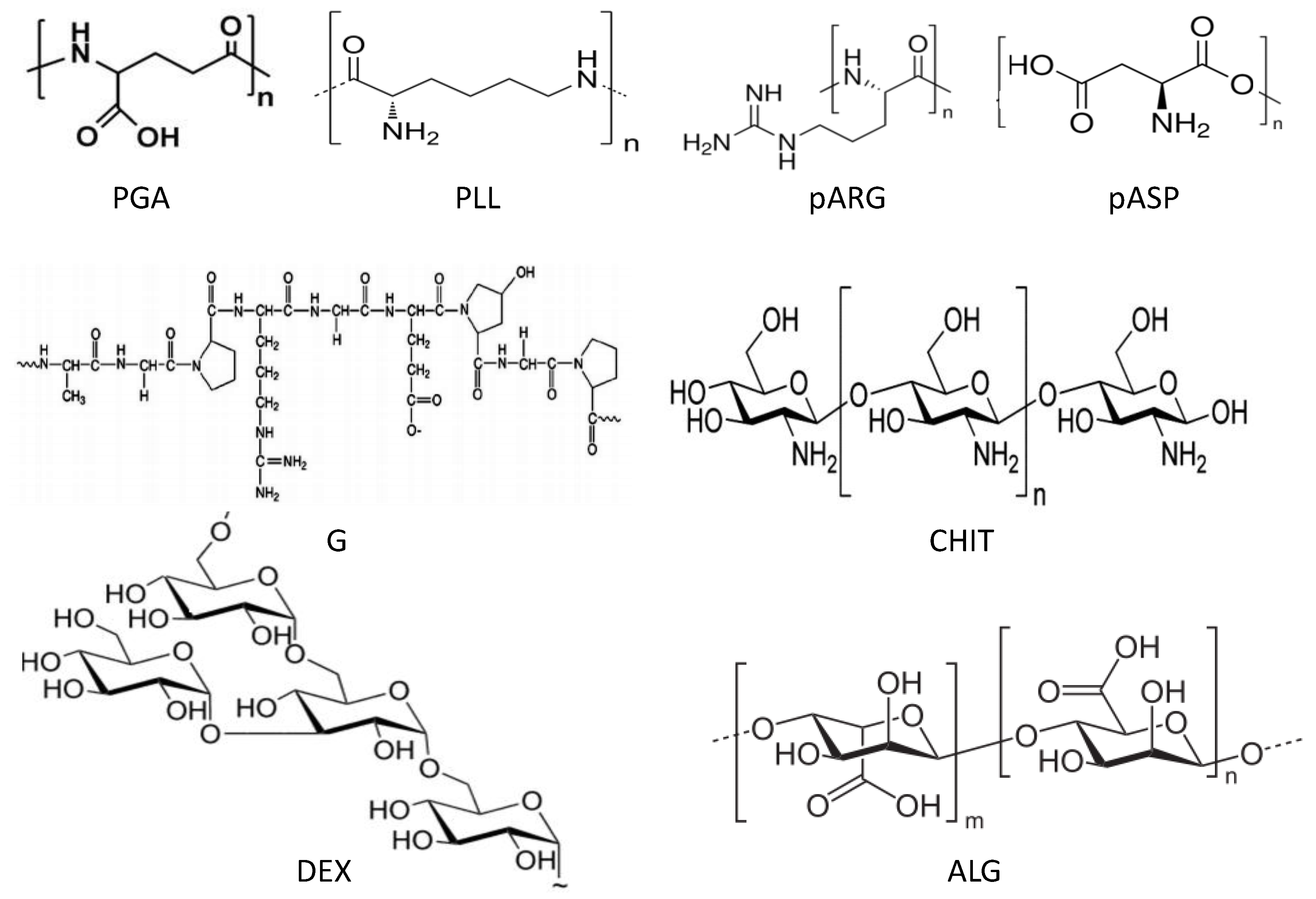
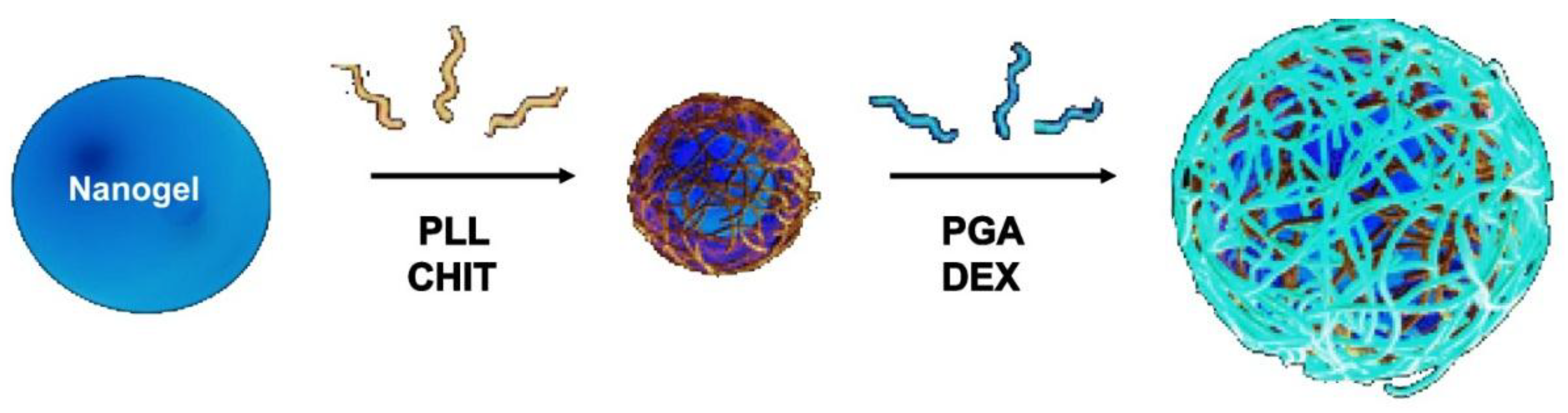
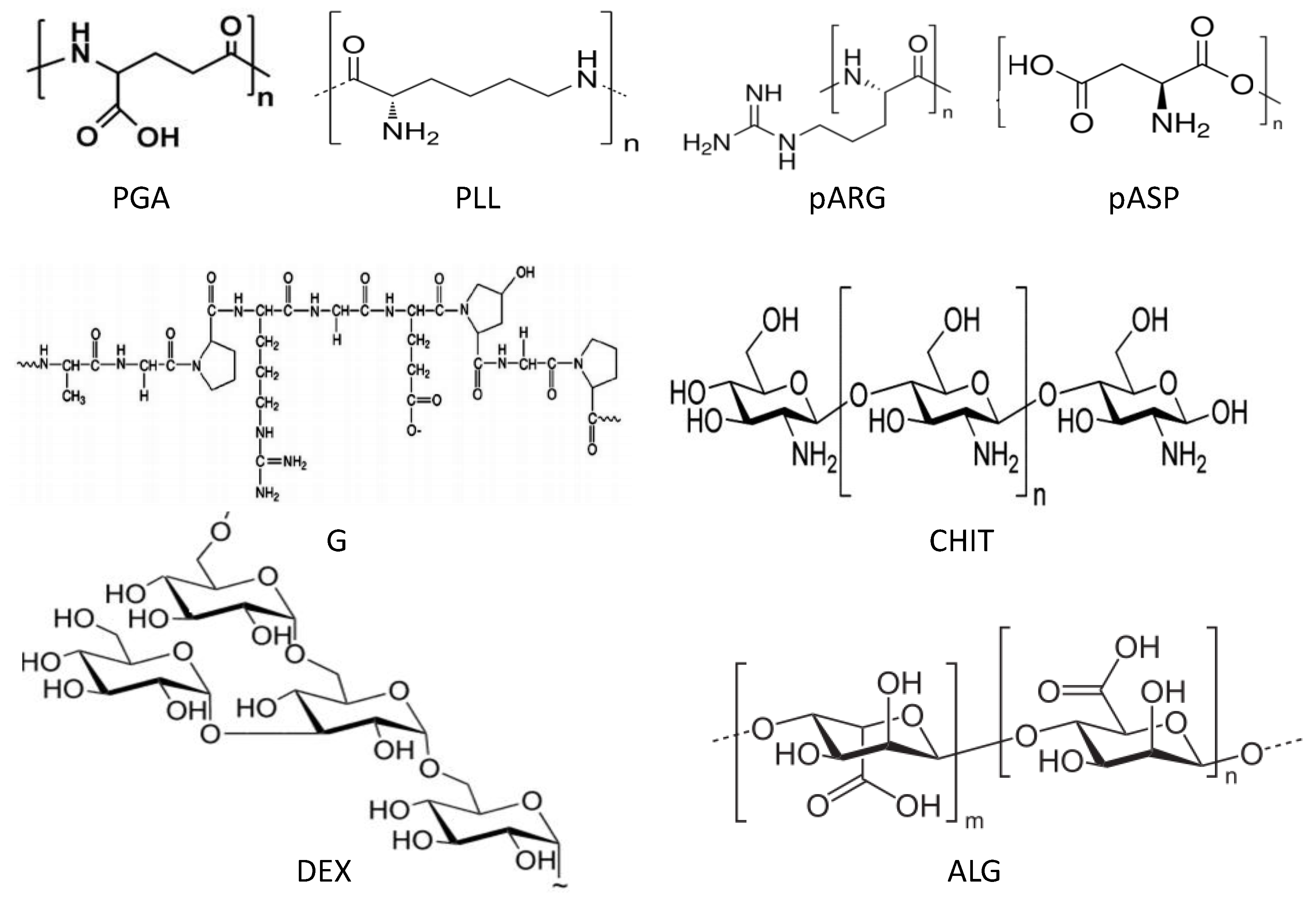
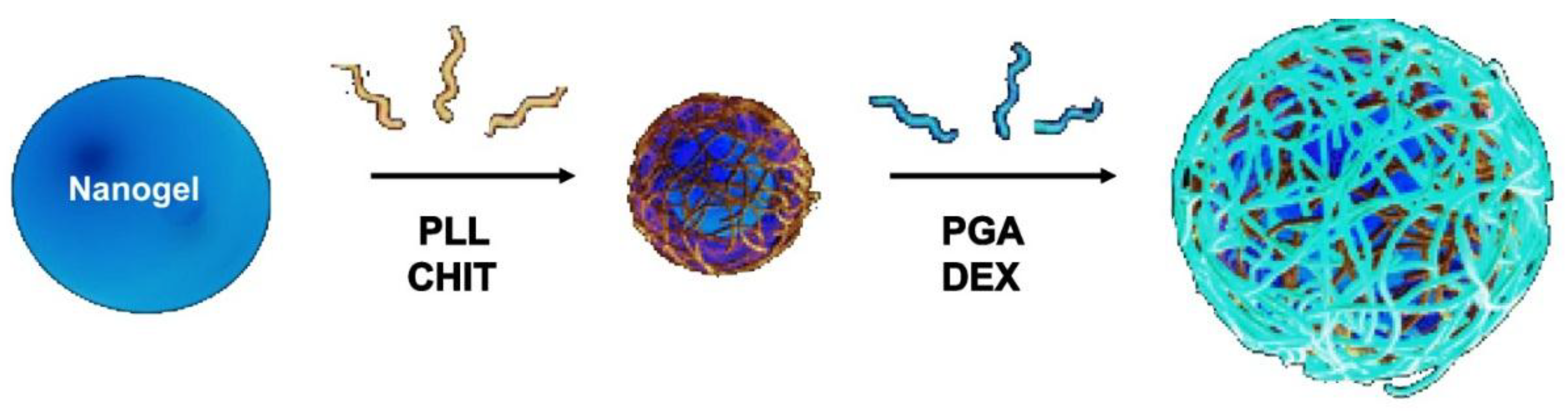
6.1. Morphological Characterization
The morphology of uncoated and coated-MGs (or NGs) in the hydrated state can be observed directly by ESEM. Diez-Pascual and Wong [
8] analysed the morphology of PNIPAM-
co-MAA coated with polypeptides and polysaccharides, as illustrated in
Figure 1. The image of the pure NG (
Figure 1a) showed soft, rough and porous nanospheres interconnected by bridges. In contrast, the PLL- and CHIT-coated NGs (
Figure 1b,c, respectively) were more separated and presented more individual particles with few connections; analogous morphology has been described for all coated NGs [
8]. The comparison of NG/PLL and NG/CHIT images revealed that polypeptide-coated systems partially maintain the rough surface of the uncoated NG, while those covered with polysaccharides exhibit a smoother structure. This behaviour could be related to differences in layer thickness, as reported for biopolymers assembled onto flat substrates [
37]. Polypeptides have more propensity to rearrange and form complexes with both PLL and PGA in β sheet antiparallel conformation, with extensive interpenetration and strong ionic interaction between biopolymers from adjacent layers, which lead to a highly compact structure that fits easily onto the NG surface, thus retaining its rough morphology. This is consistent with the images reported by De Geest
et al. [
6] for pARG-coated DEX-HEMA MGs showing very rough microspheres attached to each other.
Figure 1.
Typical environmental scanning electron microscopy (ESEM) micrographs of: (
a) bare NG; (
b) NG/(PLL/PGA) and (
c) NG/(CHIT/dextran sulfate (DEXS)) in the hydrated state. Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 1.
Typical environmental scanning electron microscopy (ESEM) micrographs of: (
a) bare NG; (
b) NG/(PLL/PGA) and (
c) NG/(CHIT/dextran sulfate (DEXS)) in the hydrated state. Reproduced with permission from [
8], copyright 2010, Elsevier.
Though, as can be seen in
Figure 1 the PNIPAM-
co-MAA systems presented small evenly sized spherical particles, the average diameter of which, for the pure NG, NG/CHIT and NG/PLL was 692, 462 and 590 nm, and those of the bi-coated samples, NG/(CHIT/DEXS) and NG/(PLL/PGA) were 538 and 676 nm, respectively, in agreement with measurements obtained from DLS experiments in the swollen state [
8]. The decrease in diameter upon the addition of the initial polycation layer and
vice versa increasing with addition of the second polyanion coating is not untypical and has been seen for a number of systems below the VPTT temperature, such as PLL and PGA coated-PNIPAM-
co-MAA MGs [
27] as well as NGs coated with synthetic polymers such as PDACMAC or PPS [
19]. The electrostatic attraction between the negatively charged NG and the positively charged polycation layer induces a contraction, whilst the addition of the next polyanion layer affectively shares the positive charge with the NG, weakening the interaction and results in a relaxation in the structure, and hence, increase in diameter. Overall, SEM images not only provide evidence of the success of the LbL assembly process, but also demonstrate that the morphology of the NGs can be tuned by surface adsorption of the PE layers.
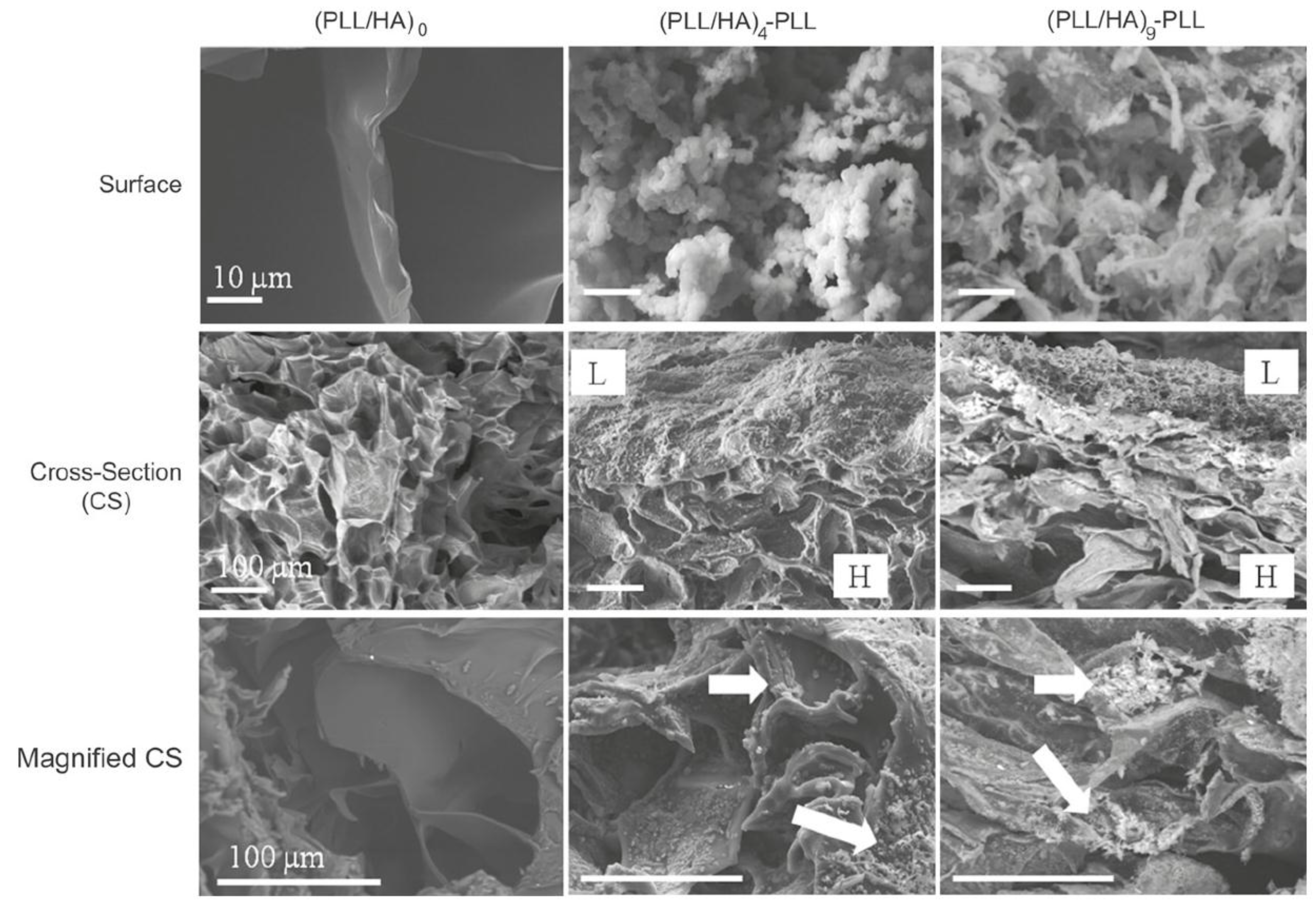
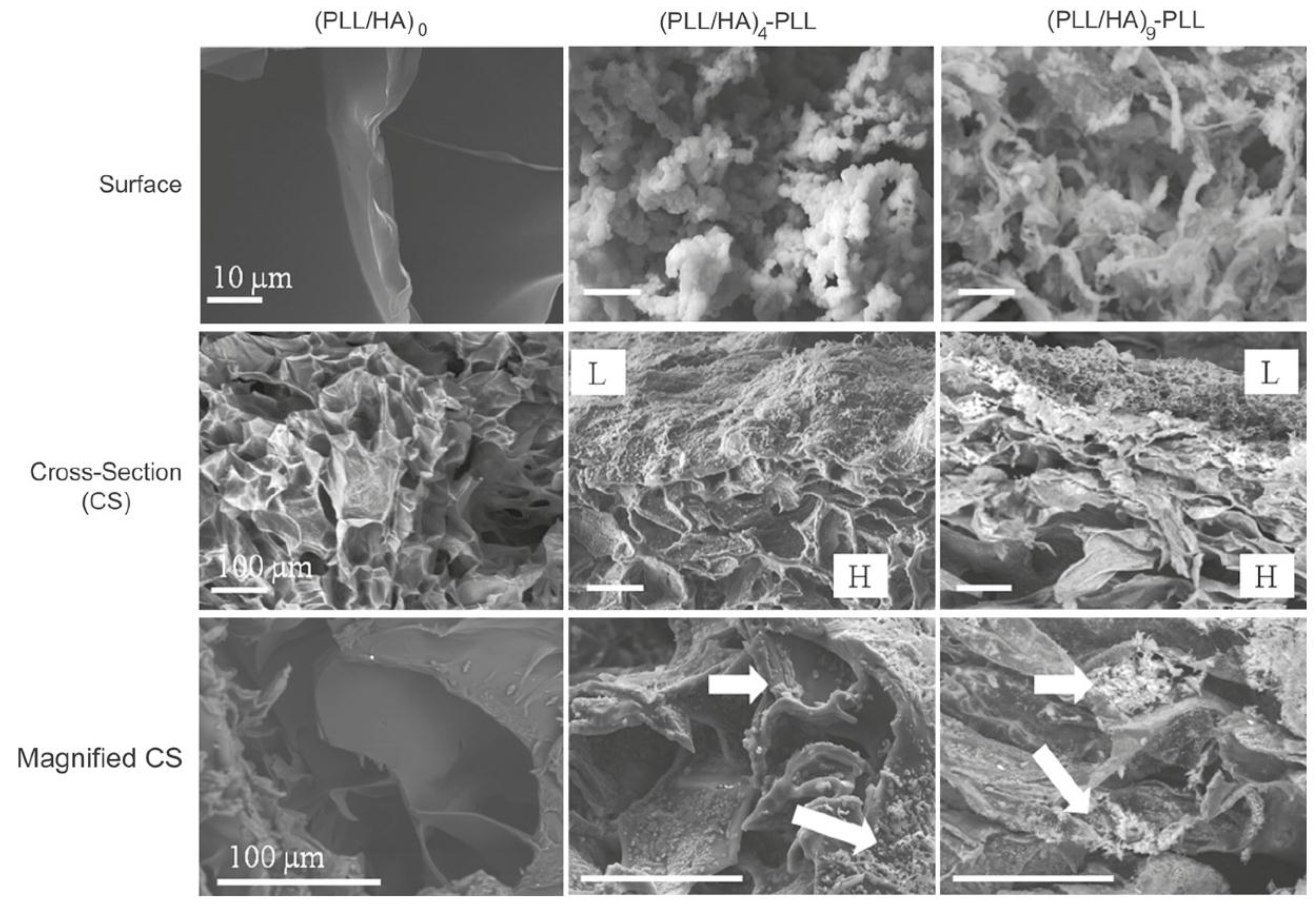
The morphology of HA MGs LbL coated with PLL/HA layers was investigated by Yamanlar
et al. [
37] (
Figure 2). SEM images of the bare MG (left column) revealed a smooth flaky surface, in contrast to the morphology found for PNIPAM based gels [
8,
19,
27]. However, systems with four and nine PLL/HA bilayers (middle and right columns, respectively), displayed a rough surface with granular or hair-like structures. The outer surface of the coated MGs showed formation of the LbL film (labelled as L in the micrographs), and the film thickness was higher for the assembly with (PLL/HA)
9-PLL as compared to (PLL/HA)
4-PLL. Moreover, granular features were visible in the bulk region of the hydrogel (marked as H) indicating diffusion of the PEs into the gel (indicated by arrows). When the HA MG was brought into contact with the PLL, some of the polycations diffused into the bulk gel, likely forming the initial phase of multilayer growth inside the intrinsic pores due to negatively charged HA. However, as the deposition steps increased, the layers were formed on the surface masking the MG pores as validated by the SEM images (top row).
