the subsequent pyrolysis.
Binary systems, comprising silica and one metal oxide, may be complicated by the formation of multiple silicate phases, each with a specific molar balance between silica and the metal oxide. A fundamental example is given by silica coupled with CaO, leading to many silicates with a different CaO/SiO2 molar ratio, such as CaO·SiO2 (=CaSiO3), 3CaO·2SiO2, 2CaO·SiO2 and 3CaO·SiO2, in their polymorphic variants, useful mostly for applications in the field of biomaterials [32,35]. Local concentrations of CaO, released from a filler, such as CaCO3, may lead to the development of a silicate with a higher CaO/SiO2 ratio than the desired one; as an example, micro-sized CaCO3, in a filler/silicone formulation theoretically yielding wollastonite (CaO·SiO2), was found to favor the formation of di-calcium silicate (2CaO·SiO2), whereas nano-sized CaCO3, in the same mixing conditions, led to almost pure wollastonite [35]. The different molecular structure of the polymeric precursors, in the case of calcium silicates, was found to affect also the polymorphism: the use of H62C instead of MK led to traces of additional phases (including α-phase or “pseudowollastonite”), in addition to the β-phase, normally obtained at the adopted processing temperature (900–1100 °C [32,35]). Ring-structured silicate variants (such as the α-phase) [127], instead of chain-structured ones (such as the β-phase), might be produced, because of the availability of short Si-O fragments, enhanced by the use of a polymer with a lower molecular weight, such as H62C.
Other complex binary systems correspond to the interaction of silicones with MgO and Y2O3. Enstatite contaminations (MgO·SiO2 = MgSiO3) can be produced in a formulation theoretically yielding forsterite (2MgO·SiO2 = Mg2SiO4), to be used as a biomaterial [128] and a high-performance dielectric [129]. Silicones filled with nano-sized MgO are highly reactive, since forsterite can be obtained already at 800 °C, but the characteristic poor ionic interdiffusion in silicates impedes the complete dissolution of MgO, with consequent development of enstatite in MgO-poor zones. Nano-sized titania, forming a solid solution (Mg2Si0.9Ti0.1O4), besides providing an improvement in the densification, as previously mentioned, is fundamental in removing both un-reacted MgO and enstatite contaminations [112].
Yttria and silica can combine into two silicates, i.e., they form mono- (Y2O3·SiO2 = Y2SiO5, or Y-MS) and di-silicates (Y2O3·2SiO2 = Y2Si2O7, or Y-DS (yttrium di-silicate)) [109]. Y-MS is practically monophasic, being subjected only to a dislocative (i.e., martensitic) transformation between the X1 phase (low temperature phase) and the X2 phase (high temperature phase) [130]. Y-DS, on the other hand, features many polymorphs (y, α, β, γ, δ, z) [131,132]; all the forms are stable in a certain temperature range, but high temperature polymorphs can be retained at room temperature, due to extremely slow phase transformations [130]. A particular polymorph may be associated with a specific processing procedure [132]: for instance, whereas sol-gel processing favors the formation of the α-phase [133,134], hydrothermal synthesis is known to promote the development of the y-phase [135]. Using MK silicone containing Y2O3 nano-powders [109], both silicates can be obtained at particularly low temperatures (1000–1350 °C), in analogy with the results of the more complicated sol-gel processing; Y-DS is obtained in its most stable variant (γ-phase). Contaminations of Y-MS, in Y-DS, are quite difficult to remove, but they do not represent an issue, especially for some applications; in fact, Y-DS is applied (due to the similarity in the coefficient of thermal expansion) in anti-oxidation coatings on SiC ceramics (see below), and Y-MS may combine with SiO2, from the partial oxidation of SiC, to yield additional Y-DS [136].
Ternary systems pose difficulties, besides the possible formation of many silicate phases, according to multiple combinations among silica and oxides (e.g., the CaO-MgO-SiO2 system may lead to CaMgSi2O6, known as diopside, or Ca2MgSi2O7, known as akermanite, and so on), also for the possible formation of silica-free compounds. As an example, cordierite (2MgO·2Al2O3·5SiO2, or Mg2Al4Si5O18) and gehlenite (2CaO·Al2O3·SiO2, or Ca2Al2SiO7) may be accompanied by Mg and Ca aluminates, respectively. The formation of silica-free compounds impedes the complete incorporation and reaction of oxides with the polymer-derived silica, with the risk of the formation of cristobalite. The contaminations may be removed by adjusting the firing temperatures (MgAl2O4 disappears in polymer-derived cordierite fired at 1350 °C [113]) or by enhancing the ionic interdiffusion, by the formation of solid solutions (i.e., more “open” crystal structures). In particular, the partial replacement of Ca2+ ions with Eu3+, aimed at developing luminescent materials, with corresponding tuning of the Al/Si ratio (the formation of Ca2−2xEu2xAl(Al1+2xSi1−2xO7) solid solutions), is effective in suppressing Ca-aluminate phases [114].
(Silicon) oxynitrides are intrinsically challenging to produce from preceramic polymers and fillers, due to the very complex reaction paths possible, involving carbothermal reduction and nitridation. C in SiCO or SiCN ceramic residues, deriving from polysiloxanes and polysilazanes, respectively, when treated in nitrogen, must be removed as CO. These interactions may be summarized by the following general reaction schemes:
where MxOy stands for a metal oxide, inserted as a nano-sized powder. Ideally, the adoption of SiCO or SiCN would be conditioned only by the x/y ratio of the nano-sized filler and by the stoichiometry of the desired oxynitride. However, there are at least four fundamental issues to consider:
The balance of Si, O, N and C atoms in the ceramic residues of preceramic polymers is not strictly defined; it is highly sensitive to the processing conditions, especially for polysilazanes (for which significant oxygen contaminations in the SiCN residue should be taken into account);
Silicate formation may precede the formation of oxynitride phases; in particular, silicones and nano-sized alumina lead to mullite (3Al2O3·2SiO2), subsequently subjected to carbothermal reduction and nitridation, with the separation of corundum in addition to the desired SiAlON phase (3Al2O3·2SiO2 + 6C + 2N2 → Si2Al4O4N4 + 6CO + Al2O3) [30];
Reducing conditions may lead to the formation of other by-products, e.g., SiO gas; the escape of SiO has, obviously, an impact on the stoichiometry of the obtained oxynitride;
The ceramic residue does not react as a homogeneous mixture of Si, O, N and C atoms; e.g., a SiC separate phase may first form, with an impact on the stoichiometry of the obtained oxynitride and on secondary reactions (e.g., 2SiO2 + SiC → 3SiO + CO) [137,138].
The development of many gaseous species impedes the obtainment of monolithic samples. This finding, however, does not compromise the potential of the approach for the development of porous bodies, ceramic joints (e.g., the joining of pre-shaped SiAlON pieces by ceramization of the silicone/nano-alumina interlayer [36]) and powders, to be used as a sintering aid for Si3N4 or as phosphors, in the case of doping with rare-earth ions [39,110]. The contaminations may be suppressed by the reformulation of polymer/filler ratios and/or insertion of additional fillers: in particular, in β-SiAlON ceramics, the Si and N content may be adjusted by the use of amorphous Si3N4 or of polymers with an optimized yield of these elements (perhydropolysilazane) [110]. Some specific systems, finally, follow a more straightforward reaction path: for instance, we recently found that Y-Si-O-N phases can be obtained by direct reaction, i.e., with no formation of silicates and the separation of SiC [39].
5. Products and Selected Case Studies
Table 1 reports the many ceramic systems investigated so far, with some remarks referring to the possible applications. Detailed information can be found in the cited literature, but we can list four general types of products, each associated with some applications.
Monoliths
Mullite, zircon, cordierite and SiAlON are widely appreciated as high temperature ceramics, especially for their high thermal shock resistance (in turn, associated with the relatively low coefficient of thermal expansion); in addition, silicates, such as mullite and forsterite, are generally used in high-performance electronic packaging [39]. The proposed approach could be highly attractive for the associated shaping possibilities: in fact, simple uniaxial pressing (especially warm pressing, conducted at temperatures in the range of 150 to 180 °C) leads to the formation of components with controlled geometries, high green strength and reduced green porosity; the firing may lead to defect-free ceramic parts, with uniform shrinkage, resulting from both the decomposition of the preceramic polymer and the formation of the crystalline phases. Machining can be easily accomplished when the component is still in the green state, thereby reducing the wear of the tools, and shape retention, before ceramic conversion, is guaranteed by the presence of a network of solid particles, which limits the viscous flow of the polymer [39]. The integrity of samples, after firing, can be favored, besides the choice of polymers (e.g., partial replacement of MK with H62C, as mentioned above), by the introduction of secondary fillers, not reacting with the ceramic residue of preceramic polymers, such as TiO2 (in forsterite ceramics), natural zircon (zircon “seeds”, in zircon ceramics), SiC (in SiAlON ceramics) and hydroxylapatite (in Ca-based silicates, to be used as biomaterials) micron- and nano-sized powders [35,111,112,115,139].
Cellular ceramics
Highly porous ceramics, to be used as filters or highly porous scaffolds for bone tissue engineering, may be seen as the “easiest” product from preceramic polymers [39]. While for the production of dense monoliths, several strategies (the selection of the polymer, the introduction of secondary fillers) must be applied to reduce the porosity, starting from that fundamentally provided by the gas evolution occurring upon ceramic conversion of preceramic polymers, cellular ceramics fully exploit the shaping possibilities offered by the particular raw materials. Foaming of mixtures of a preceramic polymer plus nano-sized fillers can be carried out using a variety of approaches, from direct foaming using additives that decompose at low temperature (80–350 °C) releasing a large volume of gas, to the mixing with sacrificial solid polymeric microbeads, followed by cold or warm pressing (the microbeads are eliminated during firing, leaving behind a large amount of pores of the desired size) [39]. In addition, since the preceramic polymers are soluble in a large variety of organic solvents, viscous pastes can be easily obtained and processed through syringes or, more efficiently, by automated manufacturing methods (e.g., fused deposition), leading to 3D scaffolds [108]. The macro-porosity, provided by foaming, can be coupled with micro-porosity, from ceramic conversion, thus yielding components with hierarchical porosity. It should be noted that the additives releasing gasses may be of both organic and inorganic origin; in particular, hydrated salts, like borax (as mentioned above, sodium borate decahydrate, Na2B4O7·10H2O), are interesting for a double action, i.e., they provide water vapor (useful for foaming), by decomposition, and a liquid phase upon firing (in turn, favoring the interdiffusion), as recently shown for the manufacturing of akermanite (Ca2MgSiO7) ceramics [115].
Coatings
Monolithic and cellular components often rely on the grinding of the solid residue, in turn determined by the drying of silicone-based suspensions, in large glass containers, in order to prepare “composite” powders. The grinding step can be avoided if preceramic polymers and fillers are used for coatings. Suspensions based on silicones and nano-sized fillers, as an example, were used to coat both SiC foams and SiAlON monoliths. The drying directly on the substrates determines the formation of a composite layer, later ceramized upon firing in air or nitrogen, respectively. In the first case, the coating is aimed at the development of a silicate environmental barrier coating, aimed at protecting SiC from extensive oxidation; in the second case, the coating acts as a polymeric glue, at low temperature (SiAlON pieces are joined across the coated surface), and transforms into a SiAlON joint, at high temperature [36]. Figure 5 illustrates a case study on environmental barrier coatings, concerning yttrium di-silicate (Y-DS) and zircon coating on Si-SiC foams; the formulations used in the experiments are reported in Table 2. For the realization of sufficiently thick and homogeneous depositions, an optimized multi-coating procedure was followed, as illustrated by Figure 5a. It consisted of repeated steps of deposition, solvent removal after deposition (made at 60 °C for 5 min) and coating stabilization (made at 260 °C), in order to cross-link the polymeric phase and to fix the coating on the SiC substrates. After this procedure, the final heat treatment was carried out in air with a 2 K/min heating rate. The overall procedure was repeated twice, with different maximum temperatures (1 h holding time), 1250 °C for the first cycle (aimed at the consolidation of the deposit, with no melting of Si) and 1350 °C for the second (aimed at phase development). In order to compensate for the shrinkage, secondary (inert) fillers were used in both formulations (mullite for the Y-DS coating, natural zircon for zircon coatings); kaolin clay was used as a viscosity modifier. As shown by Figure 5b, uniform coatings were formed; from SEM observations, the thickness was estimated to be ~100 μm. Although still to be improved (the coatings contain some micro-cracks), the strategy is quite promising, since the oxidation of the substrate was effectively reduced, as illustrated by the weight gains of foams heated at 1200 °C (for periods from five to 100 h), in Figure 5c.
