First-Principle Calculation of High Absorption-TlGaTe2 for Photovoltaic Application


1
Department of Computing, Mathematics and Physics, Western Norway University of Applied Sciences, Inndalsveien 28, Box 5063, 5003-5268 Bergen, Norway
2
Center for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Box 1033 Blindern N-0315, 0001-1299 Oslo, Norway
*
Author to whom correspondence should be addressed.
Materials 2019, 12(17), 2667; https://doi.org/10.3390/ma12172667
Submission received: 12 July 2019
/
Revised: 8 August 2019
/
Accepted: 17 August 2019
/
Published: 22 August 2019
Abstract
:We use first-principle calculations based on hybrid functional and the Bethe-Salpeter equation method to investigate the electronic and optical properties of dichalcogenide TlGaTe2. Based on theoretical studies, TlGaTe2 has until recently been considered as an indirect band gap material, however; by employing more accurate hybrid functional model, we showed that although TlGaTe2 has an indirect band gap of 1.109 eV, it also exhibits a fundamental direct band gap of 1.129 eV. Our finding was further confirmed by the optical studies on TlGaTe2, which show that the absorption peak is registered at a photon energy of 1.129 eV. It was also shown that TlGaTe2 has high optical absorption peaks in the visible region. Based on phonon and elastic constant calculations, it was shown that TlGaTe2 is dynamically and mechanically stable. Our findings show that TlGaTe2 is a potential candidate for photovoltaic application.
1. Introduction
Photovoltaics (PV) show great potential in meeting the needs of the world’s energy consumption. Silicon-based solar cell technologies have over 90% of the PV market today, but the research community has been yearning for new materials that are cheap, flexible and efficient in solar cells. Band gap value and absorption coefficient of the material are the two major factors that decide whether the material can be of interest for photovoltaic (PV) applications. We have earlier reported several promising non-silicon materials with intermediate band gaps for photovoltaic applications [1,2]. There is an increasing interest in the family of ternary compounds with the general formula III-III-VI2 (where III = Al, Ga, In, Tl; VI = Se, Te, S)—known as dichalcogenides [3]. In a recent published article, we carried out in-depth analysis of TlBiS2 (thallium-based) and calculated electronic and optical properties for the photovoltaic applications [4]. Thallium-bearing ternary materials have been studied in optoelectronics [5], but not extensively investigated in photovoltaics due to the fact that thallium is toxic. Researchers have shown interest to the Hall effect analysis, the photoconductivity and recombination kinetics in dichalcogenide-TlGaTe2 material [6,7]. From our earlier study on non-silicon solar cell materials [8], we identified that TlGaTe2 has a strong absorption coefficient in the visible region, but a complete study of TlGaTe2 is needed to confirm that it is potential material for PV applications.
In this article, we study structural, electronic, spectroscopic and optical properties of TlGaTe2 dichalcogenide using first-principle calculations. As the density functional theory (DFT) is considered as one of the most effective and accurate computational methods, we investigate the electronic band structure of TlGaTe2 employing DFT analysis. We report accurate electronic band structure of TlGaTe2 based on a hybrid density functional (HSE06) calculations. The calculated HSE06 band structure for this compound helps to identify the type of band gap TlGaTe2 possess and its value, authenticating the suitability for the solar cell application. We also study the structural stability and mechanical stability of TlGaTe2. To verify the Raman active (R-active) and infrared active (IR-active) modes with previously published experimental results [9], we simulated the Raman spectra and infrared spectra of TlGaTe2.
2. Computational Details
Total energies of TlGaTe2 were determined by the projected augmented plane-wave (PAW) utilization of the Vienna ab initio simulation package (VASP) [10]. We used the Perde-Burke-Ernzerhof (PBE) of the generalized gradient approximation (GGA) to achieve the structure relaxations [11]. The ionic coordinates are fully relaxed until the total energy is changed 10−6 eV per atom. Ground-state geometries were calculated by minimizing stresses and Hellman-Feynman forces using the conjugate-gradient algorithm with a force convergence threshold of 10−3 eV Å−1. We used the screened-exchange Heyd-Scuseria-Ernzerhof hybrid function (HSE06) to study the electronic properties of TlGaTe2. The screened parameter was set to 0.2 Å−1 and 30% of the screened Hartree-Fock (HF) exchange was mixed with the PBE exchange functional in the HSE06 method [12]. We applied a 10 × 10 × 10 Γ-centered Monkhorst-pack k-point mesh, which is used in HSE06 self-consistent calculations for TlGaTe2. For the calculation of vibrational properties, the software PHONOPY along with VASP, was utilized [13,14]. For vibrational properties, we used 2 × 2 × 2 supercell (64 atoms). Solving the Casida’s equation is the best approach for calculating the dielectric function [15]. We used a 16 × 16 × 16 -centered grid for both GW (green function with Coulomb interaction) and the Bethe-Salpeter equation (BSE). For all these calculations, a plane-wave cut off of 500 eV was used. The Raman activity was obtained by calculating the polarizability for the vibrational modes [16]. The IR spectra is simulated with the analytic formula of Baroni et al. (using the Born effective charge tensor) [17].
