
Critical Evaluation of Various Spontaneous Polarization Models and Induced Electric Fields in III-Nitride Multi-Quantum Wells

 , , ,
, , ,  , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. The Calculation Methods
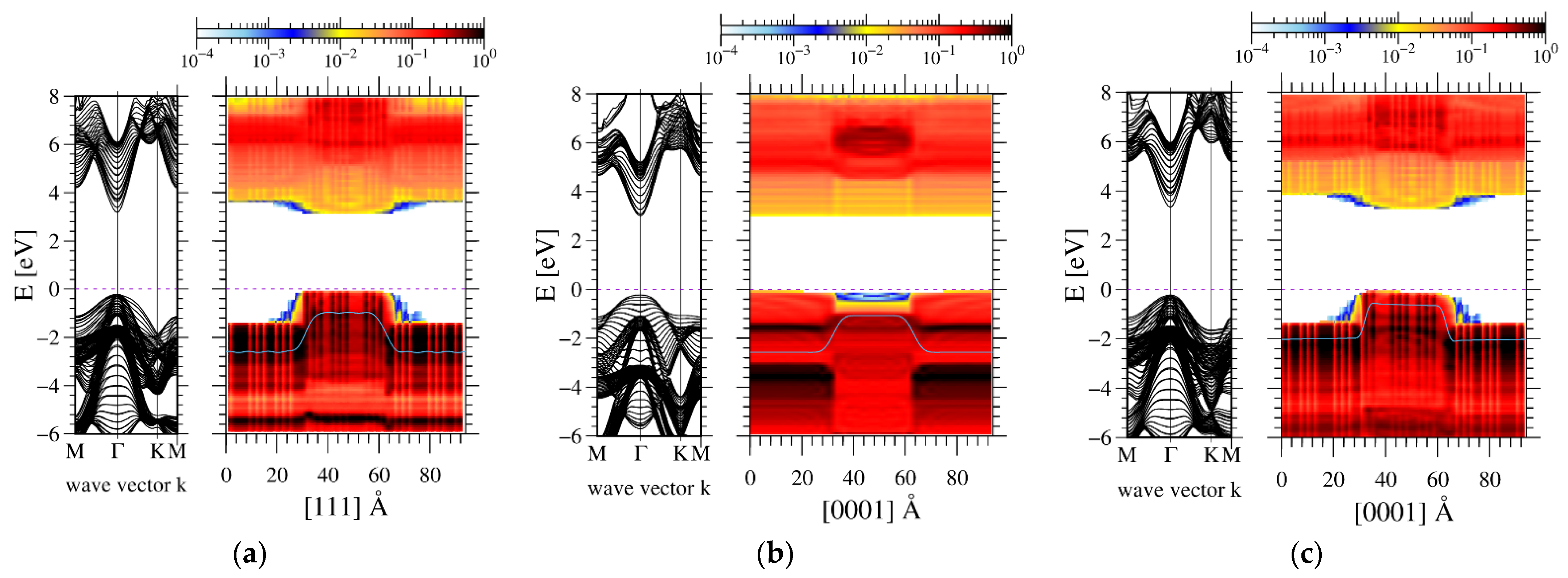
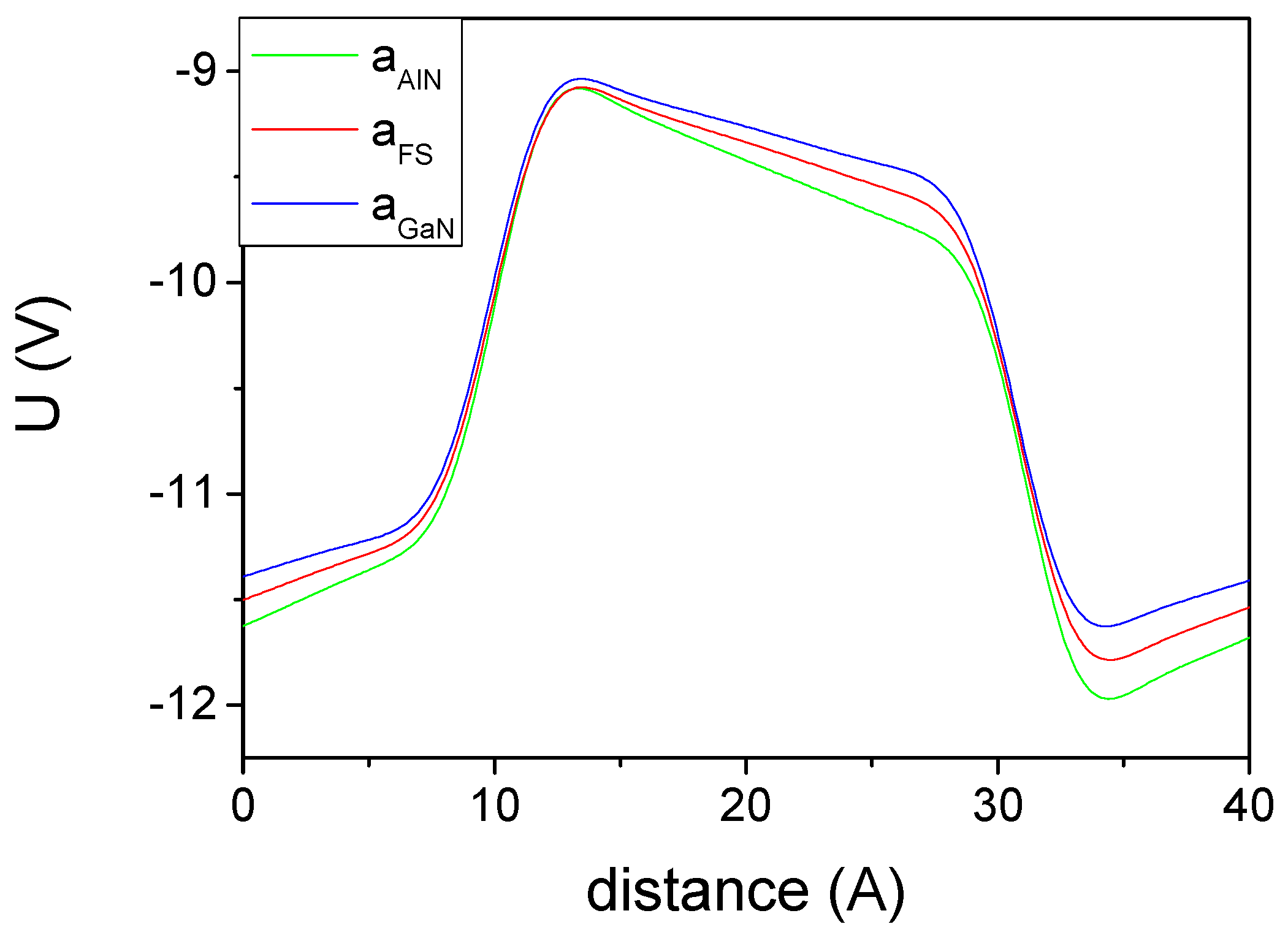
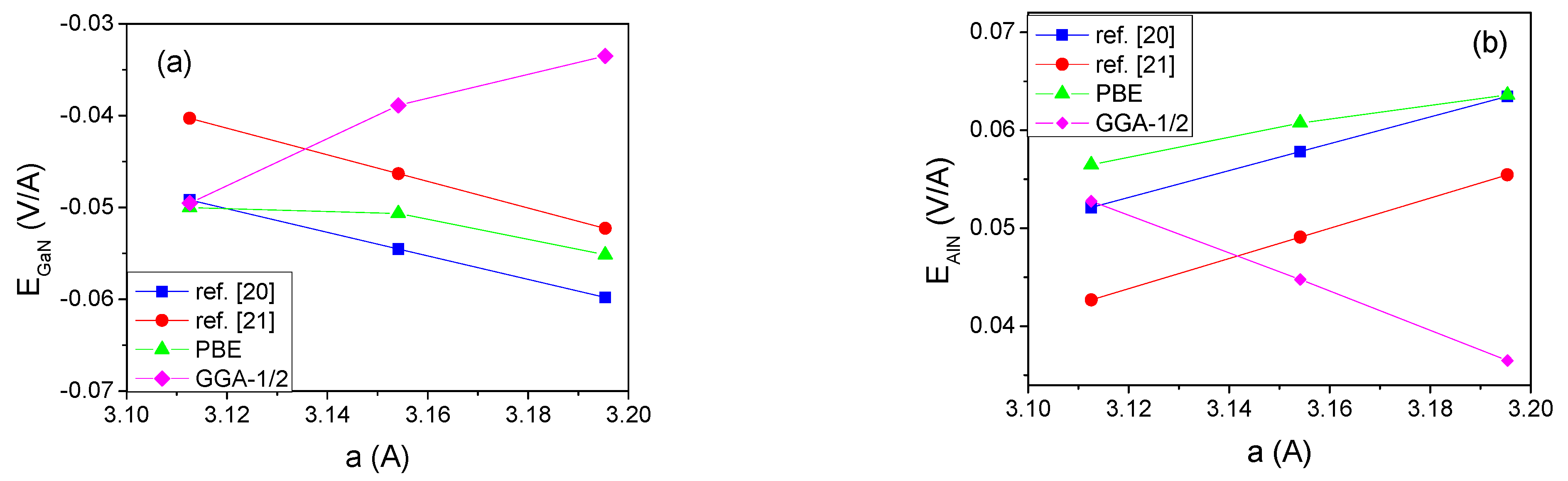
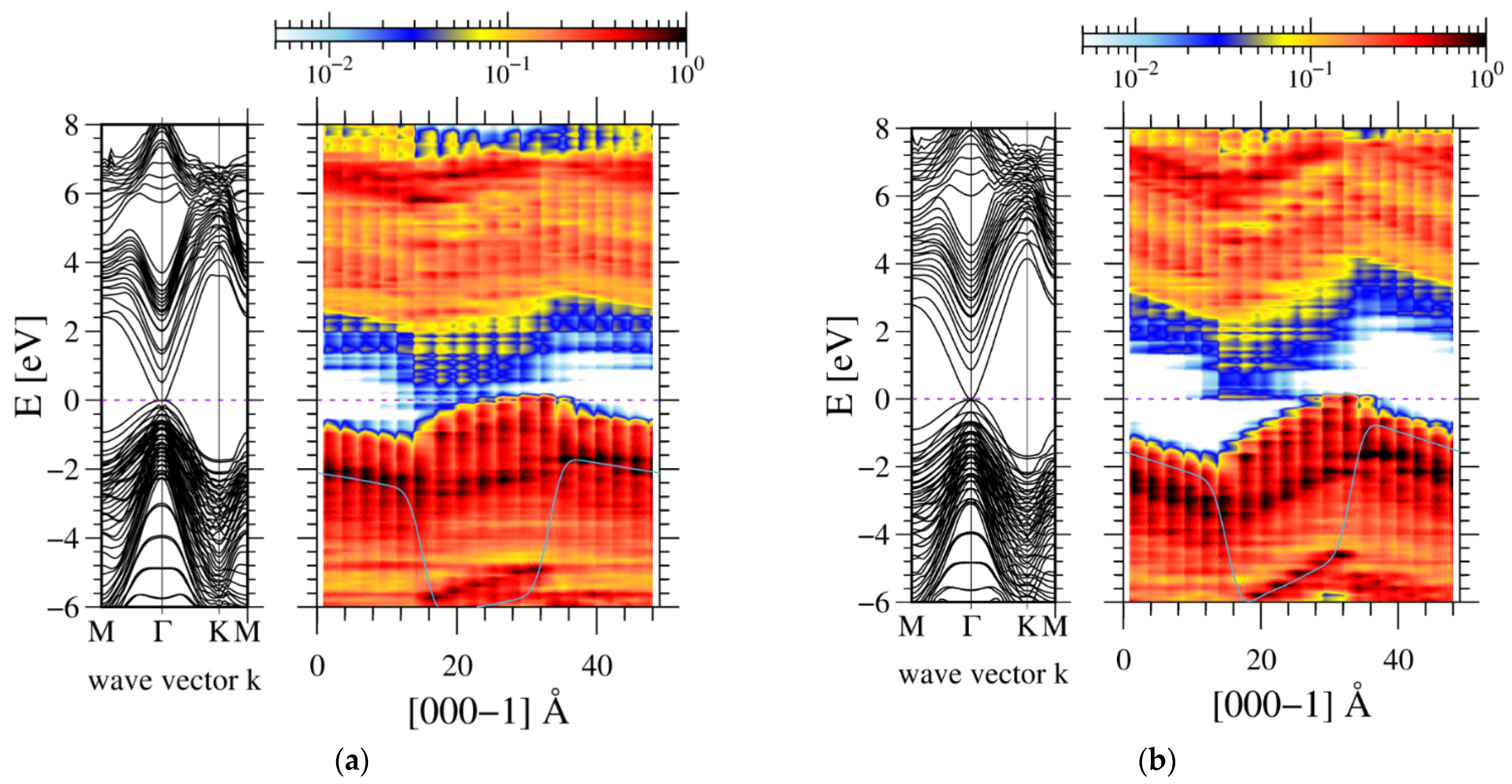
3. Results
4. The Experimental Verification
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Landau, L.D.; Lifsic, E.M. Electrodynamics of Continuous Media; Pergamon: Oxford, UK, 1984. [Google Scholar]
- Bogusławski, P.; Bernholc, J. Theoretical Approach to Polarization Effects in Semiconductors. In Polarization Effects in Semiconductors: From Ab Initio Theory to Device Applications, 1st ed.; Wood, C., Jena, D., Eds.; Springer: New York, NY, USA, 2008; pp. 2–25. [Google Scholar]
- Noguera, C.; Goniakowski, J. Polarity in Oxide nano-objects. Chem. Rev. 2013, 113, 4073–4105. [Google Scholar] [CrossRef] [PubMed]
- Spaldin, N.A. A beginner’s guide to modern theory of polarization. J. Solid State Chem. 2012, 195, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Resta, R. Macroscopic polarization in crystalline dielectrics: The geometric phase approach. Rev. Mod. Phys. 1994, 66, 899–915. [Google Scholar] [CrossRef]
