First-Principles Study on the Mechanical Properties and Electronic Structure of V Doped WCoB and W2CoB2 Ternary Borides

 ,
, 
Abstract
:1. Introduction
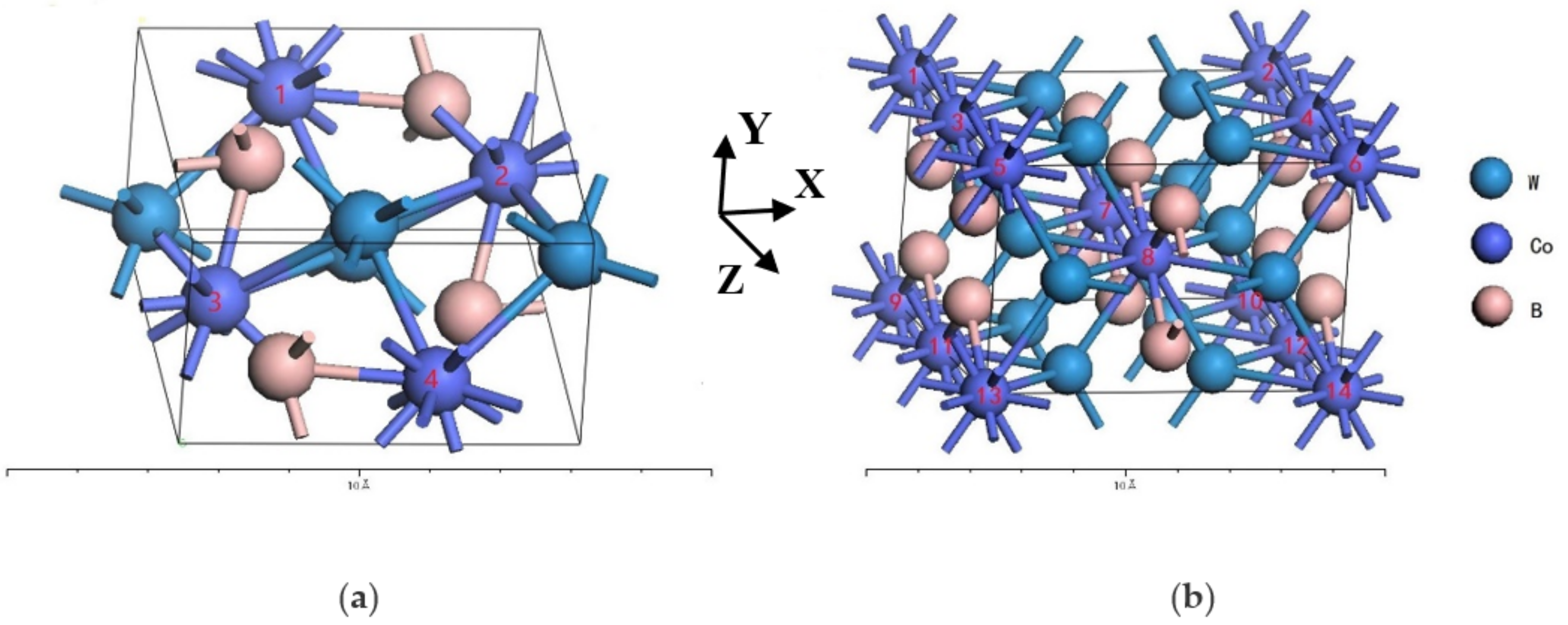
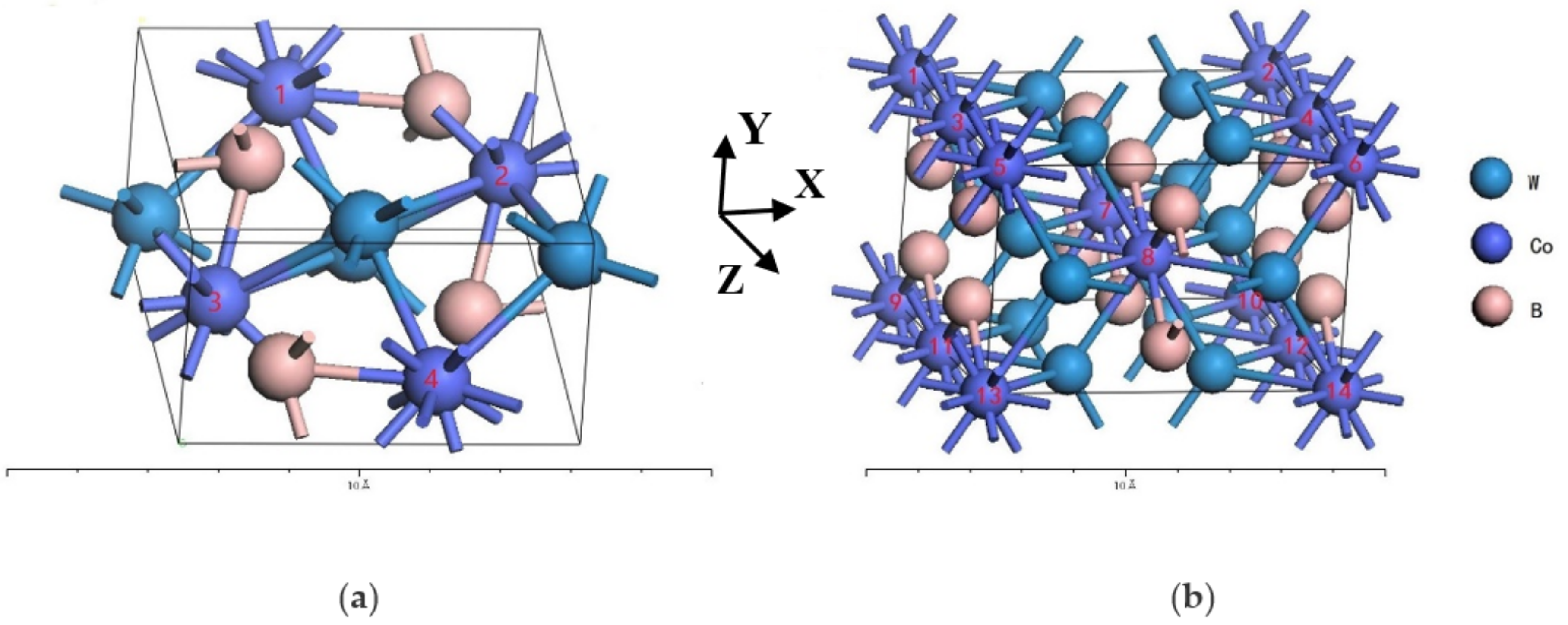
2. Crystal Structure and Calculation Method
3. Results and Discussion