Powders
A second type of “unshaped” polymer-derived product is represented by powders, from the direct firing of silicone/filler mixtures. The X-ray diffraction patterns in Figure 6a testify the easy obtainment of willemite, i.e., zinc silicate (Zn2SiO4 or 2ZnO·SiO2), from MK polymer and ZnO nano-particles. Some peaks of unreacted ZnO remain, but we should focus on the particularly low temperature (900 °C) at which the desired phase can be detected. As reported above, “nano-filled” silicones can be actually seen as an alternative to more complicated sol-gel processing. Figure 6a actually refers to Mn-doped willemite (Zn1.9Mn0.1SiO4), for which we readopted a strategy already applied for wollastonite ceramics [32]. The dopant (Mn2+) was not provided by means of MnO nanoparticles, but starting from Mn-acetate (Mn-ac). The manganese salt was used after dissolution in distilled water (10% solid content); the acetate solution was added dropwise to a silicone solution (MK in isopropyl alcohol), embedding also ZnO nanoparticles, under magnetic stirring; a non-ionic surfactant (Pluronic P 123, BASF Corporation, Florham Park, NJ), added to the silicone solution (1 g for 80 mL solution), was useful for obtaining homogeneous emulsions. The obtained Mn-doped willemite powders, after drying and ceramization, could be effectively used as green phosphors, even in the presence of some unreacted ZnO; as illustrated by Figure 6b, samples fired at 900–1100 °C exhibit a strong luminescence peak at 520–530 nm, after excitation in the UV range (250 nm), in analogy with what was reported for willemite-based phosphors obtained by conventional techniques [140].
A further case study, in the field of phosphor powders, concerns gehlenite (2CaO·Al2O3·SiO2) ceramics. This system has an impressive “tunability”, depending on the choice of dopants, on the ratios between main oxides and on the firing atmosphere. As reported above, the partial replacement of Ca2+ ions with Eu3+ could be compensated for by the tuning of the Al/Si ratio, forming the solid solution Ca1.86Eu0.14Al(Al1.14Si0.86O7), which represents the only crystal phase for treatments in air (firing at 1300 °C, for 1h), as shown by Figure 7a [114]. The obtained powders, as illustrated by Figure 7b, exhibit a strong red luminescence (main peak at ~620 nm) after excitation in the UV range, as expected by a previous paper on the tested formulation [141]; it should be highlighted, however, that the previous paper referred to conventional synthesis, helped by fluxes (e.g., boric acid). In addition, tests in nitrogen were applied, starting from a silicone/nano-fillers system, with the possibility of modifying the emission characteristics, according to reduction reactions, in turn promoted by the conversion of MK polymer into SiCO instead of pure silica. As shown by Figure 7a, gehlenite remained the main crystal phase, but some CaAl2O4 formed; while gehlenite could keep Eu3+ ions, the secondary phase is known to embed Eu2+ ions [142], made available by the presence of carbon in the ceramic residue of the polymer (Eu2O3 + C → 2 EuO + CO↑), leading to the blue emission (centered at 440–450 nm) in the spectra of Figure 7b.
Polymer-derived gehlenite is interesting also when doping with Ce3+ instead of with Eu3+. For treatments in air, the reduction of CeO2 into Ce2O3 was not complete, even at 1300 °C, as testified by the presence of un-reacted oxide (see Figure 7c). On the contrary, gehlenite was the only crystal phase for treatments in nitrogen (1400 °C), applied to both charge-compensated and not charge-compensated (NC) formulations, i.e., formulations nominally leading to Ca1.95Ce0.05Al2.05Si0.95O7 and Ca1.95Ce0.05Al2SiO7, respectively. After excitation in the near-UV (350 nm), powders from both formulations exhibited an intense blue-violet emission (peak at ~420 nm; see Figure 7d, reporting also the absorption spectra), consistent with the literature [143]. The more intense emission from the NC sample is attributable, in the authors’ opinion, to the possible partial nitridation, i.e., replacement of Si-O bonds with Si-N bonds; in fact, nitrogen-gehlenite (Ca2AlSi2O6N [144]) is feasible in the presence of a low Al/Si ratio; NC could be more prone to nitridation, owing to the lower content of Al3+ compared to Si4+. Furthermore, this hypothesis has not yet been verified by dedicated experiments, but will certainly constitute the focus of future work.
6. Concluding Remarks
Modifications at the nano-scale of preceramic polymer enable the fabrication of a vast range of advanced ceramics to be applied in numerous engineering and specialized applications. These modifications are quite simple, since: (i) preceramic polymers are in most cases easily dissolved in organic solvents, so that nano-sized fillers, active or passive, can be dispersed homogeneously; and (ii) the backbone of a preceramic polymer generally presents side groups, such as -OH, which can easily react with metallorganic compounds, with the development of metal-modified polymers.
The versatility of the technology has already been appreciated in the fabrication of components suitable for biological, high temperature and functional (e.g., phosphors) applications, but we can say that, probably, we are just at the beginning; in fact, several improvements and refinements are expected in many aspects, including:
Processing environment: composition and phase assemblage of non-oxide ceramics (oxycarbide, oxynitrides) from reactive systems (metal-modified polymers or polymers embedding active fillers) depend on the processing environment (flow rate of nitrogen or argon, type of furnace), so that reproducibility could be an issue; products should be associated with a specific protocol, to be carefully defined;
Availability of preceramic polymers: from Table 1, it can be observed that only a limited number of polymers was used for many types of ceramics; it would be very interesting to test more of the various silicone resins currently available on the market, considering the changes in densification, developed phases and integrity of samples, associated with the replacement of MK with H62C (which could be seen as only the first example);
Ceramic compositions: silicates, i.e., metal oxide + silica systems, are very numerous by themselves: infinite combinations can be explored in terms of solid solutions or composites; oxycarbides and oxynitride systems have an even more pronounced variability (e.g., in SiAlON ceramics, the balance among constituents is typically conditioned by the replacement of the Si-N bond with Al-O bond processing technologies: polymers, by themselves, offer many distinct processing strategies; bi-phasic systems or metal-modified polymers may possess a lower flowability, compared to pure polymers, so that processing conditions and additives need to be carefully selected, but nano-modified preceramic polymers remain open to a vast range of forming technologies.
Acknowledgments
The authors wish to thank Chiara Bergo, Jean François Carlotti, Alberto Cranio, Pedro Mendanha Dias, Gabriele Dal Maso, Stefano Pilati, Samuele Ruaro and Ireneo Vidali (University of Padova, Italy) for the experimental assistance, and they gratefully acknowledge the financial support of the Italy-Germany bilateral research program “Vigoni”, for research on silicate bioceramics, and of the European Community’s Seventh Framework Programme through a Marie-Curie Research Training Network (“FUnctional Nitrides for Energy Applications—FUNEA”, PITN-2010-264873). The authors are also indebted to Emanuel Ionescu (Technische Universität Darmstadt, Germany) and Günter Motz (University of Bayreuth, Germany), for gently providing figures (Figures 1 and 2) and references, and to Bert Hintzen (Eindhoven University of Technology, The Netherlands) and Dusan Galusek (Slovak Academy of Sciences), for a stimulating discussion on the possibility of producing preceramic polymer-derived luminescent materials.
Author Contributions
Laura Fiocco, Giulio Parcianello and Enrico Storti conducted the part of experiments described in the present review effectively performed at the University of Padova, under the supervision of Enrico Bernardo and Paolo Colombo. Enrico Bernardo planned the experiments and the structure of the article. The article was written and edited by Enrico Bernardo, Laura Fiocco and Paolo Colombo.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Colombo, P.; Mera, G.; Riedel, R.; Sorarù, G.D. Polymer-derived-ceramics: 40 years of research and innovation in advanced ceramics. J. Am. Ceram. Soc 2010, 93, 1805–1837. [Google Scholar]
- Polymer Derived Ceramics: From Nano-Structure to Applications; Colombo, P., Sorarù, G.D., Riedel, R., Kleebe, H.-J., Eds.; DESTech Publications: Lancaster, PA, USA, 2009.
- Mera, G.; Ionescu, E. Silicon-Containing Preceramic Polymers. In Encyclopedia of Polymer Science and Technology; Mark, E.G., Ed.; Wiley-VCH: Weinheim, Germany, 2014; Available online; (asscessed on 28 February 2014). [Google Scholar] [CrossRef]
- Renlund, G.M.; Prochazka, S.; Doremus, R.H. Silicon oxycarbide glasses: Part I. Preparation and chemistry. J. Mater. Res 1991, 6, 2716–2722. [Google Scholar]
- Wild, M.J.; Buhler, P. On the phase composition of polymethylsiloxane derived ceramics. J. Mater. Sci 1998, 33, 5441–5444. [Google Scholar]
- Kroke, E.; Li, Y.L.; Konetschny, C.; Lecomte, M.; Fasel, C.; Riedel, R. Silazane derived ceramics and related materials. Mater. Sci. Eng. R Rep 2000, 26, 97–199. [Google Scholar]
- Greil, P. Active-filler-controlled pyrolysis of preceramic polymers. J. Am. Ceram. Soc 1995, 78, 835–848. [Google Scholar]
- Greil, P. Near net shape manufacturing of polymer derived ceramics. J. Eur. Ceram. Soc 1998, 18, 1095–1014. [Google Scholar]
- Greil, P. Polymer derived engineering ceramics. Adv. Eng. Mater 2000, 2, 339–348. [Google Scholar]
- Wan, J.; Gasch, M.J.; Mukherjee, A.K. Silicon nitride-silicon carbide nanocomposites fabricated by electric-field-assisted sintering. J. Am. Ceram. Soc 2002, 86, 526–528. [Google Scholar]
- Alejandra, M.; Palencia, C.; Nistal, A.; Rubio, F.; Rubio, J.; Oteo, J.L. Dense bulk silicon oxycarbide glasses obtained by spark plasma sintering. J. Eur. Ceram. Soc 2012, 32, 3369–3378. [Google Scholar]
- Esfehanian, M.; Oberacker, R.; Fett, T.; Hoffmann, M.J. Development of dense filler-free polymer-derived SiCO ceramics by field assisted sintering. J. Am. Ceram. Soc 2008, 91, 3803–3805. [Google Scholar]
- Greil, P.; Seibold, M. Modelling of dimensional changes during polymer-ceramic conversion for bulk component fabrication. J. Mater. Sci 1992, 27, 1053–1060. [Google Scholar]
- Greil, P. Polymer-filled derived ceramics with hierarchical microstructures. Key Eng. Mater 1999, 159–160, 339–346. [Google Scholar]
- Erny, T.; Seibold, M.; Jarchow, O.; Greil, P. Microstructure development of oxycarbide composites during active-filler-controlled polymer pyrolysis. J. Am. Ceram. Soc 1993, 76, 207–213. [Google Scholar]