Figure 2.
Scanning electron microscope (SEM) images of the top surface and cross-sections of HA MGs (left column) and microgels (MGs) coated with (PLL-HA)
4-PLL, and (PLL-HA)
9-PLL films (middle and right columns, respectively). The arrows indicate diffusion of the PEs into the gel. Reprinted with permission from [
37], copyright 2011, Elsevier.
Figure 2.
Scanning electron microscope (SEM) images of the top surface and cross-sections of HA MGs (left column) and microgels (MGs) coated with (PLL-HA)
4-PLL, and (PLL-HA)
9-PLL films (middle and right columns, respectively). The arrows indicate diffusion of the PEs into the gel. Reprinted with permission from [
37], copyright 2011, Elsevier.
Kadi
et al. [
30] investigated the morphology of alkylamino hydrazide-modified HA derivatives.
Figure 3 shows AFM images of the surface of the systems containing 18 modified HA/PLL layer pairs. Two characteristic morphologies were observed: those incorporating alkylated derivatives with six and eight carbon atoms showed a smooth surface, similar to that of neat HA/PLL, with a mean roughness of 4 nm (
Figure 3a). However, films based on derivatives with 10 carbon atoms displayed a rough surface (
Figure 3b–d), particularly that with a DS of 5% and linear alkyl chains (
Figure 3d) which had an average roughness of 98.2 nm. On the other hand, “small grains” or microaggregates were visible at the surface of systems with 10 carbon atoms and linear chains. This presence of small aggregates whose size depends on the alkyl chain length and the DS is consistent with previous LbL films made of PAA and hydrophobically modified poly(ethylene oxide). Thus, it was proposed that derivatives with long chains and high DS had an irregular internal structure, with hydrophobic nanodomains coexisting with hydrophilic domains. Similar behaviour was reported for VB-g-HA/PLL, where the mean surface roughness of the systems increased from 0.3 to 9 nm when the grafting degree rose from 14% to 37% [
29].
Figure 3.
Atomic force microscopy (AFM) images of different alkylamino hydrazide-modified HA/PLL systems with 18 layer pairs: (
A) linear C6 derivative with DS of 5%; (
B) branched C10 derivative with DS of 5%; (
C) linear C10 derivative with DS of 5% and (
D) linear C10 derivative with DS of 10%. Reproduced with permission from [
30], copyright 2009, American Chemical Society.
Figure 3.
Atomic force microscopy (AFM) images of different alkylamino hydrazide-modified HA/PLL systems with 18 layer pairs: (
A) linear C6 derivative with DS of 5%; (
B) branched C10 derivative with DS of 5%; (
C) linear C10 derivative with DS of 5% and (
D) linear C10 derivative with DS of 10%. Reproduced with permission from [
30], copyright 2009, American Chemical Society.
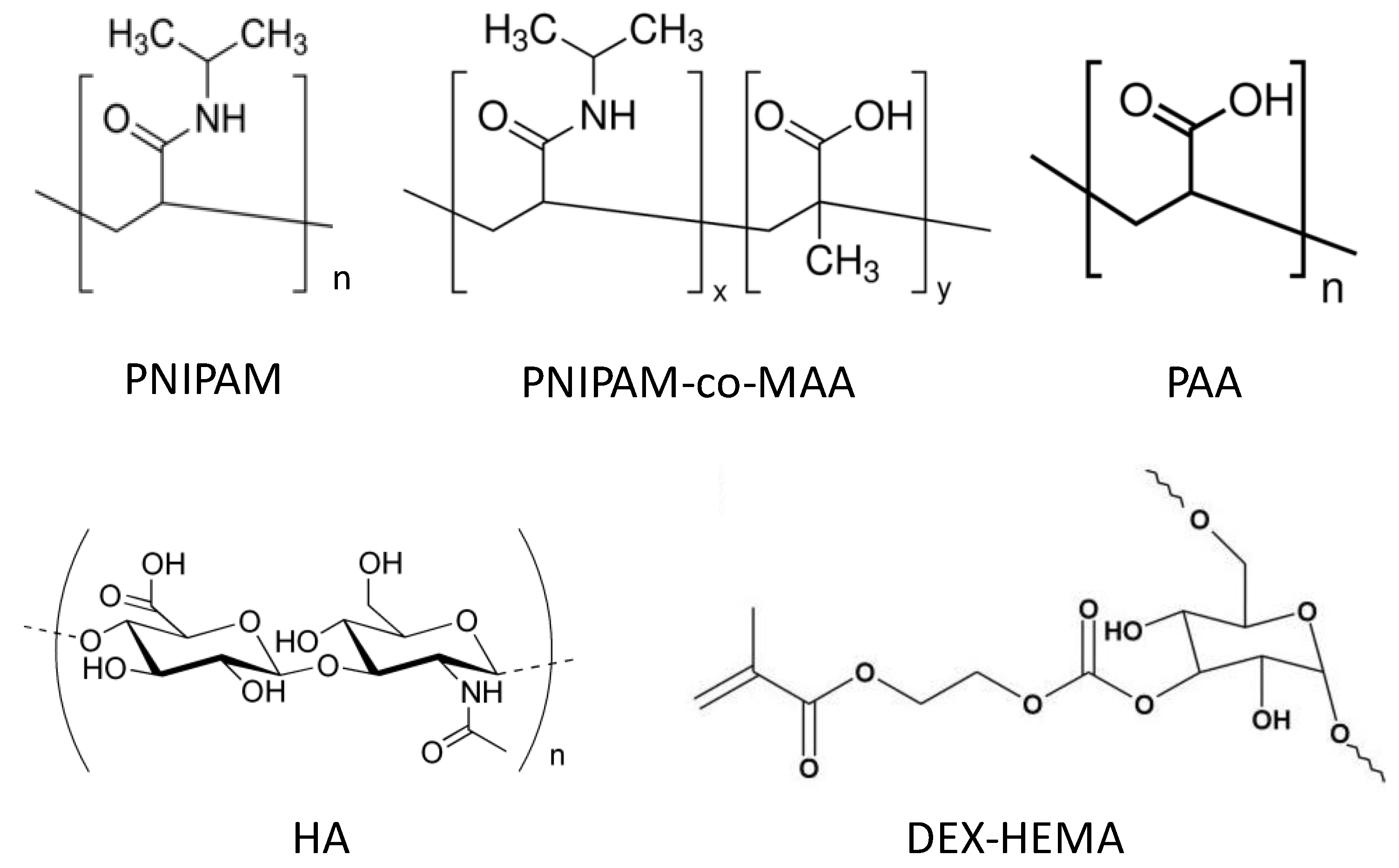
6.2. ATR-FTIR Spectra
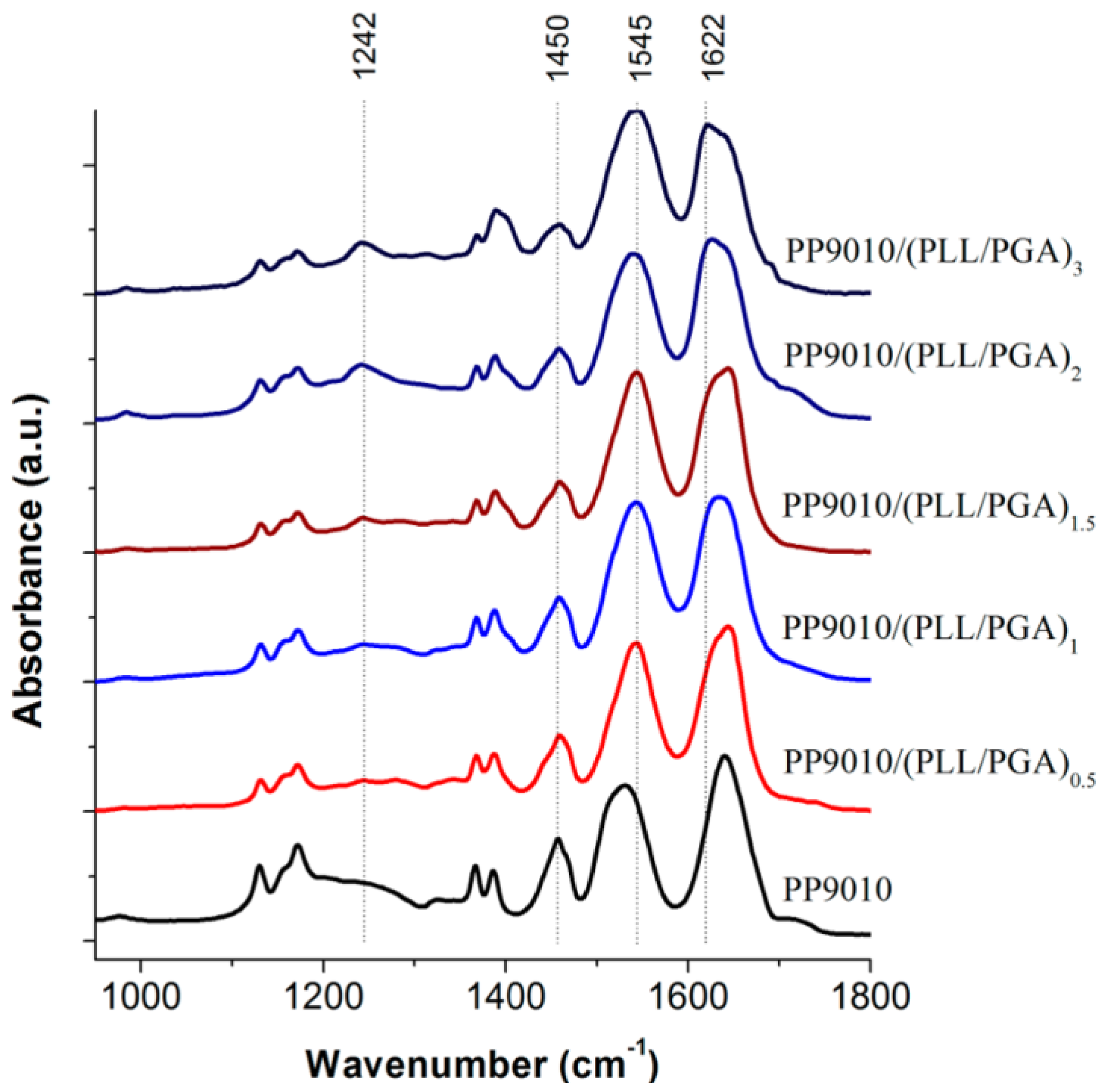
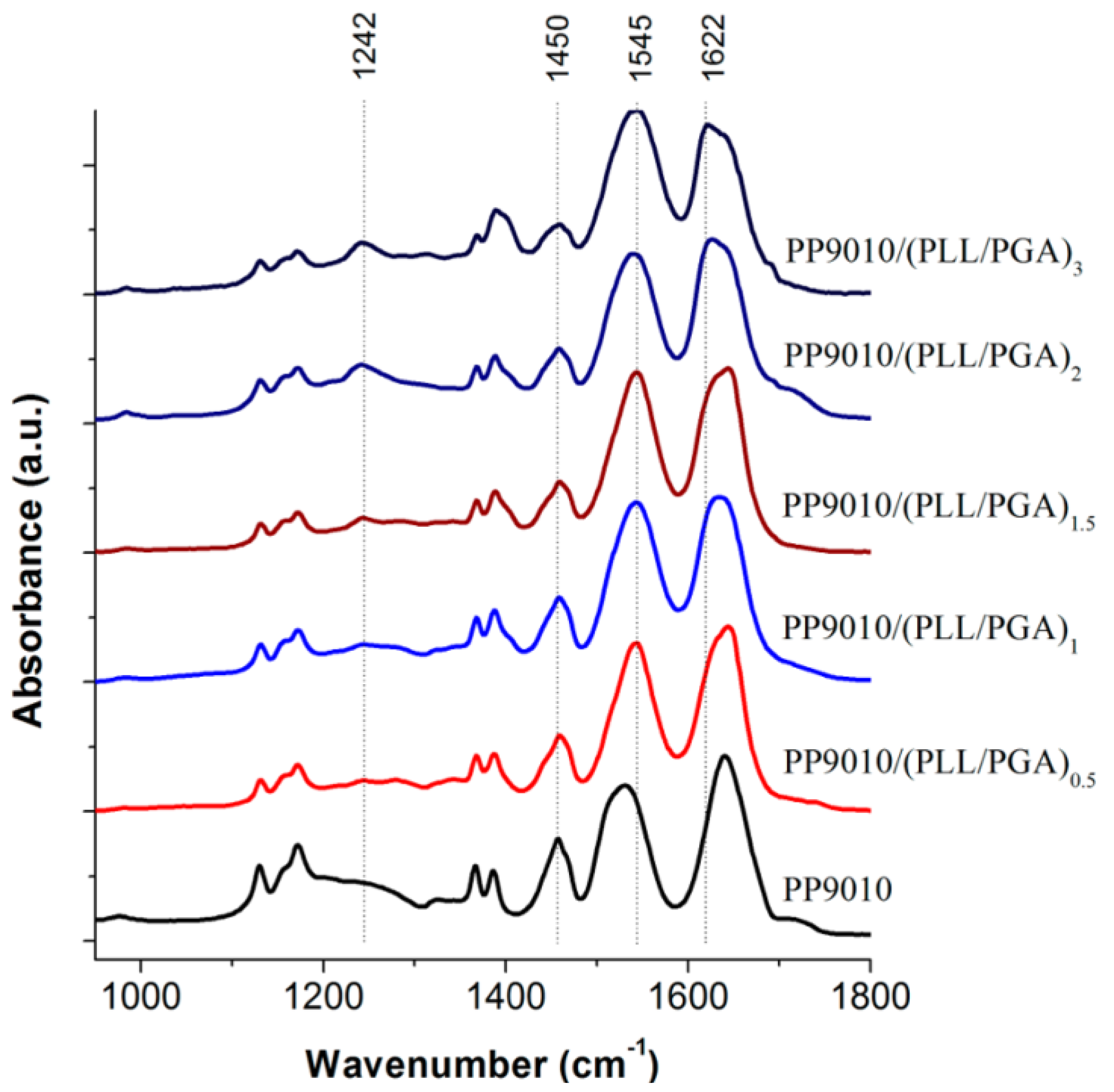
ATR-FTIR spectroscopy was used by Hammond and co-workers [
27] as shown in
Figure 4 to analyse the interactions between biopolymers and P(NIPAM-
co-MAA) MGs. Characterisation of materials is difficult due to the fact that the amide vibrations overlap with the characteristic PE bands. In addition, the polypeptide bands depend very much on their conformation (α-helix, β-sheet, or random coil), hydrogen bonding and vibrational interactions between the peptide groups. However, ATR-FTIR can be very informative, for example, in the case of the MG, it consists of two strong amide I and amide II bands at 1641 and 1531 cm
−1, and a weak amide III band at 1242 cm
−1 from the PNIPAM segments. Adsorption of the polypeptides leads to an intensity increase in the amide II band, and a decrease in the shoulder band at 1707 cm
−1, which corresponds to the C=O stretching of the unprotonated carboxylic groups of PMAA, suggesting that the PE layering process occurs primarily through ionic interactions.
Figure 4.
Attenuated total reflectance FT-IR spectroscopy (ATR-FTIR) spectra of polypeptide-coated P(NIPAM-
co-MAA) MGs. Reproduced with permission from [
27], copyright 2012, American Chemical Society.
Figure 4.
Attenuated total reflectance FT-IR spectroscopy (ATR-FTIR) spectra of polypeptide-coated P(NIPAM-
co-MAA) MGs. Reproduced with permission from [
27], copyright 2012, American Chemical Society.
Deposition of PLL on PNIPAM led to the shift in the amide bands to much higher energy states, characteristic of the vibrations from cross-linked homopolymers (δNH at 1543 cm−1, intramolecular H-bonded νC=O at 1644 cm−1 and free νC=O at 1669 cm−1). Again, the consequent adsorption of PGA altered intramolecular H-bonded νC=O (1632 cm−1) and free νC=O (1653 cm−1) to lower wavenumbers similar to that of the native NG, whereas the intensity of the free νC=O of PMAA COOH group was conserved, suggesting that the PNIPAM segments are strongly involved in H-bonding. Additionally, in the higher wavenumber region the contribution of PLL νN–H and PGA νO–H led to an increase in intensity and broadening of the N–H stretching between 3000 and 3450 cm−1 relative to the isopropyl and alkyl vibrations (2850–3000 cm−1). Subsequent PE assembly layering did not significantly alter the spectra, strengthening the previous discussion.