3.1. Structural Stability
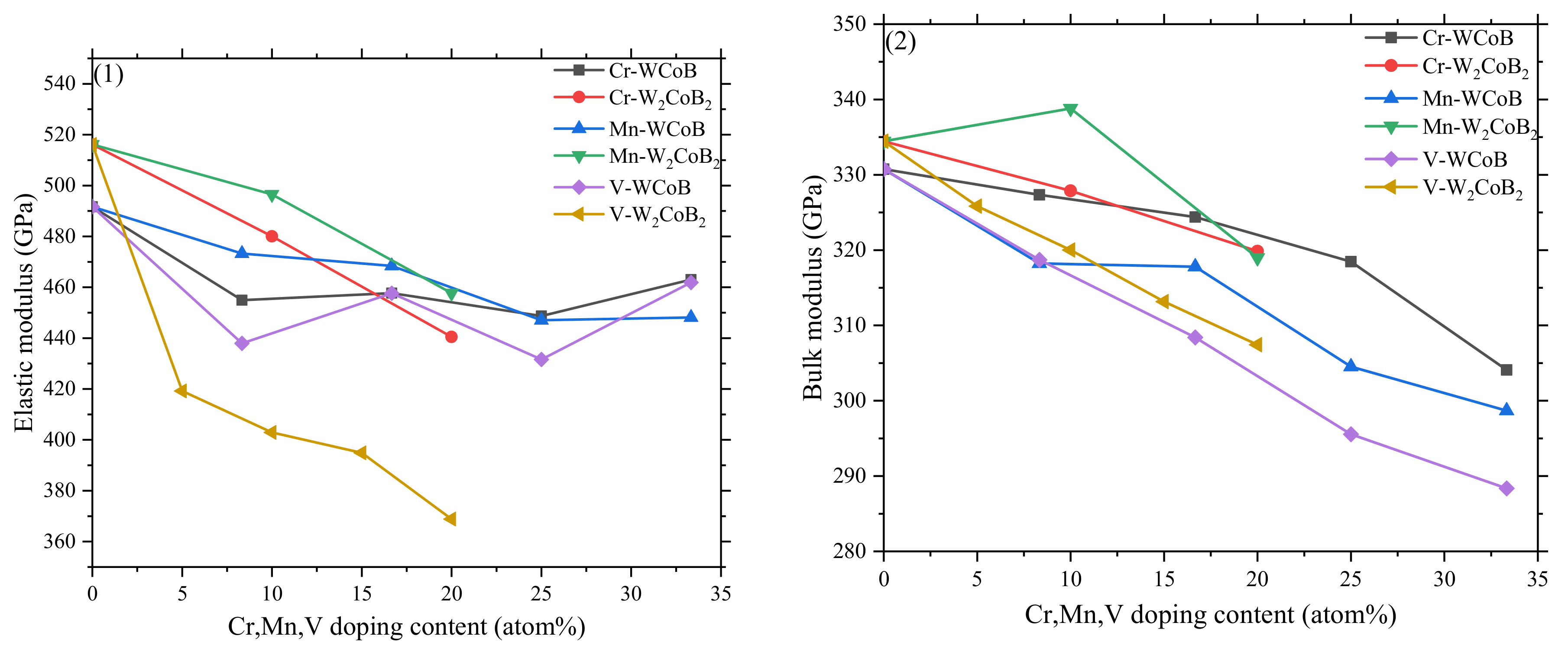
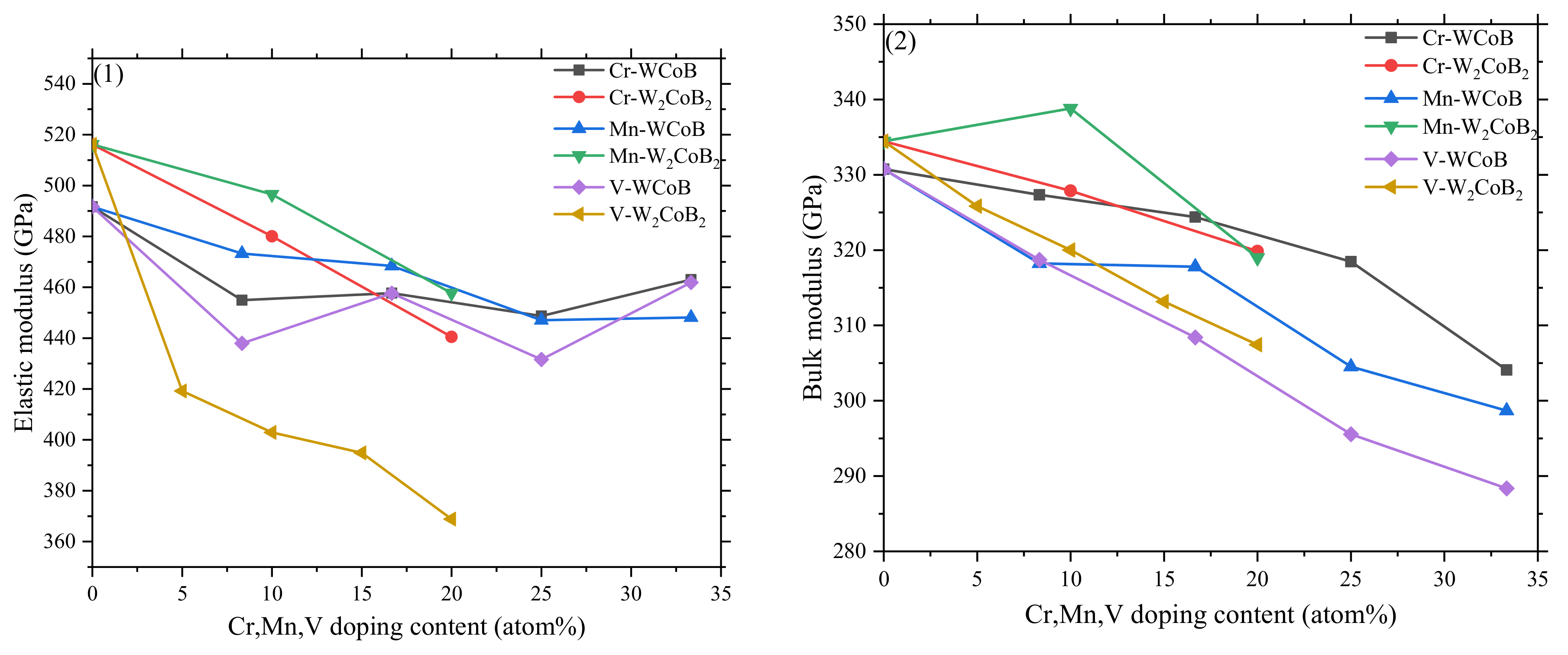
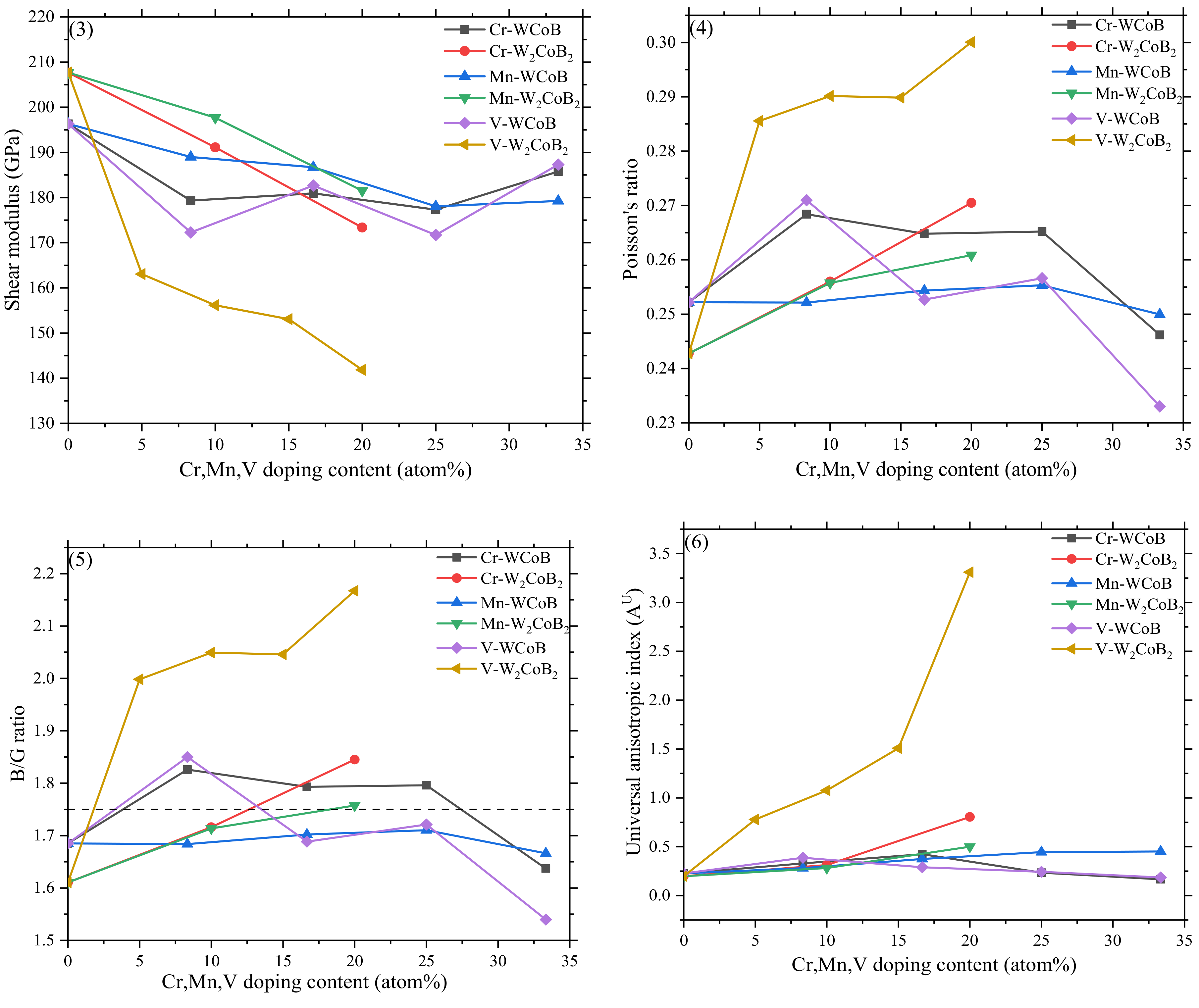
3.2. Mechanical Properties
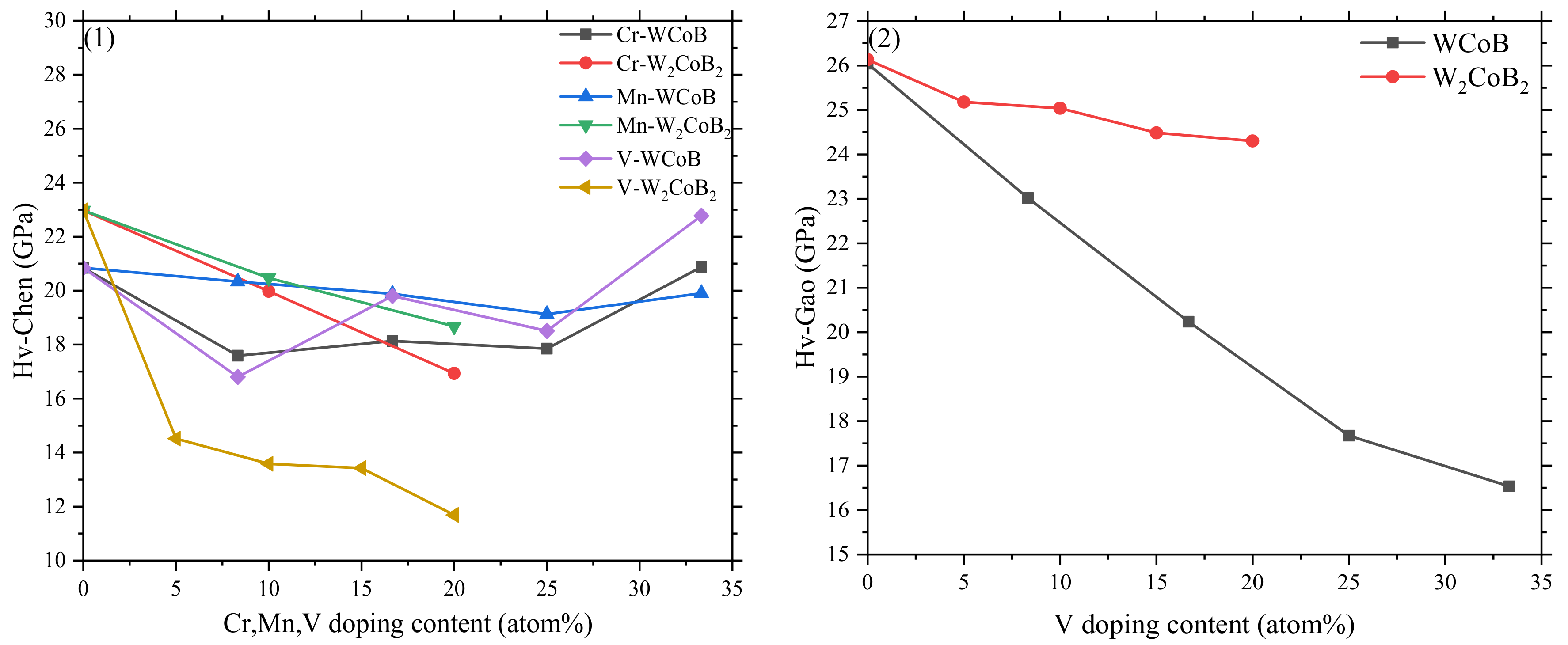
3.3. Population Analysis and Hardness
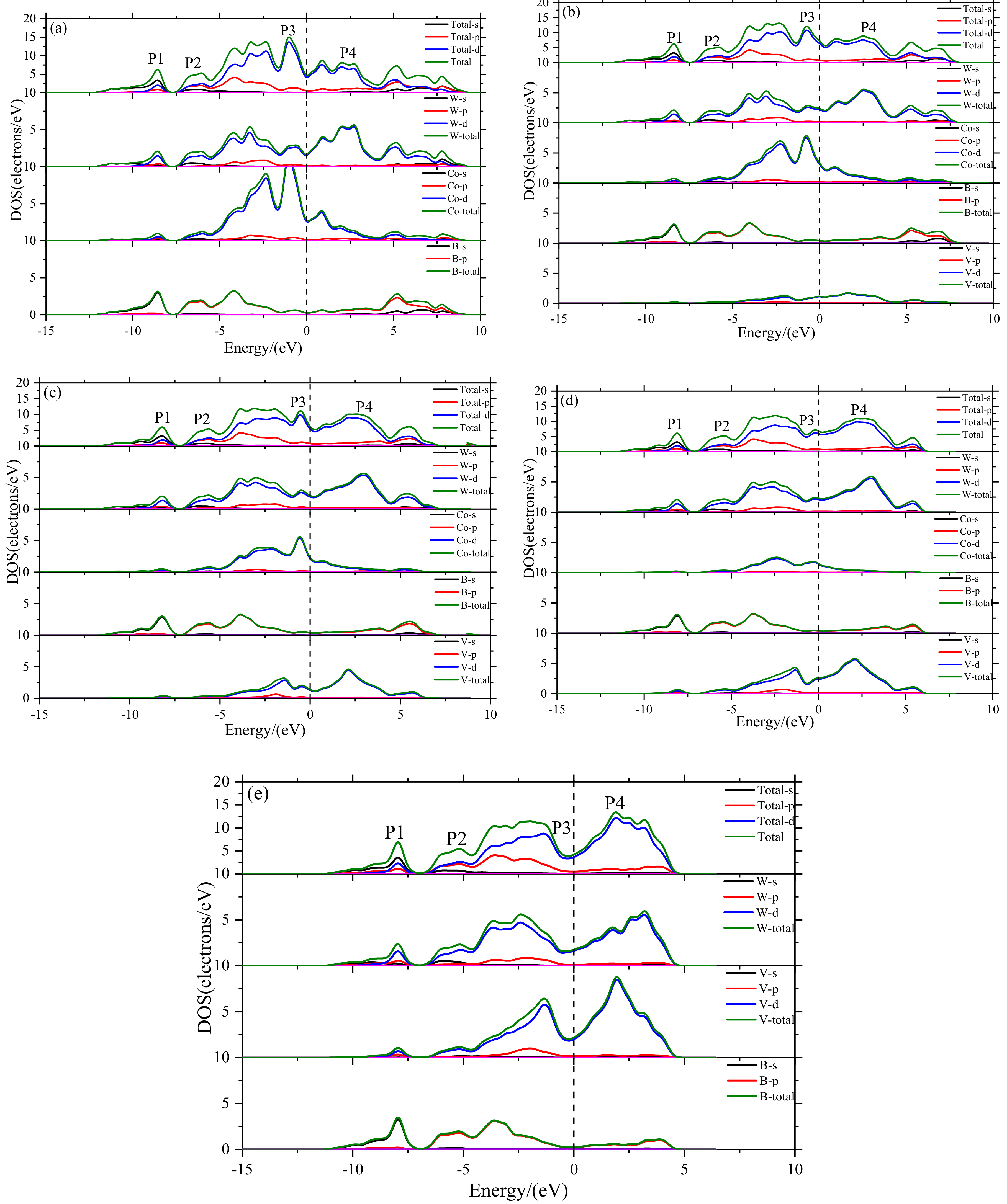
