
Energy and Electronic Properties of Nanostructures Based on the CL-20 Framework with the Replacement of the Carbon Atoms by Silicon and Germanium: A Density Functional Theory Study


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Geometry Optimization
3.2. Energy Properties
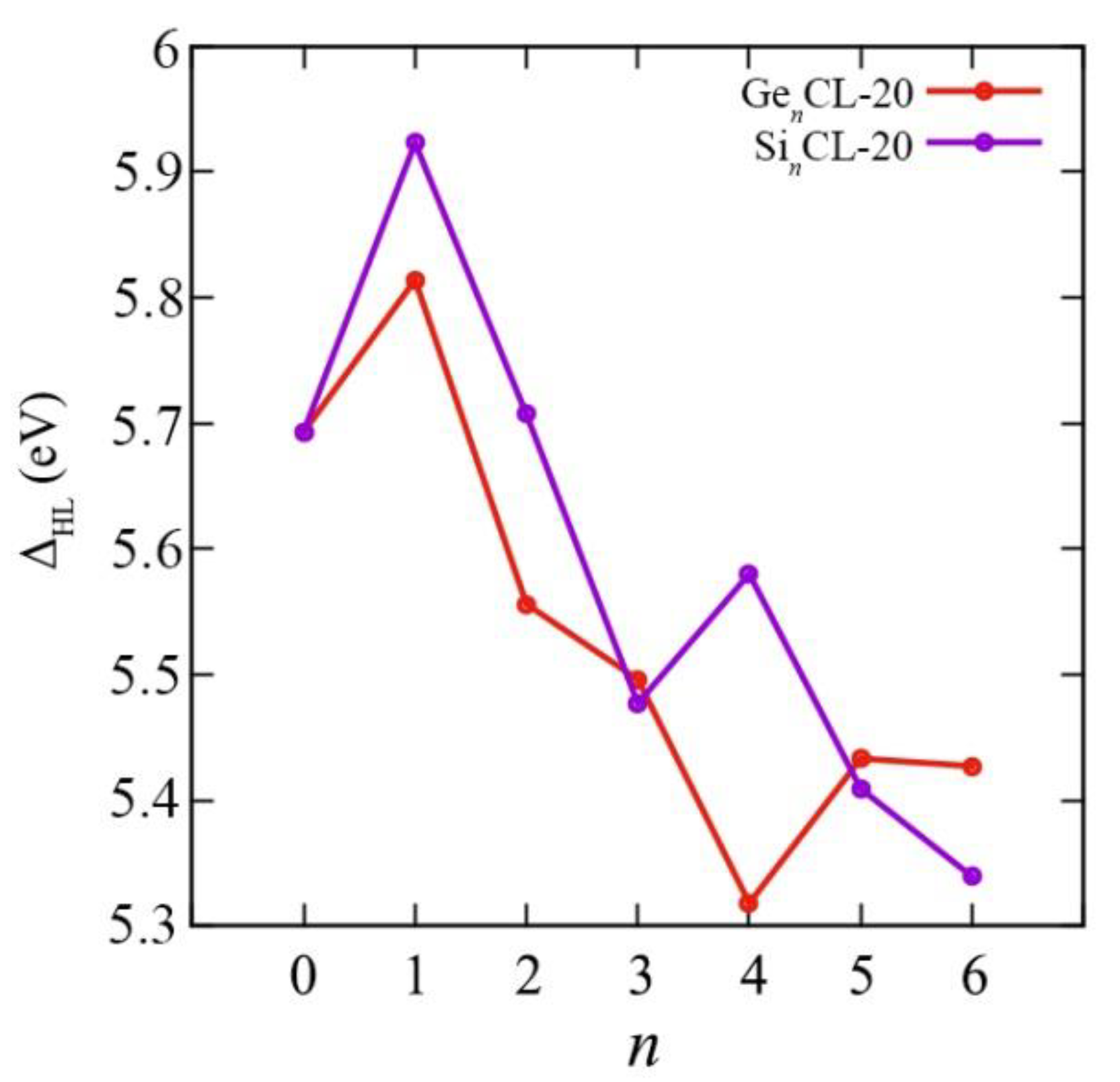
3.3. Electronic Characteristics
3.4. Chemical Reactivity Indices
3.5. The CL-20-Based Dimers
3.6. Homodesmotic Reactions and Strain Energies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sysolyatin, S.V.; Lobanova, A.A.; Chernikova, Y.T.; Sakovich, G.V. Methods of synthesis and properties of hexanitrohexaazaisowurtzitane. Russ. Chem. Rev. 2005, 74, 757–764. [Google Scholar] [CrossRef]
- Klapötke, T.M. High Energy Density Materials; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Hang, G.; Yu, W.; Wang, T.; Wang, J. Theoretical investigations into effects of adulteration crystal defect on properties of CL-20/TNT cocrystal explosive. Comput. Mater. Sci. 2019, 156, 77–83. [Google Scholar] [CrossRef]
- Tan, J.-J.; Ji, G.-F.; Chen, X.-R.; Li, Z. Structure, equation of state and elasticity of crystalline HNIW by molecular dynamics simulations. Phys. B Conden. Matter 2011, 406, 2925–2930. [Google Scholar] [CrossRef]
- Zhou, G.; Wang, J.; He, W.-D.; Wong, N.-B.; Tian, A.; Li, W.-K. Theoretical investigation of four conformations of HNIW by B3LYP method. J. Mol. Struct. THEOCHEM 2002, 589–590, 273–280. [Google Scholar] [CrossRef]
- Kozlova, S.A.; Gubin, S.A.; Maklashova, I.V.; Selezenev, A.A. Molecular-dynamic simulations of the thermophysical properties of hexanitrohexaazaisowurtzitane single crystal at high pressures and temperatures. Russ. J. Phys. Chem. A 2017, 91, 2157–2160. [Google Scholar] [CrossRef]
- McBain, A.; Vuppuluri, V.; Gunduz, I.E.; Groven, L.J.; Son, S.F. Laser ignition of CL-20 (hexanitrohexaazaisowurtzitane) cocrystals. Combus. Flame 2018, 188, 104–115. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Shi, L.-W.; Zhang, C.-Y.; Li, H.-Z.; Chen, M.-B.; Chen, W.-M. Theoretical analysis of the formation driving force and decreased sensitivity for CL-20 cocrystals. J. Mol. Struct. 2016, 1116, 93–101. [Google Scholar] [CrossRef]
- Sha, Y.; Zhang, X. Impact sensitivity and moisture adsorption on the surface of CL-20/TNT cocrystal by molecular dynamics simulation. Appl. Surf. Sci. 2019, 483, 91–97. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, S.; Gou, R.; Ren, F.; Han, G.; Zhu, S. Theoretical insight into the effect of solvent polarity on the formation and morphology of 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20)/2,4,6-trinitro-toluene(TNT) cocrystal explosive. Comput. Theor. Chem. 2018, 1127, 22–30. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, G.; Luan, J.; Chen, Z.; Su, P.; Shu, Y. Crystal structure, spectrum character and explosive property of a new cocrystal CL-20/DNT. J. Mol. Struct. 2016, 1110, 91–96. [Google Scholar] [CrossRef]
- Hang, G.; Yu, W.; Wang, T.; Wang, J.; Li, Z. Theoretical insights into effects of molar ratios on stabilities, mechanical properties and detonation performance of CL-20/RDX cocrystal explosives by molecular dynamics simulation. J. Mol. Struct. 2017, 1141, 577–583. [Google Scholar] [CrossRef]