- Karpov, S.Y. Spontaneous polarization in III-nitride materials: Crystallographic revision. Phys. Status Solidi C 2010, 7, 1841–1843. [Google Scholar] [CrossRef]
- Langer, R.; Simon, J.; Ortiz, V.; Pelekanos, N.T.; Barski, A.; Andre, R.; Godlewski, M. Giant electric fields in unstrained GaN single quantum wells. Appl. Phys. Lett. 1999, 74, 3827–3829. [Google Scholar] [CrossRef]
- Feneberg, M.; Thonke, K. Polarization fields in III-nitrides grown in different crystal orientation. J. Phys. Condens. Matter 2007, 19, 403201. [Google Scholar] [CrossRef] [PubMed]
- Im, J.S.; Kollmer, H.; Off, J.; Sohmer, A.; Scholz, F.; Hangleiter, A. Reduction of oscillator strength due to piezoelectric fields in GaN/AlxGa1−xN quantum wells. Phys. Rev. B 1998, 57, R9435–R9438. [Google Scholar]
- Kaminska, A.; Koronski, K.; Strak, P.; Sobczak, K.; Monroy, E.; Krukowski, S. Wurtzite quantum well structures under high pressure. J. Appl. Phys. 2020, 128, 050901. [Google Scholar] [CrossRef]
- Miller, D.A.B.; Chemla, D.S.; Damen, T.C.; Gossard, A.C.; Wiegmann, W.; Wood, T.H.; Burrus, C.A. Band-edge electroabsorption in quantum well structures–the quantum confined Stark effect. Phys. Rev. Lett. 1984, 53, 2173–2176. [Google Scholar] [CrossRef]
- Halsall, M.P.; Nicholls, J.E.; Davies, J.J.; Cockayne, B.; Wright, P.J. CdS/CdSe intrinsic Stark superlattices. J. Appl. Phys. 1992, 71, 907–915. [Google Scholar] [CrossRef]