6.3. Surface Charge Density
Electrophoretic mobility (μ
e) measurements can be employed to monitor the subsequent deposition of PEs onto gels after each adsorption step. µ
e values at 20 °C of PNIPAM-
co-MAA NGs coated with polypeptides and polysaccharides as a function of the number of layers deposited are compared in
Figure 5, with an analogous trend found for other temperatures tested [
8]. µ
e of the bare PNIPAM-
co-MAA NG in aqueous solution is −2.7 × 10
−8 m
2 V
−1 s
−1 (negatively charged), due to the deprotonation of most of the acid groups of MAA (pH = 4.4). After adsorption of each polycation and polyanion layer, the surface charge becomes positive and negative, respectively, consistent with the behaviour observed by other authors dealing with similar biopolyelectrolytes and different substrates [
38,
39]. Nevertheless, the surface charge is not completely compensated in terms of absolute values, in contrast to what is observed for LbL assembly on flat surfaces or rigid particles. This suggests that interactions between the adsorbed PE films and the NG take place, and might be related to incomplete coverage of the NG surface and/or interpenetration with the PEs. A similar trend of charge reversal after each PE deposition was found for PLL/PGA-coated PNIPAM-
co-MAA MGs [
27], and confirms the possibility of the LbL assembly of biopolyelectrolytes onto thermoresponsive gels. The values of µ
e obtained are quite high, consistent with those found by Haidar
et al. [
40] for liposomes coated by biopolymers, and demonstrates the high stability of these systems.
Figure 5 reveals that µ
e behaviour is similar for both types of PEs, although the larger charge reversal (+4.3 × 10
−8 m
2 V
−1 s
−1) is obtained after deposition of the first CHIT layer compared to PLL (+3.9 × 10
−8 m
2 V
−1 s
−1). This could be understood by considering that CHIT has more functional groups and higher charge density since it is assembled from solutions with low pH and ionic strength, and tries to adopt an extended rod-like configuration, leading to a larger excess of positive charges onto the NG surface. Interestingly, on assembly of the first polyanion layer, the absolute
µe value is higher for DEXS-terminated NG compared to PGA-terminated NG, likely due to their dissimilar charge density, provoked by the different assembly conditions. As indicated in the introduction, when these weak PEs are incorporated into the multilayer system, their degree of ionization can vary considerably from the solution value owing to the influence of the local environment via electrostatic and hydrophobic effects. Therefore, when DEXS is adsorbed, the presence of many oppositely charged cationic groups will augment the degree of ionization of this polyanion compared to the solution value, resulting in a high surface charge density. However, when PGA is adsorbed, there are less cationic groups on the surface, because some of the amine groups of PLL are compensated by salt counterions, consequently reducing the magnitude of the electrostatic effects. Furthermore, the apparent pK
a of PGA can be increased and that of PLL reduced due to hydrophobic effects, which would diminish the degree of ionization of these PEs in the multilayer. Thus, the pK
a of the carboxylic groups of PGA has been reported to fall slightly upon incorporation in the multilayer [
41], though it increases up to ~4.5 in the presence of salt. Similarly, a pK
a drop from ~10.5 in solution to 9.8 was detected for the amine groups of PLL, interpreted in terms of a proton release from the NH
3+ due to the interaction with the negatively charged groups of PGA. All the above mentioned changes in the degree of ionization of the different groups should have an effect on the charge balance during the LbL assembly.
Figure 5 also shows a systematic drop in µ
e with increasing number of layers deposited onto the NG, regardless of the type of PE. This is a key difference with the LbL assembly on hard and rigid particles [
38], where the magnitude of charge reversal does not change. LbL films can be considered as ordered non-equilibrium arrangements in which the PE segments have a lot of freedom for mobility [
42]. The soft and porous nature of the gels allows penetration of the PEs to a certain extent and mutual interactions that are not feasible with solid and rigid substrates. PEs can diffuse in and out, thereby neutralizing charges. Rupture and formation of ion pairs can occur not only between polycations and polyanions but also between polycations and gels; all these facts account for the fall in µ
e with increasing number of layers. This behaviour indicates that the electrostatic interactions and the intrinsic mechanism of charge compensation primarily contribute to the multilayer formation during the adsorption of the initial layers, whereas the secondary interactions become the driving force of the assembly with increasing number of deposition stages, which is reflected in a reduction in µ
e. However, a different trend was reported for HA/PLL multilayer films, where the magnitude of the zeta potential (ζ) even slightly increased with the deposition steps, likely because HA acts both as template and polyanion in this system [
28].
Figure 5.
Electrophoretic mobility μ
e at 20 °C
vs. number of layers deposited on PNiPAM-
co-MAA NGs: (
a) NG/(PLL/PGA) and (
b) NG/(CHIT/DEXS). Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 5.
Electrophoretic mobility μ
e at 20 °C
vs. number of layers deposited on PNiPAM-
co-MAA NGs: (
a) NG/(PLL/PGA) and (
b) NG/(CHIT/DEXS). Reproduced with permission from [
8], copyright 2010, Elsevier.
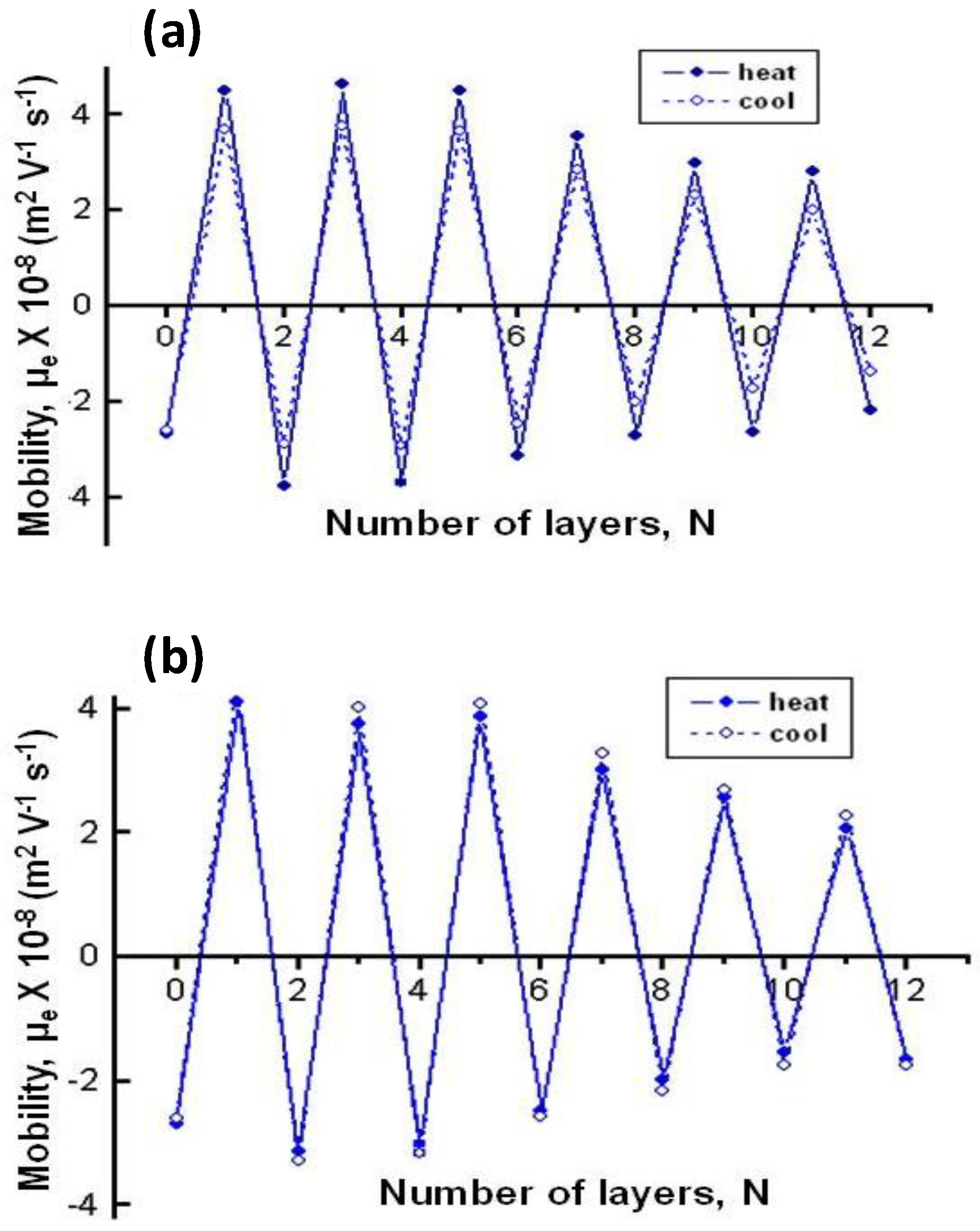
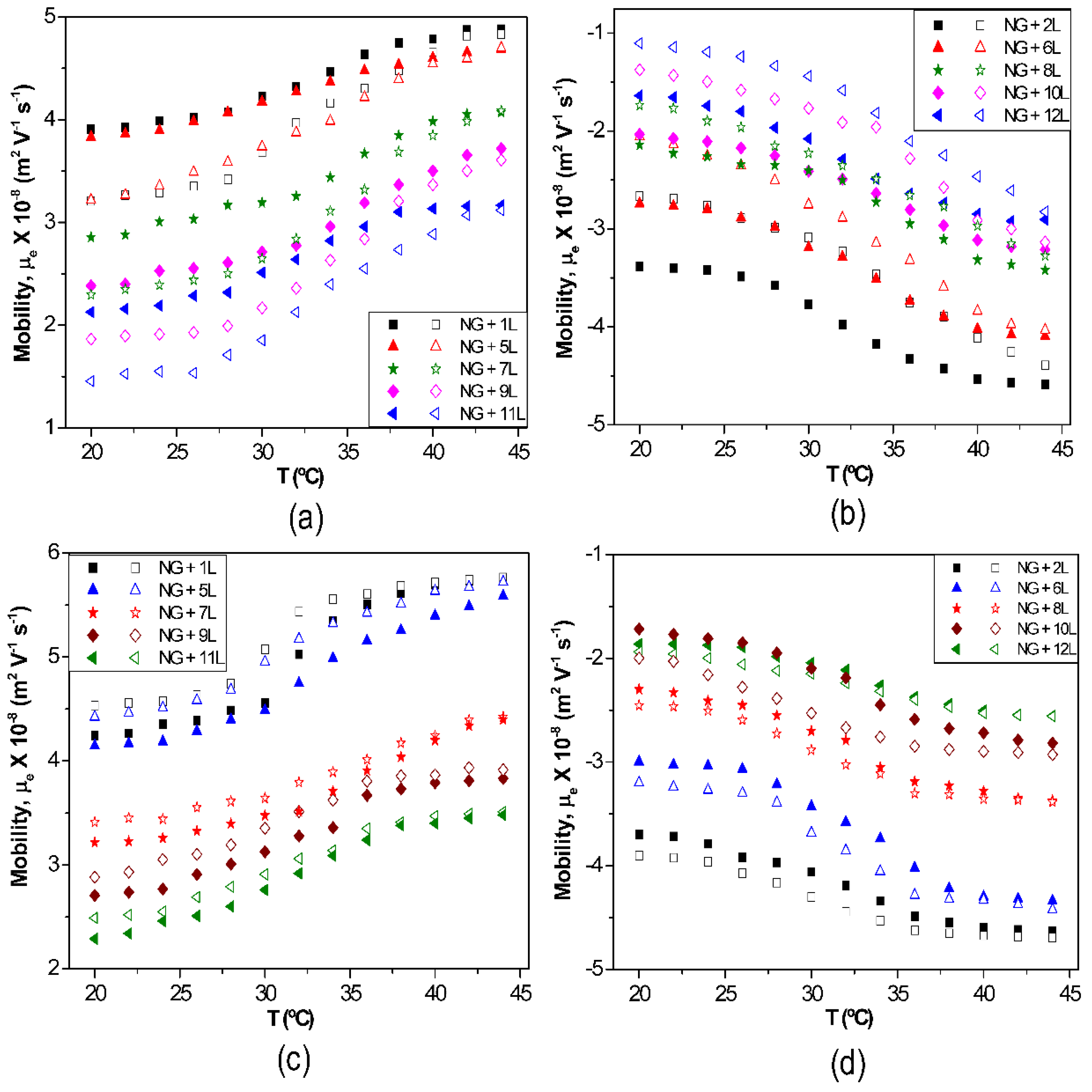
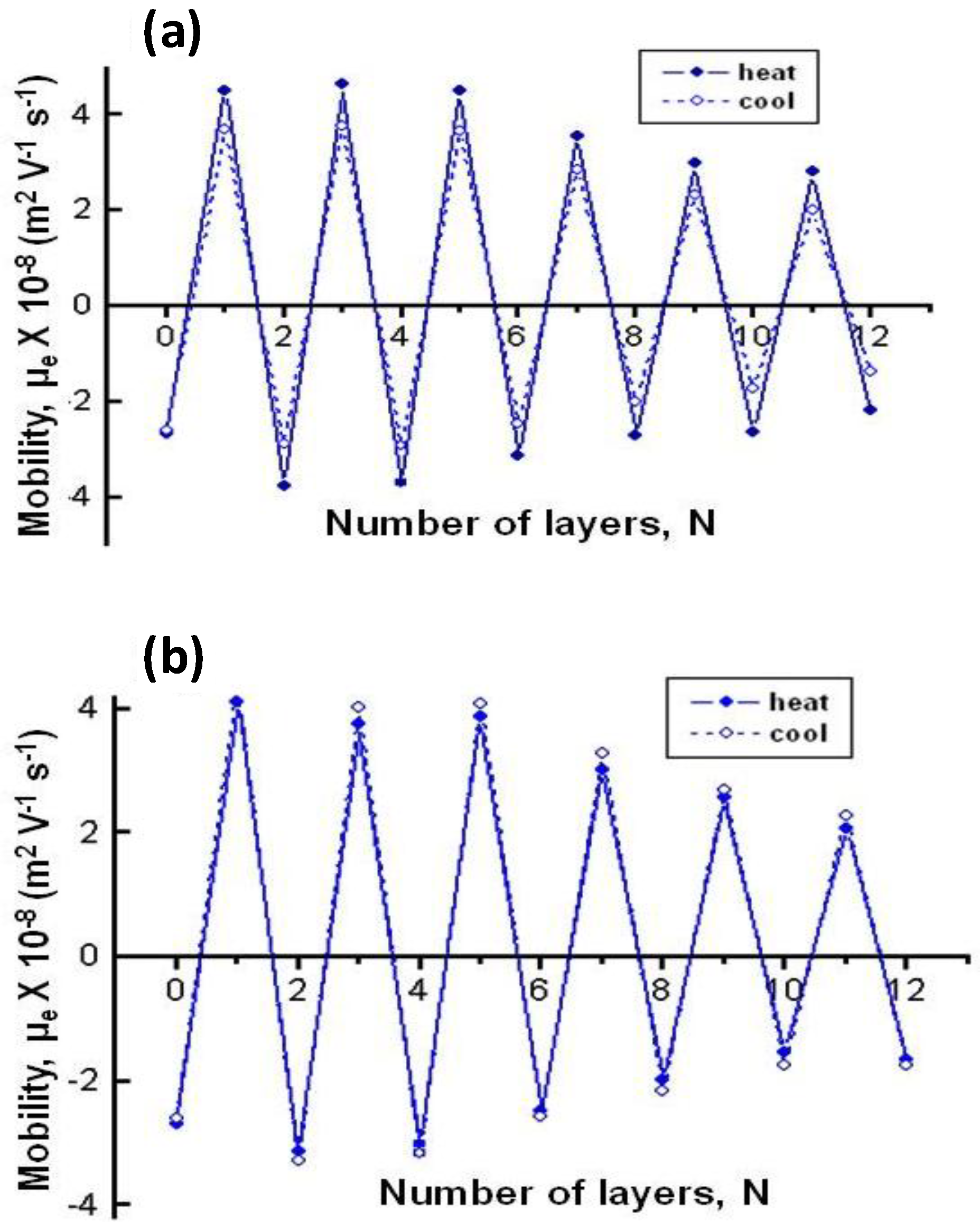
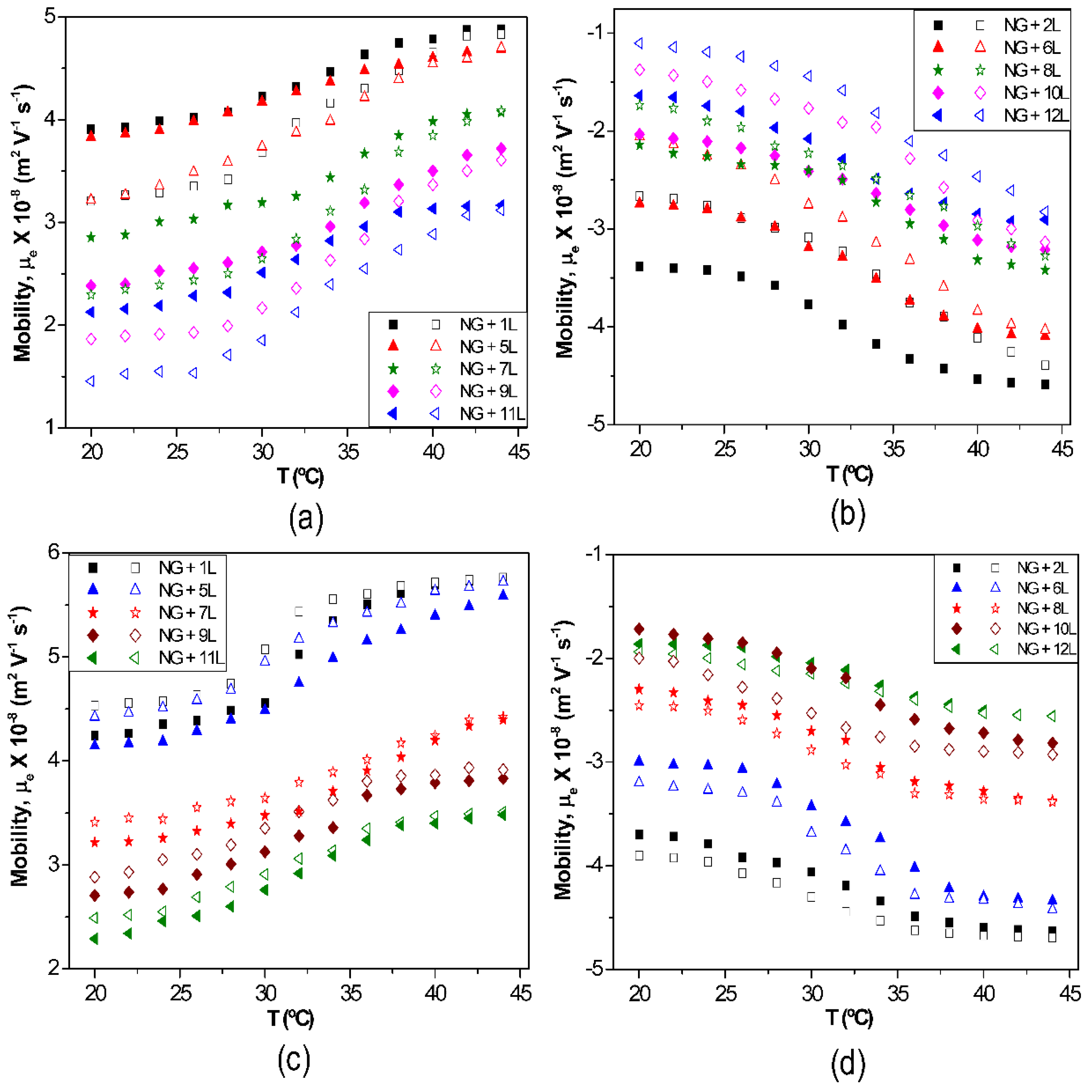
The effect of temperature on µ
e for biopolymer-coated PNIPAM-
co-MAA NGs is shown in
Figure 6: (a) PLL, (b) PGA, (c) CHIT and (d) DEXS [
8]. For all the systems, the absolute value of µ
e rises with increasing temperature owing to the inherent thermoresponsivity of the NG. This is explained by considering that the gel swelling is entropically driven and depends on the formation of hydrogen bonds between the amide groups of PNIPAM and the water molecules. The rise in temperature breaks these stabilizing H-bonds, hence the NG shrinks, pushing the charges out, and provoking an increase in the surface charge density. Since the NG has water inside, in the collapsed state, on cooling some trapped segments are able to reorganize as water flows into the gel, and this could account for the hysteresis found in µ
e between the heating and the cooling cycles. As can be observed from
Figure 6, the aforementioned hysteresis is more significant for NGs coated with polypeptides compared to those coated with polysaccharides, indicating a higher extent of internal rearrangement in the former biopolymers. This behaviour could also be ascribed to the high ionic strength of the PLL and PGA dipping solutions. As indicated previously, salt counter ions occluded within the multilayer, swell the ensemble by screening the electrostatic forces that join the layers, thereby provoking higher mobility of the polymer chains, hence restructuration of the entire ensemble, leading to a noticeable hysteresis. However, in the case of high
Mw polysaccharides assembled from water solutions, less rearranging is allowed, albeit they also undergo small conformational changes between different helical structures caused by the swelling and collapsing of the NG. For the same number of layers and temperature, polysaccharides display higher µ
e values, attributed to their larger number of functional groups and charges, and try to adopt a stretched conformation, they thus have more charges at the NG surface, while polypeptides adopt a more coiled configuration, exposing fewer charges on the surface.
