Polyacylurethanes as Novel Degradable Cell Carrier Materials for Tissue Engineering

Abstract
:1. Introduction
2. Results and Discussion
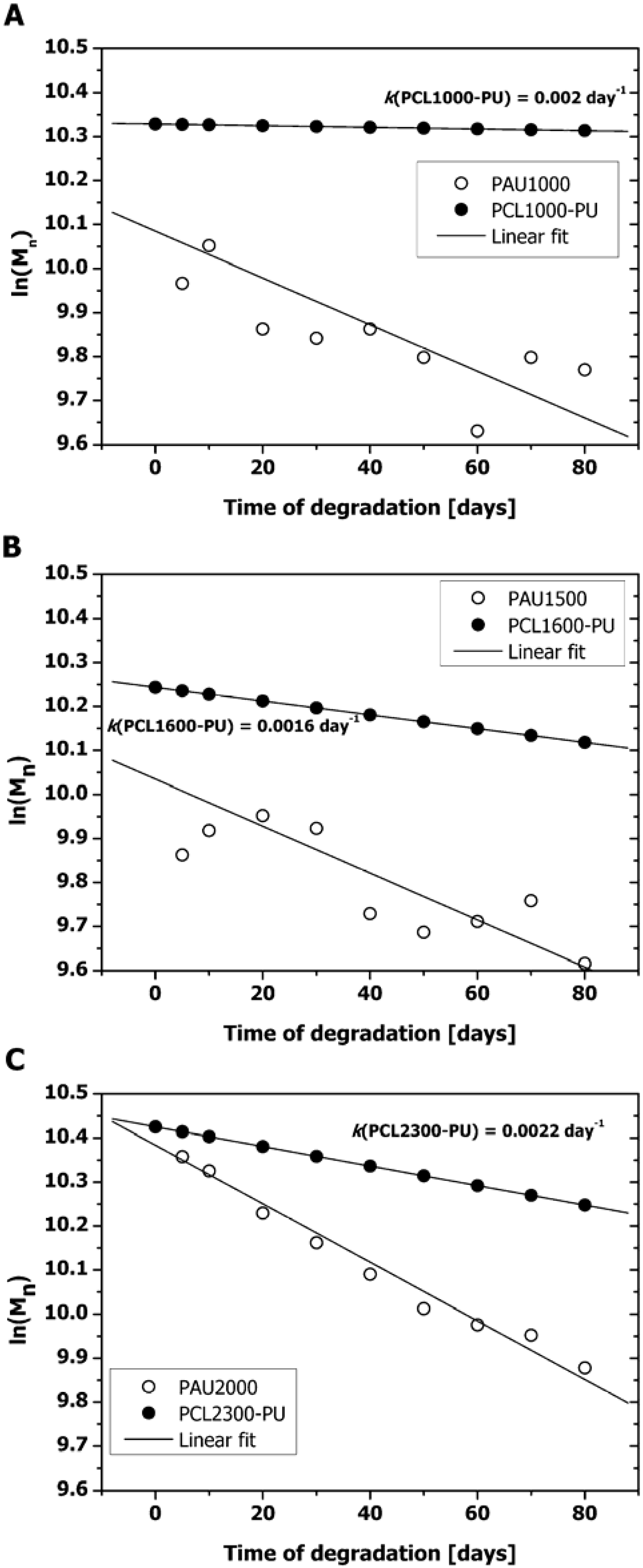
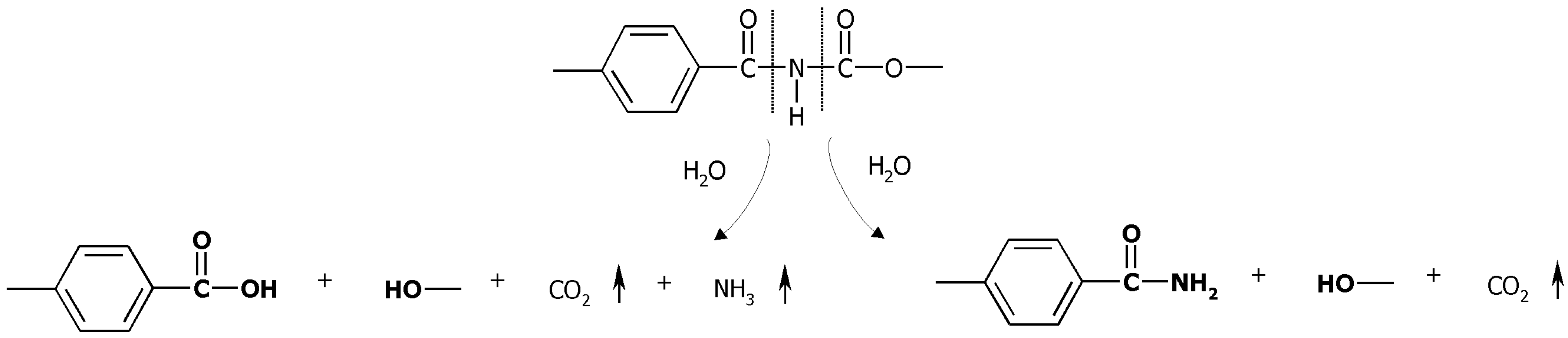
2.1. Mass Loss and Molar Mass Change

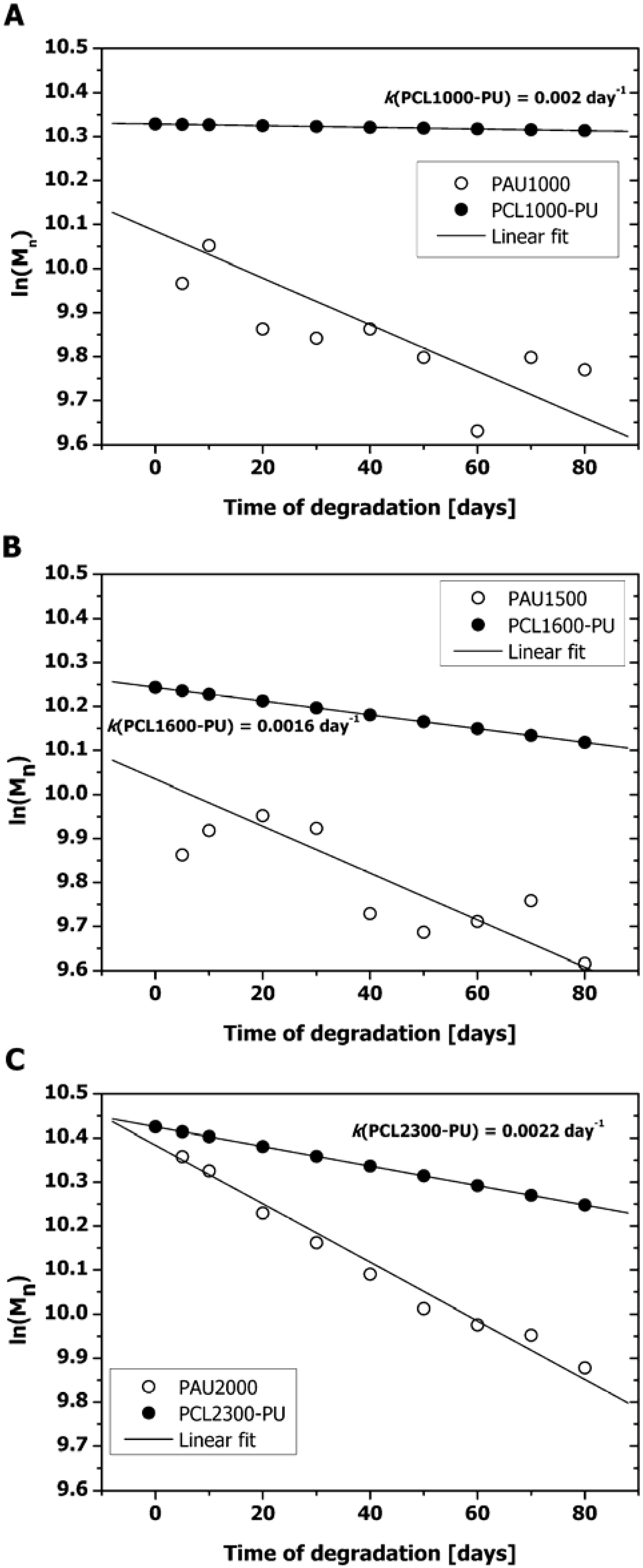
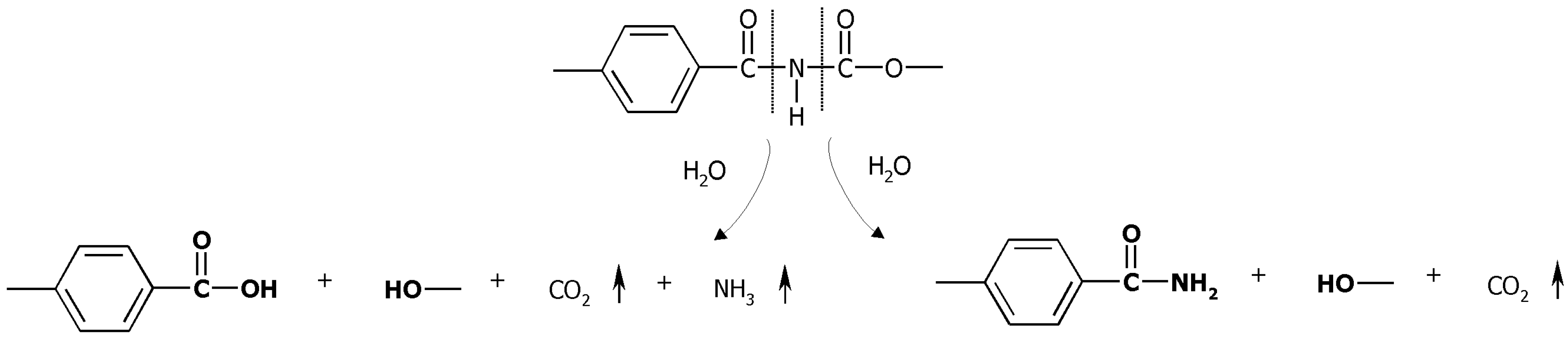
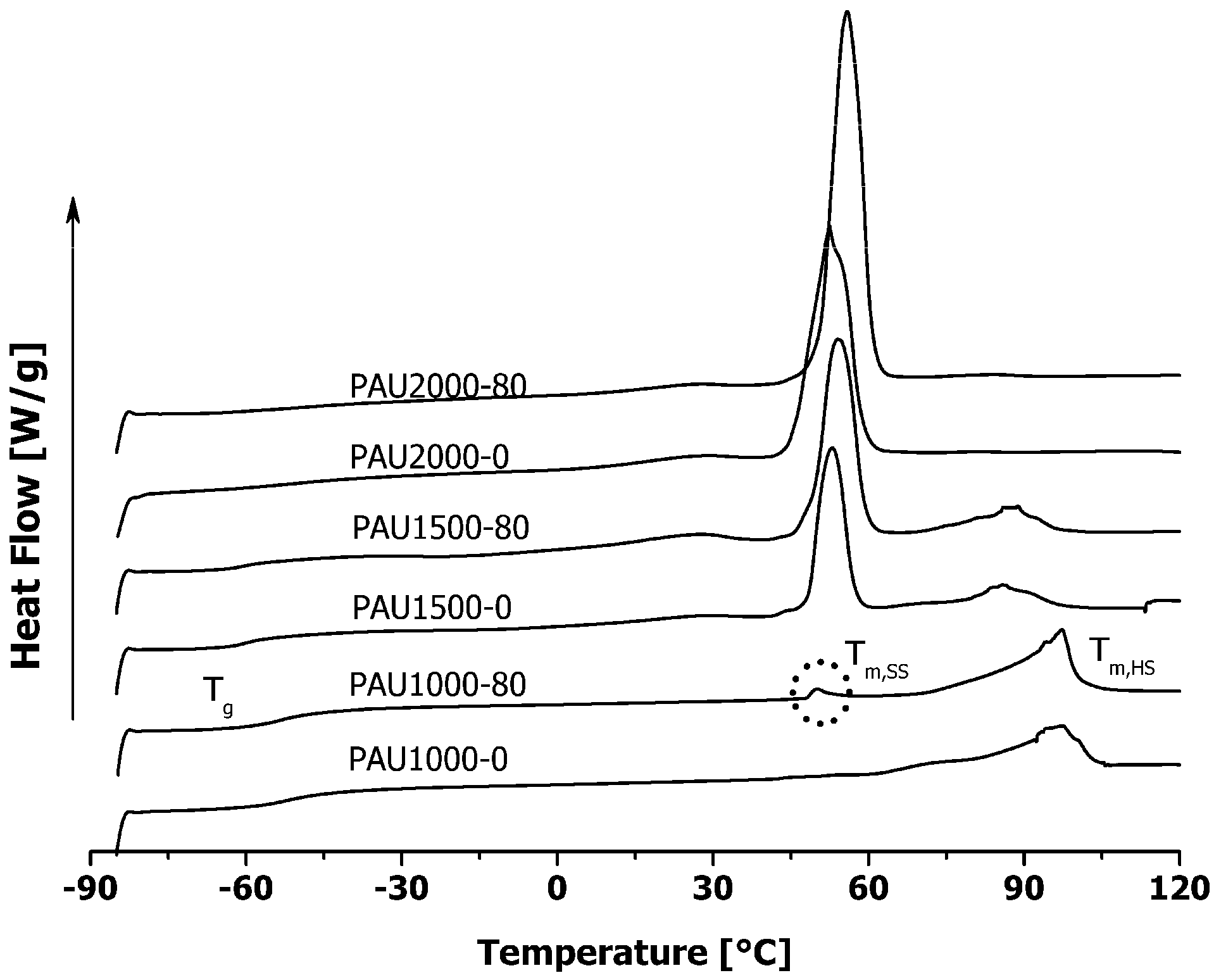
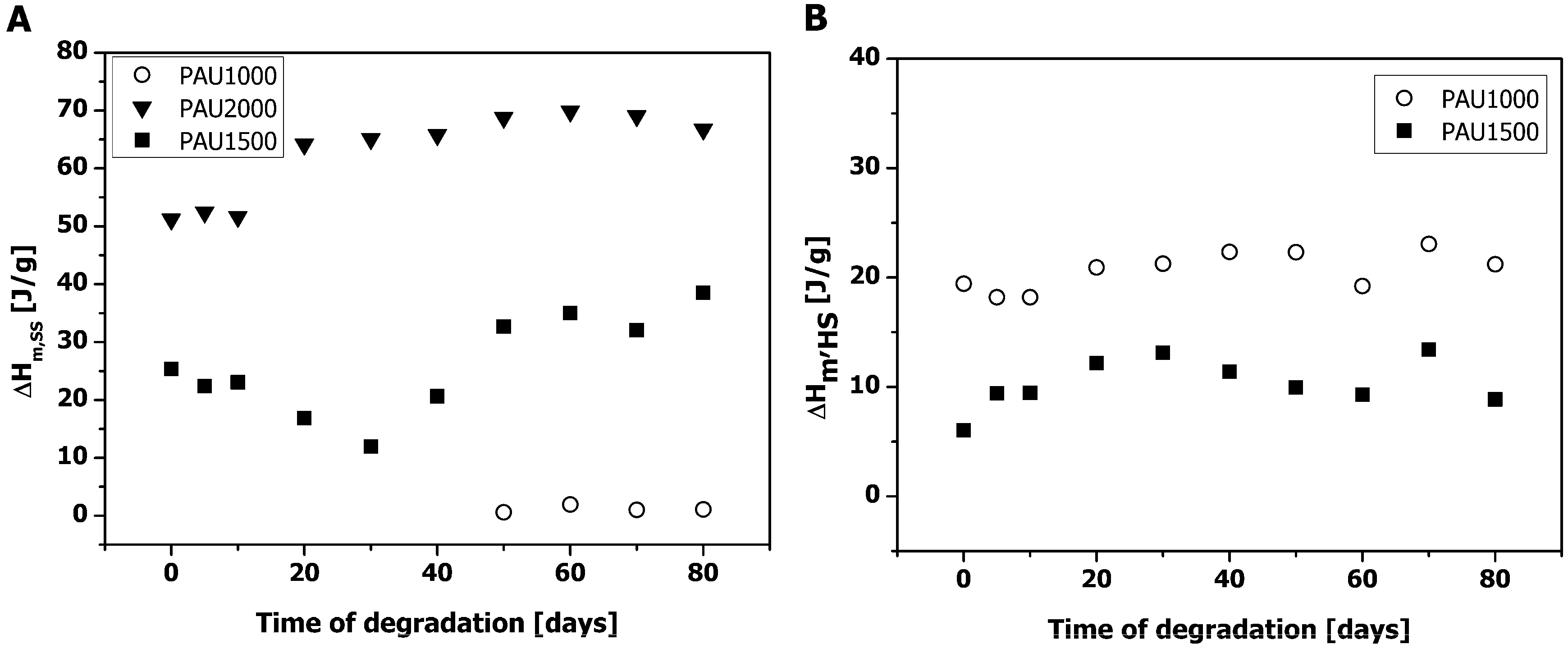
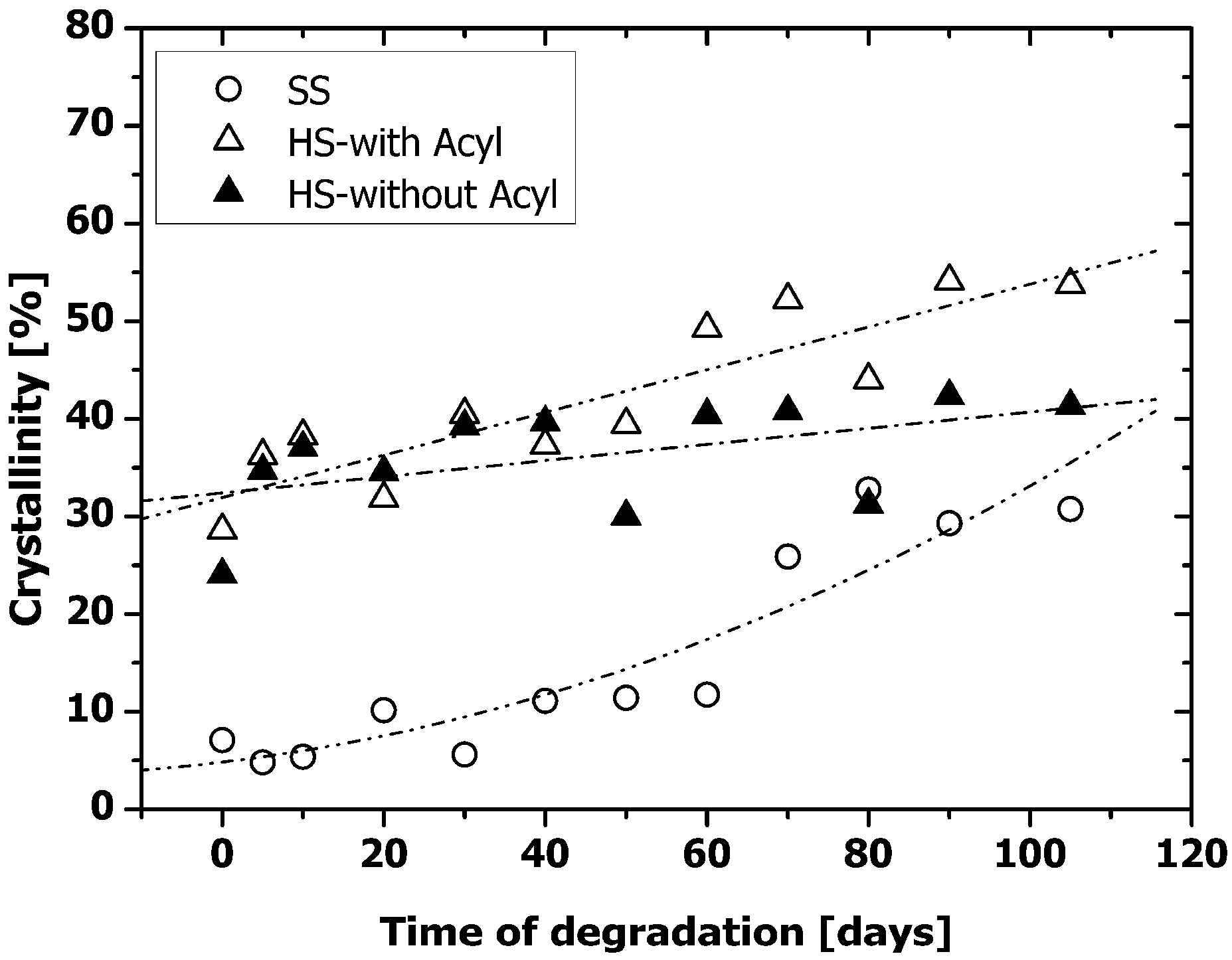
2.2. Thermal Properties of PAUs Upon Degradation
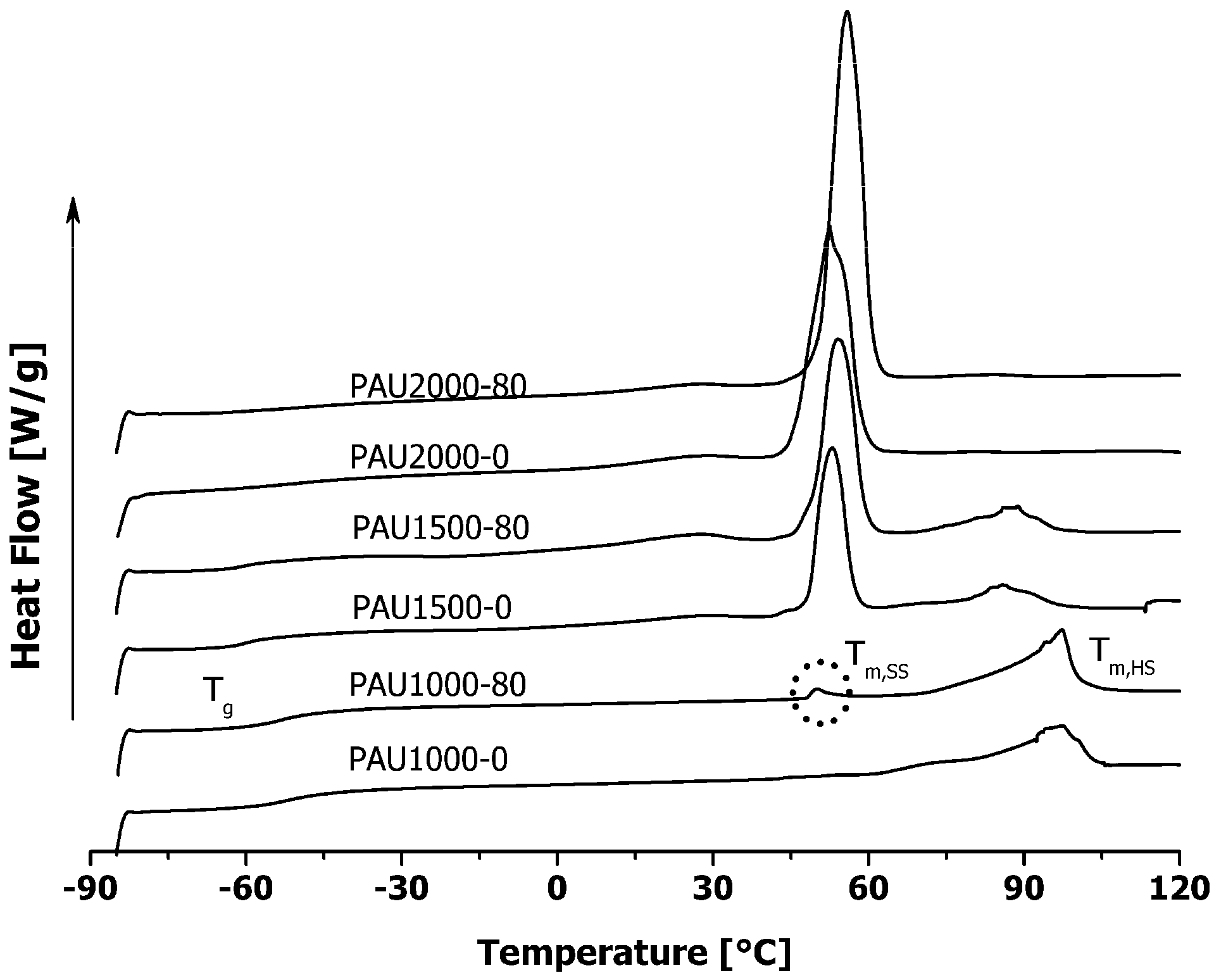
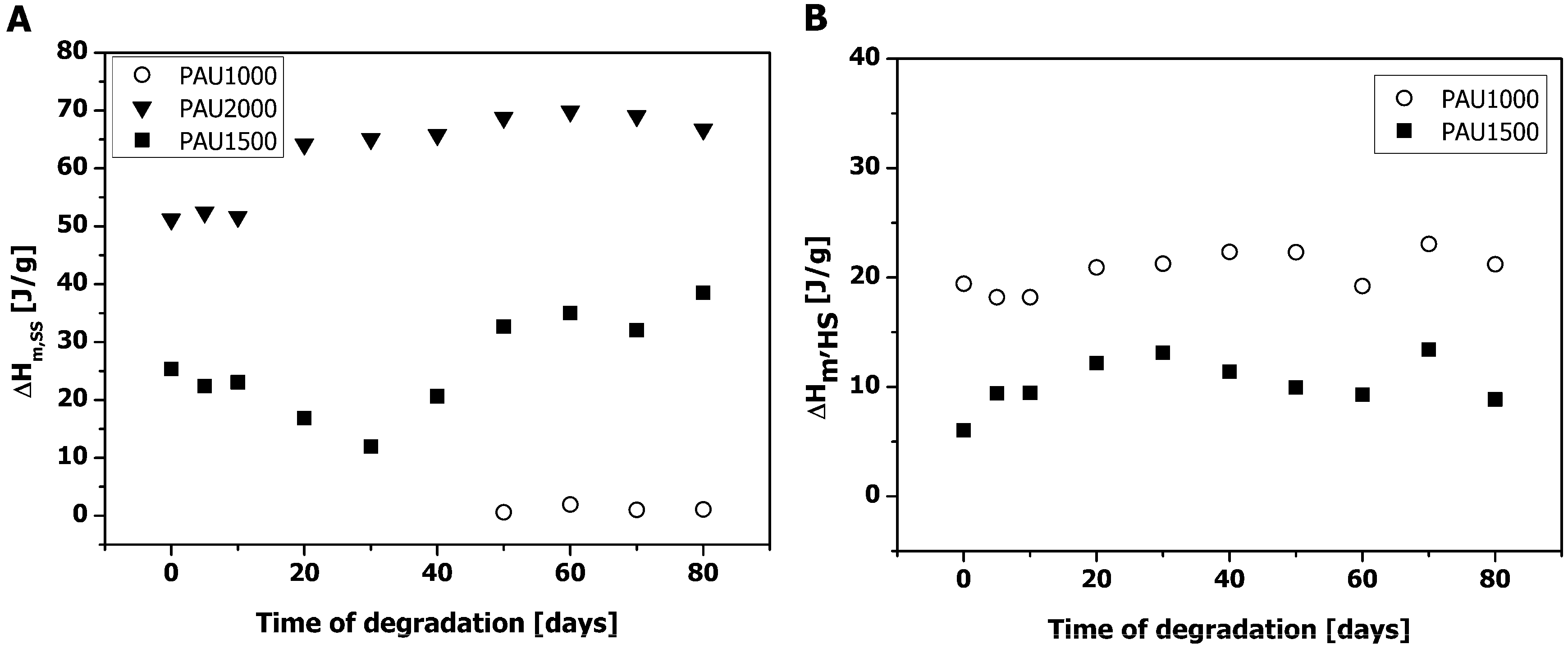
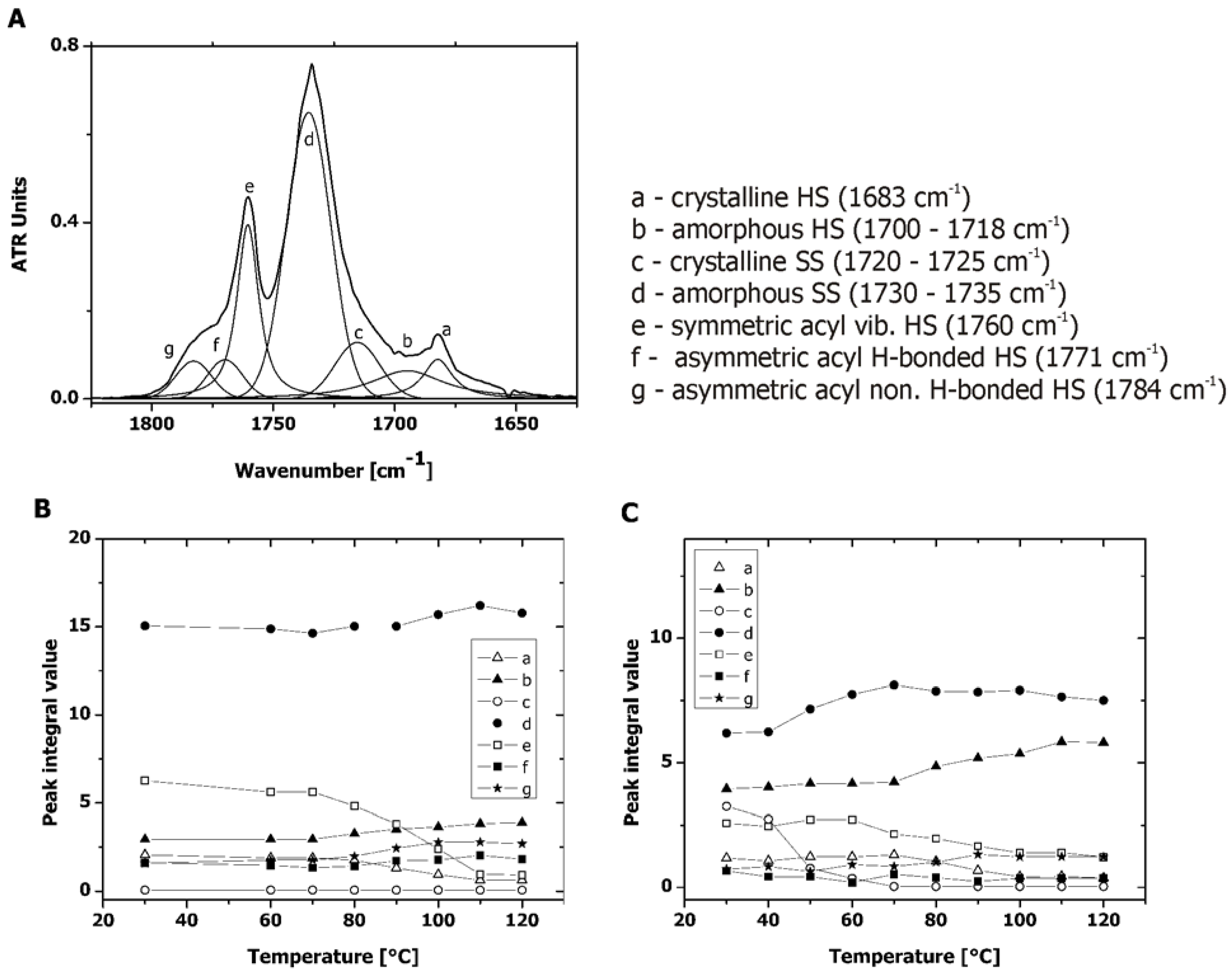
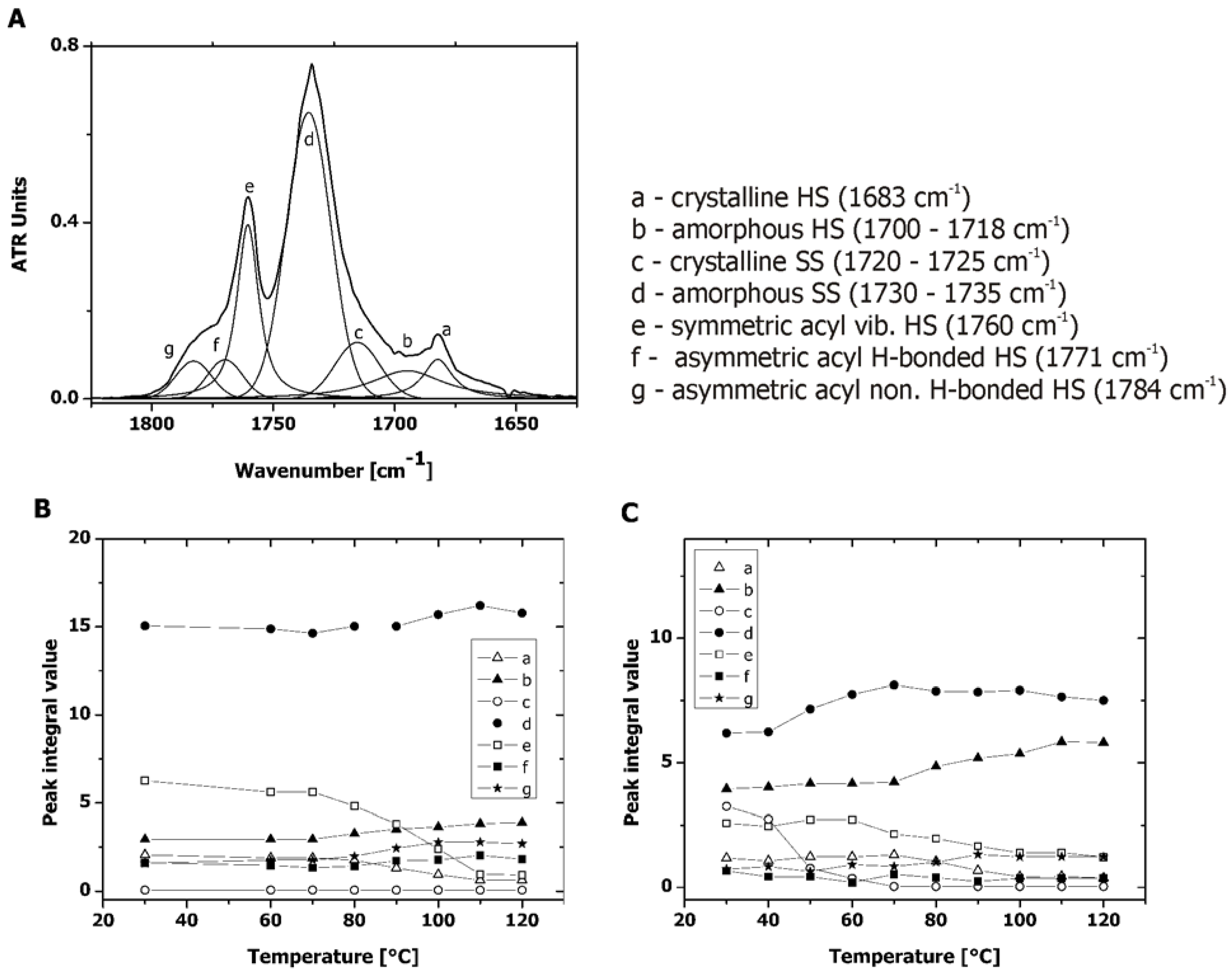
2.3. Microphase Separation of PAUs Prior to Degradation (Trans-FTIR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | HS cryst* (%) | SS cryst** (%) |
|---|---|---|
| PAU1000-0 | 57.5 | 0.4 |
| PAU1500-0 | 41.3 | 34.4 |
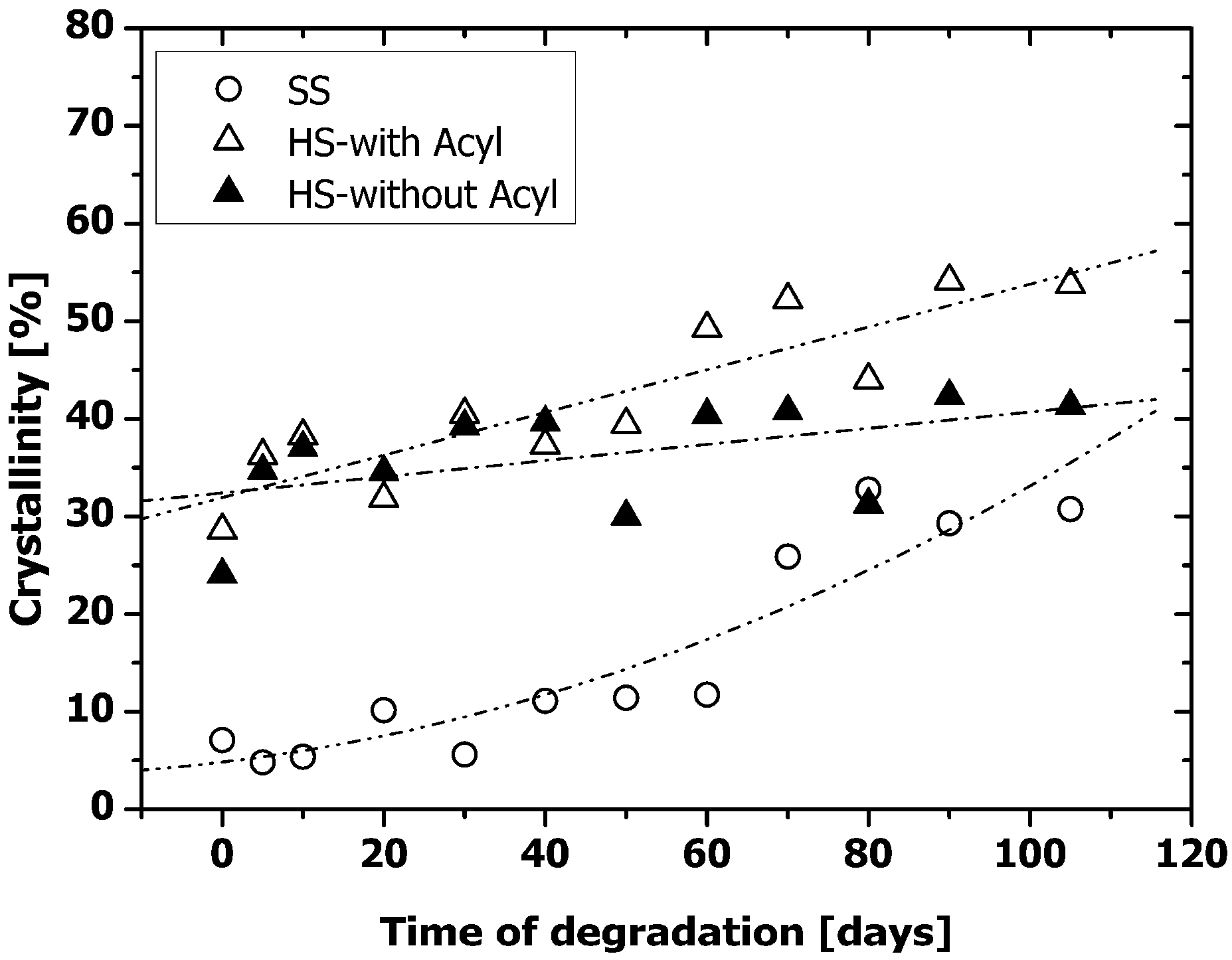
2.4. Microphase Separation of PAUs Upon Degradation (ATR-FTIR)
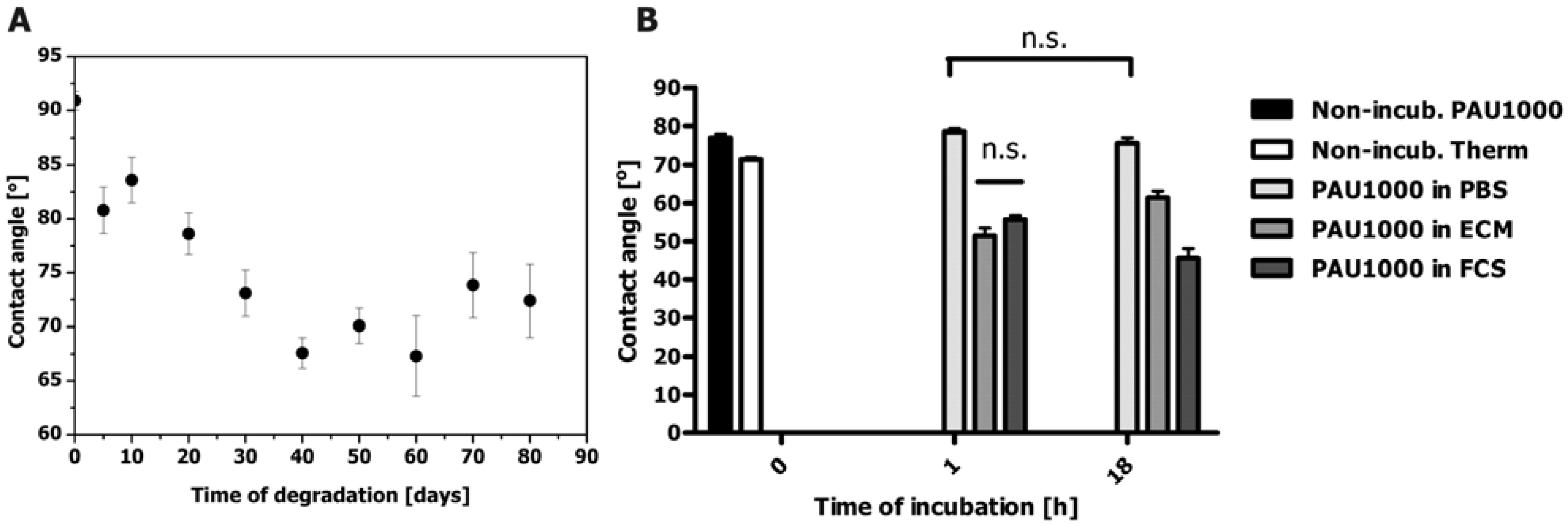
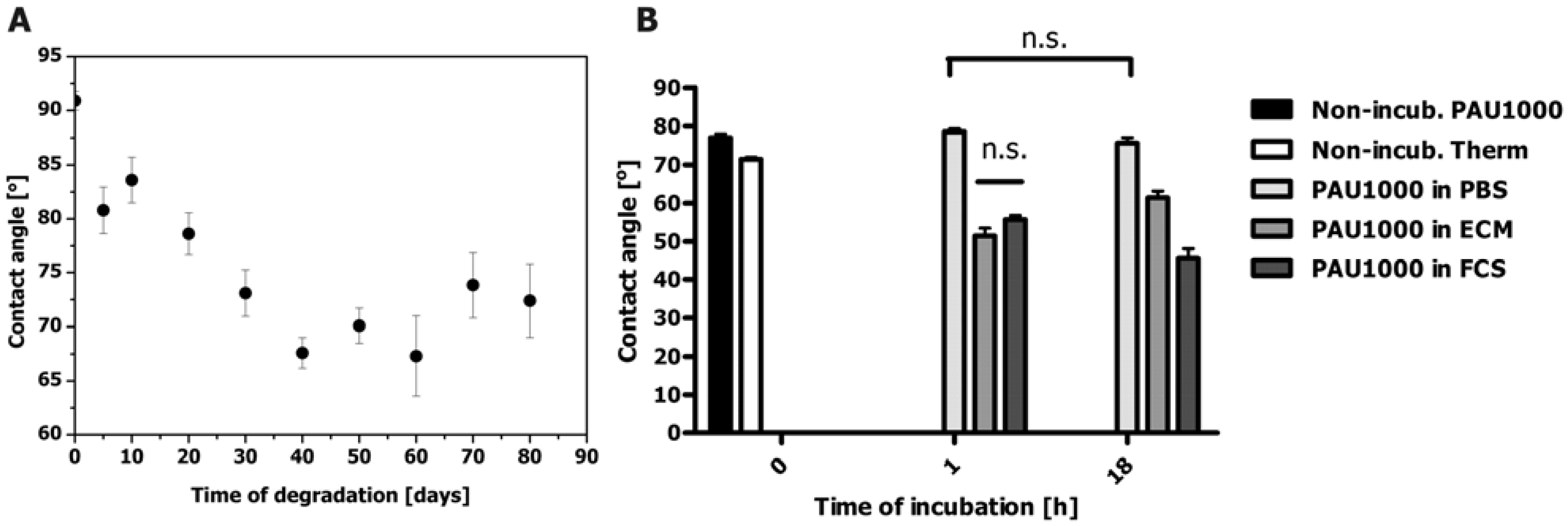
2.5. Contact angle Measurements
2.6. Cytotoxicity (MTS Assay)
| [Test material | Cell survival [%] |
|---|---|
| PAU1000 | 86 ± 6 |
| Pellethane® | 91 ± 6 |
| Latex | 38 ± 1 |
| Medium | 104 ± 13 |
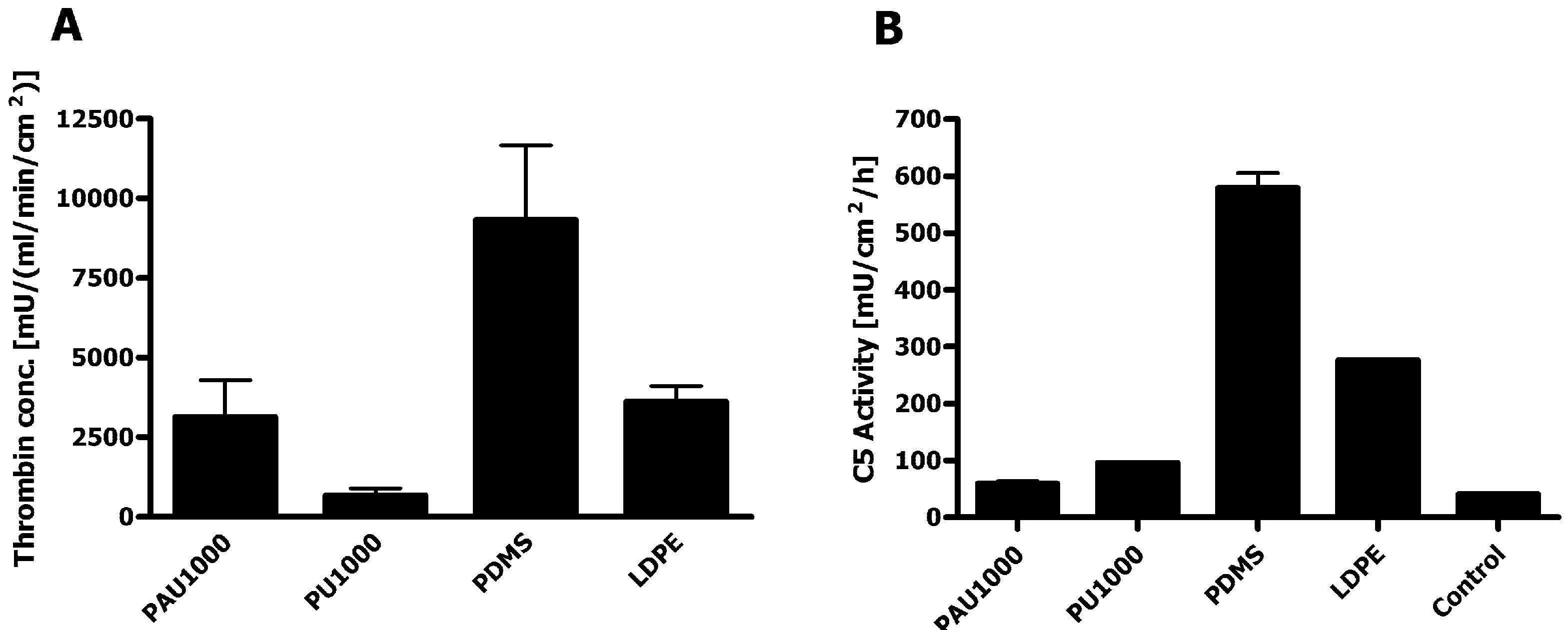
2.7. Haemocompatibility