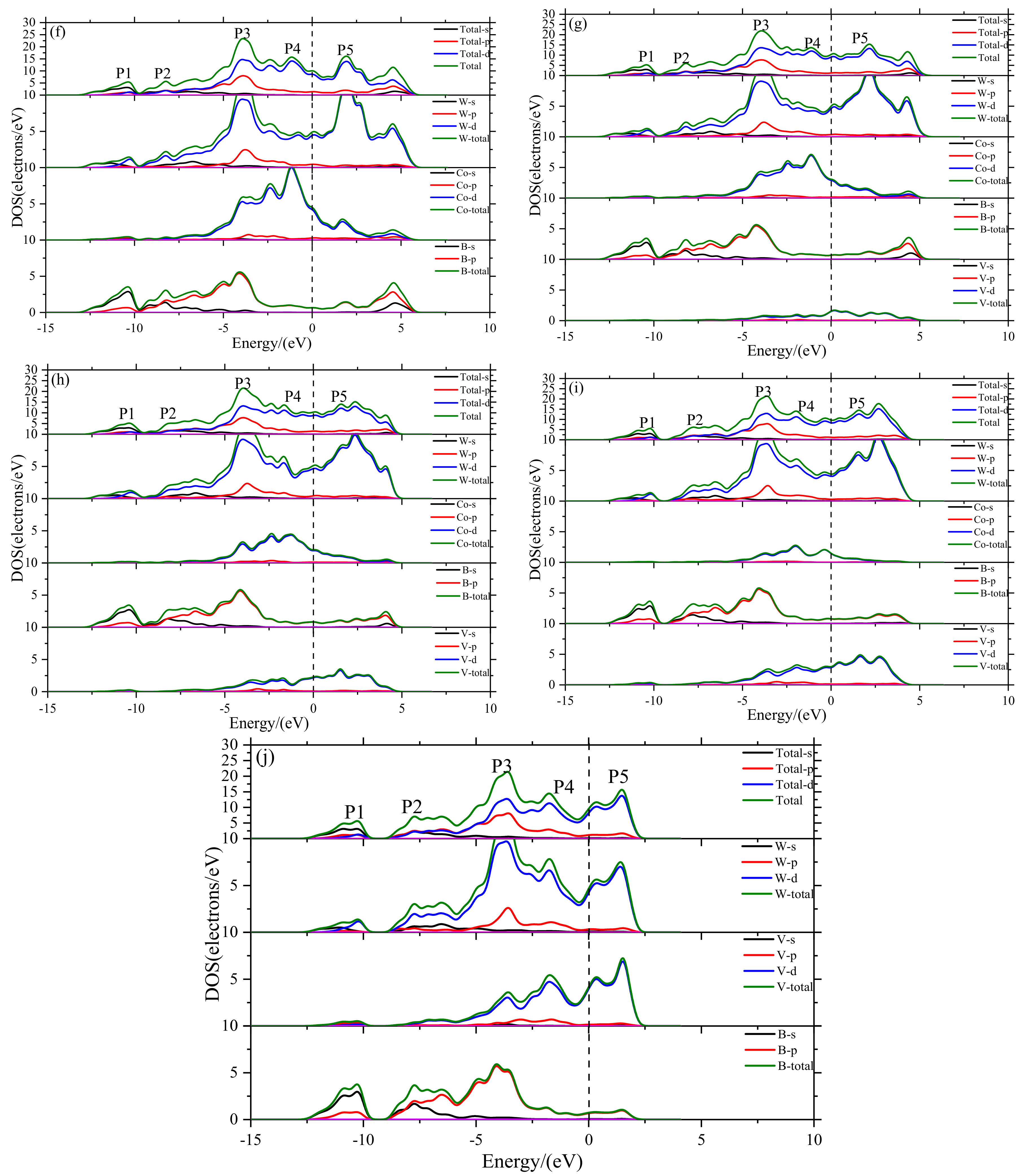
3.4. Density of States
3.5. Charge Density Difference
4. Conclusions
- (1)
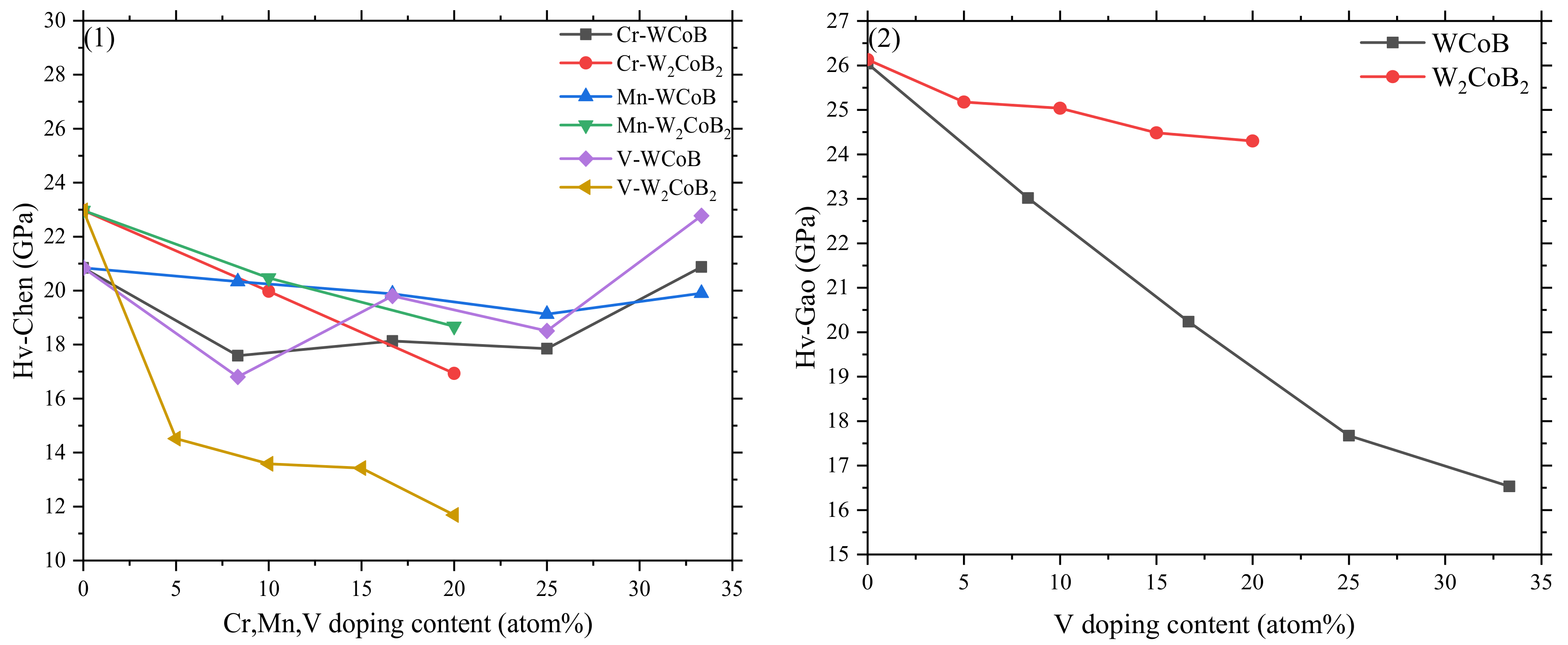
- By analyzing the cohesive energy, formation energy, we can see that all V doped structures are stable. With increasing V doping content, stability increases slightly when the V doping content is up to 16.67 at.% for WCoB and 20 at.% for W2CoB2. However, the formation energy shows V doped WCoB and W2CoB2 ternary boride are harder to form except for the W4V4B4 structure. The larger atom radius of V atoms leads to the increase of lattice constants.