Figure 6.
Electrophoretic mobility μ
e vs. temperature for different biopolymer-terminated PNIPAM-
co-MAA: (
a) PLL; (
b) PGA; (
c) CHIT and (
d) DEXS. Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 6.
Electrophoretic mobility μ
e vs. temperature for different biopolymer-terminated PNIPAM-
co-MAA: (
a) PLL; (
b) PGA; (
c) CHIT and (
d) DEXS. Reproduced with permission from [
8], copyright 2010, Elsevier.
The sequential LbL deposition onto MGs has also been monitored by contact angle measurements. For instance, Yamanlar
et al. [
37] studied the multilayer growth of PLL and HA onto HA MGs. The contact angle of uncoated HA was 36.5°, and increased to 44° upon deposition of the first PLL layer. Subsequent deposition of each HA and PLL layers led to similar results (
i.e., low and high contact angles for HA and PLL, respectively) indicating the multilayer buildup on the outer surface of the MG. Therefore, the polycationic PLL layers were more hydrophobic than the polyanionic HA ones. Similarly, Kolasinska
et al. [
43] reported that PLL terminated multilayers were more hydrophobic than PGA terminated ones. The authors suggested that the electric charge density is lower when the polycation forms the outermost layer. Another proposed reason was the more favourable orientation of water molecules at the negatively charged surfaces. For the first three layer pairs, there was little change in the contact angle values. However, when the number of the layers grew, the contact angle in each deposition increased, ascribed to the interlayer mixing during PLL/HA film growth, since the polypeptide can diffuse in and out of the layers.
6.4. Hydrodynamic Size
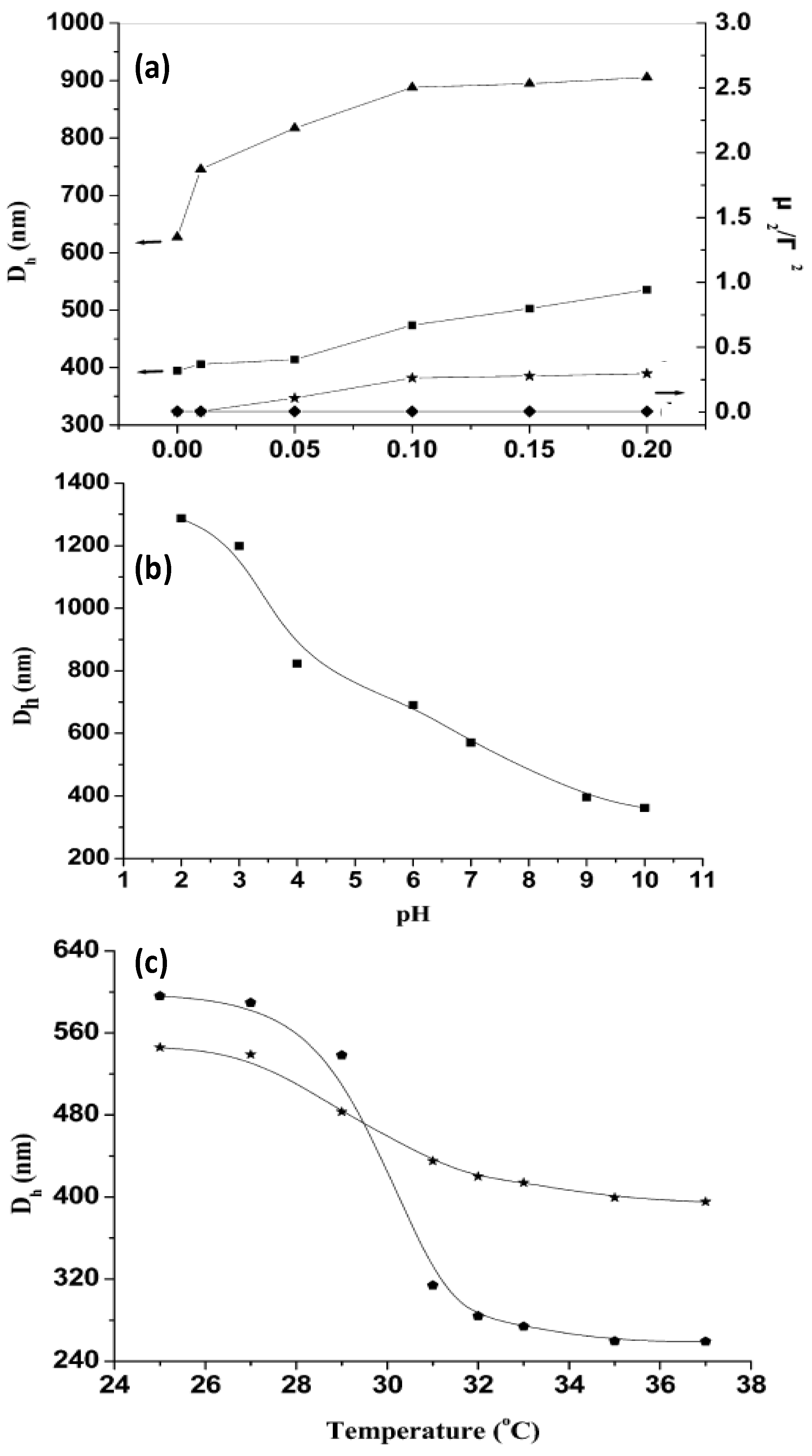
The thermoresponsive behaviour of uncoated and coated gels can be monitored by DLS. Mu
et al. [
44] investigated the influence of the ionic strength, pH and temperature on the hydrodynamic diameter (
Dh) of PNIPAM-grafted (CHIT/ALG)
4 hollow microspheres (
Figure 7). For comparison, the size of (CHIT/ALG)
4 microspheres was also investigated.
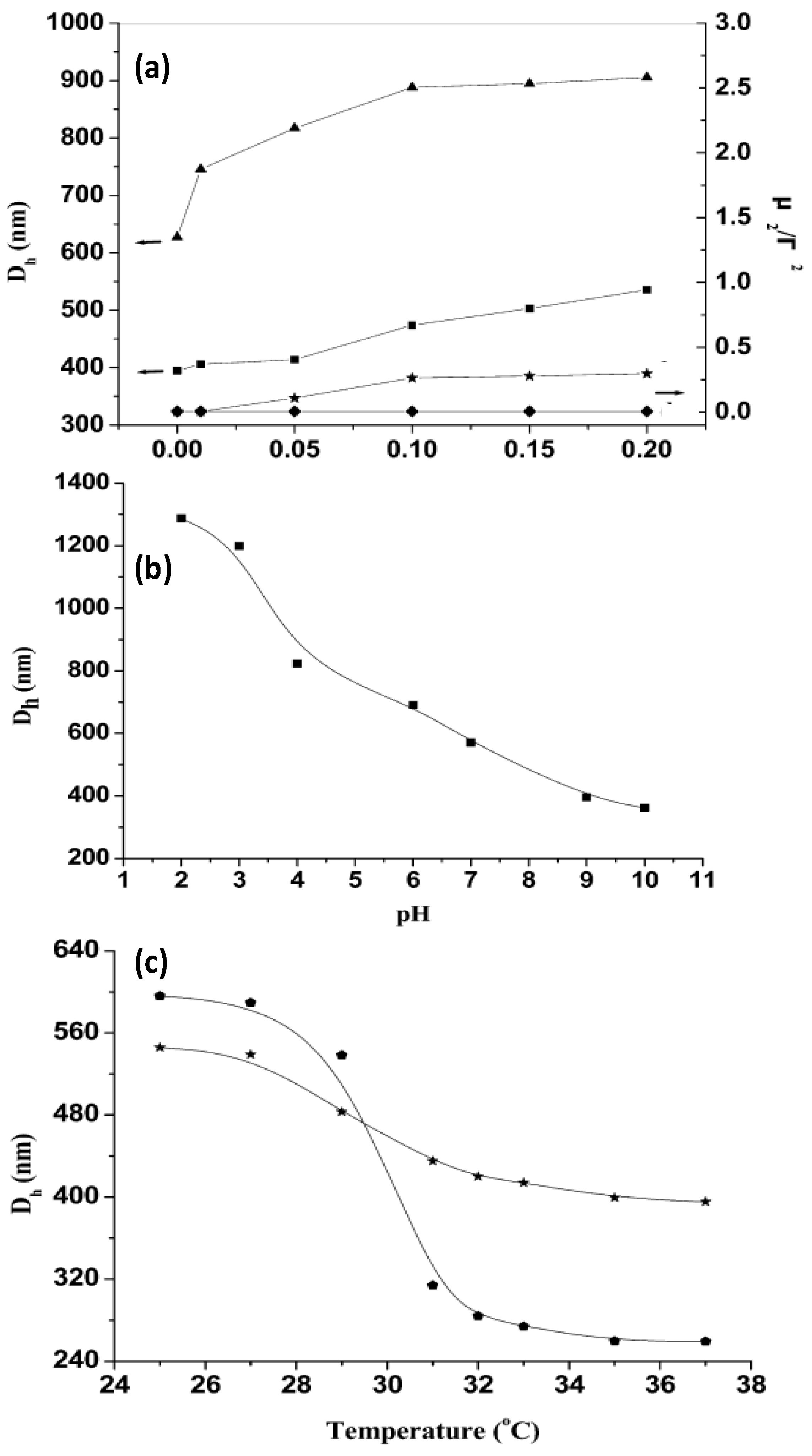
Figure 7.
(
a) Influence of ionic strength on the hydrodynamic diameter
Dh and polydispersity index (μ
2/Г
2) of PNIPAM-grafted (CHIT/ALG)
4 hollow microspheres (▲ and ♦) and (CHIT/ALG)
4 hollow microspheres (■ and ★); (
b) pH dependence of
Dh of PNIPAM-grafted (CHIT/ALG)
4 hollow microspheres at 25 °C; (
c) Temperature dependence of
Dh of PNIPAM-grafted (CHIT/ALG)
4 microspheres with low (★) and high (
![Materials 07 07472 i001]()
) grafting degree. Reproduced with permission from [
44], copyright 2012, American Chemical Society.
Figure 7.
(
a) Influence of ionic strength on the hydrodynamic diameter
Dh and polydispersity index (μ
2/Г
2) of PNIPAM-grafted (CHIT/ALG)
4 hollow microspheres (▲ and ♦) and (CHIT/ALG)
4 hollow microspheres (■ and ★); (
b) pH dependence of
Dh of PNIPAM-grafted (CHIT/ALG)
4 hollow microspheres at 25 °C; (
c) Temperature dependence of
Dh of PNIPAM-grafted (CHIT/ALG)
4 microspheres with low (★) and high (
![Materials 07 07472 i001]()
) grafting degree. Reproduced with permission from [
44], copyright 2012, American Chemical Society.
It was found that the increase in the ionic strength from 0 to 0.20 mol/L NaCl increased
Dh of the polysaccharides multilayers from ~400 to 540 nm, whilst that of PNIPAM-grafted (CHIT/ALG)
4 increased from ~630 to 910 nm (
Figure 7a). This behaviour was explained by considering that the presence of salt weakens the electrostatic interaction between ALG and CHIT, hence the PE chains are stretched and the size of the hollow microspheres rises. Moreover, the polydispersity index (μ
2/Γ
2) of the microspheres composed only of polysaccharides increased steadily with increasing ionic strength, indicative of aggregation during the measurements, whilst that of the assemblies with PNIPAM remained almost constant, demonstrating that the grafted PNIPAM prevents flocculation among the microspheres in solutions with higher salt concentration.
On the other hand,
Dh of PNIPAM-grafted (CHIT/ALG)
4 decreased from ~1290 to 360 nm with increasing pH (
Figure 7b). At low pH (<4), CHIT was hardly ionized, hence was scarcely crosslinked with ALG, leading to a big PE shell. Between pH 4 and 7, there is a strong electrostatic attraction between both PEs, resulting in a reduction in
Dh. The ionization of the amine groups of CHIT decreased greatly when the solution pH increased above 6.0 (close to the pK
a of CHIT) and at pH > 7.5 less than 10% of its amine groups were ionized, provoking the shrinkage of the polysaccharide chains, and hence a remarkable reduction in size. The results demonstrate that the PE shells of the hollow microspheres were pH-sensitive.
The influence of temperature on
Dh was also analysed (
Figure 7c). Clearly, the VPTT occurs at around 32 °C. Upon heating from 25 to 37 °C,
Dh of the grafted ensemble with lower degree of grafting decreased from 545 to 400 nm, whilst that of the system with higher grafting degree dropped from 600 to 260 nm, corroborating that the higher PNIPAM content, the larger the thermosensitivity of the system. Overall, the hollow microspheres retained the pH, ionic strength and thermosensitivity, and they did not disintegrate with variations in these parameters.
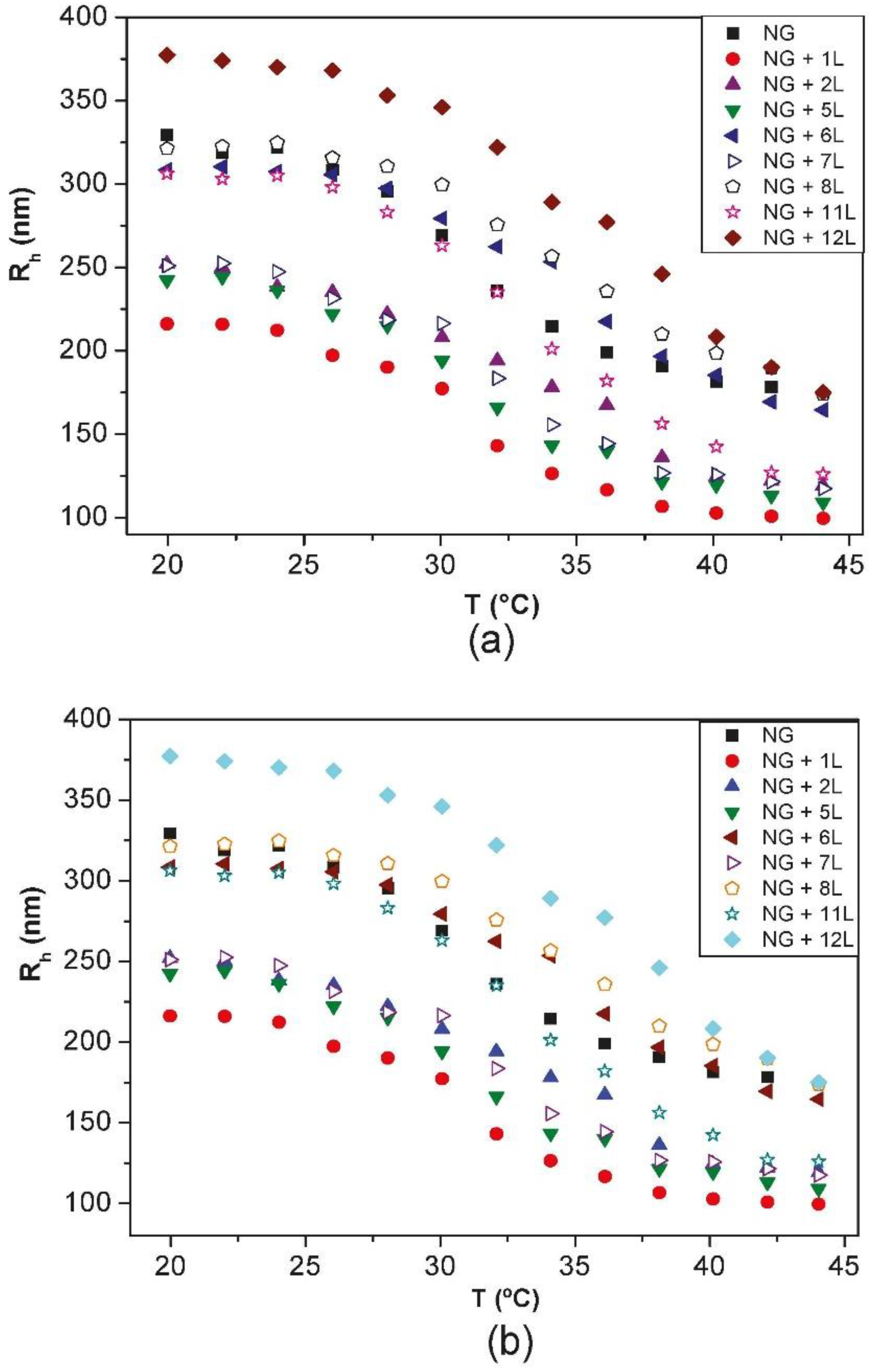
Diez-Pascual and Wong [
8] compared the change in the hydrodynamic radius
Rh with increasing temperature for PNIPAM-
co-MAA NGs coated with different layers of PLL/PGA and CHIT/DEXS (
Figure 8). The bare NG in water (pH = 4.4) possesses a
Rh of 330 nm at 20 °C. During heating, it collapses to ~180 nm, and on cooling it recovers the original size. Upon adsorption of the first polycation layer there is a strong decrease in the NG size, albeit the coated NGs maintain the thermoresponsive behaviour, and on heating they still undergo the transition to a collapsed state. The deposition of the second layer provokes an increase in size, and the temperature sensitivity of the coated NG is also retained. This “odd-even” effect was corroborated by ESEM images. The size of one-layer polysaccharide coated NG in the collapsed state is ~100 nm, whereas PLL-coated NG shrinks to ~170 nm. To understand the differences between the behaviour of NGs coated with polypeptides and polysaccharides, the PE charge density and secondary cooperative interactions that contribute to the film formation have to be considered. The polysaccharide displays a larger number of ionized functional groups that can shield the negative charges of the NG, causing a rearrangement of the gel in order to adopt a more coiled and compact conformation, resulting in a smaller
Rh. Further, upon deposition of CHIT, strong H-bonds can be formed between its hydroxyl groups and the solvent at the expense of breaking stabilizing interactions between the amide groups of PNIPAM and the water molecules, resulting in a decrease in size. Upon approach of the DEXS layer, the H-bonds of the confined CHIT are shared between the solvent and the polyanion; this weakens the interactions with the solvent, leaving some water molecules mobile again and able to penetrate the NG, causing the assembly to expand. This explains how PNIPAM-
co-MAA/(CHIT/DEXS) presents the mentioned “odd-even” effect in both swollen and collapsed states.
Figure 8.
Hydrodynamic radius
Rh vs. temperature for PNIPAM-
co-MAA with different layers of: (
a) PLL/PGA and (
b) CHIT/DEXS. For clarity, only the heating cycles are shown. Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 8.