- Katin, K.P.; Maslov, M.M. Toward CL-20 crystalline covalent solids: On the dependence of energy and electronic properties on the effective size of CL-20 chains. J. Phys. Chem. Solids 2017, 108, 82–87. [Google Scholar] [CrossRef]
- Gimaldinova, M.A.; Maslov, M.M.; Katin, K.P. Electronic and reactivity characteristics of CL-20 covalent chains and networks: A density functional theory study. CrystEngComm 2018, 20, 4336–4344. [Google Scholar] [CrossRef]
- Kan, S.B.J.; Lewis, R.D.; Chen, K.; Arnold, F.H. Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Showell, G.A.; Mills, J.S. Chemistry challenges in lead optimization: Silicon isosteres in drug discovery. Drug Discov. Today 2003, 8, 551–556. [Google Scholar] [CrossRef]
- Jones, R.G.; Ando, W.; Chojnowski, J. Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2000. [Google Scholar] [CrossRef]
- Gimaldinova, M.A.; Katin, K.P.; Salem, M.A.; Maslov, M.M. Energy and electronic characteristics of silicon polyprismanes: Density functional theory study. Lett. Mater. 2018, 8, 454–457. [Google Scholar] [CrossRef]
- Ahmadi, T.S.; Seif, A.; Rozbahani, G.M. The pristine and carbon, silicon or germanium-substituted (10,0) BN nanotube: A computational DFT study of NMR parameters. Arabian J. Chem. 2010, 3, 121–126. [Google Scholar] [CrossRef]
- Tan, B.; Huang, H.; Huang, M.; Long, X.; Li, J.; Yuan, X.; Xu, R. Computational screening of several silicon-based high-energy hexanitrohexaazaisowurtzitane-like derivatives. J. Fluor. Chem. 2014, 158, 29–37. [Google Scholar] [CrossRef]
- Katin, K.P.; Javan, M.B.; Kochaev, A.I.; Soltani, A.; Maslov, M.M. Kinetic Stability and Reactivity of Silicon and Fluorine-Containing CL-20 Derivatives. ChemistrySelect 2019, 4, 9659–9665. [Google Scholar] [CrossRef]
- Ostovari, F.; Hasanpoori, M.; Abbasnejad, M.; Salehi, M.A. DFT calculations of graphene monolayer in presence of Fe dopant and vacancy. Phys. B Condens. Matter 2018, 541, 6–13. [Google Scholar] [CrossRef]
- Kamedulski, P.; Kaczmarek-Kedziera, A.; Lukaszewicz, J.P. Influence of intermolecular interactions on the properties of carbon nanotubes. Bull. Mater. Sci. 2018, 41, 76. [Google Scholar] [CrossRef]
- Gecim, G.; Ozekmekci, M. A density functional theory study of molecular H2S adsorption on (4,0) SWCNT doped with Ge, Ga and B. Surf. Sci. 2021, 711, 121876. [Google Scholar] [CrossRef]
- Javan, M.B. Electronic transport properties of linear nC20 (n ≤ 5) oligomers: Theoretical investigation. Phys. E 2015, 67, 135–142. [Google Scholar] [CrossRef]
- Strout, H.D.L. Stability of Carbon−Nitrogen Cages in Fourfold Symmetry. J. Phys. Chem. A 2006, 110, 4089–4092. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Ufimtsev, I.S.; Martínez, T.J. Quantum Chemistry on Graphical Processing Units. 3. Analytical Energy Gradients, Geometry Optimization, and First Principles Molecular Dynamics. J. Chem. Theory Comput. 2009, 5, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.V.; Ufimtsev, I.S.; Luehr, N.; Martínez, T.J. Generating Efficient Quantum Chemistry Codes for Novel Architectures. J. Chem. Theory Comput. 2013, 9, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Kästner, J.; Carr, J.M.; Keal, T.W.; Thiel, W.; Wander, A.; Sherwood, P. DL-FIND: An Open-Source Geometry Optimizer for Atomistic Simulations. J. Phys. Chem. A 2009, 113, 11856–11865. [Google Scholar] [CrossRef] [PubMed]