- Lines, M.E.; Glass, A.M. Principles and Applications of Ferroelectrics and Related Materials; Clarendon: Oxford, UK, 1974. [Google Scholar]
- Pengelly, R.S.; Wood, S.M.; Milligan, J.W.; Sheppard, S.T.; Pribble, W.L. A Review on GaN on SiC High-Electron-Mobility Power Transistors and MMICs. IEEE Trans. Micro. Theory Tech. 2012, 60, 1764–1783. [Google Scholar] [CrossRef]
- Sheppard, S.T.; Doverspike, K.; Pribble, W.L.; Allen, S.T.; Palmour, J.W.; Kehias, L.T.; Jenkins, T.J. High-power microwave GaN/AlGaN HEMT’s on semi-insulating silicon carbide substrates. IEEE Electron Device Lett. 1999, 20, 161–163. [Google Scholar] [CrossRef]
- Kaminska, A.; Strak, P.; Borysiuk, J.; Sobczak, K.; Domagala, J.Z.; Beeler, M.; Grzanka, E.; Sakowski, K.; Krukowski, S.; Monroy, E. Correlation of optical and structural properties of GaN/AlN multi-quantum wells–Ab initio study. J. Appl. Phys. 2016, 119, 015703. [Google Scholar] [CrossRef]
- Kaminska, A.; Jankowski, D.; Strak, P.; Korona, K.P.; Beeler, M.; Sakowski, K.; Grzanka, E.; Borysiuk, J.; Sobczak, K.; Monroy, E.; et al. High pressure and time resolved studies of optical proerties of n-type doped GaN/AlN multi-quantum wells: Experimental and theoretical analysis. J. Appl. Phys. 2016, 120, 095705. [Google Scholar] [CrossRef]
- Wang, T. Topical review: Development of overgrown semi-polar GaN for high efficiency green/yellow emission. Semicond. Sci. Technol. 2016, 31, 093003. [Google Scholar] [CrossRef]