- Seibold, M.; Greil, P. Thermodynamics and microstructure development of ceramic composite formation by Active-Filler-Controlled polymer pyrolysis (AFCOP). J. Eur. Ceram. Soc 1993, 11, 105–113. [Google Scholar]
- Schwarz, K.B.; Rowcliffe, D.J. Modeling density contributions in preceramic polymer/ceramic powder systems. J. Am. Ceram. Soc 1986, 69, C106–C108. [Google Scholar]
- Seyferth, D.; Bryson, N.; Workman, D.P.; Sobon, C.A. Preceramic polymers as “reagents” in the preparation of ceramics. J. Am. Ceram. Soc 1991, 74, 2687–2689. [Google Scholar]
- Kang, K.T.; Kim, D.J.; Greil, P. Development of novel ceramic composites by active filler controlled polymer pyrolysis with tungsten. J. Korean Ceram. Soc 1998, 35, 939–944. [Google Scholar]
- Dernovsek, O.; Bressiani, J.C.; Bressiani, A.H.A.; Greil, P. Reaction bonded niobium carbide ceramics from polymer-filler mixtures. J. Mater. Sci 2000, 35, 2201–2207. [Google Scholar]
- Kaindl, A.; Lehner, W.; Greil, P.; Kim, D.J. Polymer-filler derived Mo2C ceramics. Mater. Sci. Eng 1999, A260, 101–107. [Google Scholar]
- Steinau, M.; Travitzky, N.; Gegner, J.; Hofmann, J.; Greil, P. Polymer-Derived ceramics for advanced bearing applications. Adv. Ceram. Mater 2008, 10, 1141–1146. [Google Scholar]
- Biasetto, L.; Francis, A.; Palade, P.; Principi, G.; Colombo, P. Polymer-derived microcellular SiCO foams with magnetic functionality. J. Mater. Sci 2008, 43, 4119–4126. [Google Scholar]
- Cordelair, J.; Greil, P. Electrical characterization of polymethylsiloxane/MoSi2-derived composite ceramics. J. Am. Ceram. Soc 2001, 84, 2256–2259. [Google Scholar]
- Walter, S.; Suttor, D.; Erny, T.; Hahn, B.; Greil, P. Injection moulding of polysiloxane/filler mixtures for oxycarbide ceramic composites. J. Eur. Ceram. Soc 1996, 16, 387–393. [Google Scholar]
- Colombo, P.; Gambaryan-Roisman, T.; Scheffler, M.; Buhler, P.; Greil, P. Conductive ceramic foams from preceramic polymers. J. Am. Ceram. Soc 2001, 84, 2265–2268. [Google Scholar]
- Riedel, R.; Toma, L.; Fasel, C.; Miehe, G. Polymer-derived mullite—SiC-based nanocomposites. J. Eur. Ceram. Soc 2009, 29, 3079–3090. [Google Scholar]
- Bernardo, E.; Colombo, P.; Pippel, E.; Woltersdorf, J. Novel mullite synthesis based on alumina nanoparticles a preceramic polymer. J. Am. Ceram. Soc 2006, 89, 1577–1583. [Google Scholar]
- Bernardo, E.; Colombo, P. Advanced oxide ceramics from a preceramic polymer and fillers. Soft Mater 2006, 4, 175–185. [Google Scholar]
- Bernardo, E.; Colombo, P.; Hampshire, S. SiAlON-based ceramics from filled preceramic polymers. J. Am. Ceram. Soc 2006, 89, 3839–3842. [Google Scholar]
- Griggio, F.; Bernardo, E.; Colombo, P.; Messing, G.L. Kinetic studies of mullite synthesis from alumina nanoparticles and a preceramic polymer. J. Am. Ceram. Soc 2008, 91, 2529–2533. [Google Scholar]
- Bernardo, E.; Tomasella, E.; Colombo, P. Development of multiphase bioceramics from a filler-containing preceramic polymer. Ceram. Int 2009, 35, 1415–1421. [Google Scholar]
- Bernardo, E.; Colombo, P.; Hampshire, S. Advanced ceramics from preceramic polymers and nano-fillers. J. Eur. Ceram. Soc 2009, 29, 843–849. [Google Scholar]
- Parcianello, G.; Bernardo, E.; Colombo, P. Mullite/zirconia nano-composites from a preceramic polymer and nano-sized fillers. J. Am. Ceram. Soc 2011, 94, 1357–1362. [Google Scholar]
- Bernardo, E.; Colombo, P.; Cacciotti, I.; Bianco, A.; Bedini, R.; Pecci, R.; Pardun, K.; Treccani, L.; Rezwan, K. Porous wollastonite-hydroxyapatite bioceramics from a preceramic polymer and micro- or nano-sized fillers. J. Eur. Ceram. Soc 2012, 32, 399–408. [Google Scholar]
- Bernardo, E.; Parcianello, G.; Colombo, P.; Jones, B.H.; Kruise, J.; Barnes, A.T.; Hellmann, J.R. SiAlON ceramics from preceramic polymers and nano-sized fillers: Application in ceramic joining. J. Eur. Ceram. Soc 2012, 32, 1329–1335. [Google Scholar]
- Suttor, D.; Kleebe, H-J.; Ziegler, G. Formation of mullite from filled siloxanes. J. Am. Ceram. Soc 1997, 80, 2541–2548. [Google Scholar]
- Anggono, J.; Derby, B. Intermediate phases in mullite synthesis via aluminum- and alumina-filled polymethylsiloxane. J. Am. Ceram. Soc 2005, 88, 2085–2091. [Google Scholar]
- Colombo, P.; Bernardo, E.; Parcianello, G. Multifunctional advanced ceramics from preceramic polymers and nano-sized active fillers. J. Eur. Ceram. Soc 2013, 33, 453–469. [Google Scholar]
- Yajima, S.; Iwai, T.; Yamamura, T.; Okamura, K.; Hasegawa, Y. Synthesis of a polytitanocarbosilane and its conversion into inorganic compounds. J. Mater. Sci 1981, 16, 1349–1355. [Google Scholar]
- Ishikawa, T.; Yamamura, T.; Okamura, K. Production mechanism of polytitanocarbosilane and its conversion of the polymer into inorganic materials. J. Mater. Sci 1992, 27, 6627–6634. [Google Scholar]
- Song, Y.; Feng, C.; Tan, Z.; Lu, Y. Structure and properties of polytitanocarbosilane as the precursor of SiC-TiC fibre. J. Mater. Sci. Lett 1990, 9, 1310–1313. [Google Scholar]
- Chollon, G.; Aldacourrou, B.; Capes, L.; Pailler, R.; Naslain, R. Thermal behaviour of a polytitanocarbosilane-derived fibre with a low oxygen content: The Tyranno Lox-E fibre. J. Mater. Sci 1998, 33, 901–911. [Google Scholar]
- Ishikawa, T.; Kohtoku, Y.; Kumagawa, K. Production mechanism of polyzirconocarbo-silane using zirconium(IV)acetylacetonate and its conversion of the polymer into inorganic materials. J. Mater. Sci 1998, 33, 161–166. [Google Scholar]
- Babonneau, F.; Sorarù, G.D.; Mackenzie, J.D. 29Si MAS-NMR investigation of the conversion process of a polytitanocarbosilane into SiC-TiC ceramics. J. Mater. Sci 1990, 25, 3664–3670. [Google Scholar]
- Sorarù, G.D.; Ravagni, A.; Campostrini, R.; Babonneau, F. Synthesis and characterization of β′-SiAlON ceramics from organosilicon polymers. J. Am. Ceram. Soc 1991, 74, 2220–2223. [Google Scholar]
- Sorarù, G.D.; Glisenti, A.; Granozzi, G.; Babonneau, F.; Mackenzie, J.D. The pyrolysis process of a polytitanocarbosilane into SiC/TiC ceramics: An XPS study. J. Mater. Res 1990, 5, 1958–1962. [Google Scholar]
- Sorarù, G.D.; Mercadini, M.; Dal Maschio, R.; Taulelle, F.; Babonneau, F. Si-Al-O-N fibers from polymeric precursor: synthesis, structural, and mechanical characterization. J. Am. Ceram. Soc 1993, 76, 2595–2600. [Google Scholar]
- Babonneau, F.; Sorarù, G.D. Synthesis and characterization of Si-Zr-C-O ceramics from polymer precursors. J. Eur. Ceram. Soc 1991, 8, 29–34. [Google Scholar]
- Sglavo, V.M.; Dal Maschio, R.; Sorarù, G.D.; Bellosi, A. Fabrication and characterization of polymer-derived Si2N2O-ZrO2 nanocomposite ceramics. J. Mater. Sci 1993, 28, 6437–6441. [Google Scholar]
- Sorarù, G.D.; Ravagni, A.; Dal Maschio, R.; Carturan, G.; Babonneau, F. Polymer-derived Si3N4-ZrO2 nanocomposite powders. J. Mater. Res 1992, 7, 1266–1270. [Google Scholar]
- Yu, Z.; Yang, L.; Zhan, J.; Zhou, C.; Min, H.; Zheng, Q.; Xia, H. Preparation, cross-linking and ceramization of AHPCS/Cp2ZrCl2 hybrid precursors for SiC/ZrC/C composites. J. Eur. Ceram. Soc 2012, 32, 1291–1298. [Google Scholar]
- Chen, Z.; Li, X.; Wang, J.; Li, W. Preparation of continuous Si-Fe-C-O functional ceramic fibers. Trans. Nonferr. Met. Soc. China 2007, 17, 987–991. [Google Scholar]
- Thorne, K.; Liimatta, E.; Mackenzie, J.D. Synthesis of SiC/TaC ceramics from tantalum alkoxide modified polycarbosilane. J. Mater. Res 1991, 6, 2199–2207. [Google Scholar]
- Yu, Z.; Fang, Y.; Huang, M.; Li, R.; Zhan, J.; Zhou, C.; He, G.; Xia, H. Preparation of a liquid boron-modified polycarbosilane and its ceramic conversion to dense SiC ceramics. Polym. Adv. Technol 2011, 22, 2409–2414. [Google Scholar]
- Fan, X.; Feng, C.; Song, Y.; Li, X. Preparation of Si-C-O-N-B ceramic fibers from polycarbosilane. J. Mater. Sci. Lett 1999, 18, 629–630. [Google Scholar]
- Riedel, R.; Kienzle, A.; Dressler, W.; Ruwisch, L.; Bill, J.; Aldinger, F. A silicoboron carbonitride ceramic stable to 2000 °C. Nature 1996, 382, 796–798. [Google Scholar]
- Hauser, R.; Nahar-Borchard, S.; Riedel, R.; Ikuhara, Y.H.; Iwamoto, Y. Polymer-derived SiBCN ceramic and their potential application for high temperature membranes. J. Ceram. Soc. Jpn 2006, 114, 524–528. [Google Scholar]
- Ravi Kumar, N.V.; Mager, R.; Cai, Y.; Zimmermann, A.; Aldinger, F. High temperature deformation behaviour of crystallized Si-B-C-N ceramics obtained from a boron modified poly(vinyl)silazane polymeric precursor. Scripta Mater 2004, 51, 65–69. [Google Scholar]
- Wideman, T.; Su, K.; Remsen, E.E.; Zank, G.A.; Sneddon, L.G. Synthesis, characterization, and ceramic conversion reactions of borazine/silazane copolymers: New polymeric precursors to SiNCB ceramics. Chem. Mater 1995, 7, 2203–2212. [Google Scholar]
- Seyferth, D.; Plenio, H. Borasilazane polymeric precursors for borosilicon nitride. J. Am. Ceram. Soc 1990, 73, 2131–2133. [Google Scholar]
- Bhandavat, R.; Singh, G. Synthesis, characterization, and high temperature stability of Si(B)CN-coated carbon nanotubes using a boron-modified poly(ureamethylvinyl)silazane chemistry. J. Am. Ceram. Soc 2012, 95, 1536–1543. [Google Scholar]
- Bernard, S.; Weinmann, M.; Gerstel, P.; Miele, P.; Aldinger, F. Boron-modified polysilazane as a novel single-source precursor for SiBCN ceramic fibers: synthesis, melt-spinning, curing and ceramic conversion. J. Mater. Chem 2005, 15, 289–299. [Google Scholar]
- Wideman, T.; Fazen, P.J.; Su, K.; Remsen, E.E.; Zank, G.A.; Sneddon, L.G. Second-generation polymeric precursors for BN and SiNCB ceramic materials. Appl. Organometal. Chem 1998, 12, 681–693. [Google Scholar]
- Schuhmacher, J.; Berger, F.; Weinmann, M.; Bill, J.; Aldinger, F.; Müller, K. Solid-state NMR and FT IR studies of the preparation of Si-B-C-N ceramics from -boron-modified polysilazanes. Appl. Organomet. Chem 2001, 15, 809–819. [Google Scholar]
- Müller, A.; Zern, A.; Gerstel, P.; Bill, J.; Aldinger, F. Boron-modified poly(propenylsilazane)-derived Si-B-C-N ceramics: Preparation and high temperature properties. J. Eur. Ceram. Soc 2002, 22, 1631–1643. [Google Scholar]
- Saha, A.; Shah, S.R.; Raj, R. Amorphous silicon carbonitride fibers drawn from alkoxide modified ceraset. J. Am. Ceram. Soc 2003, 86, 1443–1445. [Google Scholar]
- Saha, A.; Shah, S.R.; Raj, R. Oxidation behavior of SiCN-ZrO2 fiber prepared from alkoxide-modified silazane. J. Am. Ceram. Soc 2004, 87, 1556–1558. [Google Scholar]
- Papendorf, B.; Nonnenmacher, K.; Ionescu, E.; Kleebe, H.-J.; Riedel, R. Strong influence of polymer architecture on the microstructural evolution of hafnium-alkoxide-modified silazanes upon ceramization. Small 2011, 7, 970–978. [Google Scholar]
- Ionescu, E.; Papendorf, B.; Kleebe, H.-J.; Breitzke, H.; Nonnenmacher, K.; Buntkowsky, G.; Riedel, R. Phase separation of a hafnium alkoxide-modified polysilazane upon polymer-to-ceramic transformation—A case study. J. Eur. Ceram. Soc 2012, 32, 1873–1881. [Google Scholar]
- Seyferth, D.; Brodt, G.; Boury, B. Polymeric aluminasilazane precursors for aluminosilicon nitride. J. Mater. Sci. Lett 1996, 15, 348–349. [Google Scholar]
- Haluska, L.A. Process for the Preparation of Polymetallo (Disily) Silazane Polymers and the Polymers Therefrom, US Patent 4,482,689. 13 November 1984.