Hydrodynamic radius
Rh vs. temperature for PNIPAM-
co-MAA with different layers of: (
a) PLL/PGA and (
b) CHIT/DEXS. For clarity, only the heating cycles are shown. Reproduced with permission from [
8], copyright 2010, Elsevier.
On the other hand, polypeptides are assembled from neutral solutions with high ionic strength, where the charges of both the NG and the PEs are screened. Consequently, the NG-PLL electrostatic attraction is weaker than the NG-CHIT one, hence the decrease in size is less pronounced. Furthermore, secondary cooperative interactions play a key role in the construction process. After adsorption of the PGA layer, both biopolymers also interact via H-bonds and hydrophobic interactions. To attain the electrical neutrality necessary for the multilayer buildup, part of the polyanion charge is compensated by salt counterions that get blocked within the film structure. During the washing stages, the osmotic pressure acts as the driving force for the swelling of the system. The change in ionic strength leads to a difference in the chemical potential between the ions inside and outside the coated NGs, and consequently an exchange process of ionic pairs takes place between the multilayer and the solution until the chemical potential is equilibrated, and the overall result is a rise in Rh.
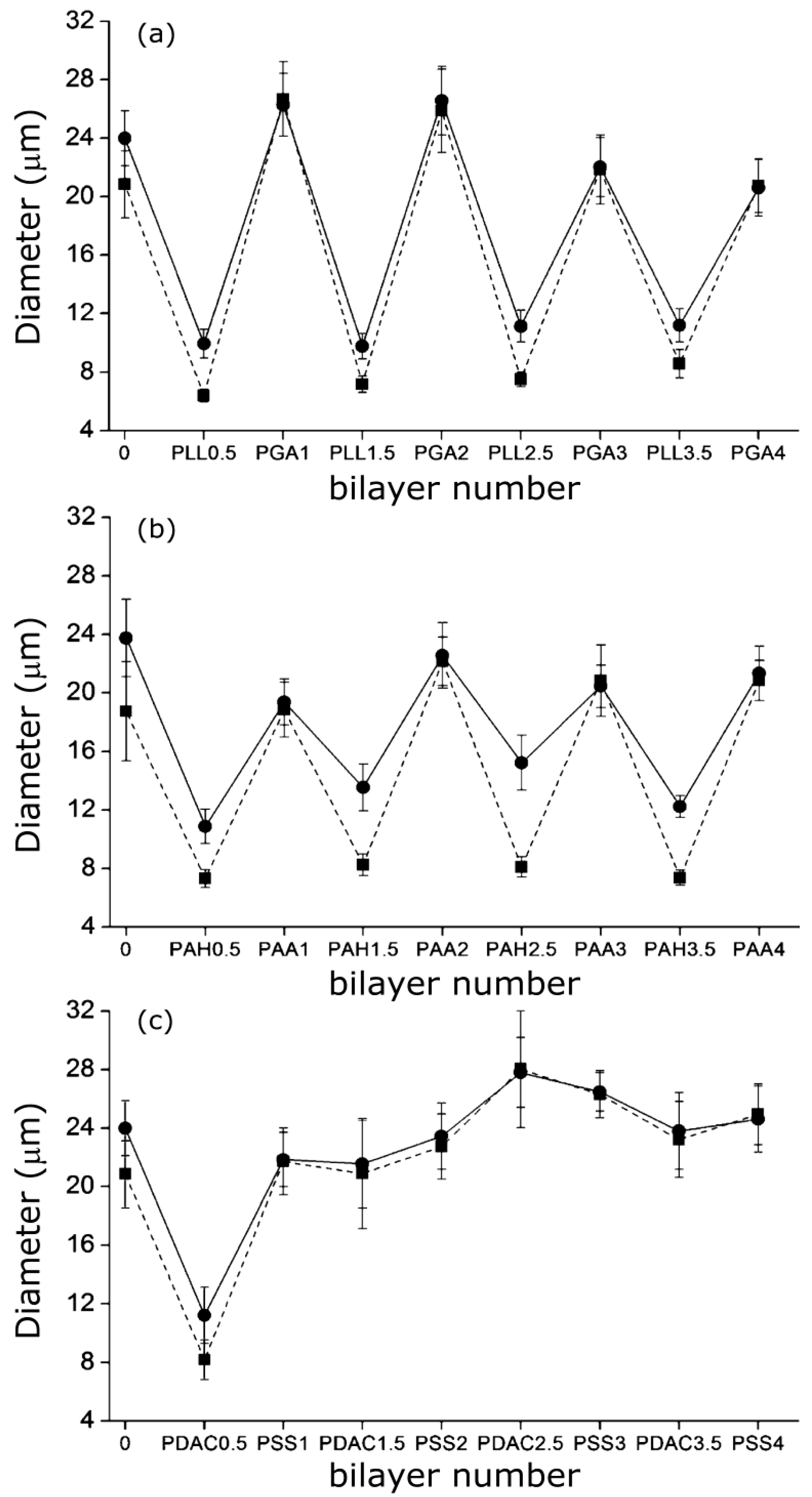
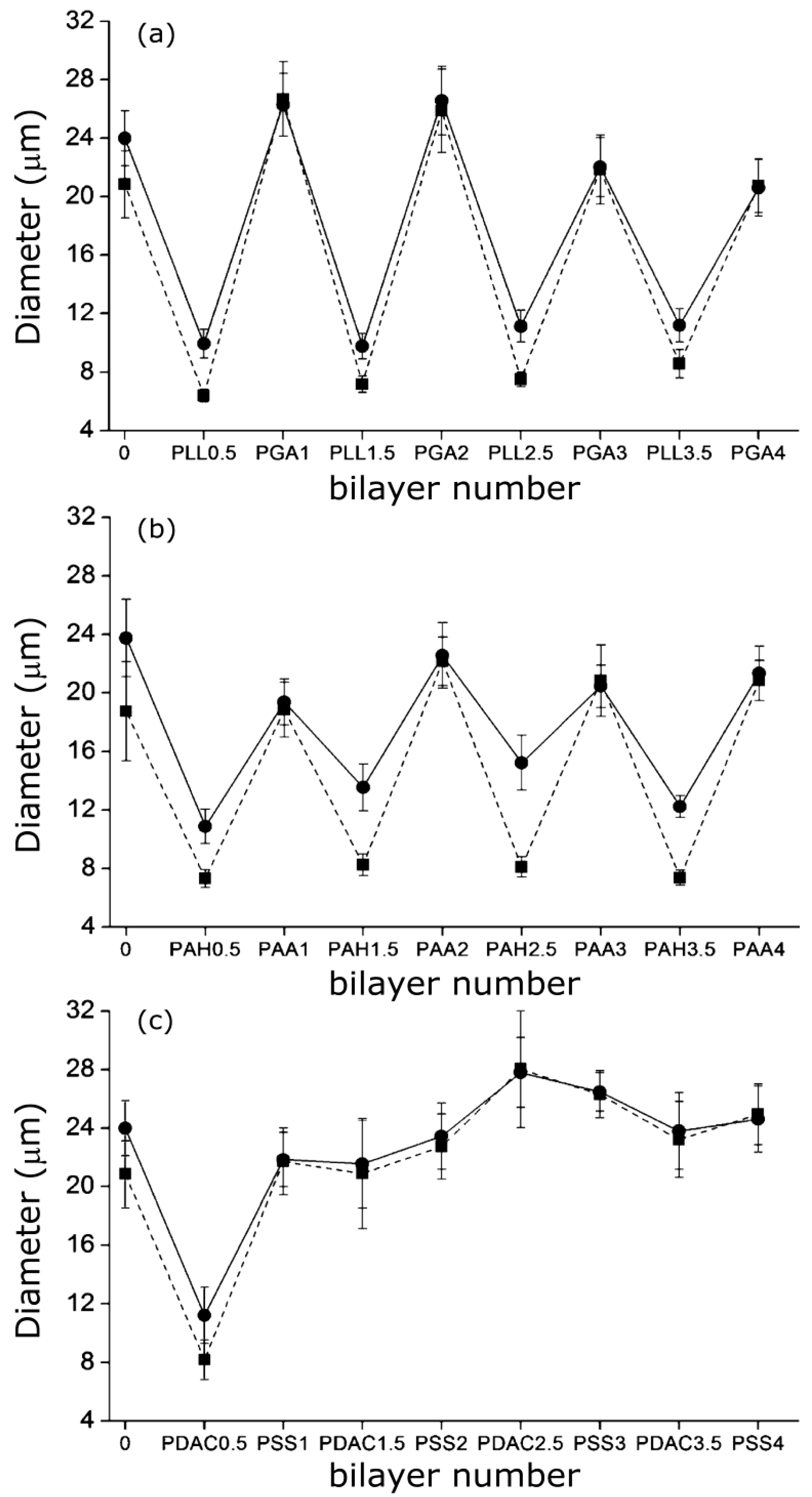
A similar “odd-even” effect both in the swollen and collapsed states was reported by Costa
et al. [
27] for PNIPAM-
co-MAA MGs. They compared the hydrodynamic diameter
vs. number of layers for MGs coated with PLL/PGA, PDACMAC/PSS and PAH/PAA (
Figure 9), and found that the size and thermoresponsive behaviour was significantly influenced by the outermost layer.
Figure 9.
Hydrodynamic diameter of PNIPAM-
co-MAA MGs upon LbL assembly of: (
a) PLL/PGA; (
b) PAH/PAA and (
c) PDACMAC/PSS at 24 °C (circles) and 37 °C (squares). Reproduced with permission from [
27], copyright 2012, American Chemical Society.
Figure 9.
Hydrodynamic diameter of PNIPAM-
co-MAA MGs upon LbL assembly of: (
a) PLL/PGA; (
b) PAH/PAA and (
c) PDACMAC/PSS at 24 °C (circles) and 37 °C (squares). Reproduced with permission from [
27], copyright 2012, American Chemical Society.
Assemblies with polypeptides showed the stronger changes in
Dh and systematically retained their thermoresponsive behaviour (
Figure 9a), ascribed to the PE inter-diffusion during film buildup, hence the polycation/polyanion effect on the gel behaviour was easily reversed. In contrast, assemblies with strong PEs only changed the MG responsiveness upon addition of the first two layers (
Figure 9c), attributable to the strength of their ionic complexes and the lower mobility of the PE chains that impair any effect of further layers on the overall responsiveness of the LbL assembled microgel. The swelling behaviour of coated gels can be characterized through the swelling ratio defined as (
Dhswollen/
Dhcollapse)
3. In the case of PNIPAM-
co-MAA MGs, this ratio is higher for biopolymer coated systems compared to those coated by synthetic polymers.
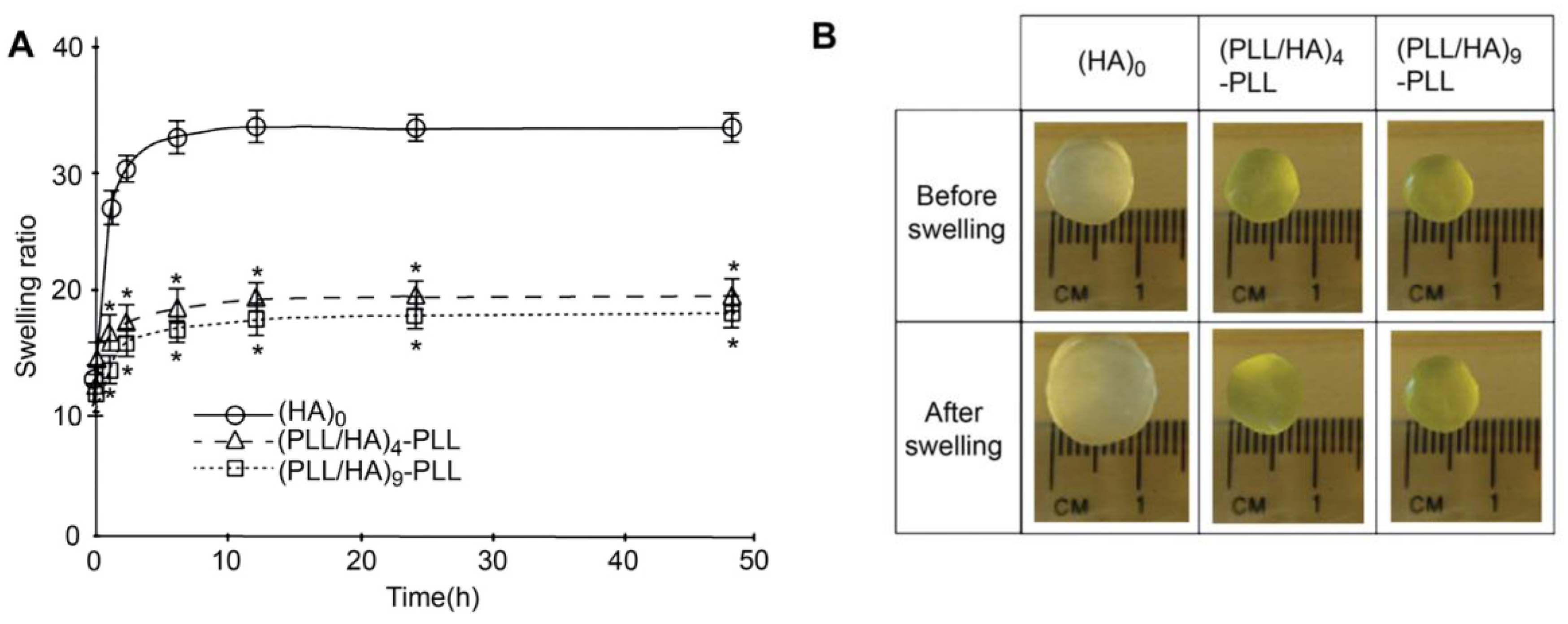
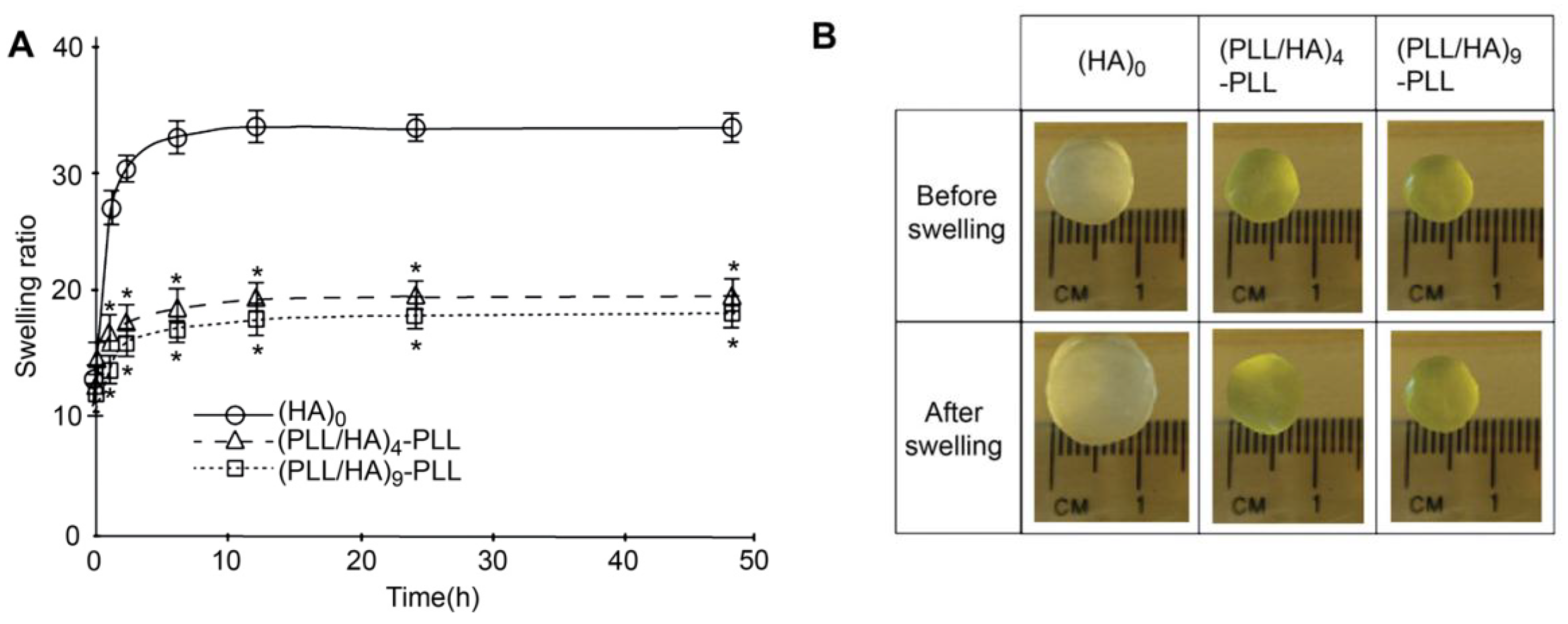
Other authors have carried out swelling studies on HA MGs coated with PLL/HA multilayers [
37]. The samples were immersed in phosphate buffered saline for predetermined times, quickly wiped to remove the residual liquid and their swollen weight recorded. Subsequently, they were dried in a vacuum oven and weighed again to determine their dry weight. It was found that coated gels displayed a lower degree of swelling compared to the unadulterated MG (
Figure 10a), explainable in terms of PE diffusion inside the pores as well as increased hydrophobicity of the surfaces. According to SEM images (
Figure 2), interpenetration of the PEs inside the pores occurs during multilayer formation, causing their partial or complete blockage, likely affecting the swelling properties of the coated gels. Further, as mentioned earlier, contact angle measurements suggest that PLL increased the surface hydrophobicity. Thus, the increased hydrophobicity and reduced permeability should result in lower swelling for the coated MGs. Indeed, when hydrophobic forces dominate, the tendency to diminish the polymer/water interface leads to reduced swelling. On the other hand, there was hardly any effect of the number of PE layers on the swelling ratio, as can be deduced from the comparison of the photographs of (PLL/HA)
4-PLL and (PLL/HA)
9-PLL before and after immersion in the buffer (
Figure 10b).
Figure 10.
(
A) Swelling ratio of bare HA MGs, (PLL/HA)
4-PLL and (PLL/HA)
9-PLL and (
B) Photographs of the uncoated and coated microgels before and after immersion in phosphate buffered saline. Reproduced with permission from [
37], copyright 2011, Elsevier.
Figure 10.
(
A) Swelling ratio of bare HA MGs, (PLL/HA)
4-PLL and (PLL/HA)
9-PLL and (
B) Photographs of the uncoated and coated microgels before and after immersion in phosphate buffered saline. Reproduced with permission from [
37], copyright 2011, Elsevier.