3. Results and Discussions
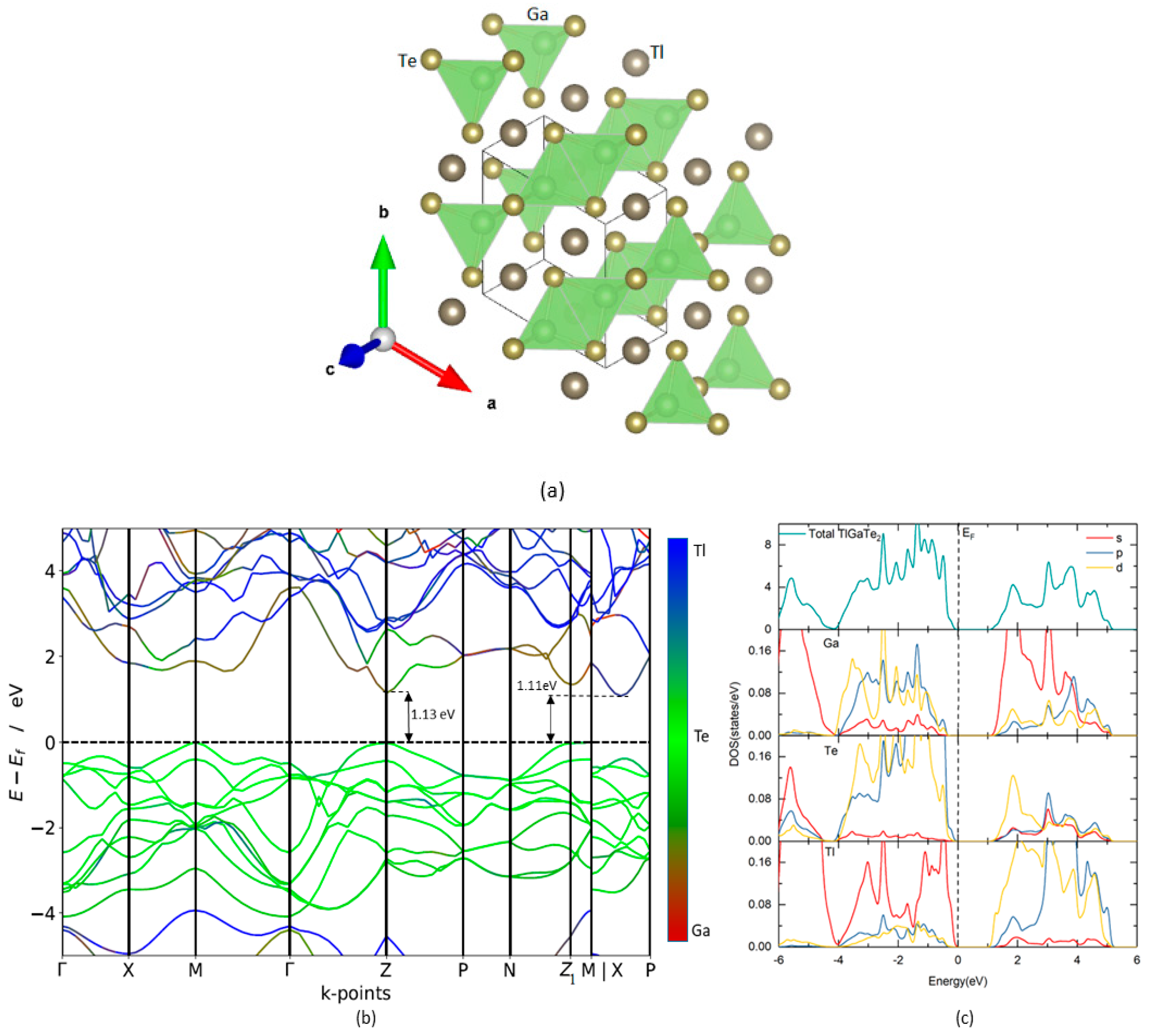
TlGaTe2 crystallizes in a tetragonal structure (I4/mcm, space group No. 140) as shown in Figure 1a. The calculated Ga-Te distance is 2.69 Å, and Tl-Tl bond length is smaller than the separation between Te and Tl atoms, which make up the GaTe4 tetrahedra (Figure 1a) [18]. The open-circuit voltage and short-circuit current of the solar cells are highly influenced by the band gap of the photoactive semiconductors. The detailed-balance limiting efficiency of an ideal solar cell is 32% for a material with the optimal band gap of 1.4 eV [19]. Band gap calculation using electronic band structure provides a promising opportunity to identify suitable PV materials. The band structure and site-projected density of states of dichalcogenide TlGaTe2 calculated with HSE06 are shown in Figure 1b,c, respectively. The valence band maximum (VBM) is located at the Z-point and Z1-point. The first conduction band minimum (CBM) is located along M-P directions and the second CBM is located at Z-point. In two different experimental studies, it was reported that the band gap energy of TlGaTe2 is 0.84 eV [20] and 1.2 eV [21]. The reason for this deviation is not known. Our calculations show that TlGaTe2 has an indirect band gap of 1.109 eV, which is higher than the mBJ calculation (0.94 eV) [22] but has a good agreement with an experimentally verified band gap value of 1.2 eV [21]. Also, we observe that TlGaTe2 exhibits a direct band gap of 1.129 eV at the Z-point and the difference between direct and indirect band gap is 20 meV. This phenomenon is verified further by a study on the optical properties of TlGaTe2 later in this article. The site-projected density of states shows that valence band maximum (VBM) is mainly due to Tl-s state with little contribution of Te-p state, while conduction band maximum (CBM) is mainly contributed by Ga-s state and Tl-d state in Figure 1c.
Since TlGaTe2 exhibits narrow indirect band gap at the Z1-point (Figure 1b), the VBM is flatter than the first CBM along M-P directions. Note that around Z-point, the VBM is somewhat flatter than the second CBM. Hence, it would appear that electrons are lighter than holes. The effective masses of holes and electrons have been calculated by the finite difference method [23]. The effective masses of TlGaTe2 calculated by HSE06 are listed in Table 1. For the indirect electronic transition, we found that the effective mass of electrons is 0.196 me (me–a mass of an electron) along the M|X-P direction and the effective mass of holes is 0.736 me along the Z1-M direction. It is interesting to note that Qasrawi et al. used the Hall effect measurement and reported the hole effective mass of TlGaTe2 to be 0.730 me [7], which is in good agreement with our calculated hole effective mass (0.736 me). For the direct transition, we noted that the EM of electrons is 0.378 me and EM of holes is 0.736 me at Z-point.