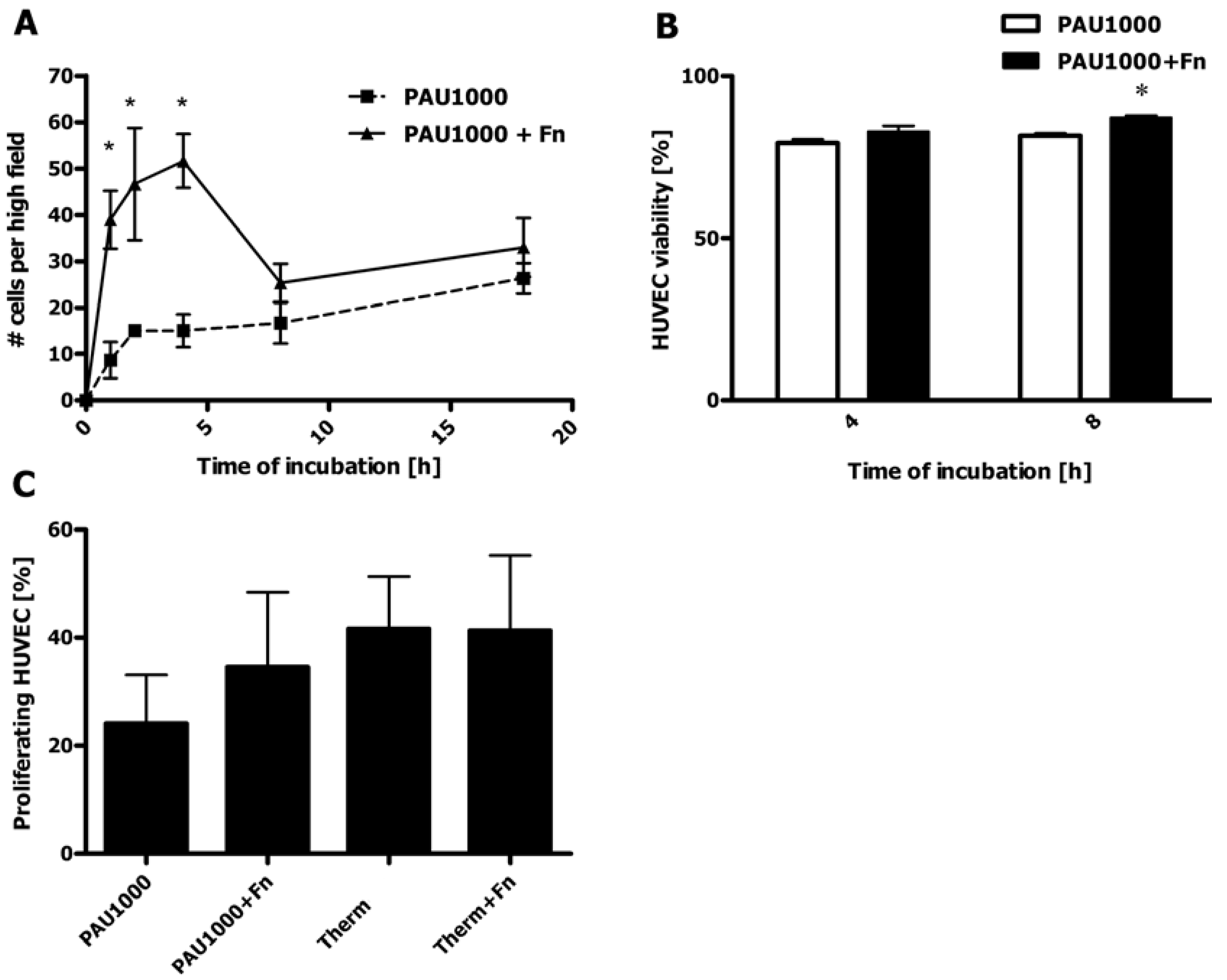
2.8. HUVEC Adhesion to PAU1000
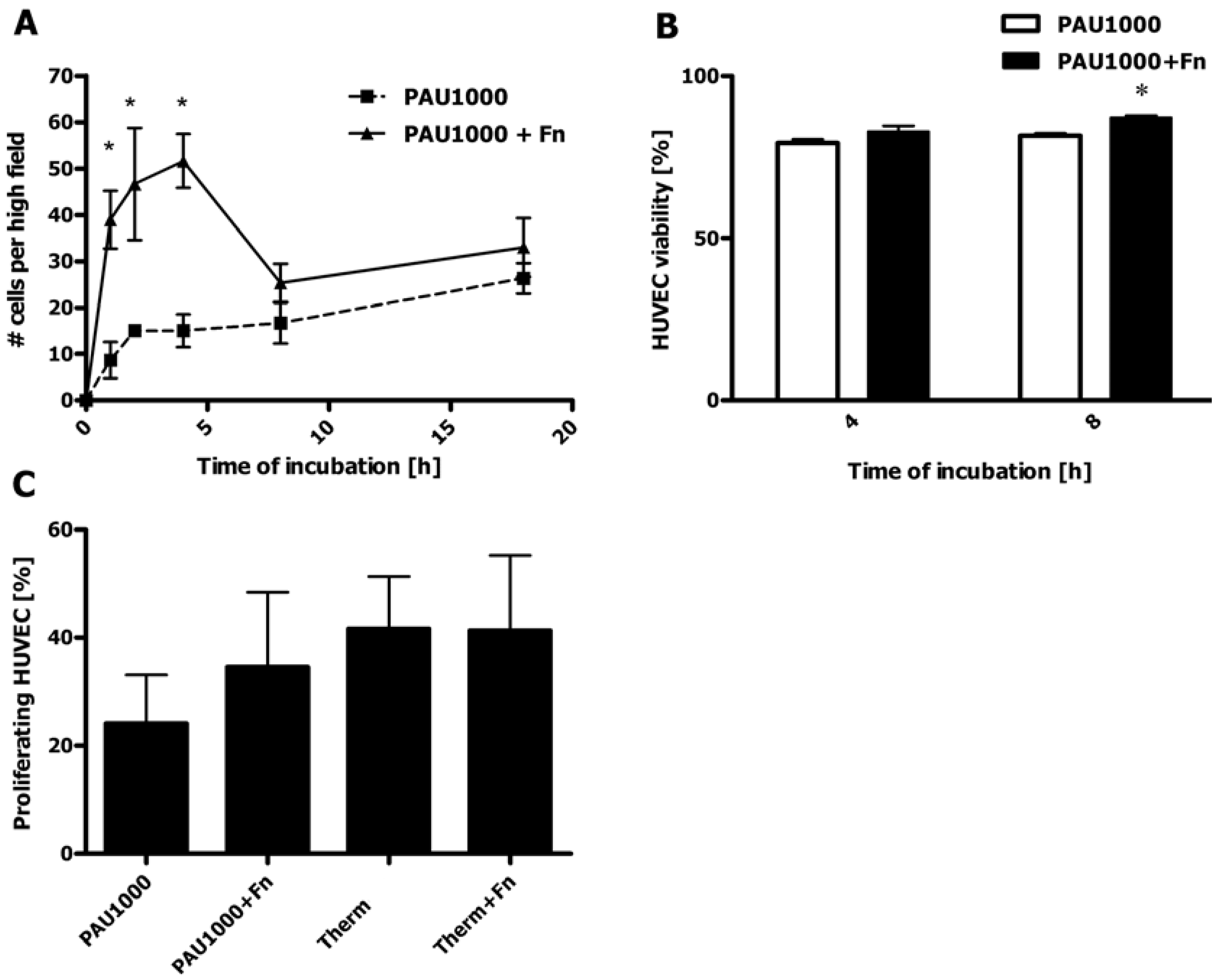
2.9. HUVEC Proliferation on PAUs
3. Experimental Section
3.1. Materials
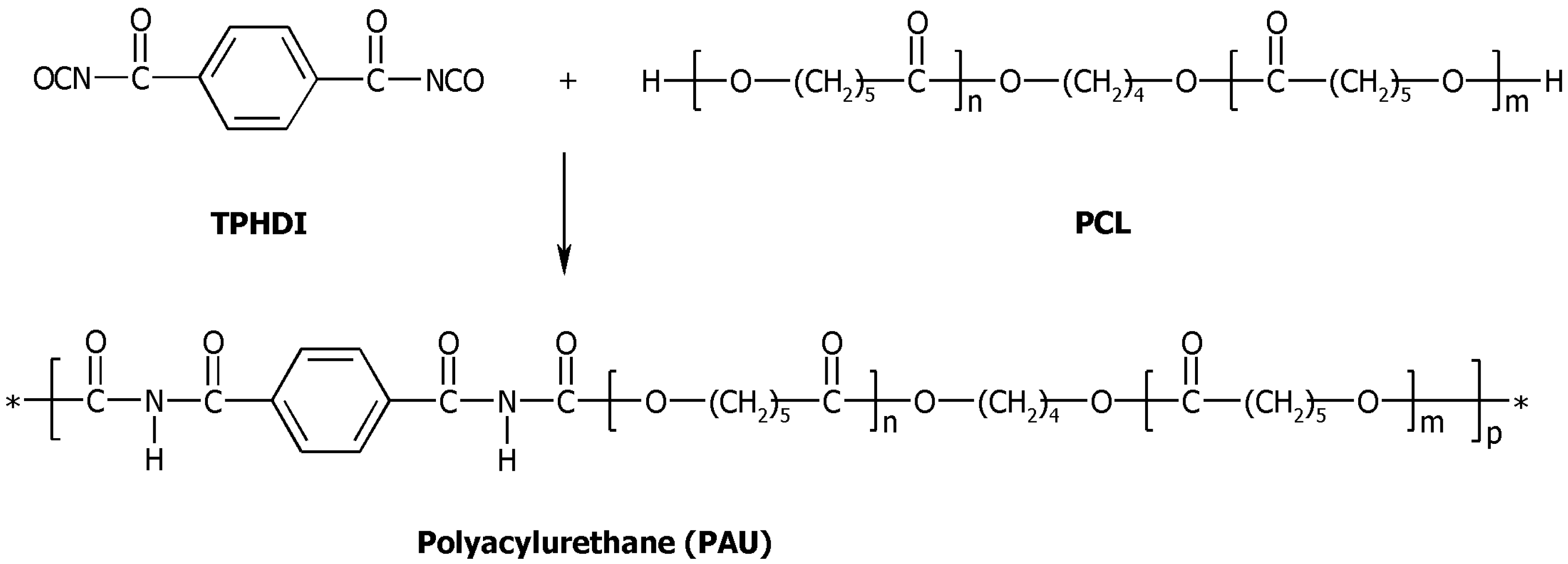
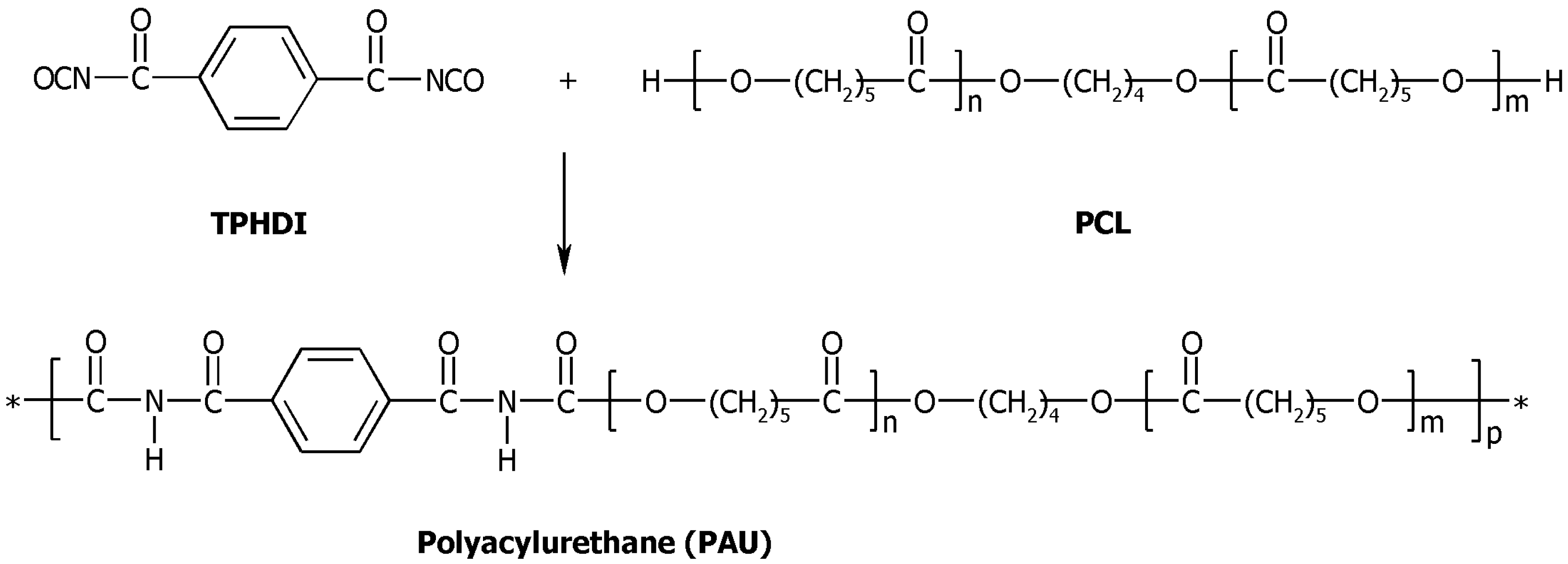
3.2. Synthesis of Polyacylurethanes
3.3. Preparation of Polymer Films
3.4. In Vitro Degradation Set-Up
3.5. Mass Loss
3.6. Molar Mass
3.7. Thermal Properties of PAUs
3.8. Fourier-Transform Infra Red Spectroscopy (FTIR)
3.9. Contact angle Measurements
3.10. MTS Cytotoxicity Assay
3.11. Haemocompatibility of PAU
3.12. HUVEC Adhesion and Viability on PAU1000
3.13. HUVEC Proliferation on PAU1000
3.14. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Pêgo, A.P.; van Luyn, M.J.A.; Brouwer, L.A.; van Wachem, P.B.; Poot, A.A.; Grijpma, D.W.; Feijen, J. In vivo behavior of poly(1,3-trimethylene carbonate) and copolymers of 1,3-trimethylene carbonate with d,l-Lactide or ε-caprolactone: Degradation and tissue response. J. Biomed. Mater. Res. Part A 2003, 67, 1044–1054. [Google Scholar] [CrossRef]
- Lam, C.X.F.; Hutmacher, D.W.; Schantz, J.-T.; Woodruff, M.A.; Teoh, S.H. Evaluation of polycaprolactone scaffold degradation for 6 months in vitro and in vivo. J. Biomed. Mater. Res. Part A 2009, 90A, 906–919. [Google Scholar] [CrossRef]