- Hardy, M.T.; Feezell, D.F.; DenBaars, S.P.; Nakamura, S. Group III-nitride lasers: A materials perspective. Mater. Today 2011, 14, 408–4015. [Google Scholar] [CrossRef]
- Bernardini, F.; Fiorentini, V.; Vanderbilt, D. Spontaneous and piezoelectric constants of III-V nitrides. Phys. Rev. B 1997, 56, R10024. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, C.E.; Janotti, A.; Van de Walle, C.G.; Vanderbilt, D. Correct Implementation of Polarization Constants in Wurtzite materials and Impact on III-Nitrides. Phys. Rev. X 2016, 6, 021038. [Google Scholar] [CrossRef]
- Strak, P.; Kempisty, P.; Sakowski, K.; Kaminska, A.; Jankowski, D.; Korona, K.P.; Sobczak, K.; Borysiuk, J.; Beeler, M.; Grzanka, E.; et al. Ab initio and experimental studies of polarization and polarization related fields in nitrides and nitride structures. AIP Adv. 2017, 7, 015027. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11185. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special point for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Leszczynski, M.; Suski, T.; Teisseyre, H.; Perlin, P.; Grzegory, I.; Jun, J.; Porowski, S.; Moustakas, T.D. Thermal expansion of gallium nitride. J. Appl. Phys. 1994, 76, 4909–4911. [Google Scholar] [CrossRef]