- (2)
- The mechanical properties, average overlap population and hardness show V doping leads to the decreasing of shear modulus and hardness, which is attributed to the weaker B–V covalent bonds and W–V metallic bonds. Two different hardness models show the shear modulus is closely related to the hardness, and V doped W2CoB2 has higher ductility.
- (3)
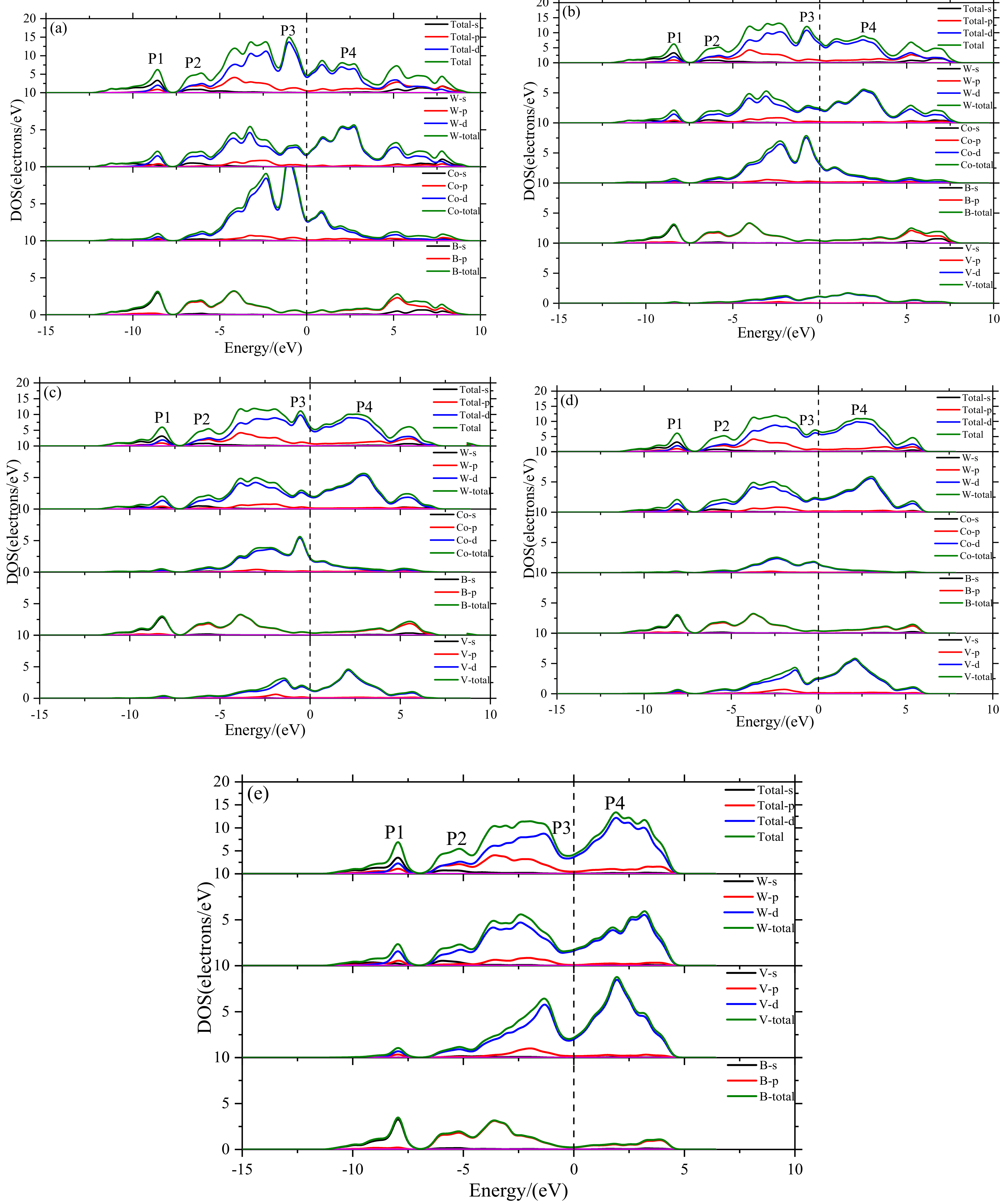
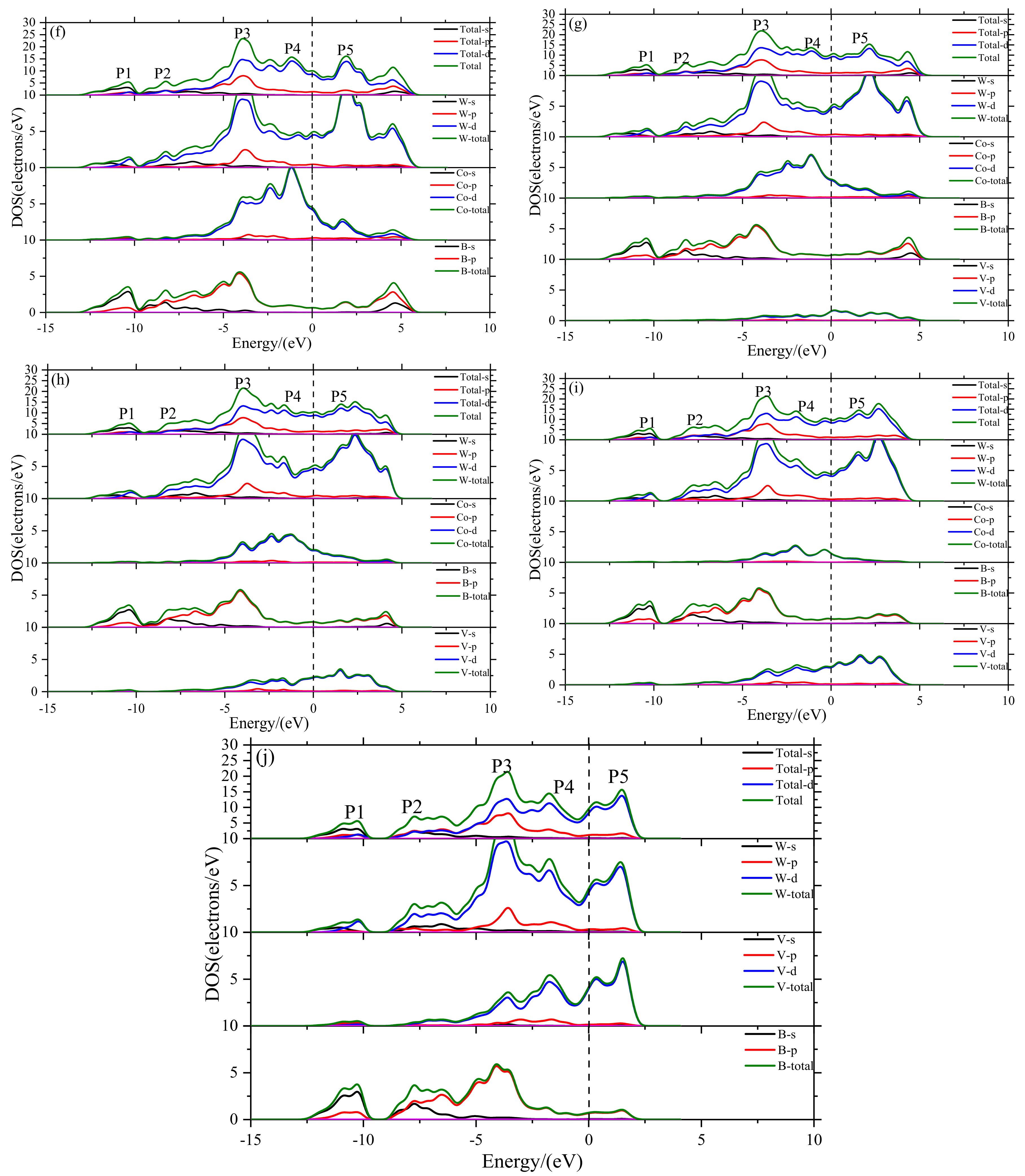
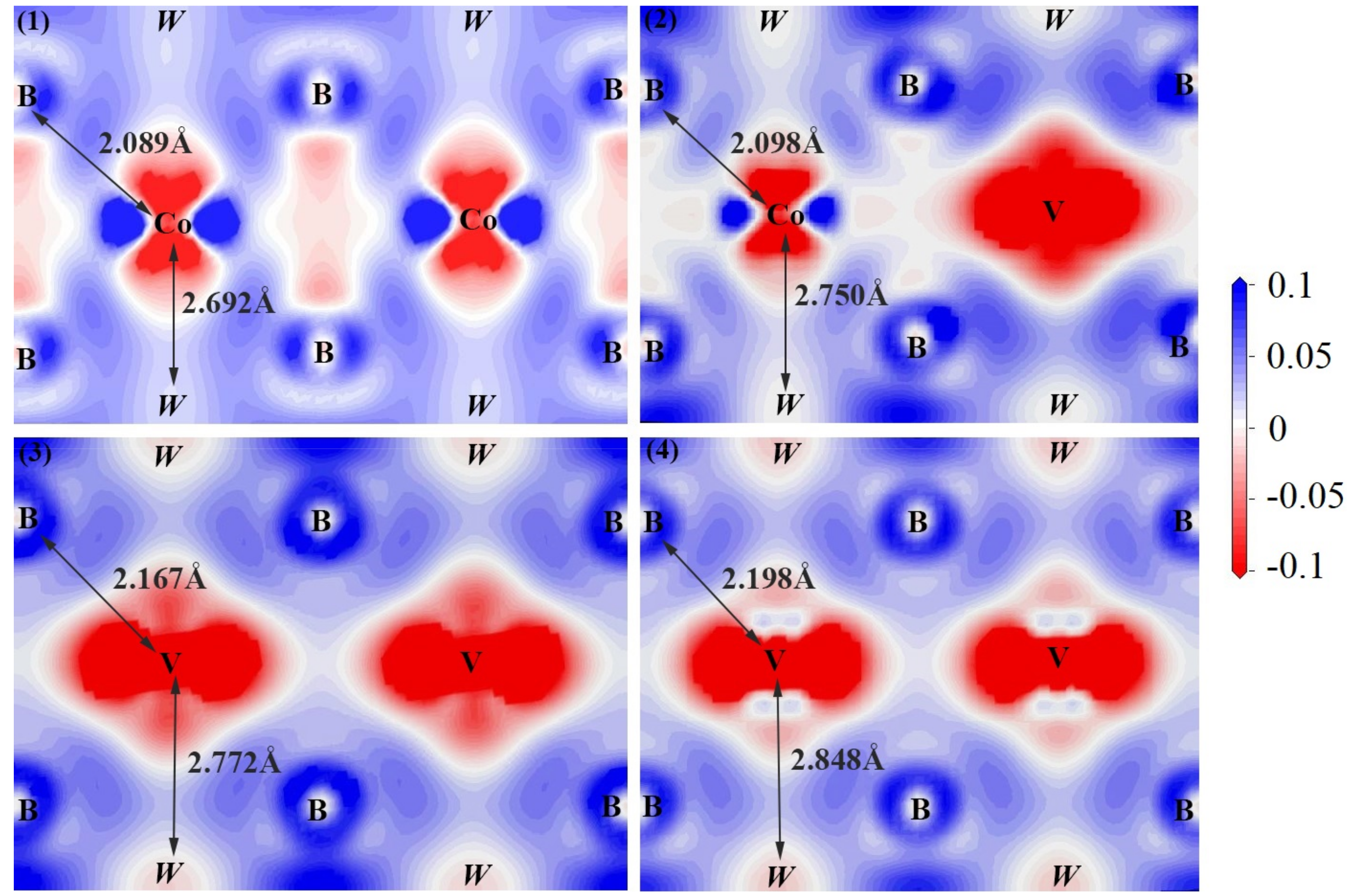
- The electronic structure of V doped WCoB and W2CoB2 ternary boride is studied by DOS, PDOS and charge density difference. The formation of weaker B–V covalent bonds and W–V metallic bonds contributes to the orbital hybridization, which is inconsistent with the analysis of mechanical properties and hardness. Moreover, the formation of W–W metallic bonds at high V doping content is harmful to the shear modulus and mechanical properties.
Author Contributions
Funding
Conflicts of Interest
References
- Takagi, K.; Ohira, S.; Ide, T.; Watanabe, S.; Kondo, Y. New multiple-boride base hard alloy. Met. Powder Rep. 1987, 42, 483–484. [Google Scholar]
- Takagi, K. Development and application of high strength ternary boride base cermets. J. Solid State Chem. 2006, 179, 2809–2818. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Uchitomi, H.; Ozaki, S.; Komai, M. Corrosion resistance of boride base cermets in a molten aluminum alloy. J. Jpn. Soc. Powder Powder Metall. 1994, 41, 994–998. [Google Scholar] [CrossRef]
- Takagi, K. Development of high strength complex boride base hard materials produced by reaction boronizing sintering. J. Jpn. Soc. Powder Powder Metall. 1998, 45, 507–514. [Google Scholar] [CrossRef]
- Tashiro, H.; Hirata, K.; Yamasaki, Y.; Takagi, K. Metal injection molding process of Mo2FeB2 boride base cermets. Mater. Sci. Forum 2007, 534, 377–380. [Google Scholar] [CrossRef]
- Wang, H.Q.; Sun, J.S.; Li, C.N.; Geng, S.N.; Sun, H.G.; Wang, G.L. Microstructure and mechanical properties of molybdenum–iron–boron–chromium cladding using argon arc welding. Mater. Sci. Technol. 2016, 32, 1694–1701. [Google Scholar] [CrossRef]
- Kuz’Ma, Y.B.; Kripyakevich, P.I.; Chepiga, M.V. Crystal structures of the compounds MoCoB, WCoB, and WFeB. J. Struct. Chem. 1968, 9, 268–269. [Google Scholar] [CrossRef]
- Zahariev, Z.T.; Marinov, M.I. Superhard boride layer deposition on a carbide-cobalt hard alloy. J. Alloys Compd. 1993, 201, 1–3. [Google Scholar] [CrossRef]
- Sáez, A.; Arenas, F.; Vidal, E. Microstructure development of WCoB–TiC based hard materials. Int. J. Refract. Met. Hard Mater. 2003, 21, 13–18. [Google Scholar] [CrossRef]
- Ide, T.; Nakano, K. Effects of Cr and Mo contents on sintering mechanisms of Mo2FeB2 base hard alloys. J. Jpn. Soc. Powder Powder Metall. 1989, 36, 38–42. [Google Scholar] [CrossRef]
- Pang, X.M.; Zheng, Y.; Wang, S.G.; Wang, Q.H. Effect of Mn on valence-electron structure and properties of hard phase in Mo2FeB2-based cermets. Int. J. Refract. Met. Hard Mater. 2009, 27, 777–780. [Google Scholar]
- Yamasaki, Y.; Uchitomi, H.; Isobe, Y.; Komai, M. Sintering mechanisms of Cr-containing Mo2NiB2 base hard alloys. J. Jpn. Soc. Powder Powder Metall. 1994, 41, 1037–1041. [Google Scholar] [CrossRef]
- Takagi, K.; Koike, W.; Momozawa, A.; Fujima, T. Effects of Cr on the properties of Mo2NiB2 ternary boride. Solid State Sci. 2012, 14, 1643–1647. [Google Scholar] [CrossRef]
- Takagi, K. Effect of Mn on the mechanical properties and microstructure of reaction sintered Mo2NiB2 boride-based cermets. Met. Mater. Int. 2003, 9, 467–471. [Google Scholar] [CrossRef]
- Ozdogan, K.; Upadhyay Kahaly, M.; Alshareef, H.N.; Schwingenschlögl, U. Anomalous enhancement of the thermoelectric figure of merit by V co-doping of Nb-SrTiO3. Appl. Phys. Lett. 2012, 100, 193110. [Google Scholar] [CrossRef]