- Bergsma, J.E.; Bruin, W.C.; Rozema, F.R.; Bos, R.R.M. Late degradation tissue response to poly(L-lactide) bone plates and screws. Biomaterials 1995, 16, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Böstman, O.; Pihlajamäki, H.K.; Partio, E.K.; Rokkanen, P.U. Clinical biocompatibility and degradation of polylevolactide screws in the ankle. Clin. Orth. Rel. Res. 1995, 320, 101–109. [Google Scholar]
- Lamba, N.M.K.; Woodhouse, K.A.; Cooper, S.L. Polyurethanes in Biomedical Applications; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Endo, T.; Kanamaru, M.; Takata, T. Synthesis of poly(N-acylurethane)s, a new class of polyurethanes. Macromolecules 1994, 27, 3694–3697. [Google Scholar] [CrossRef]
- Yabuta, M.; Urano, S. Polyacylurethane and Process for Producing the Same. Appl.: 432705. US Patent 5,556,933, 17 September 1996. [Google Scholar]
- Heijkants, R.G.J.C.; Schwab, L.W.; van Calck, R.V.; de Groot, J.H.; Pennings, A.J.; Schouten, A.J. Extruder synthesis of a new class of polyurethanes: Polyacylurethanes based on poly(ε-caprolactone) oligomers. Polymer 2005, 46, 8981–8989. [Google Scholar] [CrossRef]
- Heijkants, R.G.J.C.; Schouten, A.J.; Schwab, L.W.; Roukes, F.V.; Pennings, A.J. Polyacylurethanes Based on Diisocyanates and Polyesterpolyols. Appl.: PCT/NL2005/000528. Patent: WO/2011/009443, 26 January 2006. [Google Scholar]