- Leszczynski, M.; Teisseyre, H.; Suski, T.; Grzegory, I.; Bockowski, M.; Jun, J.; Porowski, S.; Pakula, K.; Baranowski, J.M.; Foxon, C.T.; et al. Lattice parameters of gallium nitride. Appl. Phys. Lett. 1996, 69, 73–75. [Google Scholar] [CrossRef]
- Angerer, H.; Brunner, D.; Freudenberg, F.; Ambacher, O.; Stutzmann, M.; Höpler, R.; Metzger, T.; Born, E.; Dollinger, G.; Bergmaier, A.; et al. Determination of the Al mole fraction and band gap bowing of epitaxial AlxGa1−xN films. Appl. Phys. Lett. 1997, 71, 1504–1506. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-particle Green’s Function with Application to the Electron-gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207, Erratum in 2006, 124, 211906. [Google Scholar] [CrossRef] [Green Version]
- Moses, P.G.; Van de Walle, C.G. Band bowing and band alignment in InGaN alloys. Appl. Phys. Lett. 2010, 96, 021908. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Marques, M.; Teles, L.K. Approximation to density functional theory for the calculation of band gaps of semiconductors. Phys. Rev. B 2008, 78, 125116. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Fonseca, L.R.C.; Ferreira, L.G. Accurate prediction of the Si/SiO2 interface band offset using self-consistent ab initio DFT/LDA-1/2 method. Phys. Rev. B 2009, 79, 241312. [Google Scholar] [CrossRef]