6.5. Bilayer Thickness
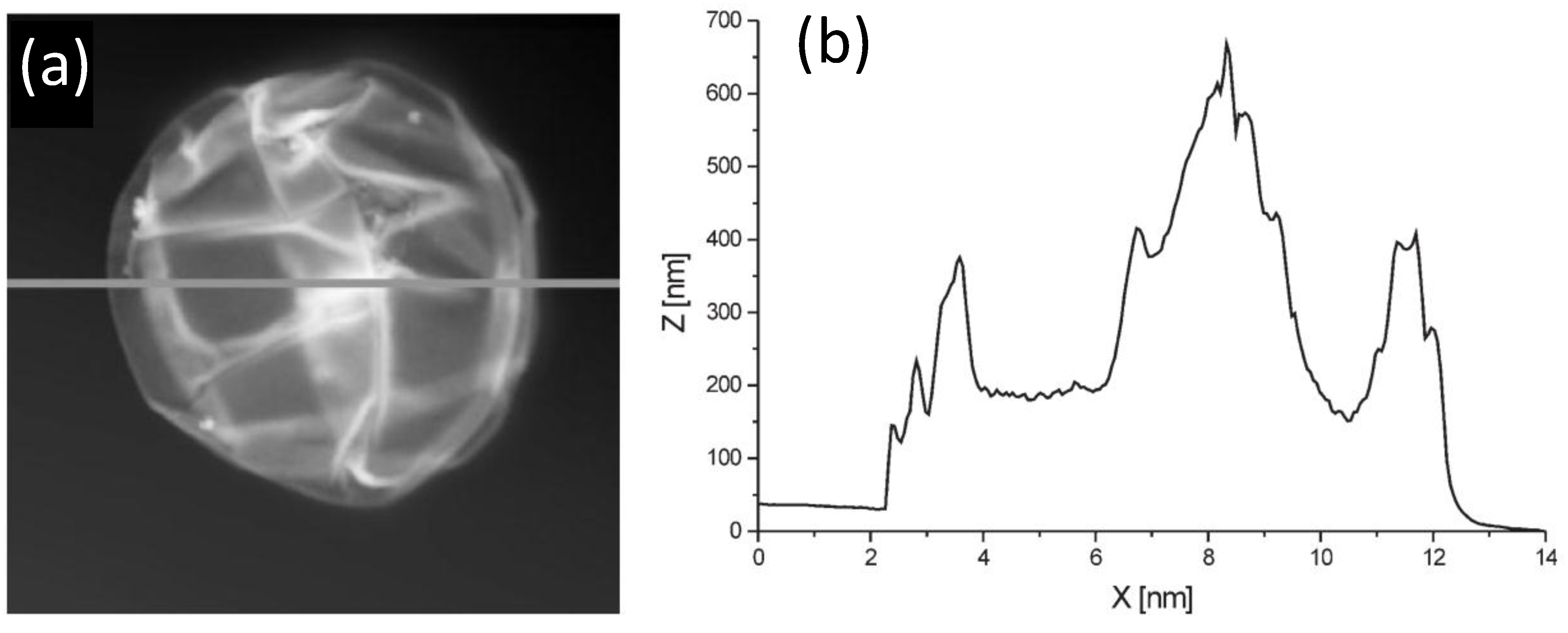
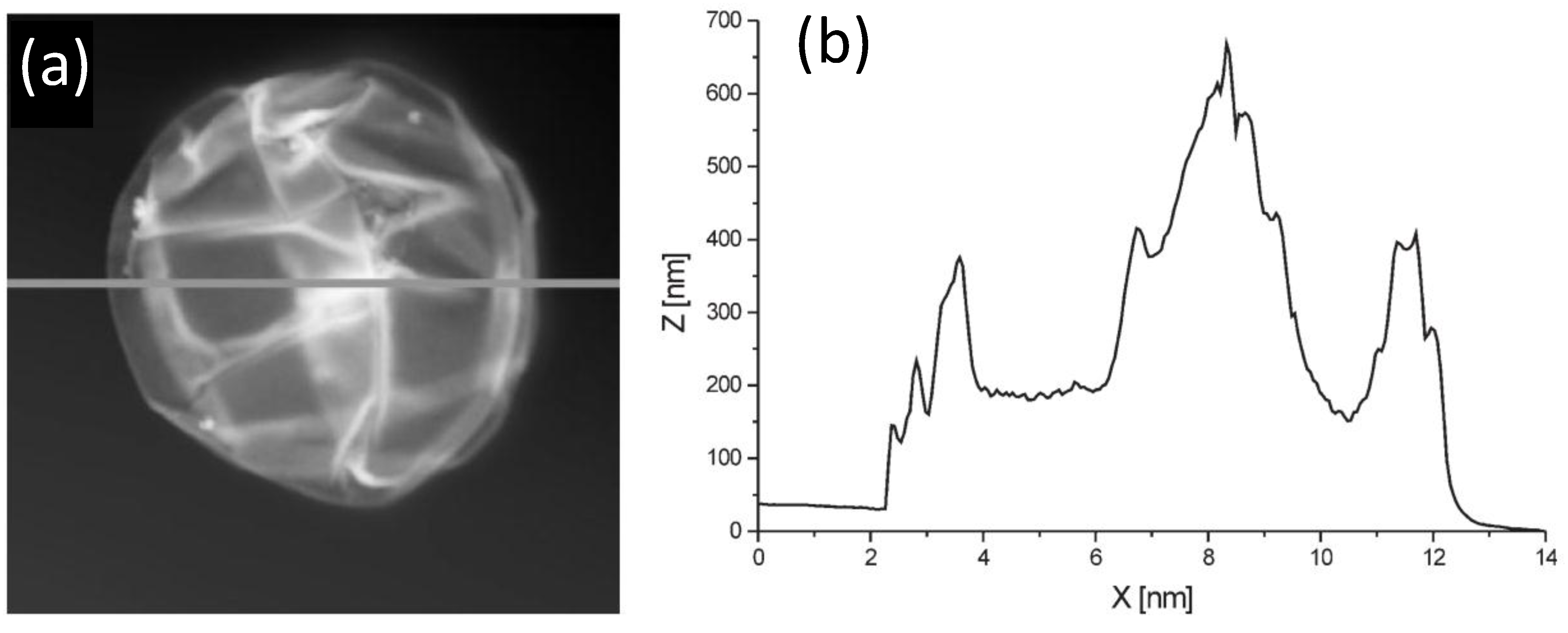
AFM can be used to estimate the bilayer thickness of PEs adsorbed onto MGs following the method described by Leporatti
et al. [
45]. De Geest and coworkers [
6] estimated the thickness of DEX-HEMA MGs coated with four bilayers of (DEXS/pARG) from the AFM height profile (
Figure 11) taking into account that the measured height is twice the thickness of the entire system. The average thickness obtained was 45 nm, ~11 nm per DEXS/pARG bilayer, which is significantly thicker than the values reported for biopolyelectrolyte films adsorbed onto rigid and flat substrates [
46].
Figure 11.
(
a) AFM image of a hollow DEX-HEMA MG coated with four (DEXS/pARG) bilayers and (
b) Height profile along the line indicated in (
a). Reproduced with permission from [
6], copyright 2007, Wiley-VCH.
Figure 11.
(
a) AFM image of a hollow DEX-HEMA MG coated with four (DEXS/pARG) bilayers and (
b) Height profile along the line indicated in (
a). Reproduced with permission from [
6], copyright 2007, Wiley-VCH.
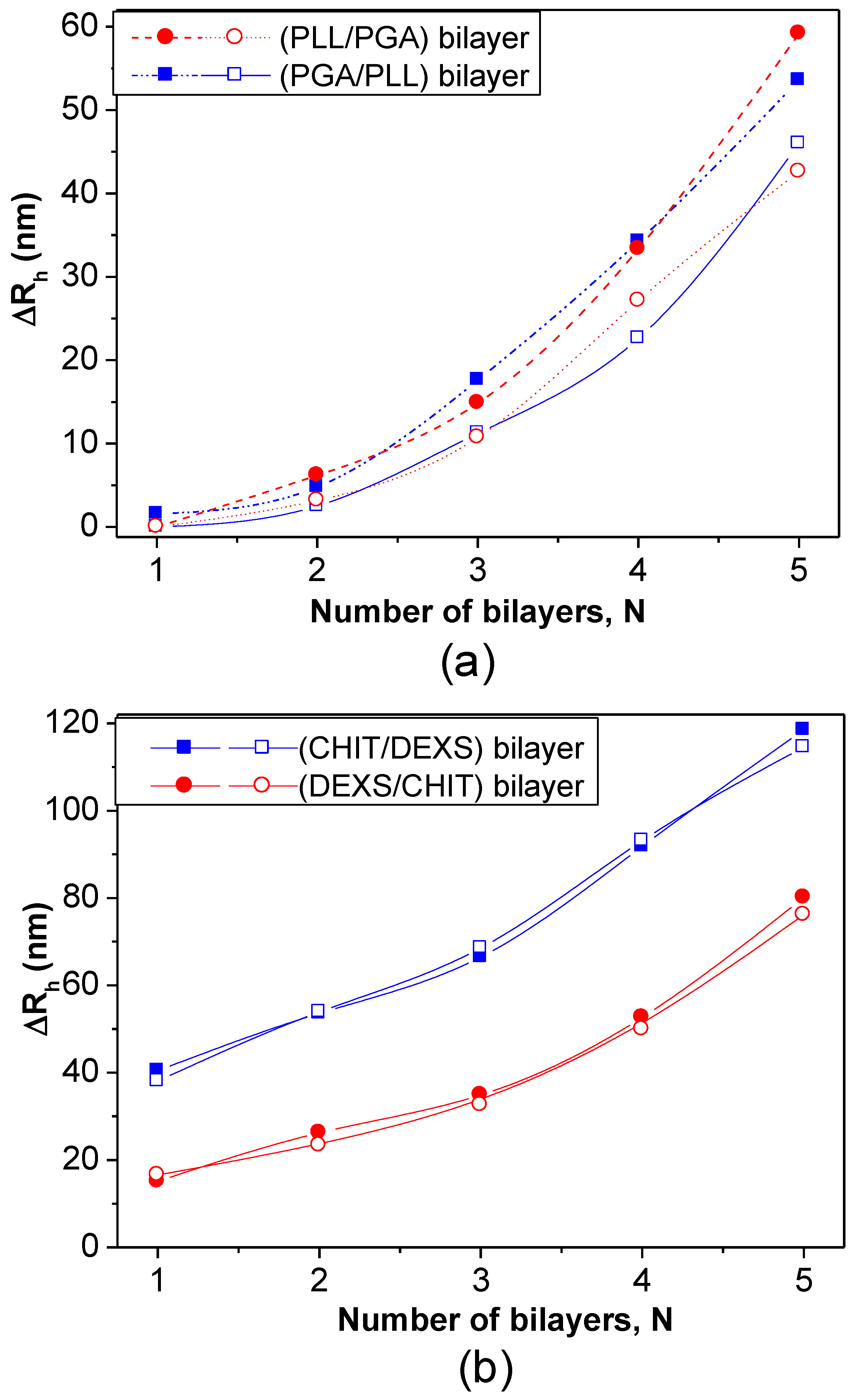
DLS measurements have also been employed to estimate the bilayer thickness of biopolymer coated PNIPAM-
co-MMA NGs in the swollen state taking the
Rh of the NG coated with the first and second layers [
8]. The increment in
Rh at 20 °C
vs. the number of (PLL/PGA) and (PGA/PLL) bilayers deposited on NG/(PLL/PGA) and NG/PLL, respectively, is shown in
Figure 12a. Likewise, the thickness of bilayers of CHIT/DEXS and DEXS/CHIT deposited on NG/(CHIT/DEXS) and NG/CHIT are plotted in
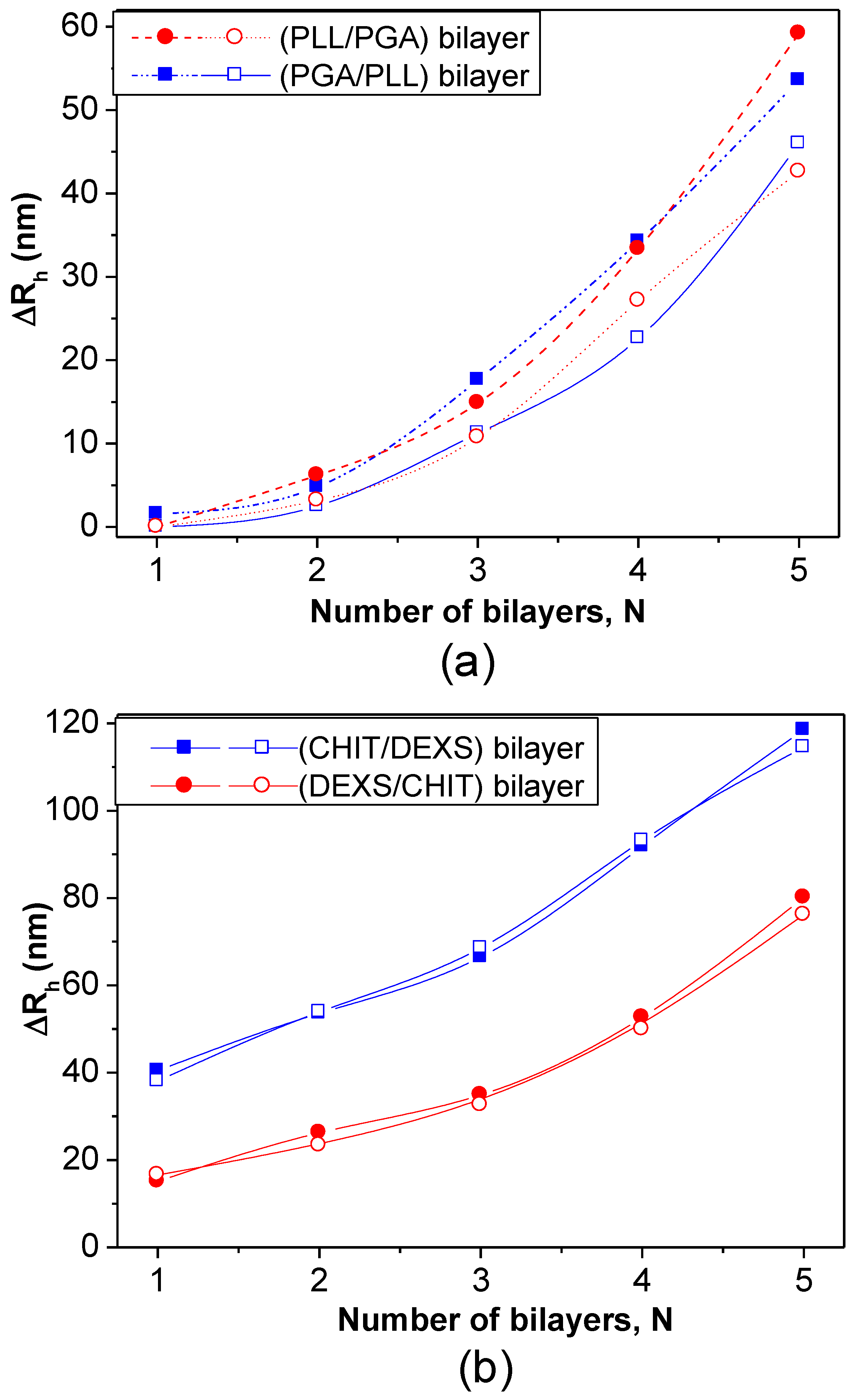
Figure 12b. PLL- and PGA-terminated films had very similar thickness: ~3 and 55 nm for the first and last bilayers deposited, consistent with values reported for the fabrication of these biopolymers onto biological substrates [
47]. For polypeptide-coated systems, values obtained from the heating curves were on average 16% higher than those derived from the cooling cycles, ascribed to the intermolecular rearrangements that polypeptides undergo when adsorbed onto NGs. In contrast, for polysaccharide-coated systems, only small differences were found between data derived from the heating and cooling curves, since their longer chains have a lower degree of freedom for reorganization. CHIT-terminated bilayers were found to be thinner than DEXS-terminated ones, indicating that the layer thickness is determined by the conformation of the biopolymer in the outermost layer. The negatively charged polysaccharide has a branched backbone and can adopt a more expanded conformation, thereby leading to thicker layers, whilst the positively charged polysaccharide displays a linear and flat structure, which therefore results in thinner layers. These bilayers are considerably thicker than those obtained upon assembly of these biopolyelectrolytes on rigid substrates [
46,
48], which can be rationalized by considering that the NG is in aqueous solution, and the PE layers are fully swollen and bound to a swollen substrate [
8]. Surprisingly, the polysaccharide bilayers were noticeably thicker than the polypeptide ones, associated with several factors influencing the relative layer thickness, such as charge density, hydrophilicity,
Mw, residue size,
etc. Despite this, polysaccharides tend to adopt a stretched conformation to minimize the repulsion between charges, which leads to thinner layers as they are more hydrophilic, adsorbing more water, leading to increased layer thickness. In addition, a raise in the polyelectrolyte,
Mw and chain length usually causes an increase in the amount of material adsorbed.
Figure 12.
Bilayer thickness for different polyelectrolyte layers at 20 °C: (
a) NG/(PLL/PGA) and (
b) NG/(CHIT/DEXS). The solid and empty symbols correspond to values obtained before and after the heating-cooling cycle is applied, respectively. Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 12.
Bilayer thickness for different polyelectrolyte layers at 20 °C: (
a) NG/(PLL/PGA) and (
b) NG/(CHIT/DEXS). The solid and empty symbols correspond to values obtained before and after the heating-cooling cycle is applied, respectively. Reproduced with permission from [
8], copyright 2010, Elsevier.
As can be observed from
Figure 12, both polypeptide- and polysaccharide-coated PNIPAM-
co-MMA NGs seem to exhibit a nonlinear growth of the layer thickness with the number of deposition stages. Exponential growth was found for PLL/PGA [
49] and HA/CHIT [
50] multilayers assembled on solid substrates, whilst quasi linear growth was reported for PSS/PAH multilayers [
51]. To explain the nonlinear buildup mechanism, different models have been proposed. McAloney
et al. [
52] suggested that such behaviour is caused by the increase in the film roughness with increasing number of deposited layers. Another viable explanation could be the ability of at least one PE to diffuse “in” and “out” of the whole structure during each bilayer deposition [
46,
53,
54].
Other authors estimated the PE thickness using fluorescently labelled species, in particular PLL
FTIC. Thus, the effect of dipping time on HA MGs coated with (PLL/HA)
4-PLL
FITC was investigated [
37]. As the adsorption time increased, the thickness of the band corresponding to PLL
FITC increased remarkably. Further, by keeping the adsorption time constant, it was found that the film thickness grew strongly with the number of layers, from 19.8 μm for a (PLL/HA)
4-PLL
FITC to 35.8 μm for a (PLL/HA)
9-PLL
FITC.