- Ionescu, E.; Kleebe, H.-J.; Riedel, R. Silicon-containing polymer-derived ceramic nanocomposites (PDC-NCs): Preparative approaches and properties. Chem. Soc. Rev 2012, 41, 5032–5052. [Google Scholar]
- Ionescu, E.; Terzioglu, C.; Linck, C.; Kaspar, J.; Navrotsky, A.; Riedel, R. Thermodynamic control of phase composition and crystallization of metal-modified silicon oxycarbides. J. Am. Ceram. Soc 2013, 96, 1899–1903. [Google Scholar]
- Ionescu, E.; Papendorf, B.; Kleebe, H.-J.; Poli, F.; Müller, K.; Riedel, R. Polymer-derived silicon oxycarbide/hafnia ceramic nanocomposites. Part I: Phase and microstructure evolution during the ceramization process. J. Am. Ceram. Soc 2010, 93, 1174–1182. [Google Scholar]
- Ionescu, E.; Papendorf, B.; Kleebe, H.-J.; Riedel, R. Polymer-derived silicon oxycarbide/hafnia ceramic nanocomposites. Part II: Stability toward decomposition and microstructure evolution at T ≫ 1000 °C. J. Am. Ceram. Soc 2010, 93, 1783–1789. [Google Scholar]
- Kleebe, H.-J.; Nonnenmacher, K.; Ionescu, E.; Riedel, R. Decomposition-coarsening model of SiCO/HfO2 ceramic nanocomposites upon isothermal anneal at 1300 °C. J. Am. Ceram. Soc 2012, 95, 2290–2297. [Google Scholar]
- Ionescu, E.; Linck, C.; Fasel, C.; Müller, M.; Kleebe, H.-J.; Riedel, R. Polymer-derived SiOC/ZrO2 ceramic nanocomposites with excellent high-temperature stability. J. Am. Ceram. Soc 2010, 93, 241–250. [Google Scholar]
- Harshe, R.; Balan, C.; Riedel, R. Amorphous Si(Al)OC ceramic from polysiloxanes: Bulk ceramic processing, crystallization behavior and applications. J. Eur. Ceram. Soc 2004, 24, 3471–3482. [Google Scholar]
- Ischenko, V.; Harshe, R.; Riedel, R.; Woltersdorf, J. Cross-linking of functionalised siloxanes with alumatrane: Reaction mechanisms and kinetics. J. Organomet. Chem 2006, 691, 4086–4091. [Google Scholar]
- Bai, H.W.; Wena, G.; Huang, X.X.; Han, Z.X.; Zhong, B.; Hu, Z.X.; Zhang, X.D. Synthesis and structural characterization of SiBOC ceramic fibers derived from single-source polyborosiloxane. J. Eur. Ceram. Soc 2001, 31, 931–940. [Google Scholar]
- Schiavon, M.A.; Armelin, N.A.; Valéria, I.; Yoshida, P. Novel poly(borosiloxane) precursors to amorphous SiBCO ceramics. Mater. Chem. Phys 2008, 112, 1047–1054. [Google Scholar]
- Yajima, S.; Okamura, K.; Hayashi, J.; Shishido, T. Borosiloxane Polymers and a Method for Producing the Same, US Patent 4,152,509. 1 May 1979.
- Dire, S.; Babonneau, F.; Sanchez, C.; Livage, J. Sol-gel synthesis of siloxane oxide hybrid coatings [Si(CH3)2O·MOx-M = Si, Ti, Zr, Al] with luminescent properties. J. Mater. Chem 1992, 2, 239–244. [Google Scholar]
- Dire, S.; Ceccato, R.; Babonneau, F. Structural and microstructural evolution during pyrolysis of hybrid polydimethylsiloxane-titania nanocomposites. J. Sol Gel. Sci. Technol 2005, 34, 53–62. [Google Scholar]
- Peña-Alonso, R.; Sorarù, G.D. Synthesis and characterization of hybrid borosiloxane gels as precursors for Si-B-O-C fibers. J. Sol Gel. Sci. Technol 2007, 43, 313–319. [Google Scholar]
- Schiavon, M.A.; Gervais, C.; Babonneau, F.; Sorarù, G.D. Crystallization behavior of novel silicon boron oxycarbide glasses. J. Am. Ceram. Soc 2004, 87, 203–208. [Google Scholar]
- Katsuda, Y.; Gerstel, P.; Narayanan, J.; Bill, J.; Aldinger, F. Reinforcement of precursor-derived Si-C-N ceramics with carbon nanotubes. J. Eur. Ceram. Soc 2006, 26, 3399–3405. [Google Scholar]
- Kokott, S.; Heymann, L.; Motz, G. Processability of multi-walled carbon nanotubes-ABSE polycarbosilazane composites. J. Eur. Ceram. Soc 2008, 28, 1015–1021. [Google Scholar]