- Goumans, T.P.M.; Catlow, C.R.A.; Brown, W.A.; Kästner, J.; Sherwood, P. An embedded cluster study of the formation of water on interstellar dust grains. Phys. Chem. Chem. Phys. 2009, 11, 5431–5436. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Guner, V.; Khuong, K.S.; Leach, A.G.; Lee, P.S.; Bartberger, M.D.; Houk, K.N. A Standard Set of Pericyclic Reactions of Hydrocarbons for the Benchmarking of Computational Methods: The Performance of ab Initio, Density Functional, CASSCF, CASPT2, and CBS-QB3 Methods for the Prediction of Activation Barriers, Reaction Energetics, and Transition State Geometries. J. Phys. Chem. A 2003, 107, 11445–11459. [Google Scholar] [CrossRef]
- Amin, E.A.; Truhlar, D.G. Zn Coordination Chemistry: Development of Benchmark Suites for Geometries, Dipole Moments, and Bond Dissociation Energies and Their Use To Test and Validate Density Functionals and Molecular Orbital Theory. J. Chem. Theory Comput. 2008, 4, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Foresman, J.B.; Frisch, A.E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Lewars, E.G. Computational Chemistry: Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Norwell, MA, USA, 2003. [Google Scholar]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Putz, M.V.; Russo, N.; Sicilia, E. Atomic radii scale and related size properties from density functional electronegativity formulation. J. Phys. Chem. A 2003, 107, 5461–5465. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Koopmans, T. Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms. Physica 1933, 1, 104–113. [Google Scholar] [CrossRef]
- Tan, B.; Long, X.; Li, J. The cage strain energies of high-energy compounds. Comput. Theor. Chem. 2012, 993, 66–72. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Williams, J.E.; Blanchard, K.R. Evaluation of strain in hydrocarbons. The strain in adamantane and its origin. J. Amer. Chem. Soc. 1970, 92, 2377–2386. [Google Scholar] [CrossRef]
- Wiberg, K.B. The concept of strain in organic chemistry. Angew. Chem. Int. Ed. 1986, 25, 312–322. [Google Scholar] [CrossRef]
- Zhao, M.; Gimarc, B.M. Strain energies in cyclic oxygen On, n = 3–8. J. Phys. Chem. 1993, 97, 4023–4030. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R.; Pople, J.A. Ab Initio Molecular Orbital Theory; Section 6.5.6; Wiley-Interscience: New York, NY, USA, 1985. [Google Scholar]
- Walker, J.E.; Adamson, P.A.; Davis, S.R. Comparison of calculated hydrocarbon strain energies using ab initio and composite methods. J. Mol. Struct. THEOCHEM 1999, 487, 145–150. [Google Scholar] [CrossRef]
- Fan, X.-W.; Qiu, L.; Ju, X.-H. Cage strain in nitro-substituted 1,3,5,7-tetraazacubanes. Struct. Chem. 2009, 20, 1039–1042. [Google Scholar] [CrossRef]