- Lelah, M.D.; Cooper, S.L. Polurethanes in Medicine; CRC Press Inc.: Boca Raton, FL, USA, 1986. [Google Scholar]
- Cheav, S.L.; Foussard-Blanpin, O. Edude comparée de la toxicité cyanés et/ou amides aromatiques. Ann. Pharmaceutiques Françaises 1990, 48, 23–31. [Google Scholar]
- Akkapeddi, M.K.; Gervasi, J.A. Chain Extension of Polyethylene Terephthalate with Polyacyllactams. Appl.: 07/161927. US Patent 4857603, 15 August 1989. [Google Scholar]
- Kanamaru, M.; Takata, T.; Endo, T. Sequence change of poly(N-acylurethane)s based on transesterification. Macromol. Chem. Phys. 1996, 197, 1795–1804. [Google Scholar] [CrossRef]
- Castner, D.G.; Ratner, B.D. Biomedical surface science: Foundation to frontiers. Surface Sci. 2002, 500, 28–60. [Google Scholar] [CrossRef]
- Horbett, T.A. Principles underlying the role of adsorbed plasma proteins in blood interactions with foreign materials. Cardiovasc. Pathol. 1993, 2, 137S–148S. [Google Scholar] [CrossRef]
- Gõpferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef] [PubMed]
- von Burkersroda, F.; Schedl, L.; Gõpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef] [PubMed]
- Heijkants, R.G.J.C. Polyurethane Scaffolds as Meniscus Reconstruction Materials. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2004. Available online: http://irs.ub.rug.nl/ppn/270337644 (accessed on 20 December 2004). [Google Scholar]