- Li, Y.; Fernandez-Recio, L.; Gerstel, G.; Srot, V.; van Aken, P.A.; Kaiser, G.; Burghard, M.; Bill, J. Chemical modification of single-walled carbon nanotubes for the reinforcement of precursor-derived ceramics. Chem. Mater 2008, 20, 5593–5599. [Google Scholar]
- Shibuya, M.; Sakurai, M.; Takahashi, T. Preparation and characteristics of a vapor-grown carbon fiber/ceramic composite using a methyl silicone precursor. Comp. Sci. Technol 2007, 67, 3338–3344. [Google Scholar]
- Rahman, A.; Singh, A.; Harimkar, S.P.; Singh, R.P. Spark plasma sintering and characterization of graphene reinforced silicon carbide nanocomposites. In Composite Materials and Joining Technologies for Composites, Proceedings of the 2012 Annual Conference on Experimental and Applied Mechanics; Patterson, E., Backman, D., Cloud, G., Eds.; Springer: New York, NY, USA, 2013; 7, pp. 139–146. [Google Scholar]
- Ji, F.; Li, Y.-L.; Feng, J.-M.; Su, D.; Wen, Y.-Y.; Feng, Y.; Hou, F. Electrochemical performance of graphene nanosheets and ceramic composites as anodes for lithium batteries. J. Mater. Chem 2009, 19, 9063–9067. [Google Scholar]
- Papendorf, B.; Ionescu, E.; Kleebe, H.-J.; Linck, C.; Nonnenmacher, K.; Riedel, R. High-temperature creep behavior of dense sioc-based ceramic nanocomposites: Microstructural and phase composition effects. J. Am. Ceram. Soc 2013, 96, 272–280. [Google Scholar]
- Linck, C.; Ionescu, E.; Papendorf, B.; Galuskova, D.; Galusek, D.; Ŝajgalík, P.; Riedel, R. Corrosion behavior of silicon oxycarbide-based ceramic nanocomposites under hydrothermal conditions. Int. J. Mater. Res 2012, 103, 31–39. [Google Scholar]
- Ionescu, E. Technische Universität Darmstadt; Darmstadt: Germany, Unpublished work; 2014. [Google Scholar]
- Hojamberdiev, M.; Prasad, R.M.; Fasel, C.; Riedel, R.; Ionescu, E. Single-Source-Precursor synthesis of soft magnetic Fe3Si- and Fe5Si3-containing SiOC ceramic nanocomposites. J. Eur. Ceram. Soc 2013, 33, 2465–2472. [Google Scholar]
- Bill, J.; Aldinger, F. Precursor-derived covalent ceramics. Adv. Mater 1995, 7, 775–787. [Google Scholar]
- Schulz, M. Polymer derived ceramics in MEMS/NEMS—A review on production processes and application. Adv. Appl. Ceram 2009, 108, 454–460. [Google Scholar]
- Motz, G. University of Bayreuth; Bayreuth: Germany, Unpublished work; 2014. [Google Scholar]
- Colombo, P.; Bernardo, E. Macro- and micro-cellular porous ceramics from preceramic polymers. Compos. Sci. Technol 2003, 63, 2353–2359. [Google Scholar]
- Colombo, P.; Bernardo, E.; Biasetto, L. Novel microcellular ceramics from a silicone resin. J. Am. Ceram. Soc 2004, 87, 152–154. [Google Scholar]
- Shibuya, M.; Takahashi, T.; Koyama, K. Microcellular ceramics by using silicone preceramic polymer and PMMA polymer sacrificial microbeads. Compos. Sci. Technol 2007, 67, 119–124. [Google Scholar]
- Geim, A.; Novoselov, K. The rise of graphene. Nat. Mater 2007, 6, 183–191. [Google Scholar]
- Kim, H.; Abdala, A.; Macosko, C. Graphene/polymer nanocomposites. Macromolecules 2010, 43, 6515–6530. [Google Scholar]
- Lee, C.; Wei, X.; Kysar, J.; Hone, J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 2008, 321, 385–388. [Google Scholar]
- Cai, D.; Song, M. Recent advance in functionalized graphene/polymer nanocomposites. J. Mater. Chem 2010, 20, 7906–7915. [Google Scholar]
- Bernardo, E.; Colombo, P.; Dainese, E.; Lucchetta, G. Novel 3D wollastonite-based scaffolds from preceramic polymers containing micro- and nano-sized reactive particles. Adv. Eng. Mater 2012, 14, 269–274. [Google Scholar]
- Bernardo, E.; Parcianello, G.; Colombo, P. Novel synthesis and applications of yttrium silicates from a silicone resin containing oxide nano-particle fillers. Ceram. Int 2012, 38, 5469–5474. [Google Scholar]