- Fan, X.-W.; Ju, X.-H. Theoretical studies on four-membered ring compounds with NF2, ONO2, N3, and NO2 groups. J. Comput. Chem. 2007, 29, 505–513. [Google Scholar] [CrossRef]
- Lewis, L.L.; Turner, L.L.; Salter, E.A.; Magers, D.H. Computation of the conventional strain energy in oxaziridine. J. Mol. Struct. THEOCHEM 2002, 592, 161–171. [Google Scholar] [CrossRef]
- George, P.; Trachtman, M.; Bock, C.W.; Brett, A.M. An alternative approach to the problem of assessing destabilization energies (strain energies) in cyclic hydrocarbons. Tetrahedron 1976, 32, 317–322. [Google Scholar] [CrossRef]
- Lewars, E.G. Modeling Marvels. Computational Anticipation of Novel Molecules; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2008; p. 242. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | 0 | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|---|
| SinCL-20 | 3.208 | 3.363 | 3.498 | 3.646 | 3.803 | 3.960 | 4.125 |
| GenCL-20 | 3.401 | 3.556 | 3.743 | 3.934 | 4.142 | 4.357 |
| n | C–C, Å | C–N, Å | C–Si, Å | Si–Si, Å | Si–N, Å |
|---|---|---|---|---|---|
| 0 | 1.590 | 1.461 | - | - | - |
| 1 | 1.591 | 1.461 | 1.938 | - | 1.778 |
| 2 | 1.589 | 1.465 | 1.941 | - | 1.778 |
| 3 | - | 1.469 | 1.944 | - | 1.779 |
| 4 | - | 1.467 | 1.940 | 2.393 | 1.787 |
| 5 | - | 1.467 | 1.948 | 2.372 | 1.792 |
| 6 | - | - | - | 2.379 | 1.795 |
| n | C–C, Å | C–N, Å | C–Ge, Å | Ge–Ge, Å | Ge–N, Å |
|---|---|---|---|---|---|
| 0 | 1.590 | 1.461 | - | - | - |
| 1 | 1.589 | 1.460 | 2.029 | - | 1.892 |
| 2 | 1.590 | 1.455 | - | 2.474 | 1.902 |
| 3 | 1.593 | 1.459 | 2.012 | 2.473 | 1.903 |
| 4 | 1.604 | 1.462 | - | 2.445 | 1.914 |
| 5 | - | 1.452 | 2.017 | 2.476 | 1.903 |
| 6 | - | - | - | 2.481 | 1.903 |
| The Dimer | εH, eV | εL, eV | ΔHL, eV | ||
|---|---|---|---|---|---|
| D1-(1) | 4.156 | −7.454 | −2.797 | 4.657 | |
| Si | Si-D2-(1) | 3.843 | −7.489 | −3.031 | 4.458 |
| Si-D2-(2) | 3.843 | −7.478 | −3.061 | 4.462 | |
| Si- D2-(3) | 3.846 | −7.609 | −2.293 | 4.715 | |
| Ge | Ge-D3-(1) | 3.579 | −7.181 | −2.771 | 4.410 |
| Ge-D3-(2) | 3.579 | −7.124 | −2.780 | 4.345 | |
| Ge-D3-(3) | 3.582 | −7.167 | −2.793 | 4.374 | |
| Molecule | CL-20 | Si6CL-20 | Ge6CL-20 |
|---|---|---|---|
| Es, eV | 2.843 | 1.474 | 2.039 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gimaldinova, M.A.; Maslov, M.M.; Katin, K.P. Energy and Electronic Properties of Nanostructures Based on the CL-20 Framework with the Replacement of the Carbon Atoms by Silicon and Germanium: A Density Functional Theory Study. Materials 2022, 15, 6577. https://doi.org/10.3390/ma15196577
Gimaldinova MA, Maslov MM, Katin KP. Energy and Electronic Properties of Nanostructures Based on the CL-20 Framework with the Replacement of the Carbon Atoms by Silicon and Germanium: A Density Functional Theory Study. Materials. 2022; 15(19):6577. https://doi.org/10.3390/ma15196577
Chicago/Turabian StyleGimaldinova, Margarita A., Mikhail M. Maslov, and Konstantin P. Katin. 2022. "Energy and Electronic Properties of Nanostructures Based on the CL-20 Framework with the Replacement of the Carbon Atoms by Silicon and Germanium: A Density Functional Theory Study" Materials 15, no. 19: 6577. https://doi.org/10.3390/ma15196577