For comparison, the effective mass of electron such as silicon (Si) and gallium arsenide (GaAs) are 0.26 and 0.12 me [24], respectively. The effective mass (EM) of the electron is low for TlGaTe2 compared to Si. Thus, TlGaTe2 has high electron mobility than silicon, which would be useful for an efficient solar cells [25].
In order to understand the dynamical stability of the TlGaTe2, we carried out the phonon calculations. The calculated vibrational spectrum curves together with the corresponding site projected density of states are plotted in Figure 2. The positive phonon frequencies suggest that TlGaTe2 is dynamically stable. The phonon frequencies in the TlGaTe2 have a separation between the low and high-frequency modes (the so-called phonon band gap). The vibrational modes are spread over from 0 to 1735 cm−1. As expected, based on the relationship between masses and frequencies given by equation [26], the heavier element thallium is normally connected with the lower frequencies. The lattice vibrational modes for Tl, Ga, and Te are present in the 0 to 450 cm−1 range, 1167 to 1735 cm−1 range and 0 to 767 cm−1 range, respectively.
To validate the mechanical stability of tetragonal-TlGaTe2, we calculated the single-crystal elastic constant tensor using the finite strain technique. This structure has six independent elastic constants namely C11 = 32.205, C12 = 15.894, C13 = 11.959, C33 = 58.188, C44 = 10.237, and C66 = 12.009. The mechanical stability criteria for the tetragonal (type I) phase are given by [27]:
C11 − C12 > 0
2(C11 + C12) + C33 + 4C13 > 0
C44 > 0, C66 > 0
C11 + C12 − 2C13 > 0
All four conditions for the mechanical stability of tetragonal-TlGaTe2 given in Equations (1)–(4) are satisfied, and this finding indicates that tetragonal-TlGaTe2 phase is mechanical stable. From the calculated elastic constants, the bulk (Bv, BR) and shear moduli (Gv, GR) are calculated from Voigt–Reuss–Hill approximations [28,29]. Our calculated values of bulk moduli (B) and shear moduli are 22 GPa and 20.143 GPa from the elastic constants, respectively. The critical G/B value that differs the ductile and brittle materials is 0.5 [30]. As known, when the G/B value of materials is lesser than 0.5, then those materials are ductile—otherwise, they are brittle. The value of G/B for TlGaTe2 is 0.916, which shows that TlGaTe2 is brittle.