- Kan, W.H.; Truong, L.; Thangadurai, V. Effect of V-doping on the structure and conductivity of garnet-type Li5La3Nb2O12. Ionics 2015, 21, 373–379. [Google Scholar] [CrossRef]
- Mi, X.; Zhang, D.; Wu, Q.; Wang, Z.H. Effect of V doping on magnetic and electrical transport properties of La0.8Sr0.2Co1−xVxO3. Ceram. Int. 2016, 42, 2670–2675. [Google Scholar] [CrossRef]
- Thongbai, P.; Pongha, S.; Yamwong, T.; Maensiri, S. Effects of Fe, Ti, and V doping on the microstructure and electrical properties of grain and grain boundary of giant dielectric NiO-based ceramics. Appl. Phys. Lett. 2009, 94, 022908. [Google Scholar] [CrossRef]
- Takahashi, M.; Noguchi, Y.; Miyayama, M. Effects of V-doping on mixed conduction properties of bismuth titanate single crystals. Jpn. J. Appl. Phys. 2003, 42, 6222. [Google Scholar] [CrossRef]
- Tegel, M.; Schmid, T.; Stürzer, T.; Egawa, M.; Su, Y.; Senyshyn, A.; Johrendt, D. Possible magnetic order and suppression of superconductivity by V doping in Sr2VO3FeAs. Phys. Rev. B 2010, 82, 140507. [Google Scholar] [CrossRef]
- Joshi, R.; Kumar, P.; Gaur, A.; Asokan, K. Structural, optical and ferroelectric properties of V doped ZnO. Appl. Nanosci. 2014, 4, 531–536. [Google Scholar] [CrossRef]
- Hua, N.; Wang, C.; Kang, X.; Wumair, T.; Han, Y. Studies of V doping for the LiFePO4-based Li Ion batteries. J. Alloys Compd. 2010, 503, 204–208. [Google Scholar] [CrossRef]
- Hu, B.; Pan, Y.; Wang, Q.; Zhou, H.; Xu, M. Influence of addition element Cr and V on microstructure and properties of Mo2FeB2 based cermet. Heat Treat. Met. 2011, 36, 29–32. [Google Scholar]
- Yu, H.; Zheng, Y.; Liu, W.; Zheng, J.; Xiong, W. Effect of V content on the microstructure and mechanical properties of Mo2FeB2 based cermets. Mater. Des. (1980–2015) 2010, 31, 2680–2683. [Google Scholar] [CrossRef]
- Shiota, Y.; Miyajima, Y.; Fujima, T.; Takagi, K. Effect of double addition of V and Cr on the properties of Mo2NiB2 ternary boride-based cermets. J. Phys. Conf. Ser. 2009, 176, 012046. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Nishi, N.; Takagi, K. Effect of Mo content on microstructures and mechanical properties of Mn and V containing Mo2NiB2 base hard alloys. J. Jpn. Soc. Powder Powder Metall. 2002, 49, 312–317. [Google Scholar] [CrossRef]
- Takagi, K.; Yamasaki, Y. Effects of Mo/B atomic ratio on the mechanical properties and structure of Mo2NiB2 boride base cermets with Cr and V additions. J. Solid State Chem. 2000, 154, 263–268. [Google Scholar] [CrossRef]
- Yamasaki, Y.; Nakano, K.; Okada, M. Microstructures and mechanical properties of V-containing Mo2NiB2 hard alloys. J. Jpn. Soc. Powder Powder Metall. 1995, 42, 438–442. [Google Scholar] [CrossRef]
- Yang, F.; Wu, Y.; Han, J.; Meng, J. Microstructure, mechanical and tribological properties of Mo2FeB2 based cermets with Mn addition. J. Alloys Compd. 2016, 665, 373–380. [Google Scholar] [CrossRef]
- Sun, C.; Yu, H.; Liu, W. Microstructure, mechanical properties and first-principles calculations of Mo2FeB2-based cermets with Mn addition. J. Ceram. Soc. Jpn. 2017, 125, 677–680. [Google Scholar] [CrossRef]
- Wang, S.; Pan, Y.; Lin, Y.; Tong, C. Influence of doping concentration on mechanical properties of Mo2FeB2 alloyed with Cr and Ni from first-principle calculations. Comput. Mater. Sci. 2018, 146, 18–25. [Google Scholar] [CrossRef]
- Lin, Y.H.; Tong, C.C.; Pan, Y.; Liu, W.Y. Elastic properties and electronic structure of Mo2FeB2 alloyed with Cr, Ni and Mn by first-principles calculations. Mod. Phys. Lett. B. 2017, 31, 1750138. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Gao, H.; Peng, D. First-principles study on the electronic structure and elastic properties of Mo2NiB2 doped with V. Mod. Phys. Lett. B. 2018, 32, 1850065. [Google Scholar] [CrossRef]