- Pitt, C.G.; Gu, Z. Modification of the rates of chain cleavage of poly(ε-caprolactone) and related polyesters in the solid state. J. Control. Rel. 1987, 4, 283–292. [Google Scholar] [CrossRef]
- Antheunis, H.; van der Meer, J.-C.; de Geus, M.; Kingma, W.; Koning, C.E. Improved mathematical model for the hydrolytic degradation of aliphatic polyesters. Macromolecules 2009, 42, 2462–2471. [Google Scholar] [CrossRef]
- Pretsch, T.; Jakob, I.; Müller, W. Hydrolytic degradation and functional stability of a segmented shape memory poly(ester urethane). Polym. Deg. Stab. 2009, 94, 61–73. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequences: Tables and Charts, 3rd ed.; John Wiley & sons, Inc.: Hoboken, NJ, USA, 2011. [Google Scholar]
- Lee, H.S.; Wang, Y.K.; Hsu, S.L. Spectroscopic analysis of phase separation behavior of model polyurethane. Macromolecules 1987, 20, 2089–2095. [Google Scholar] [CrossRef]
- Gogolewski, S. Selected topics in biomedical polyurethanes. A review. Colloid. Polym. Sci. 1989, 267, 757–785. [Google Scholar] [CrossRef]
- Sun, S.; Tang, H.; Wu, P. Interpretation of carbonyl band splitting phenomenon of a novel thermotropic liquid crystalline polymer without conventional mesogens: Combination method of spectral analysis and molecular simulation. J. Phys. Chem. B 2010, 114, 3439–3448. [Google Scholar] [CrossRef] [PubMed]