- Parcianello, G.; Bernardo, E.; Colombo, P. Optimization of phase purity of β′-sialon ceramics produced from silazanes and nano-sized alumina. J. Am. Ceram. Soc 2012, 95, 2148–2154. [Google Scholar]
- Parcianello, G.; Bernardo, E.; Colombo, P. Low temperature synthesis of zircon from silicone resins and oxide nano-sized particles. J. Eur. Ceram. Soc 2012, 32, 2819–2824. [Google Scholar]
- Bernardo, E.; Fiocco, L.; Giffin, G.A.; Di Noto, V.; Colombo, P. Microstructure development and dielectric characterization of forsterite-based ceramics from silicone resins and oxide fillers. Adv. Eng. Mater 2014. in press. [Google Scholar]
- Parcianello, G.; Bernardo, E.; Colombo, P. Cordierite ceramics from silicone resins containing nano-sized oxide particle fillers. Ceram. Int 2013, 39, 8893–8899. [Google Scholar]
- Bernardo, E.; Fiocco, L.; Prnová, A.; Klement, R.; Galusek, D. Gehlenite:Eu3+ phosphors from a silicone resin and nano-sized fillers. Opt. Mater 2013. submitted. [Google Scholar]
- Bernardo, E.; Carlotti, J.F.; Dias, P.M.; Fiocco, L.; Colombo, P.; Treccani, L.; Hess, U.; Rezwan, K. Novel akermanite-based bioceramics from preceramic polymers and oxide fillers. Ceram. Int 2014, 40, 1029–1035. [Google Scholar]
- Long, L.H.; Chen, L.D.; Chang, J. Low temperature fabrication and characterizations of β-CaSiO3 ceramics. Ceram. Int 2006, 32, 457–460. [Google Scholar]
- Sacks, M.D.; Bozkurt, N.; Scheiffele, G.W. Fabrication of mullite and mullite-matrix composites by transient viscous sintering of composite powders. J. Am. Ceram. Soc 1991, 74, 2428–2437. [Google Scholar]
- Hong, S.H.; Messing, G.L. Anisotropic grain growth in diphasic-gel-derived titania-doped mullite. J. Am. Ceram. Soc 1998, 81, 1269–1277. [Google Scholar]
- Hong, S.H.; Cermignani, W.; Messing, G.L. Anisotropic grain growth in seeded and B2O3-doped diphasic mullite gels. J. Eur. Ceram. Soc 1996, 16, 133–141. [Google Scholar]
- Iqbal, Y.; Lee, W.E. Fired porcelain microstructures revisited. J. Am. Ceram. Soc 1999, 82, 3584–3590. [Google Scholar]
- Lu, H.J.; Wang, W.L.; Tuan, W.H.; Lin, M.H. Acicular mullite crystals in vitrified kaolin. J. Am. Ceram. Soc 2004, 87, 1843–1847. [Google Scholar]
- Schneider, H.; Schreuer, J.; Hildmann, B. Structure and properties of mullite—A review. J. Eur. Ceram. Soc 2008, 28, 329–344. [Google Scholar]
- Lührs, H.; Fischer, R.X.; Schneider, H. Boron mullite: Formation and basic characterization. Mater. Res. Bull 2012, 47, 4031–4042. [Google Scholar]
- Pyzik, A.J.; Todd, C.S.; Han, C. Formation mechanism and microstructure development in acicular mullite ceramics fabricated by controlled decomposition of fluorotopaz. J. Eur. Ceram. Soc 2008, 28, 383–391. [Google Scholar]
- Sorarù, G.D.; Kleebe, H.-J.; Ceccato, R.; Pederiva, L. Development of mullite-SiC nanocomposites by pyrolysis of filled polymethylsiloxane gels. J. Eur. Ceram. Soc 2000, 20, 2509–2517. [Google Scholar]
- Toma, L.; Fasel, C.; Lauterbach, S.; Kleebe, H.-J.; Riedel, R. Influence of nano-aluminum filler on the microstructure of SiOC ceramics. J. Eur. Ceram. Soc 2011, 31, 1779–1789. [Google Scholar]
- Mazzucato, E.; Gualtieri, A.F. Wollastonite polytypes in the CaO-SiO2 system. Phys. Chem. Min 2000, 27, 565–574. [Google Scholar]
- Ni, S.; Chou, L.; Chang, J. Preparation and characterization of forsterite (Mg2SiO4) bioceramics. Ceram. Int 2007, 33, 83–88. [Google Scholar]
- Song, K.X.; Chen, X.M. Phase evolution and microwave dielectric characteristics of Ti-substituted Mg2SiO4 forsterite ceramics. Mater. Lett 2008, 62, 520–522. [Google Scholar]