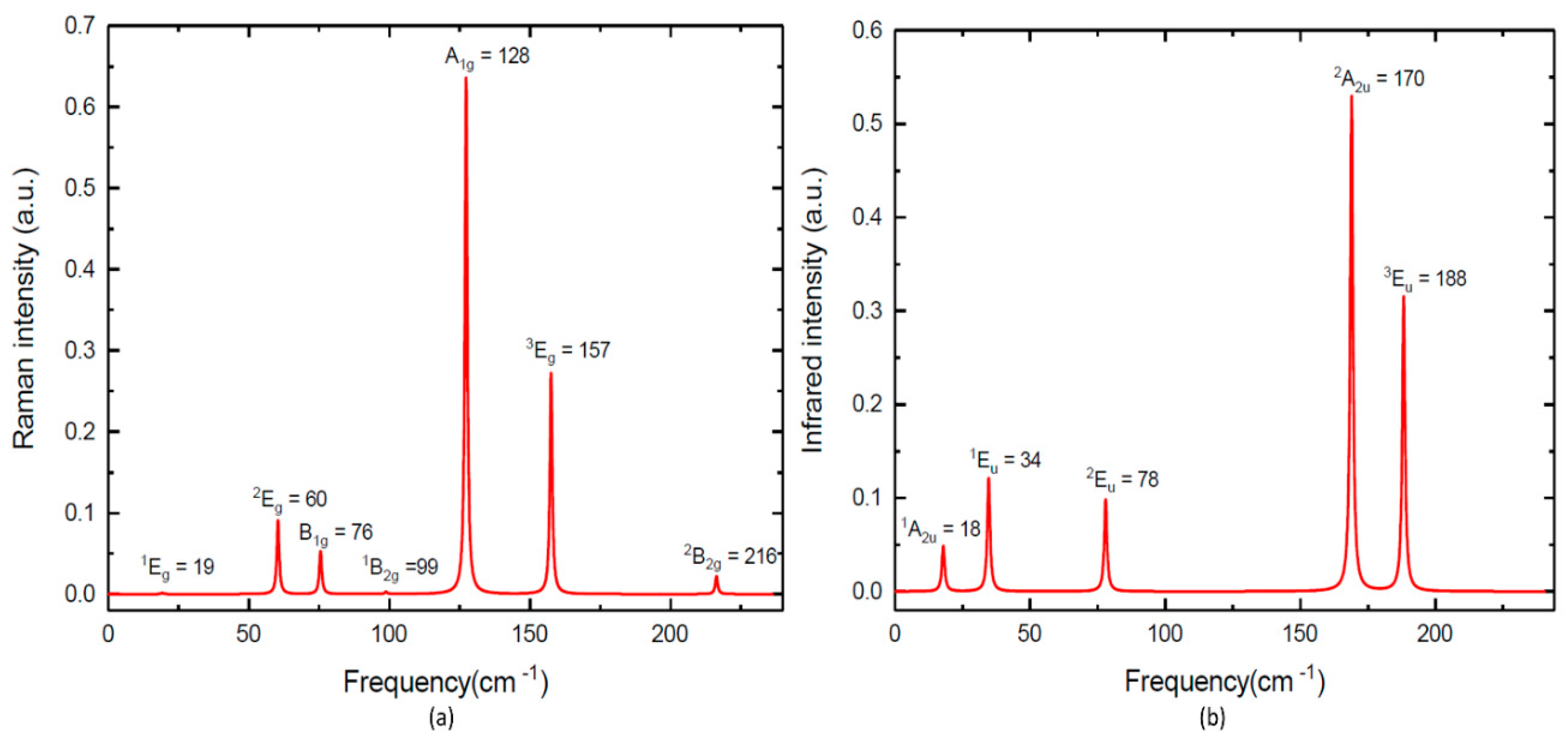
According to the crystal symmetry, we have the following modes and the selection rules for IR-active and R-active modes, Γ = A1g (R) + 2A2u (IR) + B1g (R) + B1u + 2B2g (R) + 3Eu (IR) + 3Eg (R). As seen in Figure 3a and Table 2, we observed seven R-active modes in the Raman spectra for TlGaTe2, whereas Gasanly et al. [9] found only three out of seven predicated R-active modes. Our calculated R-active modes confirm the existence of three Eg mode with frequencies of 1Eg= 19 cm−1 (16 cm−1 [31]), 2Eg = 60 cm−1 (67 cm−1 [9]) and 3Eg = 157 cm−1 (165 cm−1 [9]). Our calculated R-active modes confirm the existence of one A1g mode with a frequency of A1g = 128 cm−1 (135 cm−1 [9]) and three B1g mode with frequencies of B1g = 76 cm−1 (76 cm−1 [31]), B1g = 99 cm−1 (99 cm−1 [31]) and 2B2g = 216 cm−1 (210 cm−1 [31]).