- Heijkants, R.G.J.C.; van Calck, R.V.; van Tienen, T.G.; de Groot, J.H.; Buma, P.; Pennings, A.J.; Veth, R.P.H.; Schouten, A.J. Uncatalyzed synthesis, thermal and mechanical properties of polyurethanes based on poly(ε-caprolactone) and 1,4-butanediisocyanate with uniform hard segment. Biomaterials 2005, 26, 4219–4228. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Suska, F.; Johansson, A.; Berglin, M.; Emanuelsson, L.; Elwing, H.; Thomsen, P. Effect of molecular mobility of polymeric implants on soft tissue reactions: An in vivo study in rats. J. Biomed. Mater. Res. 2008, 84A, 652–660. [Google Scholar] [CrossRef]
- Grasel, T.G.; Cooper, S.L. Surface properties and blood compatibility of polyurethaneureas. Biomaterials 1986, 7, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ha, H.J.; Ko, Y.K.; Yoon, S.J. Correlation of proliferation, morphology, and biological responses of fibroblasts on the LDPE with different surface wettability. J. Biomater. Sci. Polym. Edn. 2007, 18, 609–622. [Google Scholar] [CrossRef]
- Lee, J.H.; Khang, G.; Lee, J.W.; Lee, H.B. Interaction of different types of cells on polymer surfaces with wettability gradient. J. Colloid. Inter. Sci. 1998, 205, 323–330. [Google Scholar] [CrossRef]
- Johansson, S.; Svineng, G.; Wennerberg, K.; Armulik, A.; Lohikangas, L. Fibronectin-integrin interactions. Front. Biosci. 1997, 2, 126–146. [Google Scholar]