- Xu, Y.; Pan, Y.; Ke, D.; Yang, L.; Wang, P. Effect of grain growth inhibitor VC on in situ formation of WCoB-TiC-Co multiphase metal-ceramics. Mater. Sci. Technol. 2017, 25, 59–65. [Google Scholar]
- Zhang, T.; Yin, H.; Zhang, C.; Qu, X.H.; Zheng, Q.J. Effect of Cr doping on the mechanical properties and electronic structure of WCoB ternary boride by first-principles calculations. Mod. Phys. Lett. B. 2018, 32, 1850240. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, H.Q.; Zhang, C.; Qu, X.-H.; Zheng, Q.-J. Effect of Mn doping on mechanical properties and electronic structure of WCoB ternary boride by first-principles calculations. Chin. Phys. B 2018, 27, 107101. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, J.; Refson, K.; Payne, M.C. First-principles methods using CASTEP. Z. Kristallogr.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Kuzma, I.B.; Chepiga, M.V. X-ray Investigation of the Tungsten-Iron-Boron and Tungsten Cobalt-Boron Systems; Lvov State University: Lviv, Ukraine, 1969. [Google Scholar]
- Rieger, W.; Nowotny, H.; Benesovsky, F. Die kristallstruktur von W2CoB2 und isotypen phasen. Monatshefte für Chemie und verwandte Teile anderer Wissenschaften 1966, 97, 378–382. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Born, M. On the stability of crystal lattices. I. Math. Proc. Cambridge Philos. Soc. 1940, 36, 160–172. [Google Scholar] [CrossRef]
- Wu, Z.J.; Zhao, E.J.; Xiang, H.P.; Hao, X.F.; Liu, X.J.; Meng, J. Crystal structures and lastic properties uperhard IrN2 and IrN3 from first-principles. Phys. Rev. B 2007, 6, 054115. [Google Scholar]
- Lewandowski, J.J.; Wang, W.H.; Greer, A.L. Intrinsic plasticity or brittleness of metallic glasses. Philos. Mag. Lett. 2005, 85, 77–87. [Google Scholar] [CrossRef]
- Aouadi, S.M. Structural and mechanical properties of TaZrN films: Experimental and ab initio studies. J. Appl. Phys. 2006, 99, 053507. [Google Scholar] [CrossRef]
- Reuss, A. Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. ZAMM-J. Appl. Math. Mech./Zeitschrift für Angewandte Mathematik und Mechanik 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Zhou, C.T.; Xing, J.D.; Xiao, B.; Feng, J.; Xie, X.J.; Chen, Y.H. First-principles study on the structural properties and electronic structure of X2B (X= Cr, Mn, Fe, Co, Ni, Mo and W) compounds. Comput. Mater. Sci. 2009, 44, 1056–1064. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275–1281. [Google Scholar] [CrossRef]
- Gao, F.; He, J.; Wu, E.; Liu, S.; Yu, D.; Li, D.; Zhang, S.; Tian, Y. Hardness of covalent crystals. Phys. Rev. Lett. 2003, 91, 015502. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.B.; Wang, W.H.; Bai, H.Y. An electronic structure perspective on glass-forming ability in metallic glasses. Appl. Phys. Lett. 2010, 96, 081902. [Google Scholar]
- Xu, W.W.; Han, J.J.; Wang, Y.; Wang, C.P.; Liu, X.J.; Liu, Z.K. First-principles investigation of electronic, mechanical and thermodynamic properties of L12 ordered Co3 (M, W) (M= Al, Ge, Ga) phases. Acta Mater. 2013, 61, 5437–5448. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WCoB/W2CoB2 | Lattice Constants (Å) | V (Å3) | Ecoh | Ef | |
|---|---|---|---|---|---|
| W4Co4B4-ex | a = 5.746, b = 3.203, c = 6.652 | 122.43 | - | - | - |
| a-W4Co4B4 | a = 5.745, b = 3.256, c = 6.624 | 123.880 | −9.311 | 0 | −0.494 |
| B–W4Co3VB4 | a = 5.830, b = 3.210, c = 6.798 | 127.213 | −9.299 | 0.149 | −0.460 |
| c-W4Co2V2B4-sy | a = 5.973, b = 3.162, c = 7.010 | 130.178 | −9.342 | −0.370 | −0.481 |