6.6. Temporal Stability
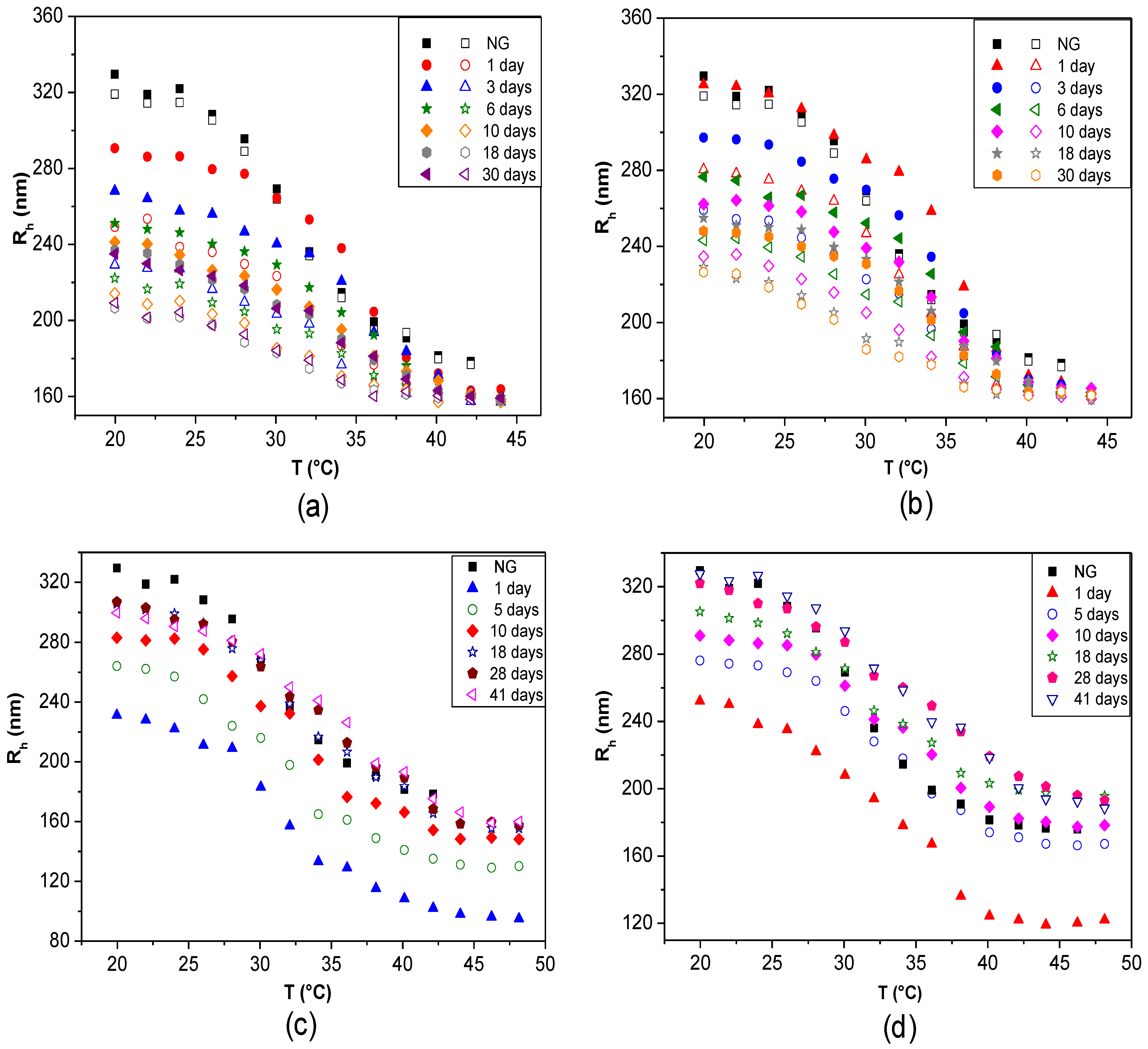
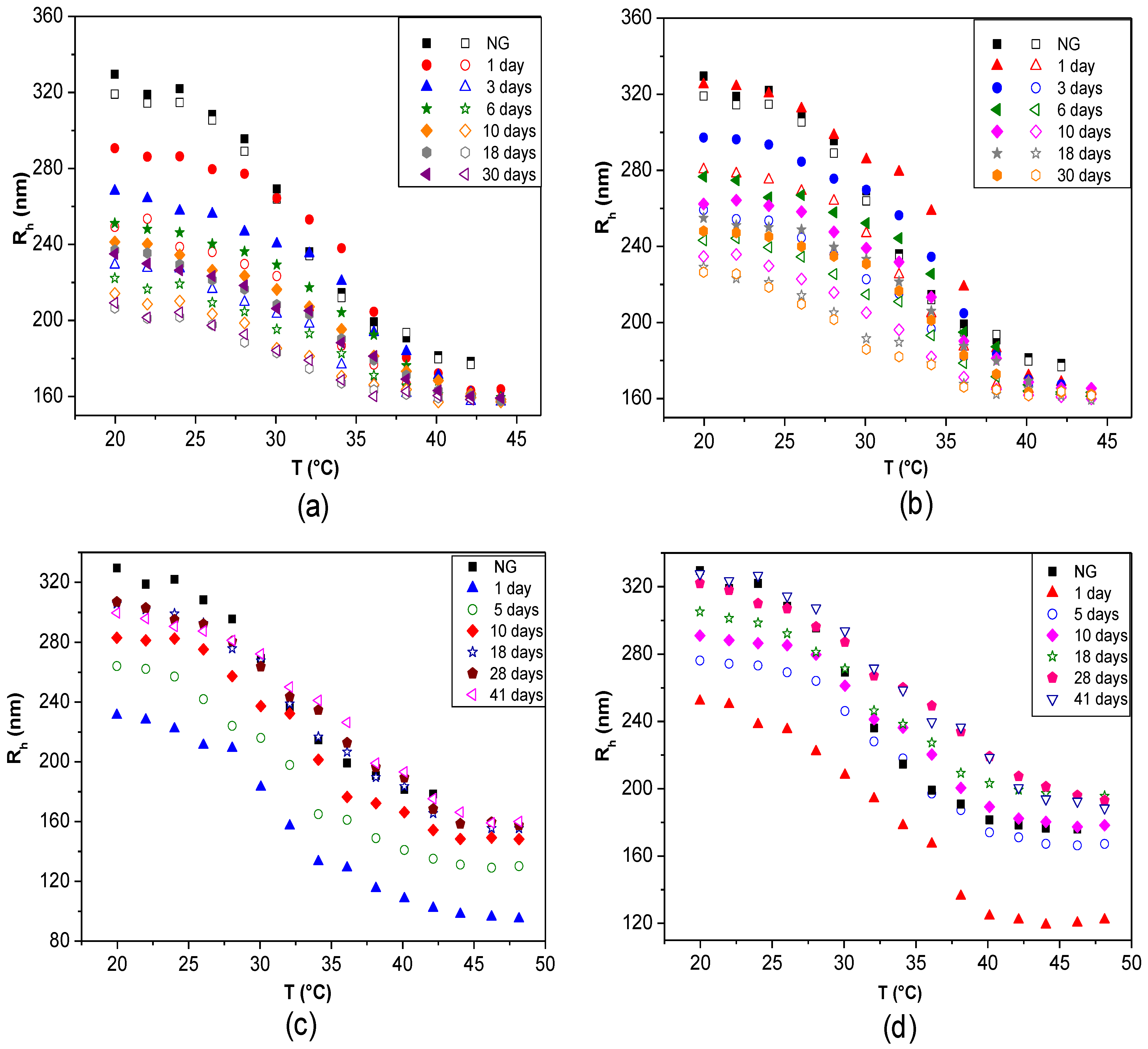
The changes in
Rh (or
Dh) and in the thermoresponsive behaviour as a function of time can be monitored to analyse the storage or temporal stability of LbL-coated NGs. Studies with fluorescently labelled PLL demonstrated that there is no desorption with time even after three months, corroborating that biopolymer-coated NGs are very stable [
42]. DLS curves over different periods of time for PNIPAM-
co-MMA NGs coated with: (a) PLL, (b) PLL/PGA, (c) CHIT and (d) CHIT/DEXS are shown in
Figure 13 [
8]. In the case of PLL-coated NG, a noteworthy decrease in
Rh with time was found, by about 9%, 15%, 17%, 19% and 21% after 3, 6, 10, 18 and 30 days, respectively (
Figure 13a). In contrast, when the coated NGs were heated, they always collapsed to nearly the same size as when they were first coated. Analogous trends were found for
Rh of systems where the outermost layer was PGA (
Figure 13b), consistent with the results reported by Pelsöczi
et al. [
47], who found a remarkable drop in the average roughness of one-month-old (PLL/PGA) films. Significant hysteresis was found between the heating and cooling cycles, as well as variations in the VPTT values, hinting towards spatial and temporal reorganization over long periods of time. Nevertheless, the differences between data obtained from the heating and cooling cycles decreased with increasing time (from ~14% to 8% over a period of one month), indicating that the NG-polypeptide interaction finally stabilizes, and it can be expected than a quasi-reversible steady state will be reached over a longer period. Consequently, it was concluded that the final conformations adopted by the polypeptides adsorbed onto the NG, caused by its thermoresponsive behaviour, are reversible. Conversely, polysaccharide-coated NGs displayed an opposite behaviour: no hysteresis was detected and
Rh rose progressively with time to approximately the initial size of the raw NG. In the case of CHIT-terminated NG (
Figure 13c), it rises by ~15%, 23%, 30%, 33% and 34% after 3, 6, 10, 18 and 30 days, respectively. Similar behaviour can be observed for DEXS-terminated systems (
Figure 13d). The NG-polysaccharide interaction likely stabilizes these systems, and after 20 days they reach almost the same size as when first coated. The discrepancies between polysaccharide and polypeptide-coated NGs were attributed to their different level of swelling. After a long period of time polysaccharide-terminated systems, which are more hydrophilic, can absorb more water, resulting in greater disentanglement of the PE chains and the NG network, thereby allowing the whole ensemble to adopt a more expanded configuration.
De Geest
et al. [
25] subjected LbL coated DEX-HEMA MGs to changes in pH to investigate their stability. Self-rupture did not occur at pH 7.4 but at pH 9, ascribed to the pH dependent permeability of the coating. Thus, at pH 7.4 the multilayers assembled were permeable to the degradation products of the MGs whilst at pH 9 they were impermeable, hence the products remained inside the ensemble and the increase in osmotic pressure caused the system to burst.
Figure 13.
Temporal stability of LbL-coated PNIPAM-
co-MMA NGs in water over different periods of time: (
a) NG/PLL; (
b) NG/(PLL/PGA); (
c) NG/CHIT and (
d) NG/(CHIT/DEXS). Closed and empty symbols correspond to heating and cooling cycles, respectively. Gels coated with polysaccharide layers (
c and
d) are reversibly thermoresponsive; for clarity, only the heating cycles are shown. Reproduced with permission from [
8], copyright 2010, Elsevier.
Figure 13.
Temporal stability of LbL-coated PNIPAM-
co-MMA NGs in water over different periods of time: (
a) NG/PLL; (
b) NG/(PLL/PGA); (
c) NG/CHIT and (
d) NG/(CHIT/DEXS). Closed and empty symbols correspond to heating and cooling cycles, respectively. Gels coated with polysaccharide layers (
c and
d) are reversibly thermoresponsive; for clarity, only the heating cycles are shown. Reproduced with permission from [
8], copyright 2010, Elsevier.
6.7. Confocal Microscopy and Fluorescence Correlation Spectroscopy
Additional proof of the successful LbL assembly of biopolymers onto gels can be obtained from confocal microscopy.
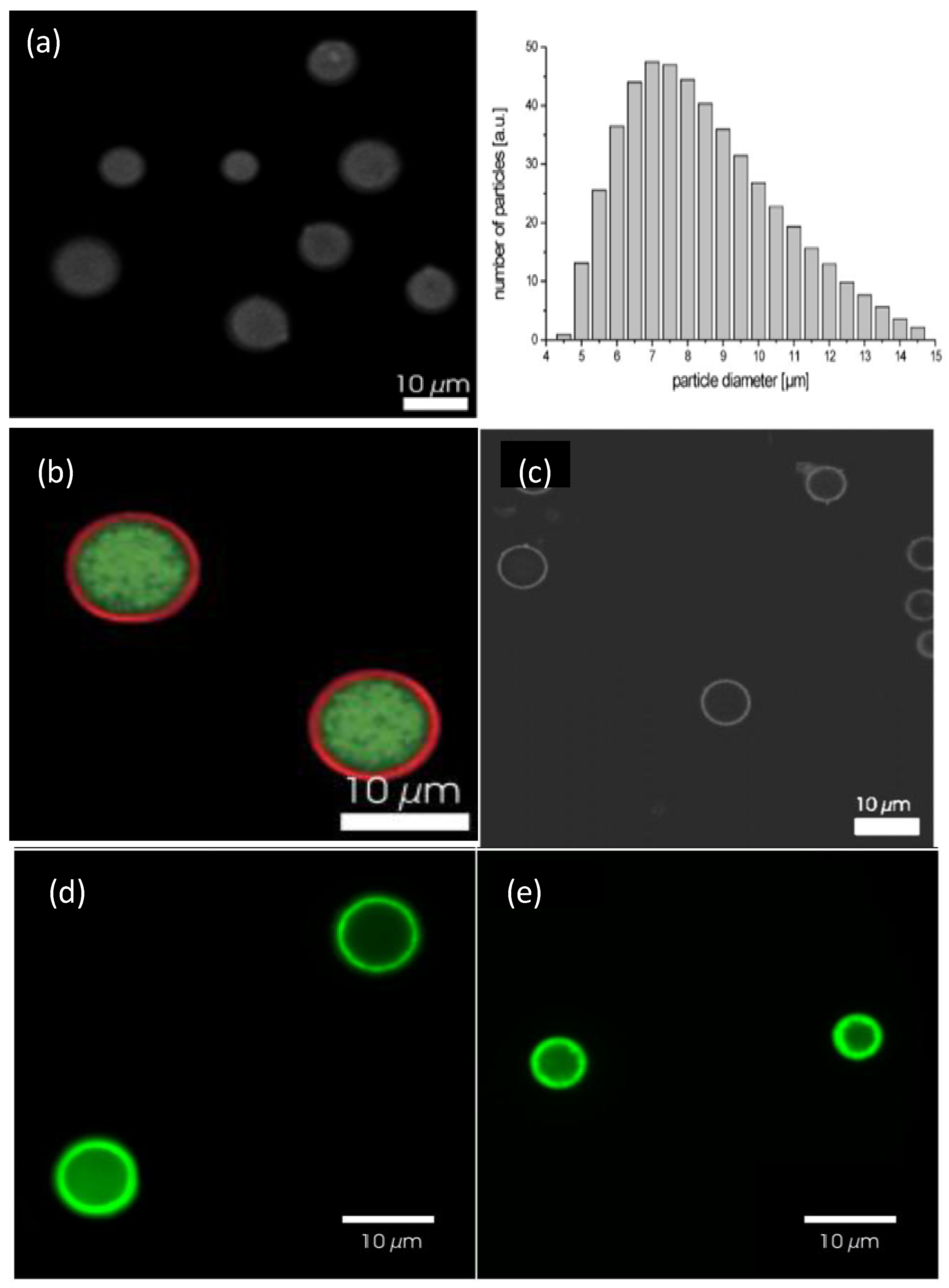
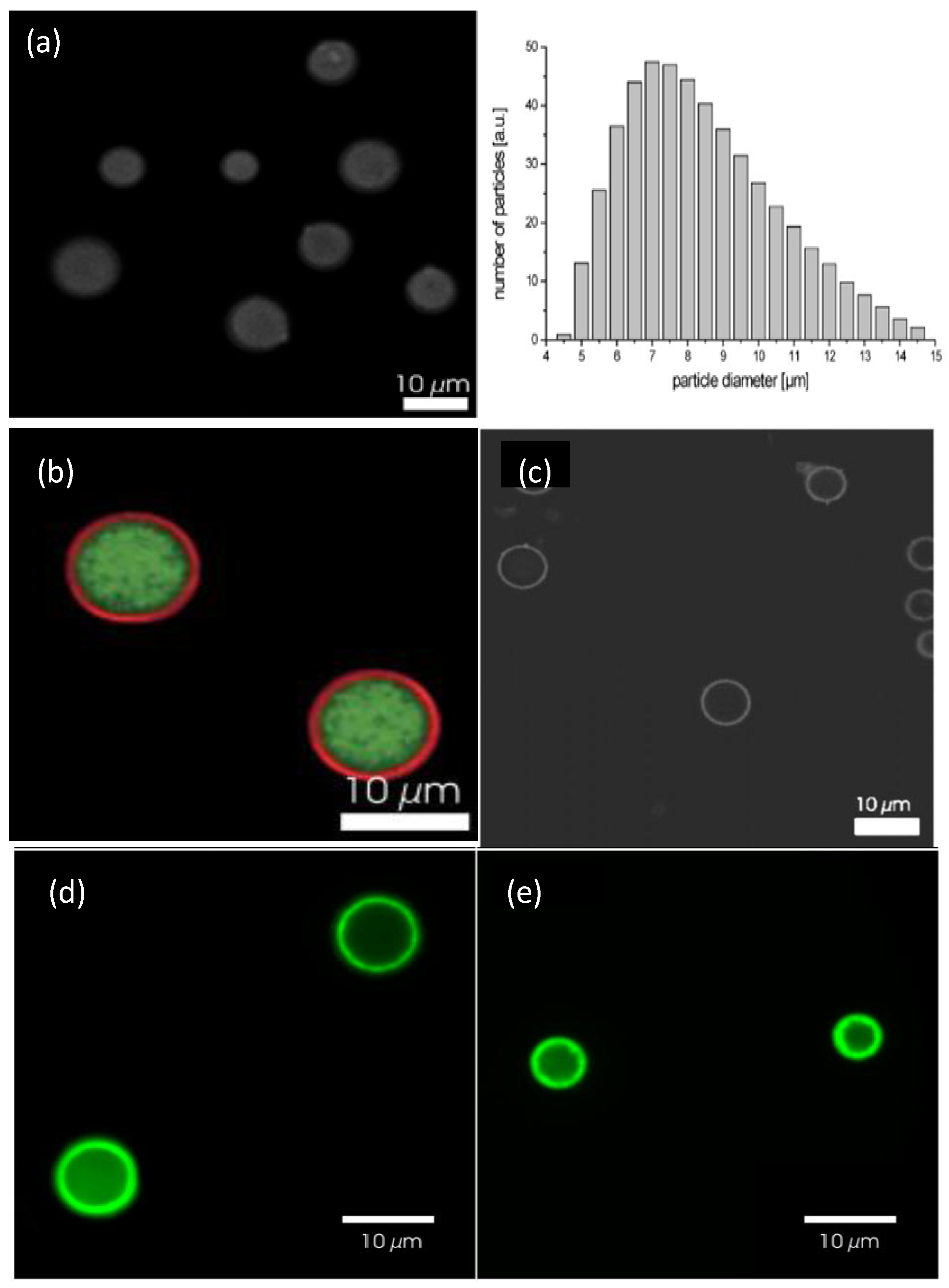
Figure 14a shows a confocal image of bare DEX-HEMA MGs and their size distribution as determined by laser diffraction [
6]. These MGs exhibit a wide size distribution, with an average diameter of 7 μm. A representative image of these MGs coated with four bilayers of (DEXS/pARG) is displayed in
Figure 14b. The MGs were fluorescently labelled with FITC-DEXS (green colour), whilst pARG was labelled with rhodamine B isothiocyanate (RITC, red colour). It can be clearly observed that a ring of red fluorescence originating from the labelled pARG surrounding the MGs is present, indicative of successful LbL coating. The DEX-HEMA core was degraded after exposure of the coated MGs to a 0.1 M phosphate buffer (pH 7.4) at 37 °C for 5 days (
Figure 14c) resulting in hollow capsules; the microcapsules did not rupture, but released the FITC-DEX since no significant fluorescence was detected within the microcapsules. The influence of the PE molecular weight on the behaviour of the core-shell ensemble was also investigated [
6]. Systems with low
Mw PGA all ruptured, while those with high
Mw PGA did not all burst. This suggests that the increase in PGA molecular weight, changes the coaction between the mechanical properties and the permeability of the biopolymer coating.
Figure 14.
(
a) Confocal microscopy images of uncoated DEX-HEMA MGs and their size distribution; (
b and
c) DEX-HEMA MGs coated with four biopolyelectrolyte bilayers before (
b) and after (
c) degradation of the MG. Reproduced with permission from [
6], copyright 2007, Wiley-VCH; (
d and
e) Confocal microscopy images of polypeptide coated P(NIPAM-
co-MAA) microgels at 24 °C (
d) and 37 °C (
e). Reproduced with permission from [
27], copyright 2012, American Chemical Society.
Figure 14.
(
a) Confocal microscopy images of uncoated DEX-HEMA MGs and their size distribution; (
b and
c) DEX-HEMA MGs coated with four biopolyelectrolyte bilayers before (
b) and after (
c) degradation of the MG. Reproduced with permission from [
6], copyright 2007, Wiley-VCH; (
d and
e) Confocal microscopy images of polypeptide coated P(NIPAM-
co-MAA) microgels at 24 °C (
d) and 37 °C (
e). Reproduced with permission from [
27], copyright 2012, American Chemical Society.
P(NIPAM-
co-MAA) coated MGs were also characterised by Costa
et al. [
27] using confocal microscopy at 24 and 37 °C (
Figure 14c,d, respectively). The fluorescent labelling demonstrated that these polyions diffuse into and distribute across the MG, with more fluorescence intensity situated on the MG shell, ascribed to the higher concentration of carboxylic acid groups in that area. Another explanation for this phenomenon might be a self-limiting adsorption process that takes place as the polycation adsorbs onto the MG surface even as it diffuses into the gel, hence producing an intrinsic barrier to the diffusion of more polymer into the MG. The presence of labelled polymer within the gel implies that PEs with higher mobility can display stronger “odd-even” effects based on size and responsive behaviour, as increased mobility offers more accessibility to the acid groups throughout the MG. A gradual rise in fluorescence was detected after each polycation deposition, following a quasi-linear regime with the bilayer number.