- Seeger, J.M.; Klingman, N. Improved endothelial cell seeding with cultured cells and fibronectin-coated grafts. J. Surg. Res. 1985, 38, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Harnett, E.M.; Alderman, J.; Wood, T. The surface energy of various biomaterials coated with adhesion molecules used in cell culture. Colloid. Surface. B 2007, 55, 90–97. [Google Scholar] [CrossRef]
- Pompe, T.; Kobe, F.; Salchert, K.; Jørgensen, B.; Oswald, J.; Werner, C. Fibronectin anchorage to polymer substrates controls the initial phase of endothelial cell adhesion. J. Biomed. Mater. Res. A 2003, 67, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Voskerician, G.; Anderson, J.M.; Ziats, M.P. High molecular weight kininogen inhibition of endothelial cell function on biomaterials. J. Biomed. Mater. Res. 2000, 51, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Imanishi, Y. Blood compatibility of polyurethanes. Crit. Rev. Biocompat. 1989, 5, 45–104. [Google Scholar]
- Lenk, T.J.; Horbett, T.A.; Ratner, B.D. Infrared spectroscopic studies of time-dependent changes in fibrinogen adsorbed to polyurethanes. Langmuir 1991, 7, 1755–1764. [Google Scholar] [CrossRef]
- Tsuge, O.; Itoh, T.; Tashiro, M. Studies of acyl isocyanate—IV: Synthesis of diol diisocyanates. Tetrahedron 1968, 24, 2583–2590. [Google Scholar] [CrossRef]