| c-W4Co2V2B4-unsy | a = 5.919, b = 3.162, c = 6.974 | 130.437 | −9.321 | −0.122 | −0.460 |
| d-W4CoV3B4 | a = 5.937, b = 3.137, c = 7.165 | 133.475 | −9.367 | −0.673 | −0.484 |
| e-W4V4B4 | a = 5.953, b = 3.113, c = 7.345 | 136.118 | −9.448 | −1.644 | −0.543 |
| W4Co2B4-ex | a = 7.075, b = 4.564, c = 3.177 | 102.59 | - | - | - |
| f-W8Co4B8 | a = 7.106, b = 4.565, c = 3.189 | 103.454 | −9.486 | 0 | −0.531 |
| g-W8Co3VB8 | a = 7.182, b = 4.643, c = 3.156 | 105.260 | −9.462 | 0.467 | −0.492 |
| h-W8Co2V2B8 | a = 7.175, b = 4.670, c = 3.166 | 106.750 | −9.469 | 0.332 | −0.483 |
| i-W8CoV3B8 | a = 7.212, b = 4.794, c = 3.140 | 108.566 | −9.472 | 0.274 | −0.470 |
| j-W8V4B8 | a = 7.189, b = 4.860, c = 3.151 | 110.094 | −9.505 | −0.386 | −0.487 |
| a-W4Co4B4 | B–W4Co3VB4 | c-W4Co2V2B4 | d-W4CoV3B4 | e-W4V4B4 | |
|---|---|---|---|---|---|
| C11 | 551.84 | 493.02 | 524.73 | 515.56 | 556.79 |
| C22 | 515.90 | 475.98 | 484.01 | 487.05 | 497.32 |
| C33 | 609.34 | 558.45 | 549.28 | 514.86 | 522.41 |
| C44 | 234.37 | 216.15 | 219.46 | 210.78 | 220.10 |
| C55 | 190.14 | 169.89 | 165.62 | 141.02 | 149.42 |
| C66 | 236.81 | 221.97 | 237.74 | 212.29 | 228.58 |
| C12 | 264.24 | 270.44 | 242.05 | 229.14 | 206.52 |
| C13 | 177.11 | 180.14 | 161.29 | 154.40 | 127.24 |
| C23 | 225.26 | 220.23 | 206.40 | 190.04 | 177.06 |
| a-W4Co4B4 | B–W4Co3VB4 | c-W4Co2V2B4 | d-W4CoV3B4 | e-W4V4B4 | |
|---|---|---|---|---|---|
| C11 | 589.86 | 544.95 | 538.71 | 517.45 | 545.80 |
| C22 | 608.81 | 561.13 | 564.37 | 565.08 | 586.25 |
| C33 | 542.51 | 490.30 | 482.64 | 482.50 | 499.95 |
| C44 | 241.93 | 241.06 | 239.58 | 244.70 | 257.87 |
| C55 | 245.71 | 228.43 | 227.96 | 244.34 | 235.65 |
| C66 | 210.82 | 95.46 | 76.37 | 63.87 | 37.31 |
| C12 | 162.49 | 158.43 | 156.86 | 156.72 | 133.52 |
| C13 | 250.69 | 261.84 | 258.16 | 258.26 | 238.41 |
| C23 | 238.90 | 248.74 | 232.46 | 212.01 | 195.72 |
| a-W4Co4B4 | B–W4Co3VB4 | c-W4Co2V2B4 | d-W4CoV3B4 | e-W4V4B4 | |
|---|---|---|---|---|---|
| B–B | - | - | - | - | - |
| B–Co | 0.307 | 0.309 | 0.303 | 0.300 | - |
| B–W | 0.530 | 0.558 | 0.565 | 0.592 | 0.603 |
| B–V | - | 0.123 | 0.157 | 0.156 | 0.157 |
| Co–Co | −0.150 | −0.140 | −0.0900 | - | - |
| Co–W | −0.055 | −0.0100 | −0.0150 | 0.0575 | - |
| Co–V | - | −0.330 | - | −0.220 | - |
| W–W | −0.055 | −0.055 | 0.0300 | 0.100 | 0.150 |
| W–V | - | −0.240 | −0.1125 | −0.0950 | −0.0425 |
| V–V | - | - | −0.530 | −0.410 | −0.33 |
| a-W8Co4B8 | B–W8Co3VB8 | c-W8Co2V2B8 | d-W8CoV3B8 | e-W8V4B8 | |
|---|---|---|---|---|---|
| B–B | 0.590 | 0.5925 | 0.575 | 0.568 | 0.580 |
| B–Co | 0.210 | 0.237 | 0.27 | 0.29 | - |
| B–W | 0.200 | 0.201 | 0.208 | 0.208 | 0.207 |
| B–V | - | 0.13 | 0.14 | 0.187 | 0.220 |
| Co–Co | - | - | - | - | - |
| Co–W | 0.0833 | 0.0933 | 0.113 | 0.127 | - |
| Co–V | - | - | - | - | - |
| W–W | −0.0833 | −0.0492 | 0 | 0.0445 | 0.0267 |
| W–V | - | −0.01 | 0.0167 | 0.04 | 0.0867 |
| V–V | - | - | - | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Yin, H.; Zhang, C.; Zhang, R.; Jiang, X.; Zheng, Q.; Qu, X. First-Principles Study on the Mechanical Properties and Electronic Structure of V Doped WCoB and W2CoB2 Ternary Borides. Materials 2019, 12, 967. https://doi.org/10.3390/ma12060967
Zhang T, Yin H, Zhang C, Zhang R, Jiang X, Zheng Q, Qu X. First-Principles Study on the Mechanical Properties and Electronic Structure of V Doped WCoB and W2CoB2 Ternary Borides. Materials. 2019; 12(6):967. https://doi.org/10.3390/ma12060967
Chicago/Turabian StyleZhang, Tong, Haiqing Yin, Cong Zhang, Ruijie Zhang, Xue Jiang, Qingjun Zheng, and Xuanhui Qu. 2019. "First-Principles Study on the Mechanical Properties and Electronic Structure of V Doped WCoB and W2CoB2 Ternary Borides" Materials 12, no. 6: 967. https://doi.org/10.3390/ma12060967