- Suzuki, M.; Uenoyama, T.; Yanase, A. First-principles calculations of effective-mass parameters of AlN and GaN. Phys. Rev. B 1995, 52, 8132–8139. [Google Scholar] [CrossRef]
- Wei, S.-H.; Zunger, A. Valence band splitting and band offsets of AlN, GaN and InN. Appl. Phys. Lett. 1996, 69, 2719–2721. [Google Scholar] [CrossRef] [Green Version]
- Vurgaftman, I.; Meyer, J.R. Band parameters for nitrogen-containing semiconductors. J. Appl. Phys. 2003, 94, 3675–3696. [Google Scholar] [CrossRef]
- Adolph, B.; Gavrilenko, V.I.; Tenelsen, K.; Bechstedt, F.; Del Sole, R. nonlocality and many-body effects in the optical properties of semiconductors. Phys. Rev. B 1996, 53, 9797–9808. [Google Scholar] [CrossRef]
- Taniyasu, Y.; Kasu, M. Origin of exciton emissions from an AlN p-n junction light-emitting diode. Appl. Phys. Lett. 2011, 98, 131910. [Google Scholar] [CrossRef]
- Kamiya, K.; Ebihara, Y.; Shiraishi, K.; Kasu, M. Structural design of AlN/GaN superlattices for deep-ultraviolet light-emitting diodes with high emission efficiency. Appl. Phys. Lett. 2011, 99, 151108. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blochl, P.E. Crystal Orbital Hamilton Populations (COHP)–Energy-resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Mahata, M.K.; Ghosh, S.; Jana, S.J.; Chakraborty, A.; Bag, A.; Mukhopadhyay, P.; Kumar, R.; Biswas, D. MBE grown AlGaN/GaN heterostructures on sapphire with ultra thin buffer. AIP Adv. 2014, 4, 117120. [Google Scholar] [CrossRef]