- Mulder, A.B.; Blom, N.R.; Smit, J.W.; Ruiters, M.H.J.; van der Meer, J.; Halie, M.R.; Boom, V.J.J. Basal tissue factor expression in endothelial cell cultures is caused by contaminating smooth muscle cells. Reduction by using chymotrypsin instead of collagenase. Thromb. Res. 1995, 80, 399–411. [Google Scholar] [CrossRef] [PubMed]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jovanovic, D.; Roukes, F.V.; Löber, A.; Engels, G.E.; Oeveren, W.v.; Seijen, X.J.G.v.; Luyn, M.J.A.v.; Harmsen, M.C.; Schouten, A.J. Polyacylurethanes as Novel Degradable Cell Carrier Materials for Tissue Engineering. Materials 2011, 4, 1705-1727. https://doi.org/10.3390/ma4101705
Jovanovic D, Roukes FV, Löber A, Engels GE, Oeveren Wv, Seijen XJGv, Luyn MJAv, Harmsen MC, Schouten AJ. Polyacylurethanes as Novel Degradable Cell Carrier Materials for Tissue Engineering. Materials. 2011; 4(10):1705-1727. https://doi.org/10.3390/ma4101705
Chicago/Turabian StyleJovanovic, Danijela, Frans V. Roukes, Andrea Löber, Gerwin E. Engels, Willem van Oeveren, Xavier J. Gallego van Seijen, Marja J.A. van Luyn, Martin C. Harmsen, and Arend Jan Schouten. 2011. "Polyacylurethanes as Novel Degradable Cell Carrier Materials for Tissue Engineering" Materials 4, no. 10: 1705-1727. https://doi.org/10.3390/ma4101705