Bysell
et al. [
10] analysed the interaction between PLL and PAA MGs (80–120 μm diameter) by confocal laser scanning microscopy. Both peptide distribution and gel de-swelling kinetics were found to be strongly influenced by the PLL molecular weight, pH and salt concentration. A slower de-swelling was observed for the PE with higher
Mw, related to the slower peptide diffusion. Further, at pH 4.5 the rate of microgel de-swelling was lower than at higher pH, explainable in terms of a complex coaction between the pH-dependence of both PLL and the PAA network, also influencing the volume of the latter. Additionally, the increase in the PE concentration resulted in a decreased de-swelling rate, although the peptide transport rate within the MG was higher. These effects will play an important role in the performance of peptide-containing MGs in drug delivery applications, particularly in release rates and loading capacity.
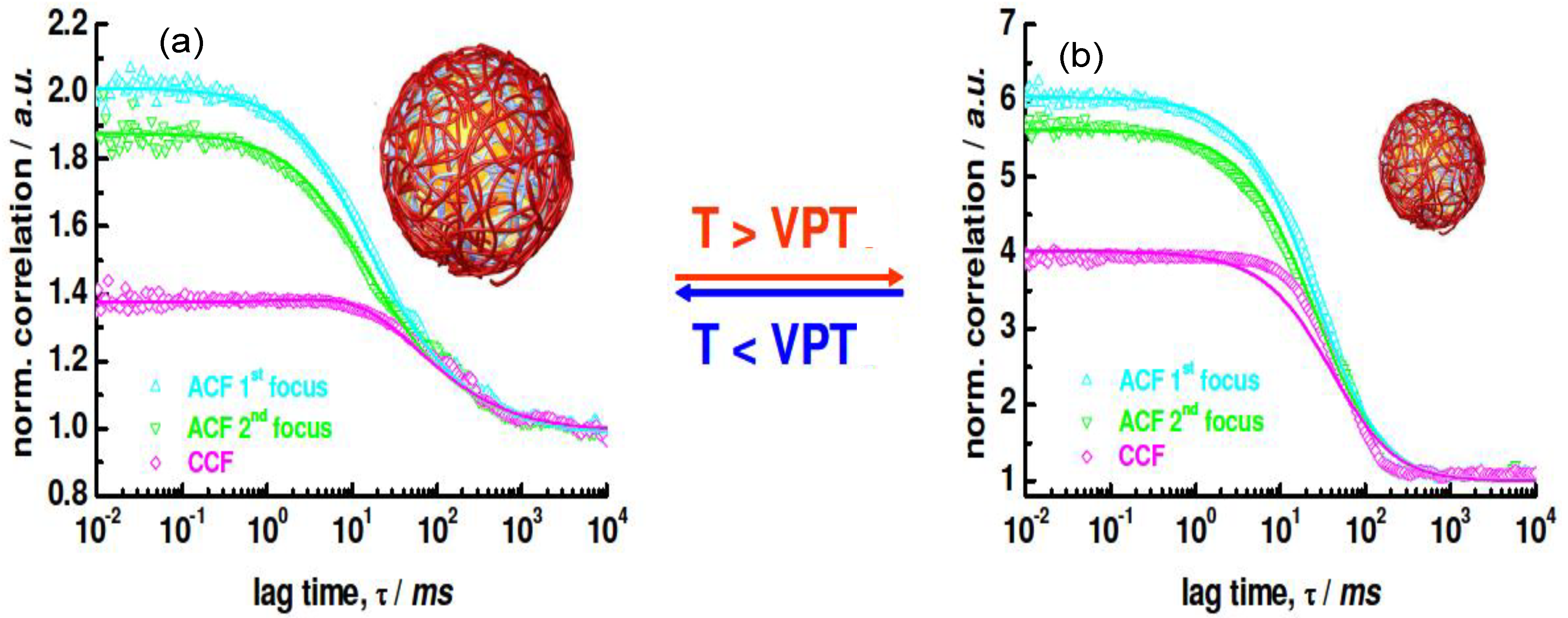
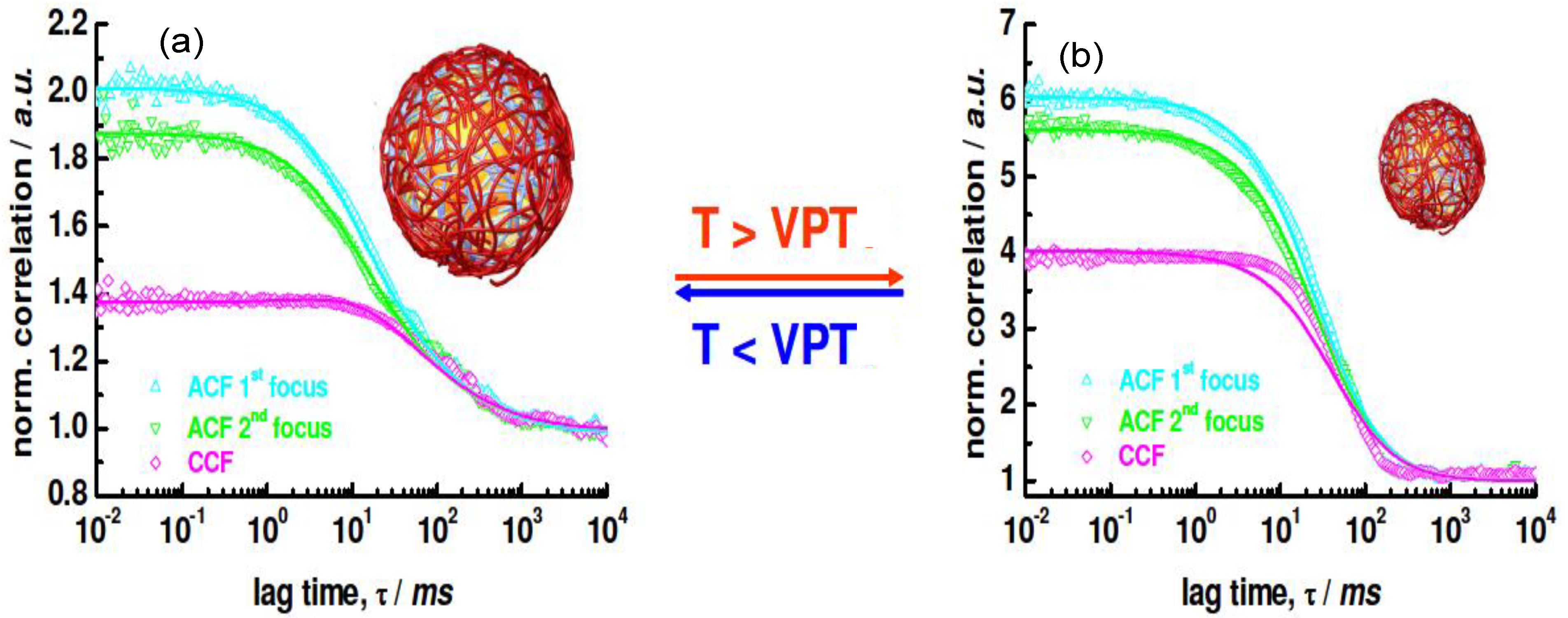
With regard to NGs, it is very difficult to visualize the exact location of the LbL coating using fluorescently-labelled species. FCS has been employed to demonstrate the successful assembly of biopolymers onto PNIPAM NGs [
42]. The 2
f-FCS curves of the uncoated NG labelled with Rhodamine at (a) 25 °C and (b) 40 °C are shown in
Figure 15.
Rh values of the swollen and collapsed state were estimated as 276 and 174 nm, respectively, using the particle size effect model, values that are in good agreement with the
Rh derived from DLS curves, which were 265 and 168 nm, respectively.
Figure 15.
2
f-FCS curves of uncoated rhodamine labeled NG detected at two different foci (ACF1 and ACF2) and cross-correlated (CCF) at (
a) 25 °C and (
b) 40 °C. Reproduced with permission from [
42], copyright 2009, American Chemical Society.
Figure 15.
2
f-FCS curves of uncoated rhodamine labeled NG detected at two different foci (ACF1 and ACF2) and cross-correlated (CCF) at (
a) 25 °C and (
b) 40 °C. Reproduced with permission from [
42], copyright 2009, American Chemical Society.
When a fluorescently-labelled biopolymer PLL
FITC was incorporated into the multilayer system, single or auto-correlated experiments carried out on each species at different layering stages provided similar curves [
42], indicating diffusion times of the same order of magnitude, thus corroborating the anchoring of the labelled PE to the NG. If the species were moving independently, the cross-correlation curve would have shown only a baseline. Images of the dried PNIPAM/PLL
FITC clearly showed that the fluorescence originated exactly from the same species, demonstrating that the PLL was bound to the gel.
6.8. Mechanical Properties
Recently, designing PEM with adjustable mechanical properties has become a key challenge from an application viewpoint. A typical method to measure the mechanical properties is to perform nano-indentation experiments using AFM technique, frequently using a colloidal probe as indenter. Several other methods [
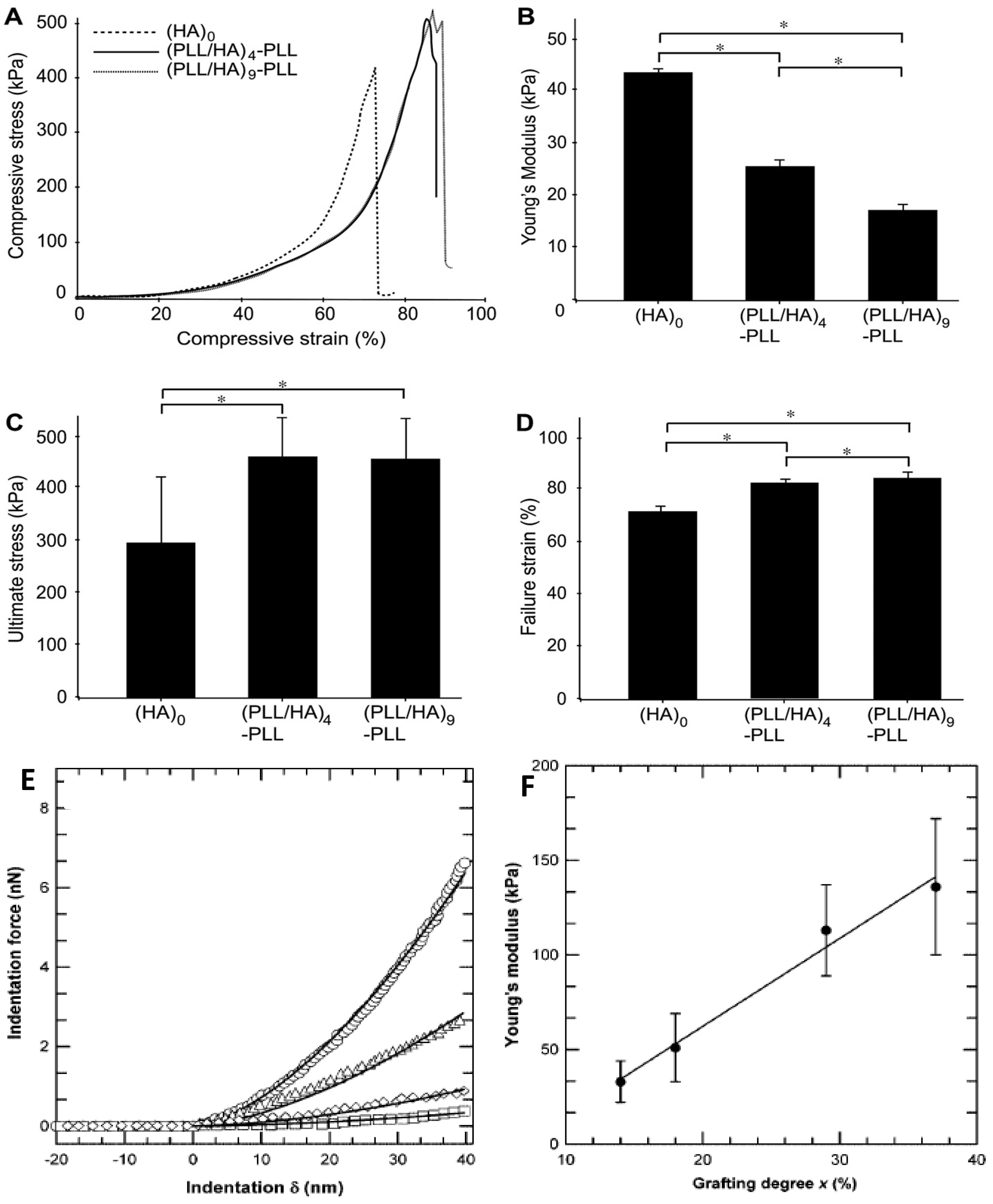
55] have been employed to characterize the thin films including quartz crystal microbalance, piezo-rheometry, the pendant drop, the capillary wave technique, as well as tensile and compression tests. Yamanlar
et al. [
37] investigated the mechanical properties of pure HA MGs and those coated with (PLL/HA)
4-PLL and (PLL/HA)
9-PLL multilayers via compression tests (
Figure 16).
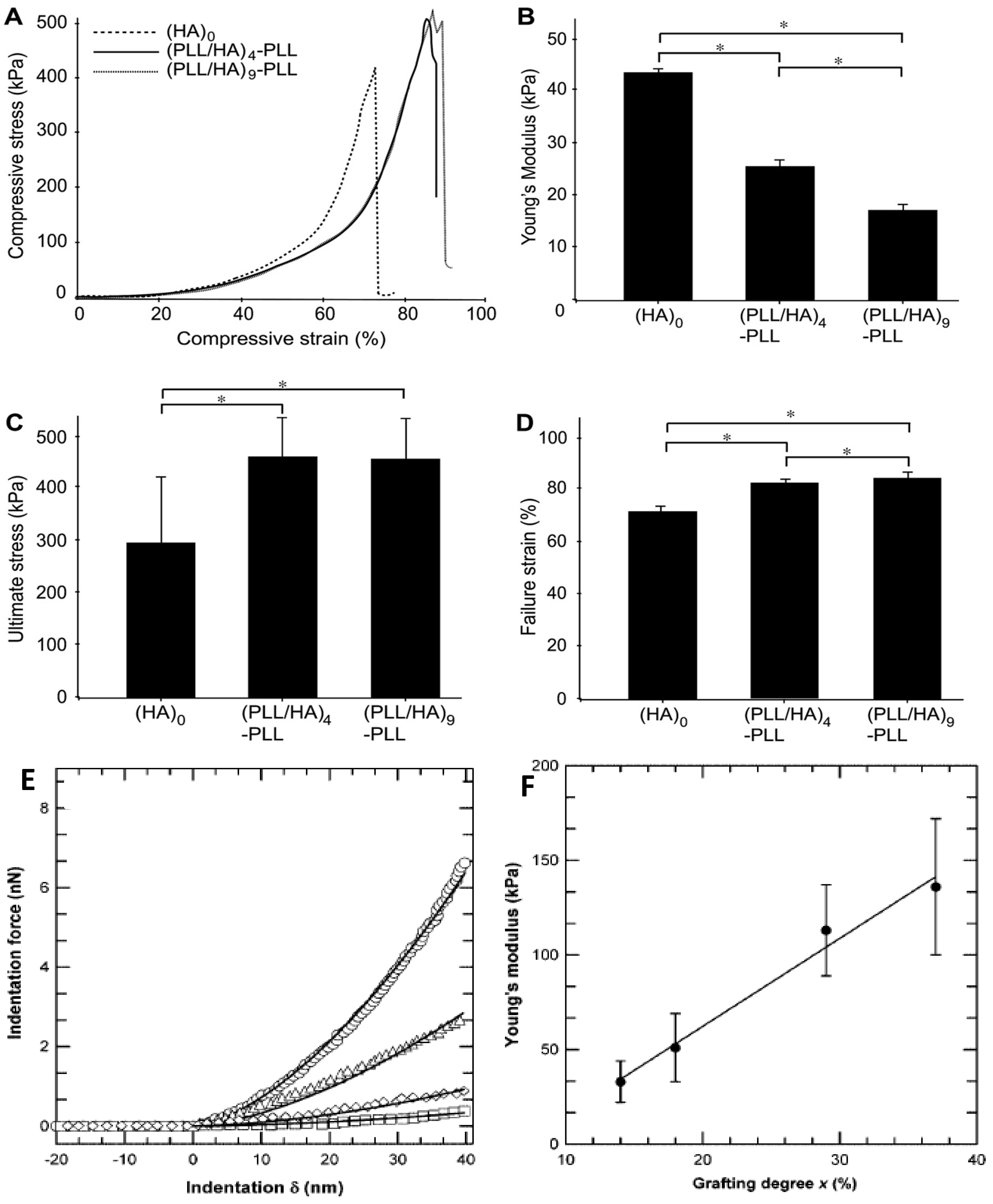
All the systems showed similar stress-strain behaviour irrespective of the number of layer deposited (
Figure 16a). However, the LbL process led to a noticeable decrease in the Young’s modulus (
Figure 16b) whilst increasing the ultimate stress and strain to failure (
Figure 16c,d, respectively). This behaviour was ascribed to the presence of PLL in the outermost layer that caused certain charge shielding, thus decreasing the chain repulsions and resulting in elastic behaviour at low compressive strain. However, at higher strains the higher network density due to the MG shrinking likely led to higher compressive strength. Overall, the stress-strain behaviour was similar for both coated and uncoated systems, suggesting that the observed changes in the mechanical properties were only related to the surface modification of the MGs without affecting their bulk properties, which was confirmed by AFM nano-indentation experiments.
The mechanical properties of VB-
g-HA/PLL films were analysed by Pozos-Vazquez
et al. [
29] using AFM nano-indentation. The comparison of force-indentation curves for samples with grafting degrees from 14% to 37% (
Figure 16e) revealed that the stiffness of the multilayers increased with the grafting degree. Young’s modulus data were estimated using the Hertz sphere model (
Figure 16f), and it was found that the modulus grew linearly with the level of grafting, attributable to the increase in the degree of crosslinking, since the rigidity of a network is inversely proportional to the average length of the segments between two crosslinks, hence proportional to the number of VB groups grafted onto HA chains. Therefore, the stiffness of the film can be precisely tuned by simply varying the grafting degree of VB-modified HA chains incorporated into the films.
Figure 16.
Mechanical properties of unmodified and PE coated HA MGs: (
A) Compressive stress as a function of the strain; (
B) Young’s modulus; (
C) ultimate stress and (
D) ultimate strain. Reproduced with permission from [
37], copyright 2011, Elsevier; (
E) Comparison of force
vs. deformation curves measured by AFM for VB-
g-HA/PLL films with grafting degrees of 14% (squares), 18% (tilted squares), 29% (triangles), and 37% (circles); (
F) Variation of Young’s modulus extracted from force-indentation profiles of VB-
g-HA/PLL
vs. the grafting degree. Reproduced with permission from [
29], copyright 2009, American Chemical Society.
Figure 16.
Mechanical properties of unmodified and PE coated HA MGs: (
A) Compressive stress as a function of the strain; (
B) Young’s modulus; (
C) ultimate stress and (
D) ultimate strain. Reproduced with permission from [
37], copyright 2011, Elsevier; (
E) Comparison of force
vs. deformation curves measured by AFM for VB-
g-HA/PLL films with grafting degrees of 14% (squares), 18% (tilted squares), 29% (triangles), and 37% (circles); (
F) Variation of Young’s modulus extracted from force-indentation profiles of VB-
g-HA/PLL
vs. the grafting degree. Reproduced with permission from [
29], copyright 2009, American Chemical Society.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


 ) grafting degree. Reproduced with permission from [44], copyright 2012, American Chemical Society.
) grafting degree. Reproduced with permission from [44], copyright 2012, American Chemical Society.