- Strak, P.; Kempisty, P.; Ptasinska, M.; Krukowski, S. Principal physical properties of GaN/AlN multiquantum well (MQWs) systems determined by density functional theory (DFT) calculations. J. Appl. Phys. 2013, 113, 193706. [Google Scholar] [CrossRef]
- Sobczak, K.; Borysiuk, J.; Strak, P.; Jakiela, R.; Koronski, K.; Kaminska, A.; Monroy, E.; Krukowski, S. Detection of Si doping in the AlN/GaN MQWs using super x-EDS measurements. Micron 2020, 134, 102864. [Google Scholar] [CrossRef] [PubMed]


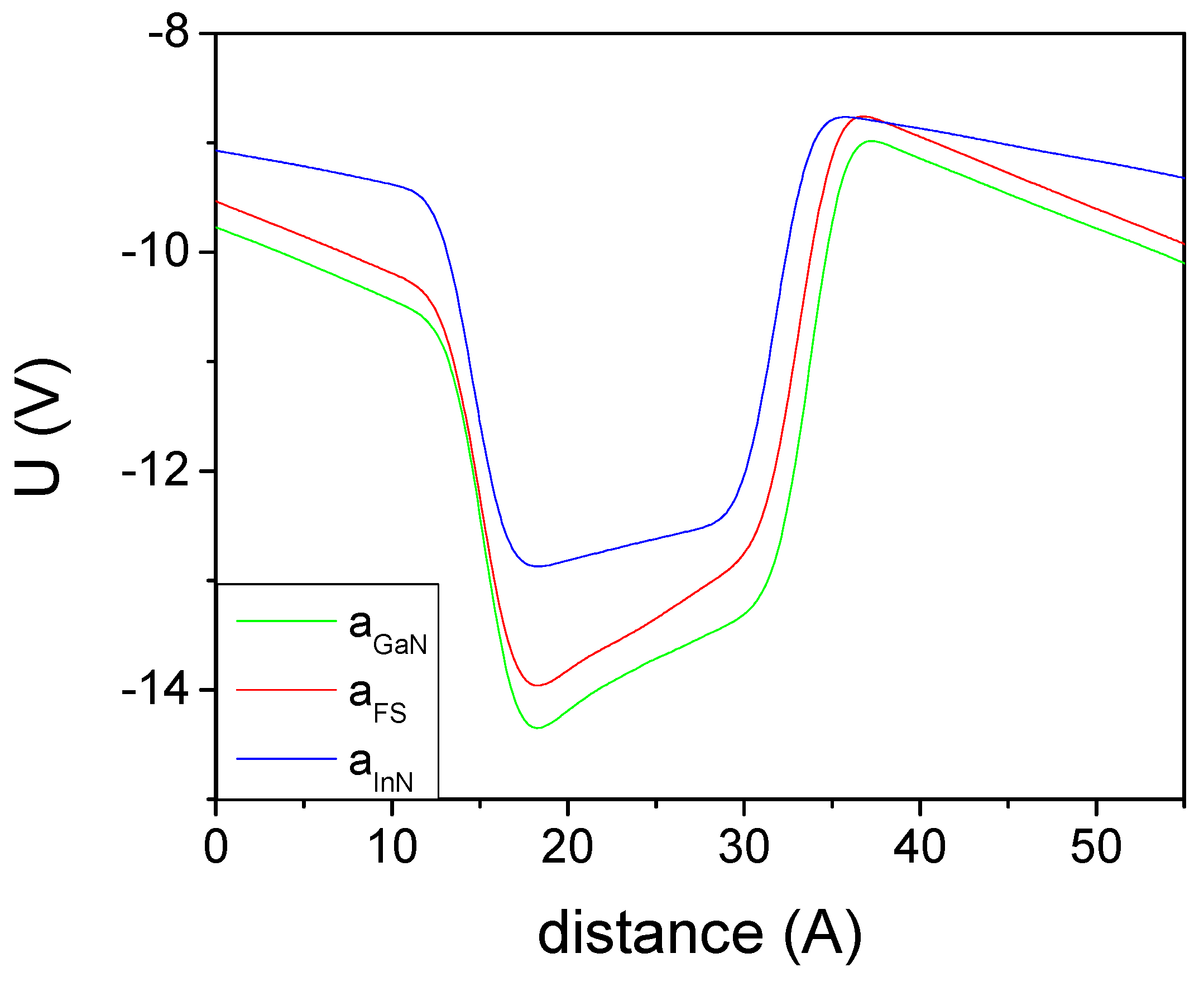

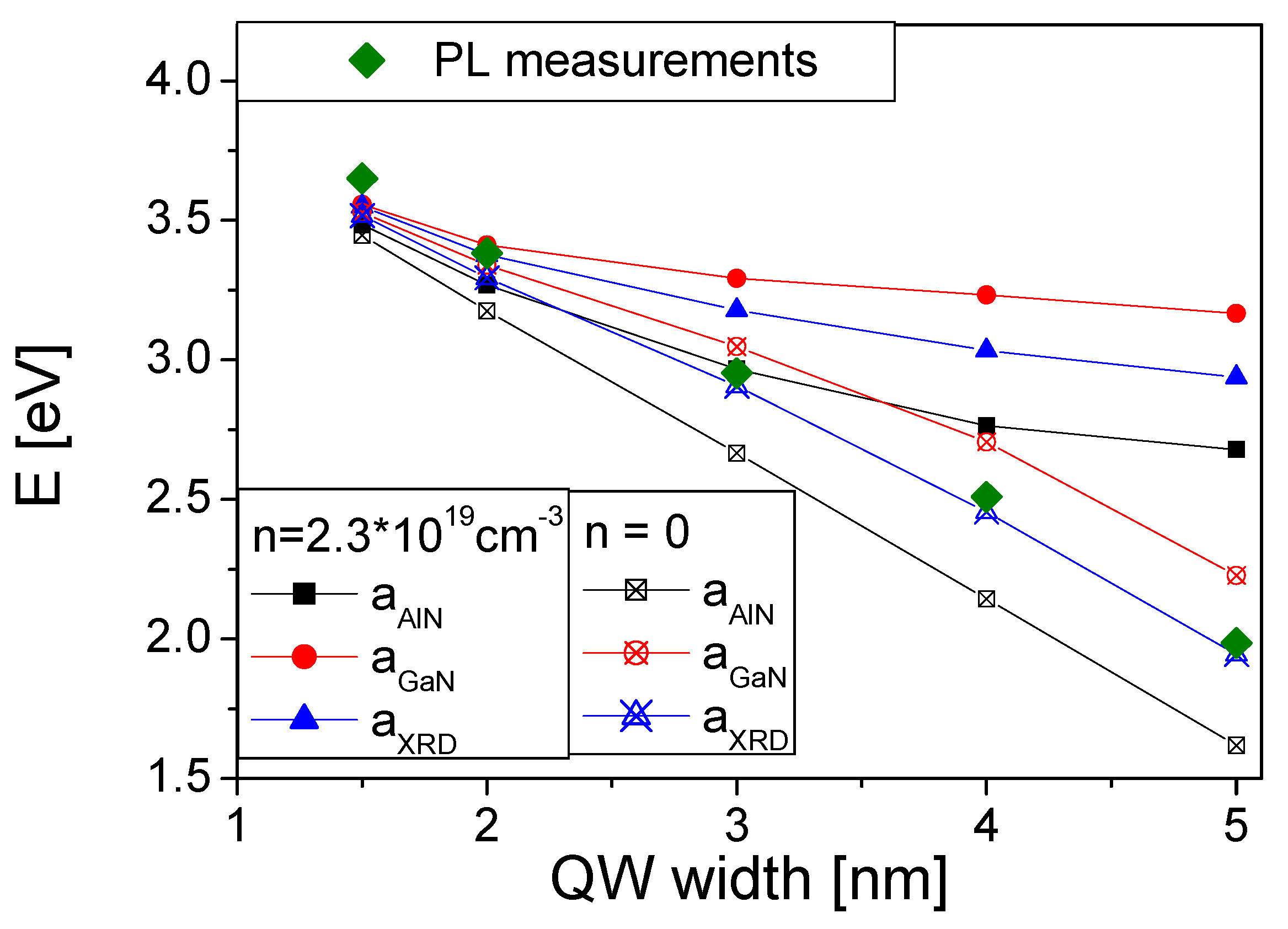

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | References | AlN | GaN | InN |
|---|---|---|---|---|
| Spontaneous polarization P3 | [20] | −0.081 | −0.029 | −0.090 |
| [21] | 1.351 | 1.312 | 1.026 | |
| [22] | −0.090 | −0.019 | −0.028 | |
| Piezo constant | [20] | −0.60 | −0.49 | −0.57 |
| [21] | −0.676 | −0.551 | −0.604 | |
| Piezo constant | [20] | 1.46 | 0.73 | 0.97 |
| [21] | 1.569 | 1.020 | 1.328 |
| Lattice Symmetryr | AlN | GaN |
|---|---|---|
| ZB lattice | −16.28 | −11.52 |
| HX lattice | −3.54 | −1.09 |
| WZ lattice | 17.09 | −11.51 |
| Lattice Symmetry | AlN | GaN |
|---|---|---|
| ZB lattice | ||
| HX lattice | ||