- Seifert, H.J.; Wagner, S.; Fabrichnaya, O.; Lukas, H.L.; Aldinger, F.; Ullmann, T.; Schmücker, M.; Schneider, H. Yttrium silicate coatings on chemical vapor deposition-SiC-precoated C/C-SiC: thermodynamic assessment and high-temperature investigation. J. Am. Ceram. Soc 2005, 88, 424–430. [Google Scholar]
- Feslche, J. The crystal chemistry of rare-earth silicates. Struct. Bonding 1973, 13, 99–101. [Google Scholar]
- Sun, Z.; Zhou, Y.; Wang, J.; Li, M. γ-Y2Si2O7, a machinable silicate ceramic: Mechanical properties and machinability. J. Am. Ceram. Soc 2007, 90, 2535–2541. [Google Scholar]
- Díaz, M.; Garcia-Cano, I.; Mello-Castanho, S.; Moya, J.S.; Rodríguez, M.A. Synthesis of nanocrystalline yttrium disilicate powder by sol-gel method. J. Non Cryst. Sol 2011, 289, 151–154. [Google Scholar]
- Moya, J.S.; Díaz, M.; Serna, C.J.; Mello-Castanho, S. Formation of nanocrystalline yttrium disilicate by an oxalate gel method. J. Eur. Ceram. Soc 1998, 18, 1381–1384. [Google Scholar]
- Becerro, A.I.; Naranjo, M.; Perdigón, A.C.; Trillo, J.M. Hydrothermal chemistry of silicates: Low-Temperature synthesis of y-yttrium disilicate. J. Am. Ceram. Soc 2003, 86, 1592–1594. [Google Scholar]
- Webster, J.D.; Westwood, M.E.; Hayes, F.H.; Day, R.J.; Taylor, R.; Duran, A.; Aparicio, M.; Rebstock, K.; Vogel, W.D. Oxidation protection coatings for C/SiC based on yttrium silicate. J. Eur. Ceram. Soc 1998, 18, 2345–2350. [Google Scholar]
- Jacobson, N.S.; Lee, K.N.; Fox, D.S. Reactions of silicon carbide and silicon (IV) oxide at elevated temperatures. J. Am. Ceram. Soc 1992, 76, 1603–1611. [Google Scholar]
- Ewing, H.; Vital, A.; Vogt, U.; Hendry, A. The nucleation and growth mechanism of Si-N-O fibres. Mater. Sci. Forum 2000, 325–326, 37–42. [Google Scholar]
- Bernardo, E.; Parcianello, G.; Colombo, P.; Hess, U.; Treccani, L.; Rezwan, K. SiAlON-bonded SiC ceramics from a silicone polymer and nano-sized fillers. Adv. Appl. Ceram 2013, 112, 158–162. [Google Scholar]
- Kang, Y.C.; Park, S.B. Zn2SiO4:Mn phosphor particles prepared by spray pyrolysis using a filter expansion aerosol generator. Mater. Res. Bull 2000, 35, 1143–1151. [Google Scholar]
- Zhang, Q.; Wang, J.; Zhang, M.; Ding, W.; Su, Q. Enhanced photoluminescence of Ca2Al2SiO7:Eu3+ by charge compensation method. Appl. Phys. A 2007, 88, 805–809. [Google Scholar]
- Park, Y.J.; Kim, Y.J. Blue emission properties of Eu-doped CaAl2O4 phosphors synthesized by a flux method. Mat. Sci. Eng. B 2008, 146, 84–88. [Google Scholar]
- Kodama, N.; Tanii, Y.; Yamaga, M. Optical properties of long-lasting phosphorescent crystals Ce3+-doped Ca2Al2SiO7 and CaYAl3O7. J. Lumin 2000, 87–89, 1076–1078. [Google Scholar]
- Hanifi, A.R.; Genson, A.; Redington, W.; Pomeroy, M.J.; Hampshire, S. Effects of nitrogen and fluorine on crystallisation of Ca-Si-Al-O-N-F glasses. J. Eur. Ceram. Soc 2012, 32, 849–57. [Google Scholar]
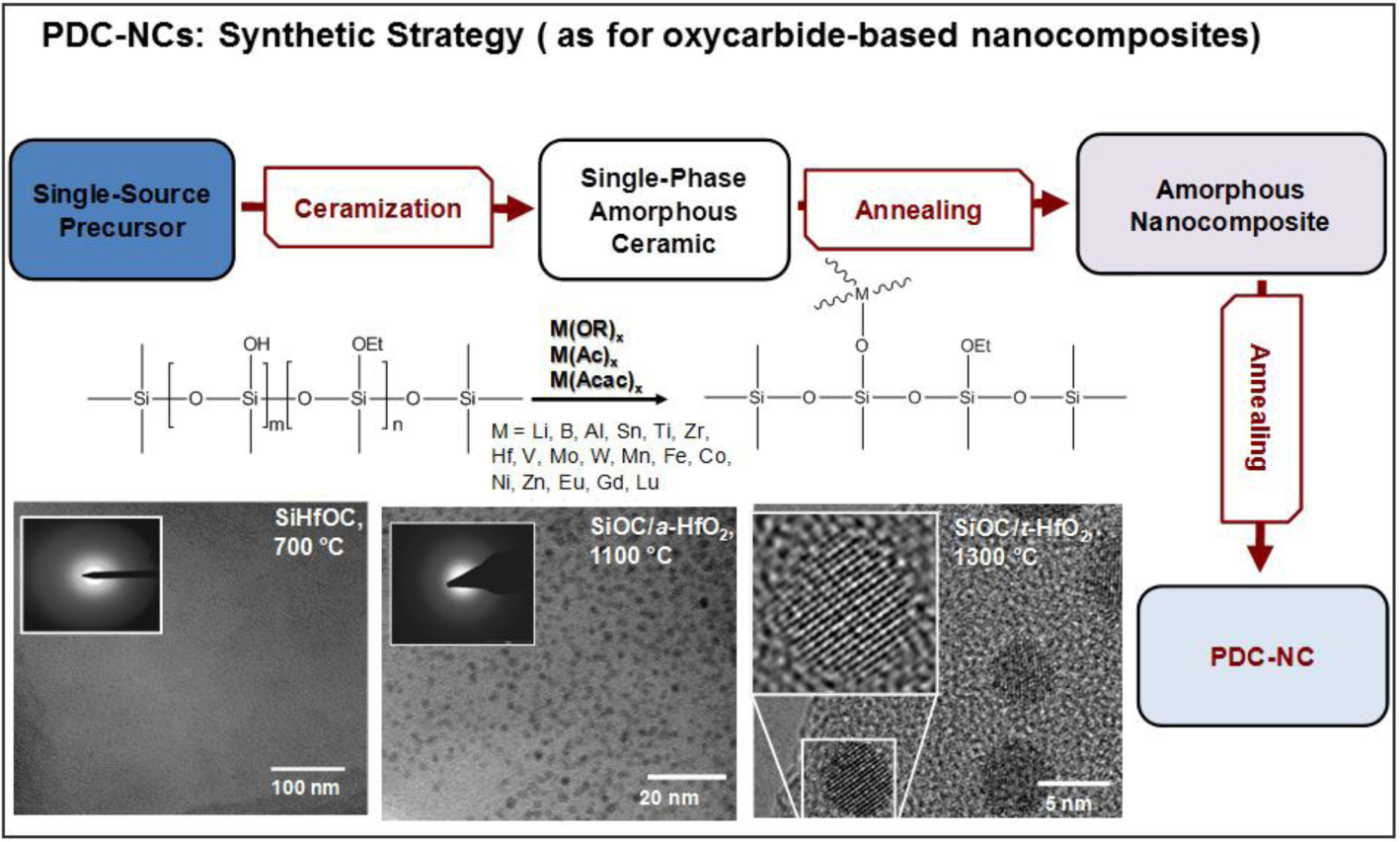

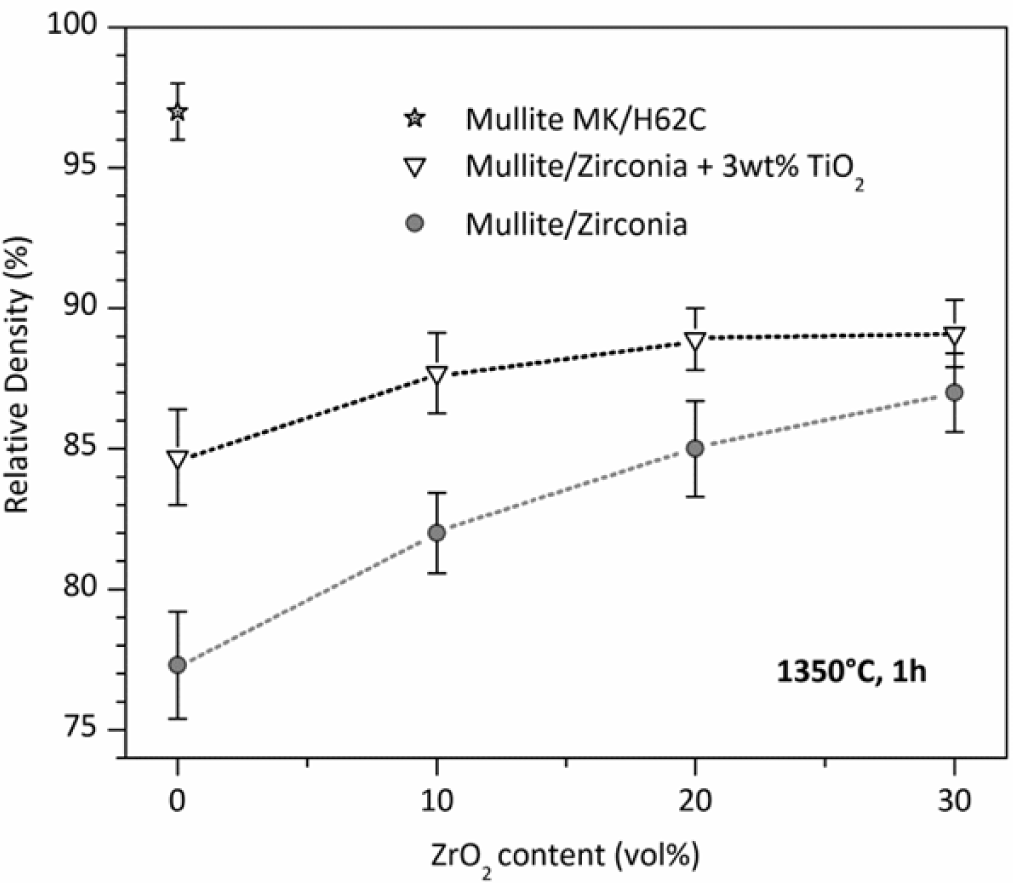

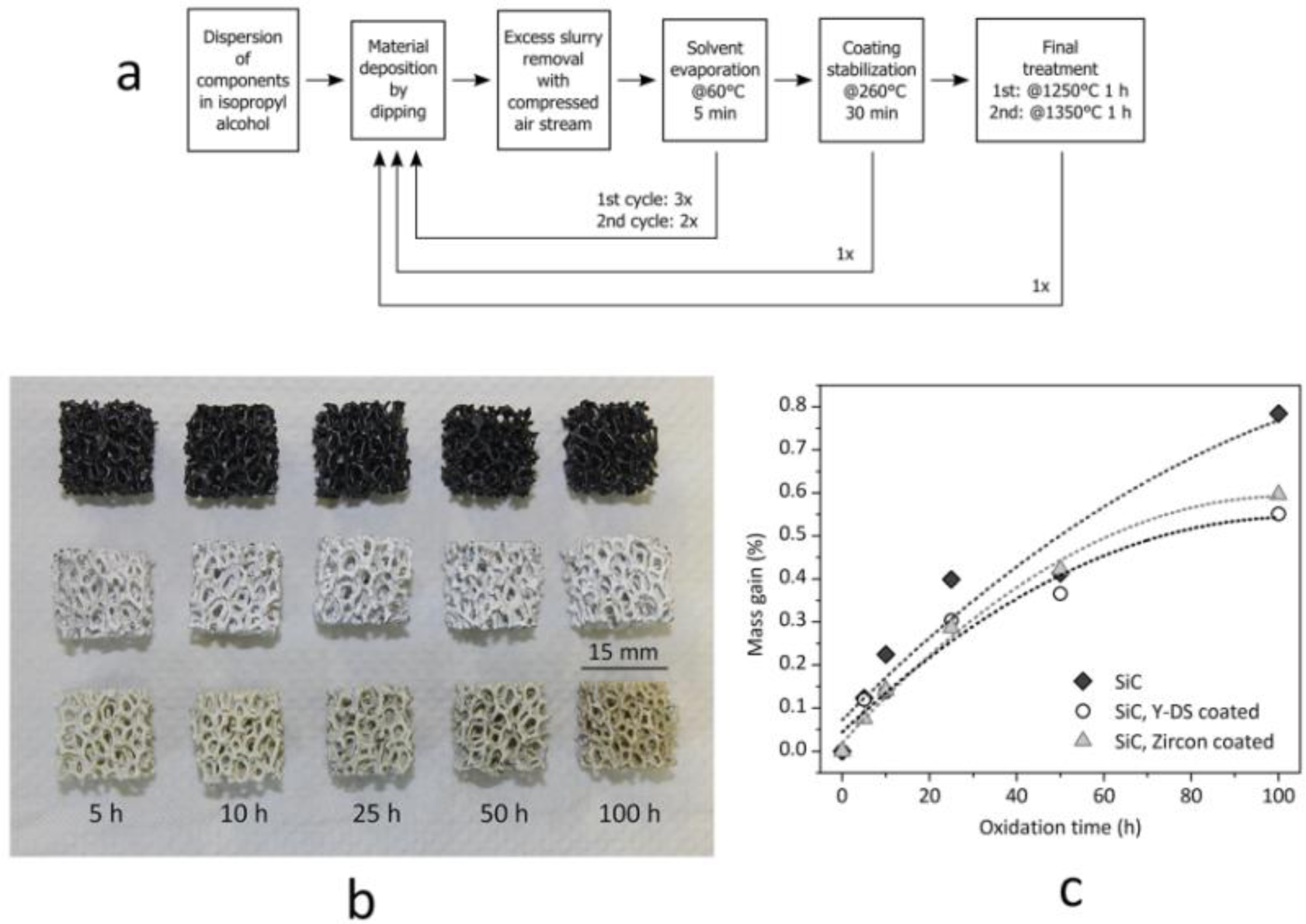
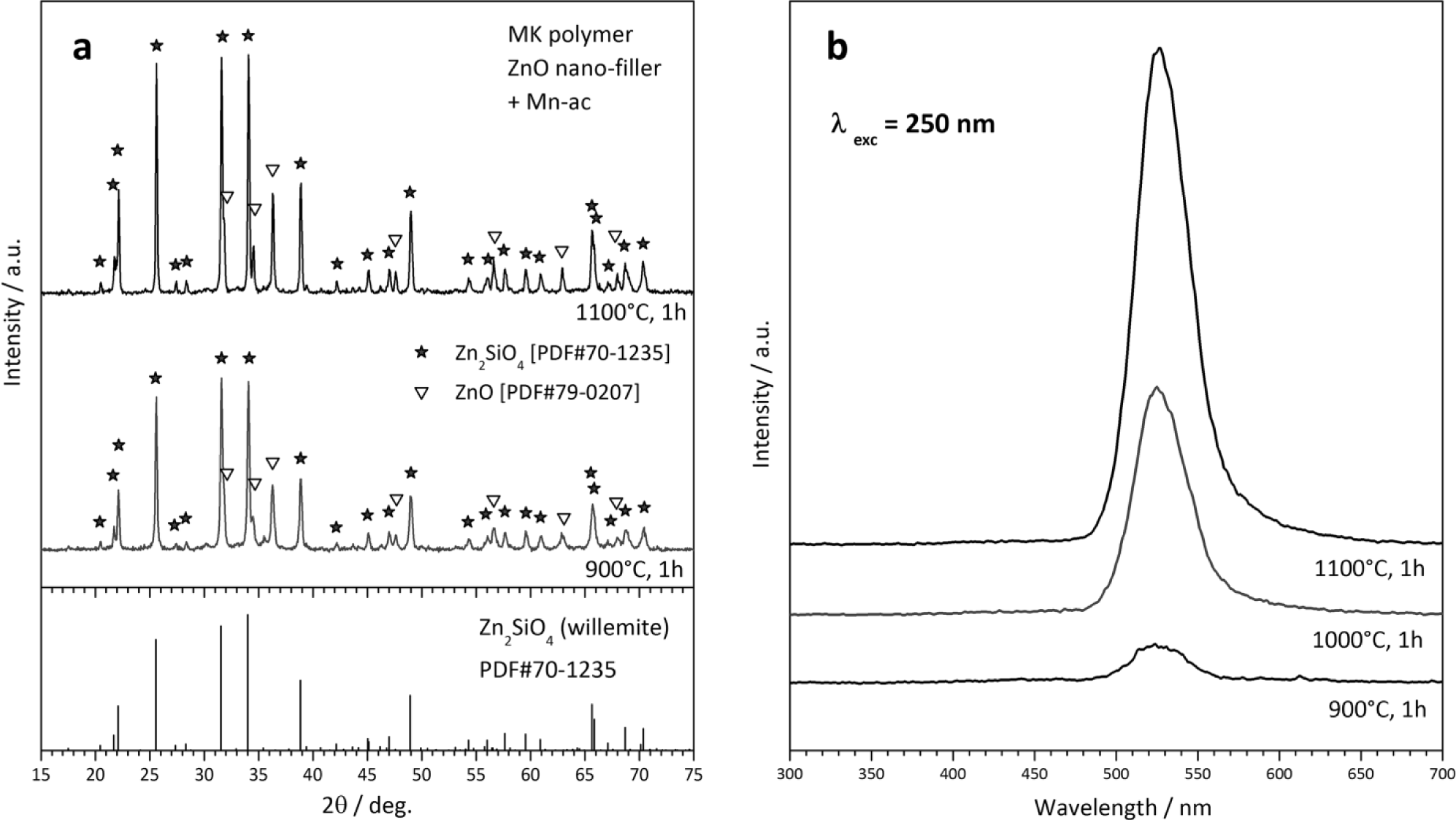
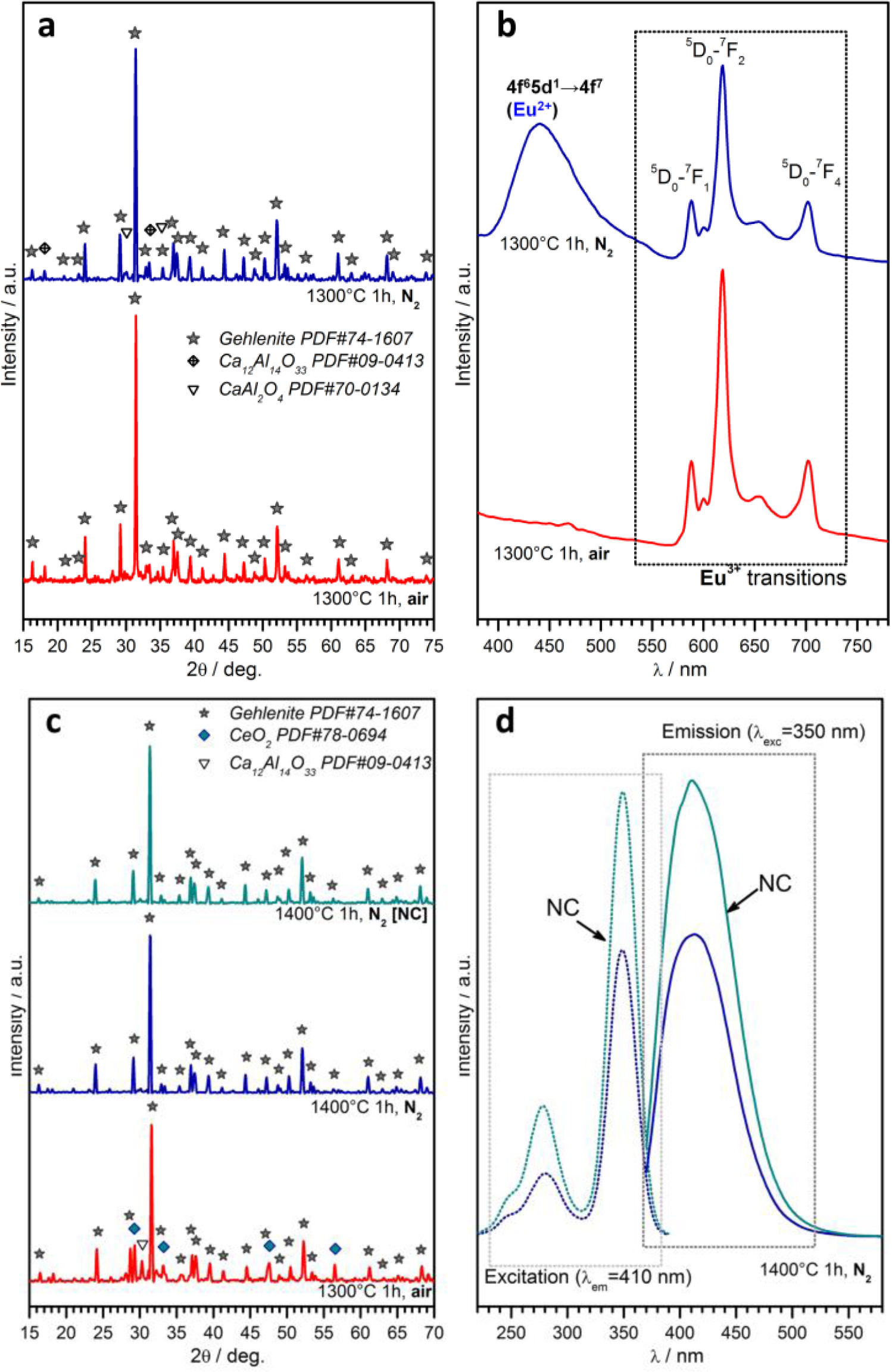

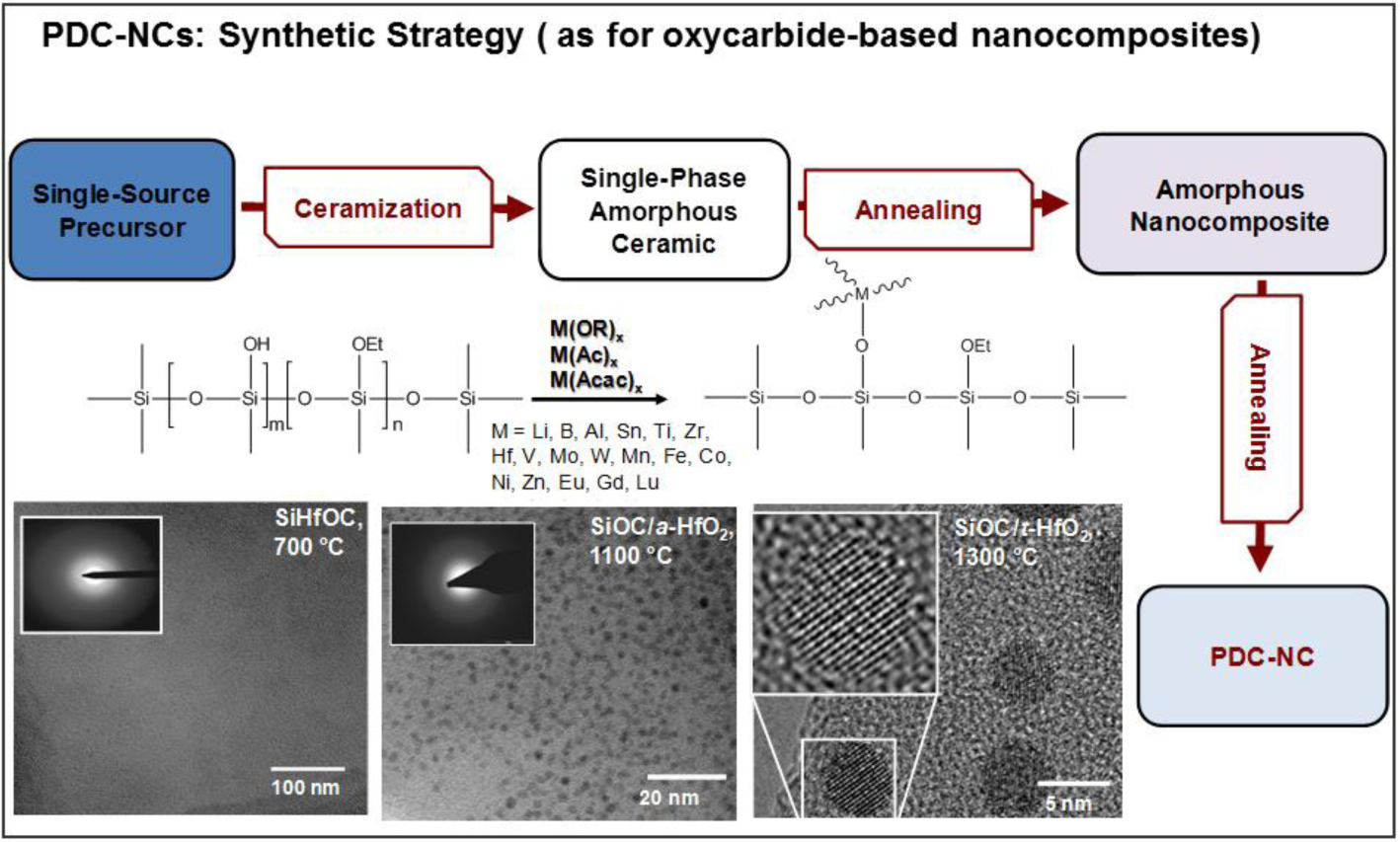{kind=link}
{kind=link}
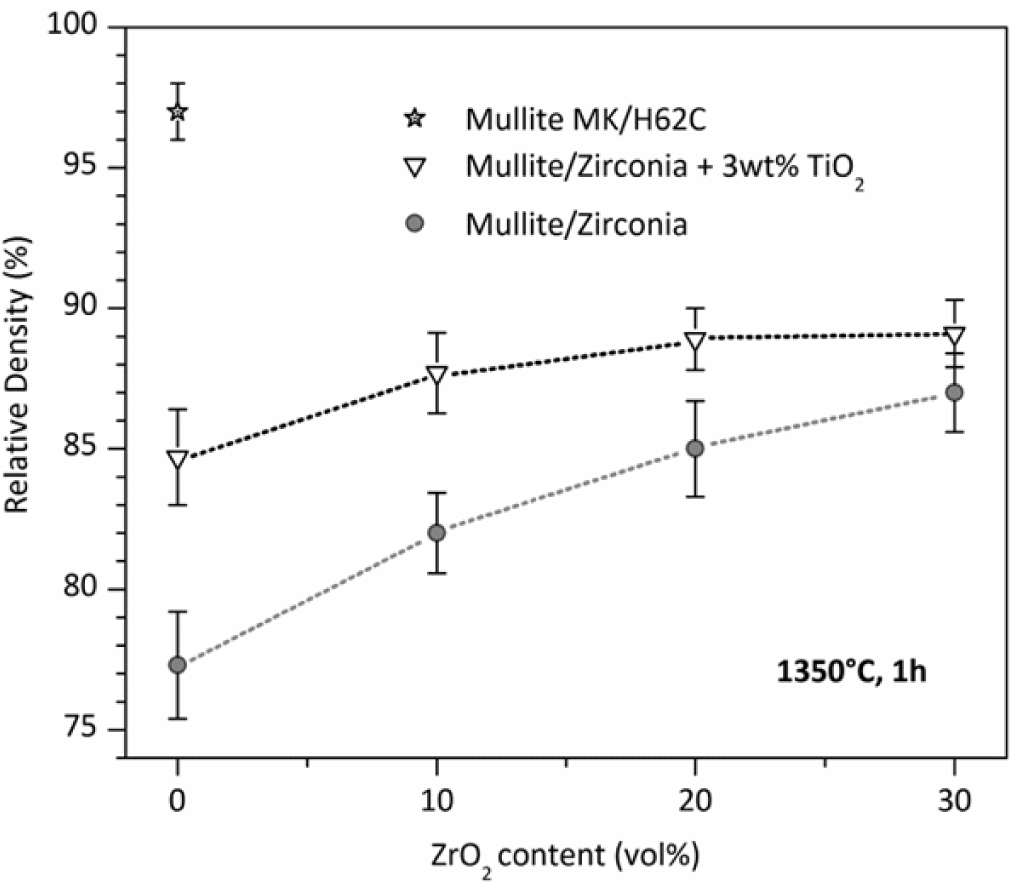{kind=link}
{kind=link}
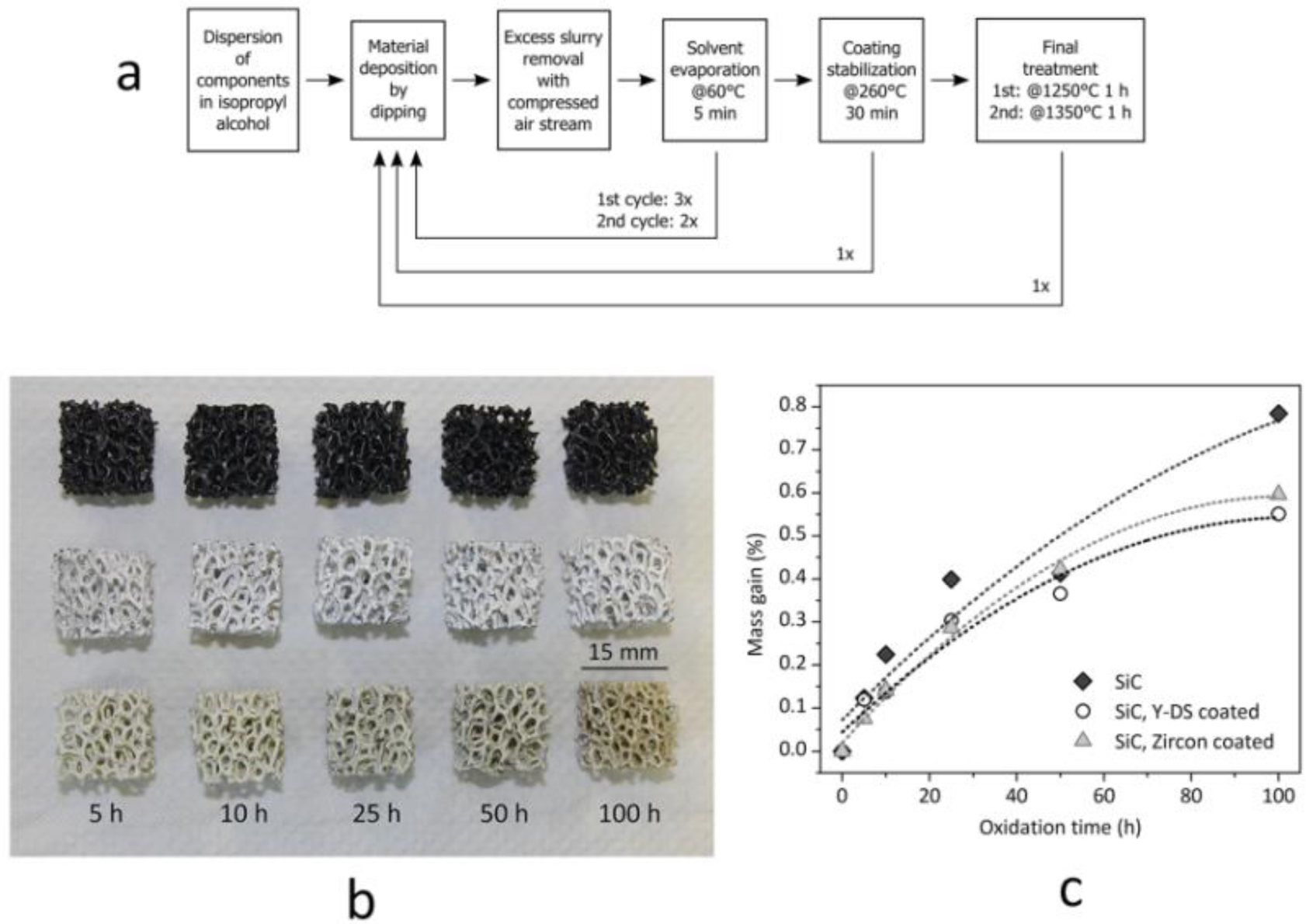{kind=link}
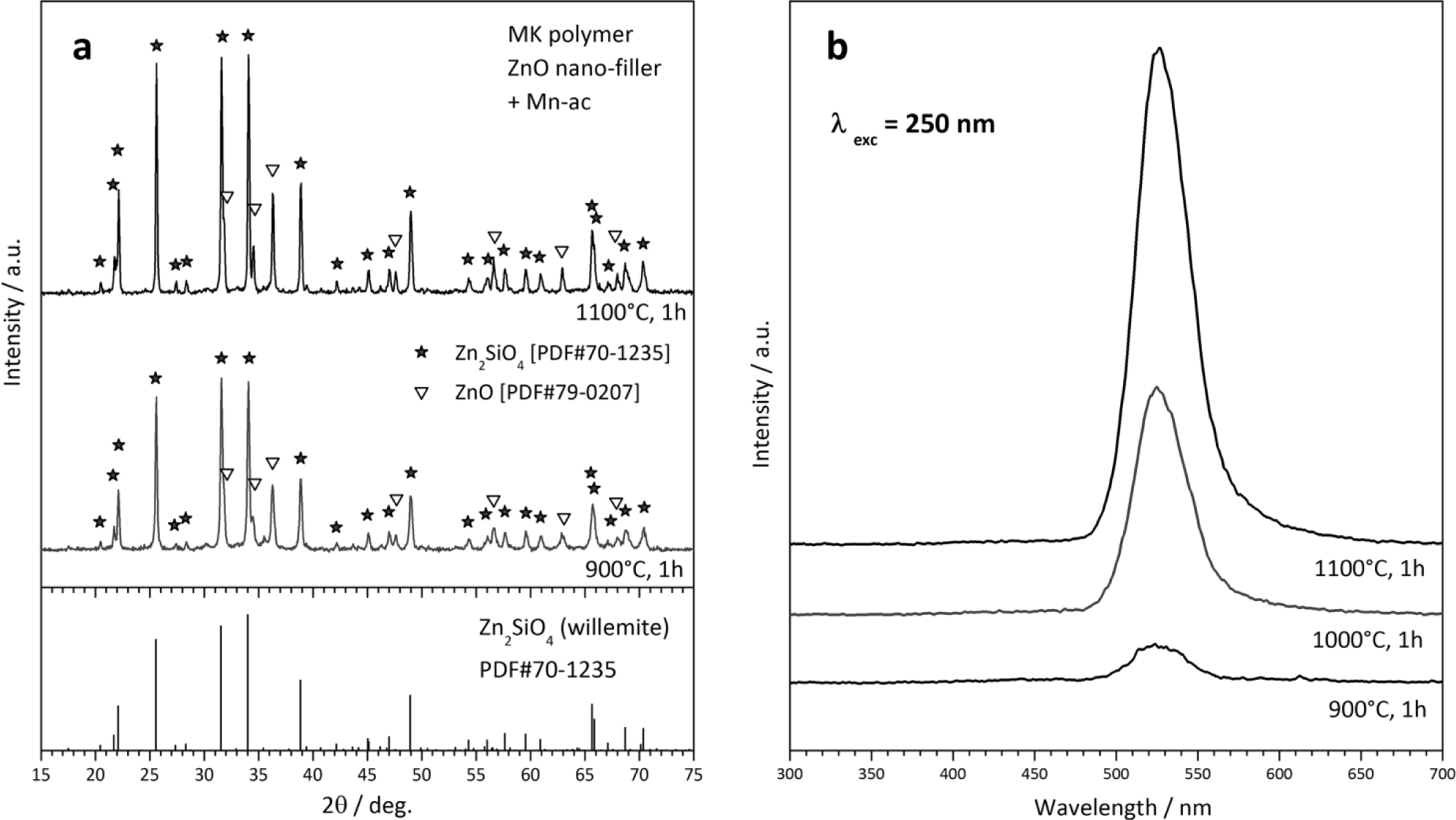{kind=link}
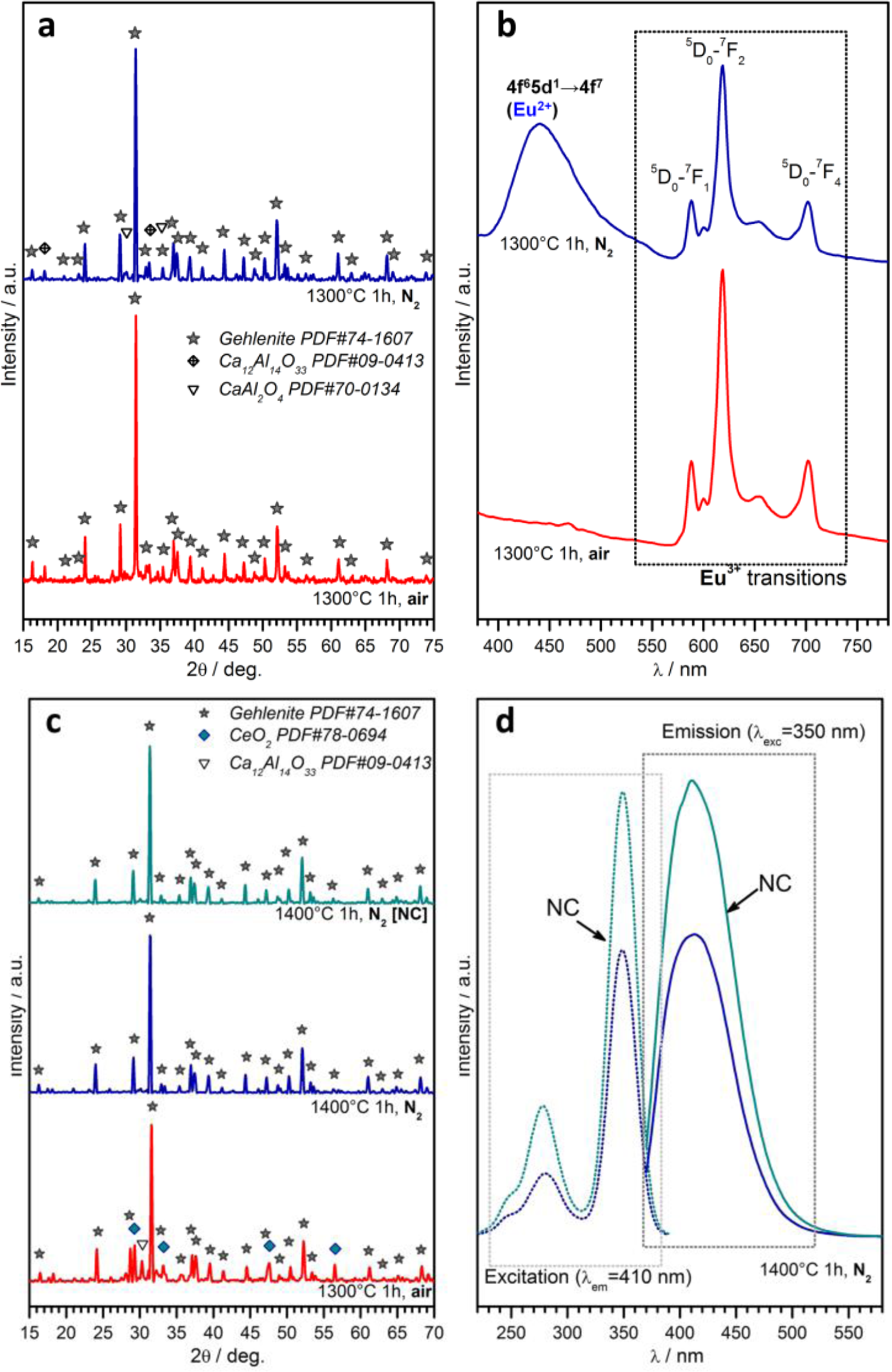{kind=link}
| Ceramic phase | Polymer | Nano-sized filler | Secondary components | Remarks | Reference |
|---|---|---|---|---|---|
| Mullite (3Al2O3·2SiO2) | MK | γ-Al2O3 (15 nm, E) | – | Monoliths grain size <300 nm | [28,31] |
| MK + H62C | Denser samples | * | |||
| H62C | Borax | Acicular mullite crystals | * | ||