From Figure 3b and Table 2, we observed five IR-active modes in infrared spectra calculation [17]. Our calculated IR-active modes confirm the existence of two A2u mode with frequencies of 1A2u = 18 (27 cm−1 [9]) and 2A2u = 170 (175 cm−1 [9]), along with three Eu mode with frequencies of 1Eu = 34 (44 cm−1 [9]), 2Eu = 78(88 cm−1 [9]) and 3Eu = 188(192 cm−1 [9]). In Table 2, we presented our calculated values for R-active and IR-active modes along with results published earlier in [31] and the experimental values [9]. Note that our calculated R-active and IR-active modes are in better agreement with the experimental results obtained in [9] than [31]. However, the calculated phonon frequencies of TlGaTe2 are lower than the corresponding experimental phonon frequencies. The exchange correlation functional used in this study is not enough to estimate the transition pressure. On the other hand, it is very difficult to obtain a pure form of TlGaTe2 at ambient conditions due to experimental limitations and metastability.
The optical properties have major influence on the solar cell materials; thus, we study the absorption properties of TlGaTe2 material by employing optical dielectric function ε(ω) = ε1(ω) + iε2(ω). The dielectric function is contingent on the wavelength of the electromagnetic radiation, and it is related to the interaction between photons and electrons. The absorption coefficient of the material is highly influenced by the imaginary part ε2(ω), and it can be determined from the inter-band optical transitions by summing over the unoccupied states using the equation [32]. The real part ε1(ω) of dielectric function can be calculated from the ε2(ω) by Kramer-Kronig relationship [33]. The calculated imaginary part of the dielectric function and absorption coefficients of the TlGaTe2 compound are presented in Figure 4. According to the directional dependency of ε1(ω) and ε2(ω), we can conclude that TlGaTe2 is an anisotropic material to the optical properties. Therefore, we present both the real and imaginary parts of the dielectric function of TlGaTe2 in Figure 4 along x and z-directions.
In Figure 4a, both the real and the imaginary part of the dielectric function are plotted against the photon energy. The absorption coefficients (α) for TlGaTe2 are calculated numerically with the inclusion of excitonic effects treated within the Bethe-Salpeter equation (BSE) method, which gives numerical results that are in better agreement with the experimental absorption spectra [34,35,36,37]. To achieve BSE calculation, we start from the DFT level, which permits one to obtain a ground state energy of the TlGaTe2. In the second stage, GW (G–Green function and W–Coulomb interactions) approximations are used to calculate the quasi-particle energies. The GW function fails to calculate the excitonic effect between electron-hole interactions. The electron-hole interactions are then included beyond GW approximations, and the BSE method is employed in order to get improved dielectric function for TlGaTe2. To shed light on the accuracy of the BSE method, we calculated the absorption coefficient for silicon and compared with the corresponding experimental results. In Figure 4b, both the numerical and experimental absorption coefficients for Si and TlGaTe2 are depicted in Figure 4b. Note that the calculated absorption coefficient based on the BSE method for silicon is almost similar to the experimental values obtained from [36].
From Figure 4a,b, it is clearly seen that TlGaTe2 exhibits absorption peaks at 1.13 eV along the x-direction, which has to be due to the direct band gap of 1.129 eV at Z-point (from electronic band structure). Figure 4b shows a high absorption coefficient in the visible region. Absorption peaks of TlGaTe2 are observed at 1.129, 2.02, 2.5 and 2.7 eV along both x and z-directions. When the photon energy is about 2.7 eV along the x-direction, and 2.5 eV along the z-direction, the absorption coefficient of TlGaTe2 reached a maximum in the visible region. The valence band maximum is mainly attributed to the states Tl-s, and thallium is a very good absorbing material in the visible region. Consequently, we observed that TlGaTe2 exhibits high absorption in the visible region. Due to the indirect band gap of silicon, the absorption occurs for the photon energies higher than 2.5 eV as shown in Figure 4b. The difference between the indirect and direct band gap in silicon is about 1.40 eV, which results in low absorption coefficients for silicon in the visible region. Our results show that TlGaTe2 has better absorption coefficients compared to silicon in the visible region.