| WZ lattice |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, A.; Strak, P.; Koronski, K.; Kempisty, P.; Sakowski, K.; Piechota, J.; Grzegory, I.; Wierzbicka, A.; Kryvyi, S.; Monroy, E.; et al. Critical Evaluation of Various Spontaneous Polarization Models and Induced Electric Fields in III-Nitride Multi-Quantum Wells. Materials 2021, 14, 4935. https://doi.org/10.3390/ma14174935
Ahmad A, Strak P, Koronski K, Kempisty P, Sakowski K, Piechota J, Grzegory I, Wierzbicka A, Kryvyi S, Monroy E, et al. Critical Evaluation of Various Spontaneous Polarization Models and Induced Electric Fields in III-Nitride Multi-Quantum Wells. Materials. 2021; 14(17):4935. https://doi.org/10.3390/ma14174935
Chicago/Turabian StyleAhmad, Ashfaq, Pawel Strak, Kamil Koronski, Pawel Kempisty, Konrad Sakowski, Jacek Piechota, Izabella Grzegory, Aleksandra Wierzbicka, Serhii Kryvyi, Eva Monroy, and et al. 2021. "Critical Evaluation of Various Spontaneous Polarization Models and Induced Electric Fields in III-Nitride Multi-Quantum Wells" Materials 14, no. 17: 4935. https://doi.org/10.3390/ma14174935
APA StyleAhmad, A., Strak, P., Koronski, K., Kempisty, P., Sakowski, K., Piechota, J., Grzegory, I., Wierzbicka, A., Kryvyi, S., Monroy, E., Kaminska, A., & Krukowski, S. (2021). Critical Evaluation of Various Spontaneous Polarization Models and Induced Electric Fields in III-Nitride Multi-Quantum Wells. Materials, 14(17), 4935. https://doi.org/10.3390/ma14174935