| ZTM (Zirconia Toughened Mullite) | MK | γ-Al2O3 (15 nm, E) | ZrO2 (13 nm, E) TiO2 (13 nm, E) | Reinforced monoliths (KC ~ 6.5 MPa·m0.5) | [34] |
| Wollastonite (CaO·SiO2) | MK | – | Ca-acetate | Monoliths and foams | [32] |
| CaO (<170 nm, D) | – | ||||
| CaCO3 (90 nm, P) | n-HAp, m-HAp | [35] | |||
| MK + H62C | CaCO3 (90 nm, P) | TEOS | 3D scaffolds | [108] | |
| Yttrium mono-silicate (Y2O3·SiO2) | MK | Y2O3 (30–50 nm, I) | Eu2O3 (45-60 nm, C) | Phosphor powders | [109] |
| Yttrium di-silicate (Y2O3·2SiO2) | – | – | Environmental barrier coatings | * | |
| Zircon (ZrO2·SiO2) | MK, H62C | ZrO2 (13 nm, E) | TiO2 (13 nm, E) Zircon seeds | Monoliths, environmental barrier coatings | [111] |
| Forsterite (2MgO·SiO2) | MK, H62C | MgO (30 nm, I) | TiO2 (13 nm, E) m-TiO2 | Monoliths for dielectric components | [112] |
| Willemite (2ZnO·SiO2) | MK | ZnO (30–50 nm, I) | Mn-acetate | Phosphor powders | * |
| Cordierite (2MgO·2Al2O3·5SiO2) | MK, H62C | γ-Al2O3 (15 nm, E) MgO (30 nm, I) | – | Monoliths and foams | [113] |
| Gehlenite (2CaO·Al2O3·SiO2) | MK | γ-Al2O3 (15 nm, E) CaCO3 (90 nm, P) | Eu2O3 (45-60 nm, C) CeO2 (20 nm, M) | Phosphors for treatment in air or in N2; Ce-doping effective in N2 | [114] |
| Akermanite (2CaO·MgO·2SiO2) | MK, H62C | CaCO3 (90 nm, P) MgO (30 nm, I) | m-HAp Borax | Monoliths and foams | [115] |
| Hardystonite (2CaO·ZnO·2SiO2) | MK | γ-Al2O3 (15 nm, E) ZnO (30–50 nm, I) | Eu2O3 (45-60 nm, C) | Phosphor powders | * |
| β′-SiAlON | MK, H44 | γ-Al2O3 (15 nm, E) | Si3N4, AlN, SiC | Monoliths, foams, ceramic joints | [30,33,36] |
| PSZ20, NN120-20 | Si3N4 (20 nm, G) Eu2O3 (45-60 nm, C) | Monoliths, phosphor powders | [39,110],* | ||
| Ca-α′-SiAlON | PSZ20 | γ-Al2O3 (15 nm, E) CaCO3 (90 nm, P) | Eu2O3 (45-60 nm, C) | Phosphor powders | * |
| Y-Si-O-Ns | MK | Y2O3 (30–50 nm, I) | Eu2O3 (45-60 nm, C) CeO2 (20 nm, M) | Phosphor powders | [39] |
Notes: Suppliers of nano-sized fillers: C = Cometox Srl, Milan, Italy; D = DGTech, Grenoble, France; E = Evonik Industries AG, Essen, Germany; G = Goodfellows, Huntingdon, U.K.; I = Inframat Advanced Materials, Manchester, CT; M = MKnano, M K Index Corp., Missisauga, Canada; P = PlasmaChem GmbH, Berlin, Germany; m-HAp: hydroxyalapatite micro-powders; b-HAp: hydroxyalapatite nano-powders; m-TiO2: titania micopowders; Polymers: MK, H44 and H62C (silicones) from Wacker Chemie AG, München, Germany; PSZ20: KiON Defence Technologies Inc., Huntingdon Valley, PA, USA;. NN120-20: Clariant AG, Sulzbach, Germany.
| Component | Amount used for Y-DS coating (wt%) | Amount used for zircon coating (wt%) |
|---|---|---|
| MK polymer | 7 | 3 |
| H62C polymer | – | 5 |
| Kaolin | 1 | 2 |
| Mullite powders | 13 | – |
| Zircon powders | – | 12 |
| Nano-Y2O3 | 12 | – |
| Nano-ZrO2 | – | 11 |
| Nano-TiO2 | – | 1 |
| Isopropyl alcohol | 67 | 66 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bernardo, E.; Fiocco, L.; Parcianello, G.; Storti, E.; Colombo, P. Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review. Materials 2014, 7, 1927-1956. https://doi.org/10.3390/ma7031927
Bernardo E, Fiocco L, Parcianello G, Storti E, Colombo P. Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review. Materials. 2014; 7(3):1927-1956. https://doi.org/10.3390/ma7031927
Chicago/Turabian StyleBernardo, Enrico, Laura Fiocco, Giulio Parcianello, Enrico Storti, and Paolo Colombo. 2014. "Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review" Materials 7, no. 3: 1927-1956. https://doi.org/10.3390/ma7031927
APA StyleBernardo, E., Fiocco, L., Parcianello, G., Storti, E., & Colombo, P. (2014). Advanced Ceramics from Preceramic Polymers Modified at the Nano-Scale: A Review. Materials, 7(3), 1927-1956. https://doi.org/10.3390/ma7031927