4. Concluding Remarks
In summary, we can mention that the electronic band structure and optical properties of TlGaTe2 with accurate first-principle calculations were investigated. The calculated HSE06 electronic structures confirmed that TlGaTe2 exhibits an indirect band gap, with a value of 1.109 eV, at the Z1-point. However, TlGaTe2 also exhibits a direct band gap, with a value of 1.129 eV at the Z-point. The first absorption peak at a photon energy of 1.129 eV confirms this finding. Until now, numerical limitations in calculating the band gap energies in the range of a few meV difference have been leading to claims stating that the fundamental gap of TlGaTe2 is indirect. Our accurate calculations from employing BSE revealed that TlGaTe2 possesses an additional direct band gap of 1.129 eV with a difference of 20 meV compared to the indirect band gap. Our comparison of the calculated absorption coefficient of silicon and TlGaTe2 shows that TlGaTe2 has a better absorption coefficient than silicon in the visible region.
Our phonon calculations show that TlGaTe2 is dynamically stable as no imaginary frequency was observed. Our elastic constant calculations illustrate that TlGaTe2 is mechanically stable. Raman and IR calculations are carried out for TlGaTe2 and compared with previous experimental and calculated results. Our results seem to be in better agreement with the experimental results than any other calculated results published previously. Our detailed studies of electronic and optical properties of the ternary dichalcogenide material suggest that TlGaTe2 is a potential candidate for photovoltaic applications.
Author Contributions
conceptualization, M.R., D.V. and P.V.; methodology, M.R., and P.V.; software, M.R., P.V. and D.V.; validation, P.V., M.R. and D.V.; formal analysis, M.R.; investigation, M.R. and P.V.; resources, M.R., P.V.; data curation, M.R. and P.V.; writing–original draft preparation, M.R.; writing–review and editing, M.R., D.V., P.V.; visualization, M.R. and P.V.; supervision, D.V., P.V.; project administration, D.V.; funding acquisition, D.V.
Funding
This research was funded by Western Norway University of Applied Sciences grant number 23300.
Acknowledgments
The authors gratefully acknowledge the Western Norway University of Applied Sciences for financially supporting the project (23300) and UNINETT Sigma2 for computing the facilities of project numbers NN2867K and NN2875K, which were used to conduct the calculations presented in this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rasukkannu, M.; Velauthapillai, D.; Vajeeston, P. Computational Modeling of Novel Bulk Materials for the Intermediate-Band Solar Cells. ACS Omega 2017, 2, 1454–1462. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Vajeeston, P. A first-principle study of the electronic, mechanical and optical properties of inorganic perovskite Cs2SnI6 for intermediate-band solar cells. Mater. Lett. 2018, 218, 233–236. [Google Scholar] [CrossRef]
- Isik, M.; Gasanly, N.; Ozkan, H. Deep Traps Distribution in TlInS2 Layered Crystals. Acta Phys. Pol. A 2009, 115, 732–737. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Ponniah, V. A promising high-efficiency photovoltaic alternative non-silicon material: A first-principle investigation. Scr. Mater. 2018, 156, 134–137. [Google Scholar] [CrossRef]
- Aliev, Z.S.; Zúñiga, F.J.; Koroteev, Y.M.; Breczewski, T.; Babanly, N.B.; Amiraslanov, I.R.; Politano, A.; Madariaga, G.; Babanly, M.B.; Chulkov, E.V. Insight on a novel layered semiconductors: CuTlS and CuTlSe. J. Solid State Chem. 2016, 242, 1–7. [Google Scholar] [CrossRef]
- Qasrawi, A.F.; Gasanly, N.M. Analysis of the Hall effect in TlGaTe2 single crystals. J. Phys. Condens. Matter 2009, 21, 235802. [Google Scholar] [CrossRef] [PubMed]
- Qasrawi, A.F.; Gasanly, N.M. Transport and recombination kinetics in TlGaTe2 crystals. Phys. Status Solidi A 2009, 206, 2555–2558. [Google Scholar] [CrossRef]
- Rasukkannu, M.; Velauthapillai, D.; Bianchini, F.; Vajeeston, P. Properties of Novel Non-Silicon Materials for Photovoltaic Applications: A First-Principle Insight. Materials 2018, 11, 2006. [Google Scholar] [CrossRef]
- Gasanly, N.M.; Goncharov, A.F.; Dzhavadov, B.M.; Melnik, N.N.; Tagirov, V.I.; Vinogradov, E.A. Vibrational Spectra of TlGaTe2, TlInTe2, and TlInSe2 Layer Single Crystals. Phys. Status Solidi B 1980, 97, 367–377. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalised gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Efficient hybrid density functional calculations in solids: Assessment of the Heyd–Scuseria–Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187. [Google Scholar] [CrossRef] [PubMed]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type andCaCl2-typeSiO2at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Paier, J.; Marsman, M.; Kresse, G. Dielectric properties and excitons for extended systems from hybrid functionals. Phys. Rev. B 2008, 78, 121201. [Google Scholar] [CrossRef]
- Fonari, A.; Stauffer, S. vasp_raman. py. Available online: https://github.com/raman-sc/VASP/ (accessed on 14 August 2018).
- Baroni, S.; De Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.; Eulenberger, G.; Hahn, H. Ternary Tl Chalcogenides with TlSe Structure. Z. Anorg. Allg. Chem. 1973, 398, 207–220. [Google Scholar]
- Shockley, W.; Queisser, H.J. Detailed Balance Limit of Efficiency of p-n Junction Solar Cells. J. Appl. Phys. 1961, 32, 510. [Google Scholar] [CrossRef]
- Nagat, A.T.; Gamal, G.A.; Hussein, S.A. Growth and Characterization of Single Crystals of the Ternary Compound TlGaTe2. Cryst. Res. Technol. 1991, 26, 19–23. [Google Scholar] [CrossRef]
- Guseinov, G.; Abdullayev, G.; Bidzinova, S.; Seidov, F.; Ismailov, M.; Pashayev, A. On new analogs of TlSe-type semiconductor compounds. Phys. Lett. A 1970, 33, 421–422. [Google Scholar] [CrossRef]
- Yücel, İ.; Çakmak, S. The Structural and Electronic Properties of TlGa1−xInxTe2 (x = 0.00, 0.25, 0.50, 0.75) Alloys. Suleyman Demirel Univ. J. Sci. 2017, 12, 30–40. [Google Scholar]
- Fonari, A.; Sutton, C. Effective Mass Calculator for Semiconductors; GitHub: San Francisco, CA, USA, 2015. [Google Scholar]
- Van Zeghbroeck, B. Principles of Semiconductor Devices; Colorado University: Boulder, CO, USA, 2004. [Google Scholar]
- García, G.; Palacios, P.; Menéndez-Proupin, E.; Montero-Alejo, A.L.; Conesa, J.C.; Wahnon, P. Author Correction: Influence of chromium hyperdoping on the electronic structure of CH3NH3PbI3 perovskite: A first-principles insight. Sci. Rep. 2018, 8, 7212. [Google Scholar] [CrossRef] [PubMed]
- Petretto, G.; Dwaraknath, S.; Miranda, H.P.; Winston, D.; Giantomassi, M.; Van Setten, M.J.; Gonze, X.; Persson, K.A.; Hautier, G.; Rignanese, G.-M. High-throughput density-functional perturbation theory phonons for inorganic materials. Sci. Data 2018, 5, 180065. [Google Scholar] [CrossRef] [PubMed]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford University Press: Oxford, UK, 1985. [Google Scholar]
- Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. ZAMM 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Jafarova, V.; Orudzhev, G.; Paucar, R.; Alekperov, O.; Shim, Y.; Wakita, K.; Mamedov, N.; Abdullayev, N.; Najafov, A. Ab initio calculations of phonon dispersion and lattice dynamics in TlGaTe 2. Phys. Status Solidi C 2015, 12, 664–667. [Google Scholar] [CrossRef]
- Gajdoš, M.; Hümmer, K.; Kresse, G.; Furthmüller, J.; Bechstedt, F. Linear optical properties in the projector-augmented wave methodology. Phys. Rev. B 2006, 73, 045112. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.-M.; Vajeeston, P.; Ravindran, P.; Fjellvåg, H.; Tilset, M. Revisiting isoreticular MOFs of alkaline earth metals: A comprehensive study on phase stability, electronic structure, chemical bonding, and optical properties of A-IRMOF-1 (A = Be, Mg, Ca, Sr, Ba). Phys. Chem. Chem. Phys. 2011, 13, 10191. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Reining, L.; Del Sole, R.; Onida, G. Ab Initio Calculation of Excitonic Effects in the Optical Spectra of Semiconductors. Phys. Rev. Lett. 1998, 80, 4510–4513. [Google Scholar] [CrossRef]
- Rohlfing, M.; Louie, S.G. Electron-Hole Excitations in Semiconductors and Insulators. Phys. Rev. Lett. 1998, 81, 2312–2315. [Google Scholar] [CrossRef]
- Bokdam, M.; Sander, T.; Stroppa, A.; Picozzi, S.; Sarma, D.D.; Franchini, C.; Kresse, G. Role of Polar Phonons in the Photo Excited State of Metal Halide Perovskites. Sci. Rep. 2016, 6, 28618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspnes, D.E.; Studna, A.A. Dielectric functions and optical parameters of Si, Ge, GaP, GaAs, GaSb, InP, InAs, and InSb from 1.5 to 6.0 eV. Phys. Rev. B 1983, 27, 985–1009. [Google Scholar] [CrossRef]
Figure 1.
(a) Crystal structures of TlGaTe2. The illustration shows the legends for the different type of atoms; (b) calculated HSE06 electronic band structure of TlGaTe2; (c) total and site projected density of states of TlGaTe2. The Fermi level is set to zero. (colour code: red-Ga, green-Te, blue-Tl).
Figure 1.
(a) Crystal structures of TlGaTe2. The illustration shows the legends for the different type of atoms; (b) calculated HSE06 electronic band structure of TlGaTe2; (c) total and site projected density of states of TlGaTe2. The Fermi level is set to zero. (colour code: red-Ga, green-Te, blue-Tl).

Figure 2.
Calculated phonon spectra and site projected phonon density of states for TlGaTe2.

Figure 3.
(a) Calculated Raman spectra of TlGaTe2; (b) calculated infrared spectra of TlGaTe2.

Figure 4.
(a) Calculated dielectric function of TlGaTe2; (b) absorption coefficient of TlGaTe2.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The calculated effective masses of TlGaTe2, where m*hh and m*e are the effective masses of heavy holes and electrons respectively. me is mass of the electron.
Table 1.
The calculated effective masses of TlGaTe2, where m*hh and m*e are the effective masses of heavy holes and electrons respectively. me is mass of the electron.
| Compounds | Effective Masses | |
|---|---|---|
| m*hh me | m*e me | |
| TlGaTe2 (indirect electronic transition 1.109 eV) | 0.736 | 0.196 |
| TlGaTe2 (direct electronic transition 1.129 eV) | 0.736 | 0.378 |
Table 2.
Calculated phonon frequencies ωcal (in this work and previous work [31]) and their experimental values ωexp [9].
| TlGaTe2-Phonon Activity | Phonon Frequencies (in cm−1) | ||
|---|---|---|---|
| ωcal [31] | ωcal (This Work) | ωexp [9] | |
| R-active 1Eg | 16 | 19 | - |
| R-active 2Eg | 60 | 60 | 67 |
| R-active 3Eg | 152 | 157 | 165 |
| R-active B1g | 76 | 76 | - |
| R-active A1g | 125 | 128 | 135 |
| R-active 1B2g | 99 | 99 | - |
| R-active 2B2g | 210 | 216 | - |
| IR-active 1A2u | 8 | 18 | 27 |
| IR-active 2A2u | 161 | 170 | 175 |
| IR-active 1E2u | 31 | 34 | 44 |
| IR-active 2E2u | 77 | 78 | 88 |
| IR-active 3E2u | 182 | 188 | 192 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rasukkannu, M.; Velauthapillai, D.; Vajeeston, P. First-Principle Calculation of High Absorption-TlGaTe2 for Photovoltaic Application. Materials 2019, 12, 2667. https://doi.org/10.3390/ma12172667
AMA Style
Rasukkannu M, Velauthapillai D, Vajeeston P. First-Principle Calculation of High Absorption-TlGaTe2 for Photovoltaic Application. Materials. 2019; 12(17):2667. https://doi.org/10.3390/ma12172667
Chicago/Turabian StyleRasukkannu, Murugesan, Dhayalan Velauthapillai, and Ponniah Vajeeston. 2019. "First-Principle Calculation of High Absorption-TlGaTe2 for Photovoltaic Application" Materials 12, no. 17: 2667. https://doi.org/10.3390/ma12172667
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.